 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect conversion of chitin into a N-containing furan derivative†
Xi
Chen
a,
Shu Ling
Chew
a,
Francesca M.
Kerton
b and
Ning
Yan
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117576, Singapore. E-mail: ning.yan@nus.edu.sg
bDepartment of Chemistry, Memorial University of Newfoundland, St. John's, NL A1B 3X7, Canada
First published on 7th January 2014
Abstract
This paper describes the direct conversion of chitin into a nitrogen-containing (N-containing) furan derivative (3A5AF) for the first time. Under optimized conditions, the yield of 3A5AF reaches 7.5% with ca. 50% chitin conversion by using boric acid and alkaline chlorides as additives, and NMP as a solvent. A variety of other compounds, including levoglucosenone, 4-(acetylamino)-1,3-benzenediol, acetic acid and chitin–humins, have been identified as side products, based on which a plausible reaction network involved in the process is proposed. Mechanistic investigation by NMR studies and poison tests confirms the formation of a boron complex intermediate during the reaction, shedding light on the promotional effects of boric acid. Kinetic studies show that the depolymerization of the chitin crystalline region is rate-determining, and therefore disruption of the hydrogen bonding in the crystalline region of chitin, either before or during the reaction, is the key to further improving the reaction yields.
1. Introduction
The imperative to reduce society's dependence on crude oil has led to significant research efforts at the conversion of biomass.1,2 So far, remarkable progress has been made in the conversion of lignocellulosic biomass into bioethanol3,4 and value-added platform chemicals.5–14 On the other hand, much less attention has been directed to the utilization of chitin,15 which is a major component of the exoskeletons of insects and crustaceans16 and the second most abundant biopolymer on earth. The chemical structure of chitin is highly similar to cellulose, consisting of N-acetyl-D-glucosamine (NAG) monosaccharide units as the building blocks linked together by β-glycosidic bonds (see Fig. 1). Chitin has an underestimated but remarkable potential for the production of renewable, value-added chemicals, especially N-containing compounds, as it comprises 7 wt% of biologically-fixed nitrogen. Nowadays, the manufacture of N-containing chemicals is typically laborious and energy intensive starting from high temperature, high pressure ammonia synthesis. Chitin is an attractive, sustainable and cheap organic nitrogen resource that holds the potential to revolutionize the production of certain chemicals bearing nitrogen. | ||
| Fig. 1 The chain structure of chitin polymer. | ||
Current utilization of chitin remains very limited. Chemical derivatizations have been frequently conducted to modify the properties of chitin.17–19 However, these are based on stoichiometric chemical reactions on the substitution groups on chitin polymer chains, and therefore are unable to produce non-polymeric chemicals. Pyrolysis or radiolysis of chitin/chitosan to produce volatile aromatic heterocyclic compounds such as pyrazines, pyridines, pyrroles and furans has been reported.20–22 Nevertheless, these methods cannot achieve a selective degradation resulting in trace amounts of products. Hydrolysis of chitin by enzymes or concentrated acids affords NAG and oligomers, but this route suffers from low efficiency, environmental issues and limited product diversity.23,24 Starting from chitin, the production of 5-(chloromethyl)furfural,25 levulinic acid (LA),26 and 5-hydroxymethylfurfural (5-HMF)27 with yields ranging from 9 to 11% was reported very recently. Unfortunately, the precious, biologically fixed nitrogen was lost during the transformation. To the best of our knowledge, direct conversion of chitin into value-added nitrogen-containing chemicals with noticeable yields has not yet been realized.
In 2012, the transformation of NAG into 3-acetamido-5-acetylfuran (3A5AF) was reported by dehydration reactions, demonstrating the feasibility of obtaining N-containing furan derivatives from chitin monomers.28,29 Encouraged by this advancement, we for the first time tested the possibility of producing 3A5AF directly from chitin, which, in principle, could be obtained by combining chitin hydrolysis and NAG dehydration. In this paper, 6 solvents, 26 additives and their combinations were evaluated. For the optimized system, detailed studies concerning the reaction kinetics, reaction pathway, effects of water as an additive, and mechanistic investigation using NMR and poison tests were conducted.
2. Experimental
2.1. Materials
Chitin was purchased from Wako Pure Chemical Industry. Boric acid was purchased from Amresco. Sodium chloride (NaCl) was purchased from Schedelco. Lithium chloride (LiCl) and dimethylacetamide (DMA) were purchased from Alfa Aesar. N-Acetyl-D-glucosamine (NAG), iron(II), tin(II), barium and cesium chloride salts (FeCl2, SnCl2, BaCl2 and CsCl, respectively), N-methyl-2-pyrrolidone (NMP) and dimethyl sulfoxide (DMSO) were purchased from Sigma Aldrich. Dimethylformamide (DMF) and glycerine (Gly) were from Fisher Scientific. Ethylene glycol (EG) was purchased from Fluka. DMSO-d6 was purchased from VWR Singapore. Other chemicals, such as copper chloride (CuCl2), tungsten oxide (WO3), cobalt chloride (CoCl2), nickel chloride (NiCl2) and calcium chloride (CaCl2), were obtained from Sinopharm Chemical Reagent (SCR). All chemicals were used as received.2.2. General procedure
Under optimized conditions, chitin (100 mg, 0.5 mmol based on the NAG monomer) was placed in a thick-wall glass tube (35 mL). Following that a magnetic stir bar, boric acid (122 mg, 2.0 mmol), NaCl (58 mg, 1.0 mmol) and anhydrous NMP (3 mL) were added. The tube was sealed by a Teflon stopper and placed into a pre-heated oil bath at 215 °C for 1 h under a stirring speed of 400 rpm. After the reaction, the reaction mixture was cooled down to room temperature. Methanol (15 mL) was added. After thorough mixing, a portion of the liquid sample (1 mL) was filtered by a PTFE syringe filter with a 0.2 μm pore size before analyzing by high-performance liquid chromatography (HPLC).It is noteworthy that all the reactions were carried out in a similar manner whilst varying the following parameters: additive (or additive amount), solvent, reaction time and temperature.
HPLC analysis was performed on an Agilent 1200 Series (Agilent Technologies, Germany) LC system by using an Agilent ZORBAX Eclipse carbon-18 column. The mobile phase was 83% water and 17% acetonitrile. The flow rate was kept at 0.5 ml min−1 with a run-time of 20 min. A UV-vis detector setting at 230 nm was used to analyze the product. 3A5AF standard was prepared following a literature method28 and the purity verified by nuclear magnetic resonance (NMR) spectroscopy. A calibration curve (Fig. S1†) was generated with a series of 3A5AF standard solutions and used for quantification of the product.
To obtain the conversion of chitin, the reaction mixture was centrifuged after the reaction. The remaining solid was washed with water three times and dried in an oven over 24 h at 70 °C and weighed. The conversion of chitin is calculated as:
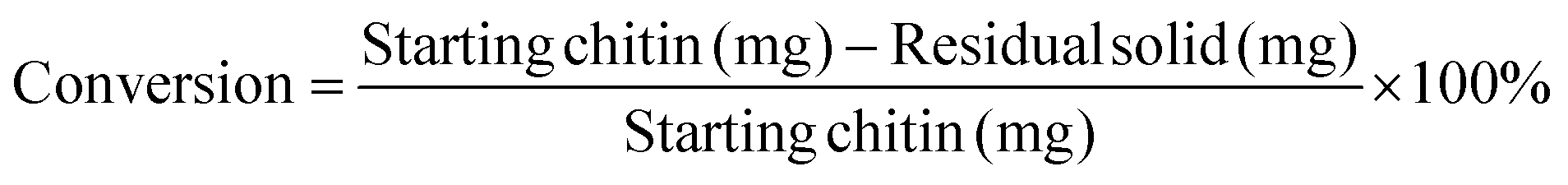
2.3. Identification of other products
After the reactions, the liquid samples from six parallel experiments were combined. Then, reduced pressure distillation was applied for the removal of NMP solvent. The Edwards Rotary Vane Pump (Model RV3) with an ultimate pressure of 0.002 mbar was used. The liquids were put into a round bottle flask and heated at about 90 °C under vacuum. After the majority of the solvent was removed, column chromatography was employed for separation using silica gel with particle size ranging from 40 to 63 μm as the stationary phase and 5% methanol and 95% dichloromethane as the mobile phase. Afterwards the collected fractions were concentrated by a rotary evaporator and analyzed by gas chromatography-mass spectrometry (GC-MS).2.4. Characterization
X-ray powder diffraction (XRD) was performed on a Bruker D8 Advanced Diffractometer with Cu Kα radiation at 40 kV. Gel permeation chromatography (GPC) analysis was carried out with a system equipped with a Waters 2410 refractive index detector, a Waters 515 HPLC pump and two Waters styragel columns (HT 3 and HT 4) using DMF as an eluent at a flow rate of 1 ml min−1 at 25 °C. The raw data were processed using narrow polydispersity polystyrene standards and calibration using the software Breeze. Fourier transform infrared spectroscopy (FTIR) was conducted on a Bio-Rad FTS-3500 ARX instrument. GC-MS was performed on an Agilent 7890A GC system with 7693 Autosampler and 5975C inert MSD with triple-axis detector. Elemental analysis (EA) was conducted using an Elementar Vario Micro Cube and the degree of acetylation (DA) was calculated according to EA analysis.30 Proton, boron and carbon NMR (1H, 11B and 13C NMR) were performed on a Bruker ultrashield 400 plus spectrometer.| DA = [(C/N − 5.14)/1.72] × 100% |
3. Results and discussion
3.1. Solvent screening
Similar to cellulose,31–33 one of the major challenges for chitin conversion is its robust crystal structure with extensive inter- and intra-hydrogen bonds among the polymer chains.34,35 Solvent or solvent systems that are able to dissolve chitin should prove advantageous in its transformation. Previously, dipolar aprotic solvents combined with metal salts, such as 5–7% LiCl/NMP and LiCl/DMA systems, have been reported to dissolve chitin.36 We started by screening 6 solvents including DMA, DMF, DMSO, NMP, EG and Gly. The first four solvents are commonly used for sugar dehydration37–41 whereas EG and Gly are selected because their hydroxyl groups may disrupt the hydrogen bonding network in chitin. When chitin was dispersed in these 6 solvents at 195 or 225 °C for 1 h, no 3A5AF was detected. LiCl (5 wt%, based on solvent) was added into these solvents as a solubility enhancing agent, but the yield of 3A5AF remained at 0% in all cases. These two series of experiments indicate that chitin dehydration to 3A5AF could not be obtained by thermal decomposition under the temperatures investigated, irrespective of the solvent used.To differentiate the solvent effect in converting chitin into 3A5AF, different additives that are active in sugar dehydration, such as HCl,42–44 boric acid,45–47 and their combination with LiCl, were employed. The results are shown in Fig. S2.† By adding HCl, 3A5AF was obtained in DMA, DMF and NMP (2.3, 1.1 and 3.0% respectively). By adding boric acid, 3A5AF was obtained in DMA, DMF, DMSO and NMP. No 3A5AF was detected in EG and Gly under these conditions. The best performance was achieved in the presence of boric acid and LiCl in NMP, reaching a 3A5AF yield of 5%. From the above DMA, DMF, DMSO and NMP are all suitable solvents for chitin conversion but NMP appears to be the most effective. Previously, DMF and DMA were identified to be the best solvents for chitin monomer dehydration, and NMP was slightly less effective. The different performances may be ascribed to the structure difference of chitin and NAG. DMA and DMF are smaller in their molecular size, which may favor the solute–solvent interaction in sugar monomer conversion. On the other hand, a major challenge in chitin conversion is its low solubility, and therefore NMP is more effective considering that LiCl/NMP represents a classic solvent system for chitin dissolution. After that, the system was further optimized by conducting the reaction at different temperatures and with a different solvent to chitin ratio (Fig. S3 & S4†). The optimal temperature was identified to be 215 °C and the optimal solvent to chitin ratio to be 3 mL to 100 mg.
3.2. Additive screening
We systematically evaluated the performance of a wide range of additives for chitin conversion to 3A5AF in NMP under selected conditions. These additives included 13 metal chlorides, 6 organic/simple inorganic acids, 5 bases and 3 heteropolyacids (see Fig. 2a). Heteropolyacids, which have been reported to be excellent catalysts for glucose/cellulose dehydration,48–50 did not produce any 3A5AF. Bases were not effective either. Ba(OH)2 is the only basic additive that produced 3A5AF, but the yield was very low (0.4%). Metal chlorides are widely used as catalysts for glucose dehydration to 5-HMF,51–55 and in our experiments CrCl3 effectively promoted 3A5AF formation (2.5%). Organic acids such as phenylboronic acid and acetic acid are not able to facilitate the reaction. Among the inorganic acids, HCl is the only one effective for chitin conversion to 3A5AF with a yield of 2.2%, indicating the promotional effect of chloride ion in the reaction. This is consistent with a previous finding in NAG, glucose and cellulose conversion where chloride played an important role.28,45,56,57 It is very likely that chloride acts as a promoter to disrupt the hydrogen bonding in the substrate rather than as a catalyst, considering that the majority of metal chlorides are inactive in 3A5AF formation. Boric acid is the most effective additive for the reaction with a 3A5AF yield of 3.6%. Presumably, boric acid facilitates both the hydrolysis of chitin and the dehydration due to its acidity and this gives rise to its promotional ability. In previous studies it was proposed that boric acid coordinated with the hydroxyl groups of glucose and this enables its dehydration to generate 5-HMF.47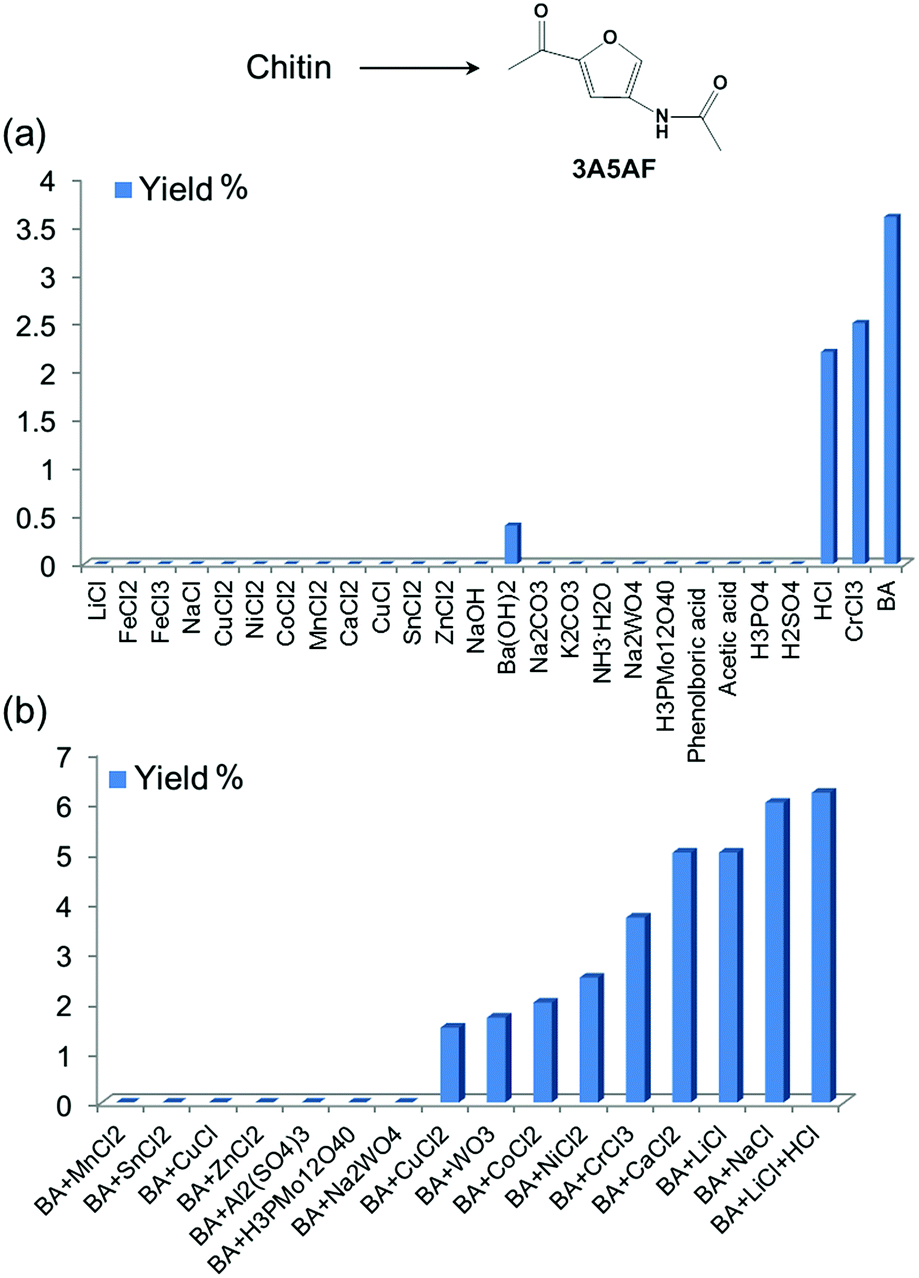 | ||
| Fig. 2 Additive screening for chitin conversion into 3A5AF. Reaction conditions: 215 °C, 1 h, NMP (3 mL), chitin (100 mg), 200 mol% additive based on chitin monomer (400 mol% for boric acid). (a) Single component screening; (b) combinations of additives (BA is short for boric acid). | ||
In an attempt to obtain higher yields, combinations of additives were investigated. Since boric acid is the most effective single component additive, the combinations are based on boric acid and another additive (see Fig. 2b). Interestingly, the combination of boric acid with some metal chlorides, such as MnCl2, SnCl4, CuCl and ZnCl2, and heteropolyacids did not afford any 3A5AF from chitin. Boric acid with CuCl2, WO3, CoCl2 and NiCl2 led to 3A5AF formation but the yield was lower than when using boric acid alone. On the other hand, the combinational use of boric acid with CrCl3, LiCl, NaCl and CaCl2 resulted in enhanced yields. Since the highest yields were obtained by using the combination of boric acid and alkali/alkaline earth metal chlorides, more detailed optimizations were conducted on a series of alkali/alkaline earth metal chlorides, or HCl, combined with boric acid. Different amounts of additives were investigated and the results are compiled in Fig. S5.† This led to the discovery of a few systems that are able to produce ca. 6% 3A5AF directly from chitin within one hour. These include: (1) the combination of 400 mol% boric acid and 200 mol% NaCl; (2) the combination of 200 mol% boric acid and 100 mol% LiCl; (3) the tri-component combination of 400 mol% boric acid, 200 mol% LiCl and 100 mol% HCl. The last combination is slightly better than the other two, reaching a 3A5AF yield of 6.2%. The addition of HCl is assumed to facilitate the hydrolysis of chitin.
3.3. Reaction kinetics
The combination of boric acid and NaCl was chosen to study the chitin conversion and the 3A5AF yield as a function of reaction time (see Fig. 3). Note that if the chitin conversion is calculated based on the recovered solids after reaction, the value will be underestimated if insoluble char or humins forms during the reaction. In the first few minutes, the product was not yet formed despite a chitin conversion of 15.5%. Normally, chitin has an amorphous and a crystalline part,16,58 and amorphous chitin is more easily converted. The crystalline index for chitin ranges from 70 to 80%. Therefore the initial conversion may be ascribed to the conversion of amorphous chitin, which depolymerized and generated soluble, low-molecular weight oligomers. As the reaction proceeded from 5 min to 2 hours, the chitin conversion and 3A5AF yield increase simultaneously. It is not unreasonable to speculate that the chitin crystalline region depolymerizes at a slower pace, because the conversion rate slows down significantly compared to the initial conversion. After 2 hours, a maximum 3A5AF yield of 7.5% was achieved with a chitin conversion of 49.7%. As the reaction further progressed, the yield dropped sharply probably due to 3A5AF decomposition. Interestingly, the chitin conversion decreased slightly after 2 hours, which is indicative of the formation of insoluble solids such as char. During the entire reaction, NAG is not observed, suggesting that NAG dehydration is not rate-determining. Since the depolymerization of crystalline chitin is slow and the products are not stable, the 3A5AF yield cannot be further improved simply by prolonging the reaction time.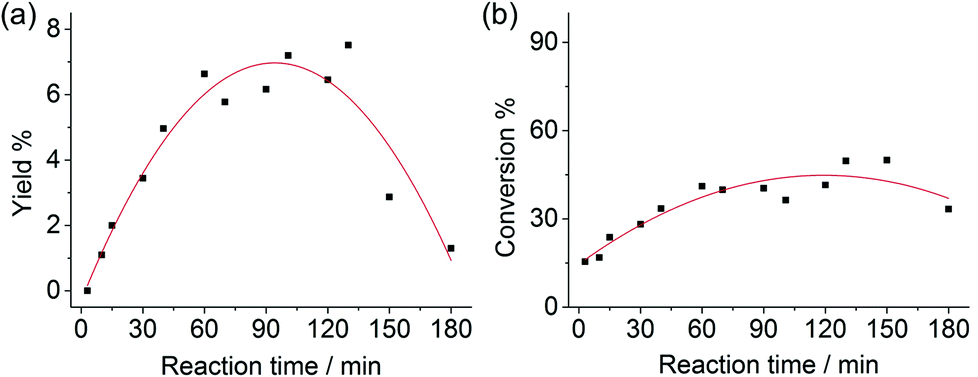 | ||
| Fig. 3 (a) 3A5AF yield as a function reaction time; (b) chitin conversion as a function reaction time. Reaction conditions: 215 °C, NMP (3 mL), chitin (100 mg), boric acid (400 mol%), NaCl (200 mol%). | ||
Chitin and the recovered solids after the reaction (215 °C, 1 h reaction time) have been characterized by FTIR, XRD and EA. As shown in the FTIR spectrum of pure chitin (see Fig. 4, dash line), the bands at 3452 and 3266 cm−1 are attributed to the OH and NH stretching, respectively. The shape and intensity of these peaks will change if the hydrogen bonding network in chitin is altered. The bands ranging from 2886 to 2961 cm−1 represent CH, CH3 symmetric stretching and CH2 asymmetric stretching. And the CH bending, symmetric CH3 deformation and CH2 wagging bands appear at 1380 and 1312 cm−1. The peaks at 1629 and 1662 cm−1 are assigned to Amide I band (two types of hydrogen bonds in a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group with the NH group of the adjacent chain and the OH group of the inter-chain). Amide II band (in-plane N–H bending and C–N stretching mode) and Amide III band (in-plane mode of the CONH group) are observed at 1558 and 1312 cm−1, respectively. The bands ranging from 1027 to 1163 cm−1 are attributed to the asymmetric bridge oxygen and C–O stretching.59,60 In the FTIR spectrum of recovered solid, the shape, position and relative intensity of all characteristic peaks are well preserved, which indicates no appreciable modifications on the chemical structure including functional groups and the hydrogen bonding network.
O group with the NH group of the adjacent chain and the OH group of the inter-chain). Amide II band (in-plane N–H bending and C–N stretching mode) and Amide III band (in-plane mode of the CONH group) are observed at 1558 and 1312 cm−1, respectively. The bands ranging from 1027 to 1163 cm−1 are attributed to the asymmetric bridge oxygen and C–O stretching.59,60 In the FTIR spectrum of recovered solid, the shape, position and relative intensity of all characteristic peaks are well preserved, which indicates no appreciable modifications on the chemical structure including functional groups and the hydrogen bonding network.
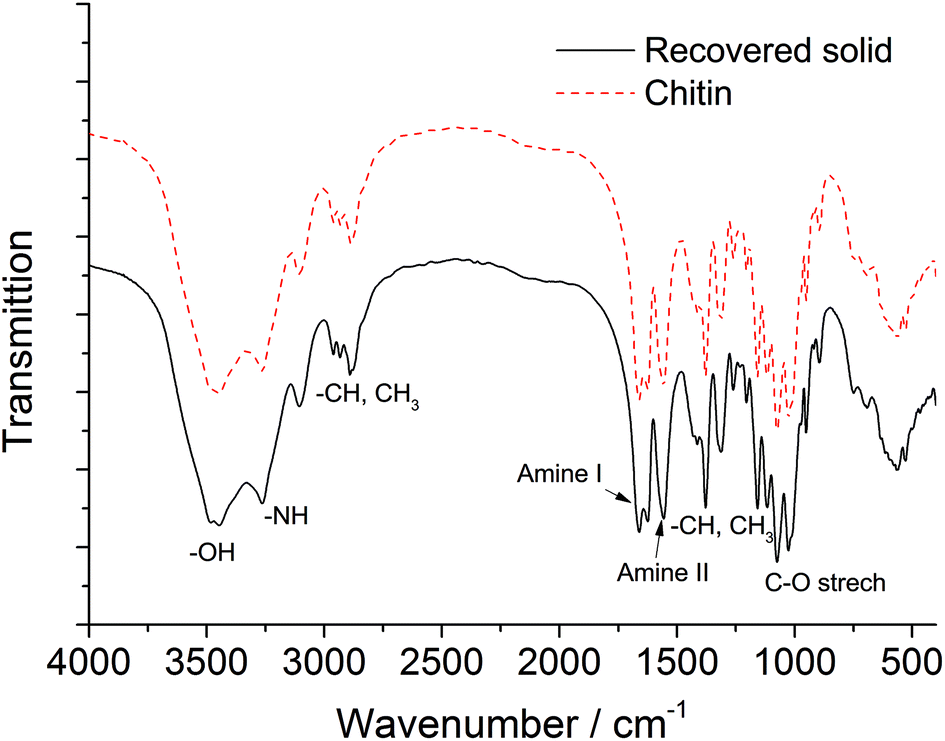 | ||
| Fig. 4 FTIR spectra of chitin and the recovered solid after the reaction. | ||
In the XRD pattern of pure chitin (see Fig. 5), the peak at 2θ = 19° represents the (110) plane of crystalline chitin,61 together with a few other peaks with much weaker intensity. The XRD pattern of the recovered solid highly resembles that of the chitin standard, suggesting that there is negligible change in the crystalline region. The EA data further corroborate with FTIR and XRD analysis, as the C, H, and N content remains at a similar level in the recovered solid (see Table S1†). According to the EA, the DA of chitin is 99% and the value remains almost unchanged for the recovered solid. From these observations, we can conclude that the unreacted solids maintain the chemical backbone, the hydrogen bonding network, and the crystalline structure of chitin. It is assumed that chitin polymer chains undergo the depolymerization into oligomers and NAG prior to any other transformations towards 3A5AF and side products.
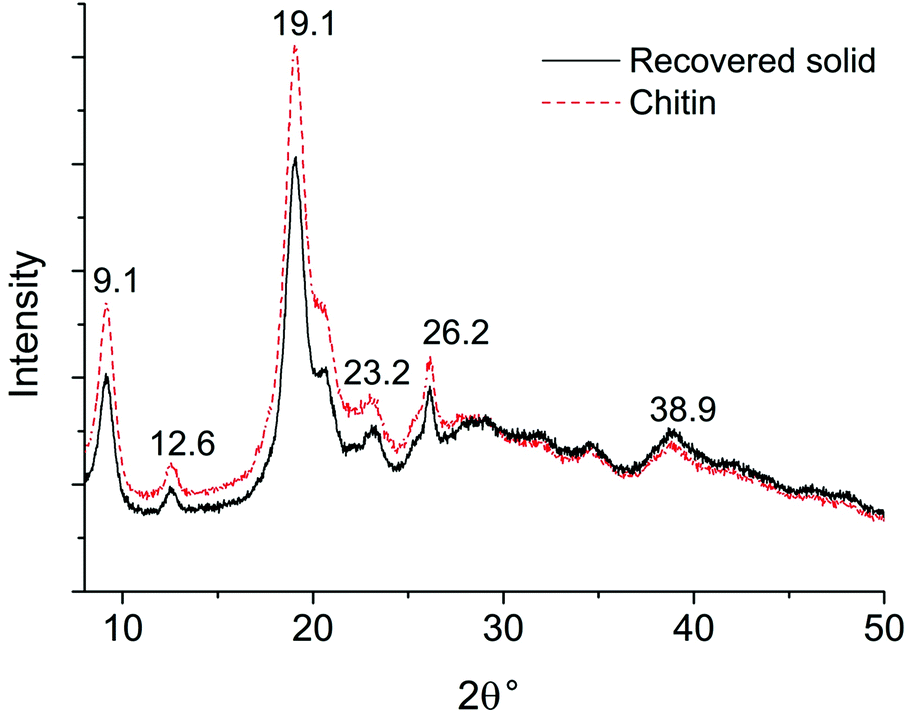 | ||
| Fig. 5 XRD patterns of chitin and the recovered solid after the reaction. | ||
3.4. Reaction pathway elucidation
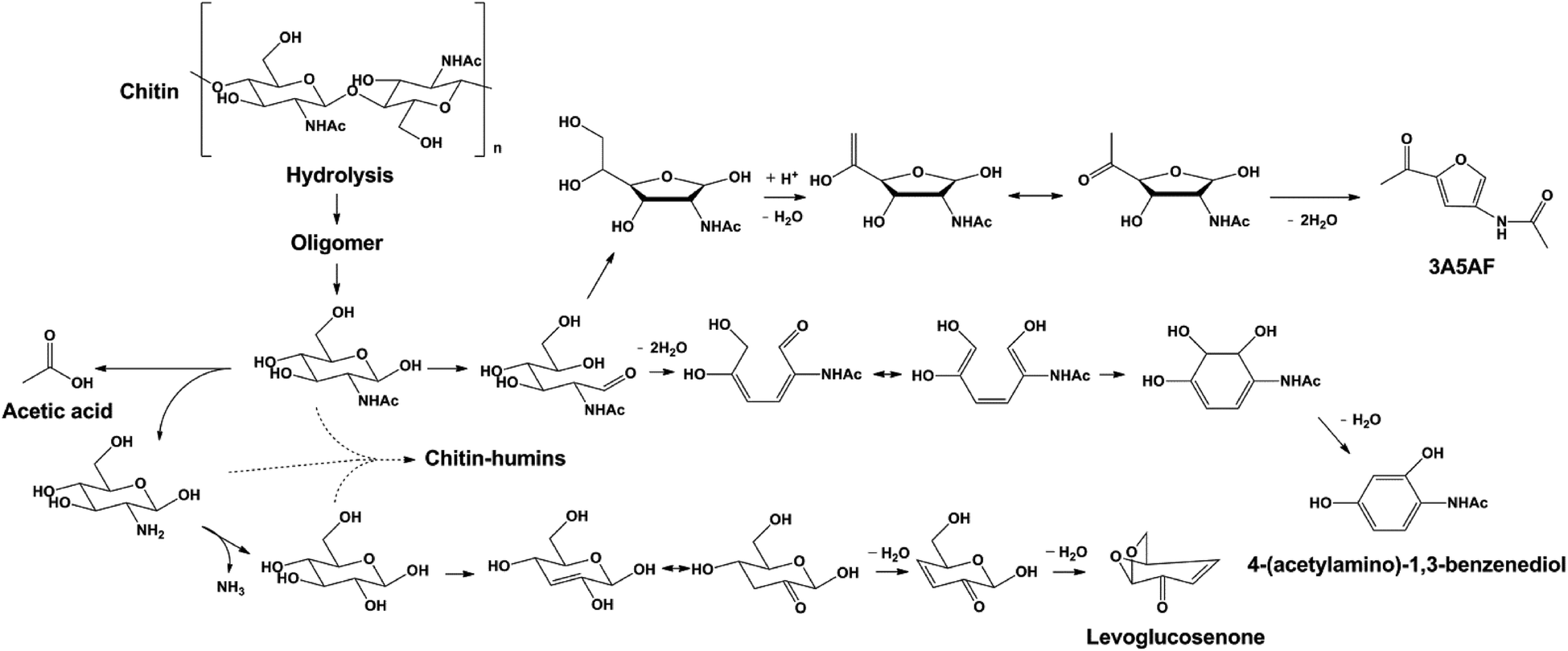
Under optimized conditions, 3A5AF was obtained with ca. 7.5% yield despite a much higher chitin conversion (50%). In the following work, significant effort was spent on the identification of other products generated from chitin, by combining column chromatography, GC-MS, HPLC, EA, GPC, NMR and FTIR analysis. The major product, 3A5AF, was further confirmed by NMR technique after column chromatography separation (see Fig. S6†). From GC-MS a variety of other products have been identified, including levoglucosenone, acetic acid and 4-(acetylamino)-1,3-benzenediol. Besides these, a black solid fraction was obtained after column separation and characterized by GPC, FTIR and EA. It shares some similarities with humins reported in glucose/cellulose conversion,62 as both are polymeric black solids and have a higher C/H ratio (see Table S1†) than their parent carbohydrates. On the other hand, the structure of the black solid from chitin is not identical with previously reported humin materials. Humins generated from glucose/cellulose conversion are assumed to form by aldol condensation between sugars and furanics, and the furanic and CC conjugating bands can be observed in FTIR spectra.63 These bands are absent in the FTIR spectrum of the black solid from chitin. Furthermore, humins from glucose/cellulose contain no nitrogen, whereas the nitrogen content in the black solid (8%) is even higher than that in chitin. We tentatively term this black solid fraction as chitin–humins. GPC analysis indicates that the average molecular weight of chitin–humins is at around 1 kDa (see Fig. S7†). Interestingly, GPC analysis of the raw reaction mixture revealed another fraction of products with MW of 3–4 kDa, in addition to small molecules and chitin–humins (see Fig. S8†). This fraction may be the partially depolymerized and solubilized part of chitin generated in the reaction.
Based on these we propose a plausible reaction pathway for the formation of 3A5AF and other products (see Scheme 1). Hydrolysis of chitin is the first step in the reaction, leading to the partially depolymerized chitin, NAG oligomers and eventually NAG. NAG generated in situ undergoes three parallel reaction pathways. In the first pathway, 3A5AF is generated via a five-membered ring formed from the open ring aldose. The subsequent enolization and dehydration afford the N-containing furan derivative, similar to that proposed in an earlier study.28 Alternatively, NAG dehydrates to hexatriene, after which keto–enol tautomerism, electrolytic rearrangement and dehydration take place to form a six-membered ring product. The formation of the aromatic compound is not unusual in biomass conversion. For example, 1,2,4-benzenetriol is widely reported in glucose conversion via 5-HMF as an intermediate.64–66
 | ||
| Scheme 1 Proposed reaction pathway of the formation of other identified products. | ||
The difference is that 1,2,4-benzenetriol forms via a five-membered furan ring whereas 4-(acetylamino)-1,3-benzenediol more likely forms via a six-member ring intermediate. Finally, hydrolysis of acetyl amide in NAG affords acetic acid and amino-sugar. With further deamination and dehydration, levoglucosenone can be generated. The formation pathway of chitin–humin is not fully understood but it should involve condensation reactions between NAG and amino-sugar considering its high nitrogen content.
3.5. Mechanism investigation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to the substrate) were added. For chitin conversion (see Fig. 6), ethanol did not affect the product yield. However, upon the addition of EG or 1,3-PG, the 3A5AF yield decreased from ca. 4% to 1% and 0.2%, respectively. 1,3-PG seems to be a stronger poison compared with EG. The poison experiments were conducted using NAG as the starting material (see Fig. S10†), and exactly the same trend was observed. Afterward, isopropanol (IPA), 1,2-PG, glycerol (Gly), 1,3-butanediol and 2,3-butanediol were added. IPA had no observable effect on the reaction whereas 1,2-PG, Gly, 1,3- and 2,3-butanediol all inhibited the reaction significantly (data not shown). As such, the 3A5AF formation is achieved via five- and six-membered ring boron complexes, and it is likely that the six-membered ring complex is dominant.
:1 to the substrate) were added. For chitin conversion (see Fig. 6), ethanol did not affect the product yield. However, upon the addition of EG or 1,3-PG, the 3A5AF yield decreased from ca. 4% to 1% and 0.2%, respectively. 1,3-PG seems to be a stronger poison compared with EG. The poison experiments were conducted using NAG as the starting material (see Fig. S10†), and exactly the same trend was observed. Afterward, isopropanol (IPA), 1,2-PG, glycerol (Gly), 1,3-butanediol and 2,3-butanediol were added. IPA had no observable effect on the reaction whereas 1,2-PG, Gly, 1,3- and 2,3-butanediol all inhibited the reaction significantly (data not shown). As such, the 3A5AF formation is achieved via five- and six-membered ring boron complexes, and it is likely that the six-membered ring complex is dominant.
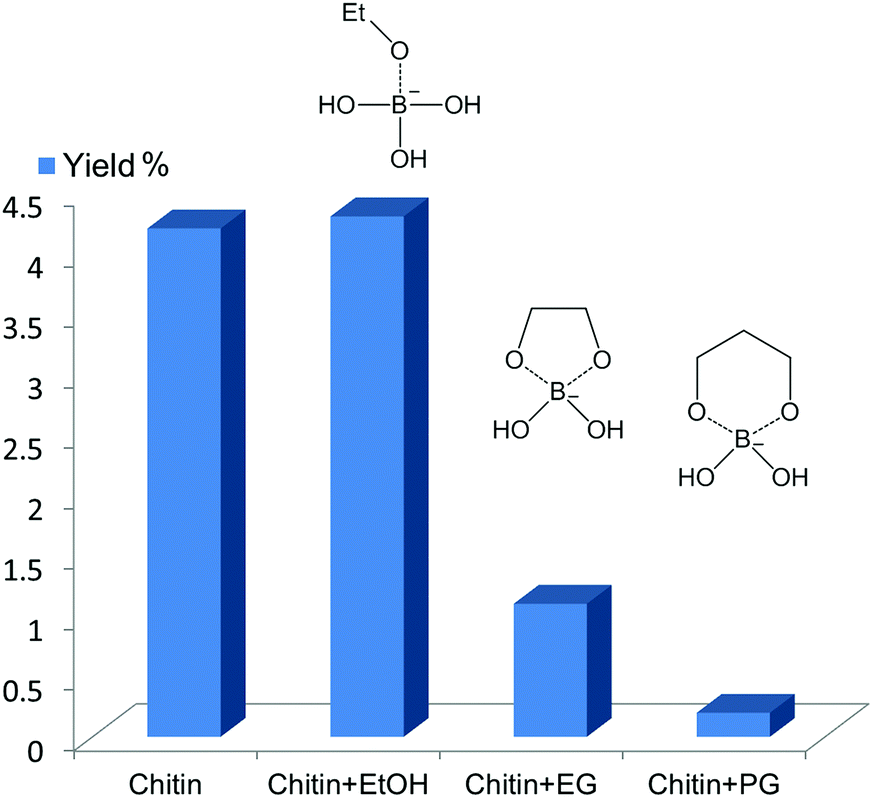 | ||
| Fig. 6 The inhibition effect of different alcohols as additives. Reaction condition: 215 °C, 1 h, NMP (3 mL), chitin (100 mg), boric acid (200 mol%), NaCl (200 mol%). | ||
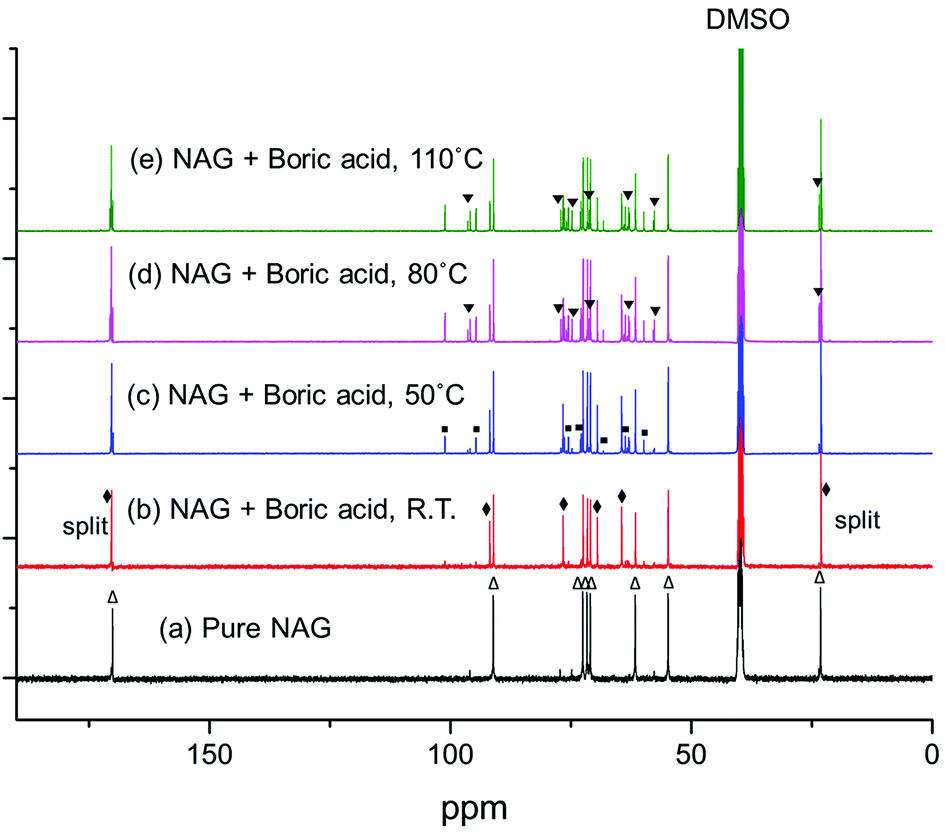 | ||
| Fig. 7 13C NMR of pure NAG and with boric acid at different temperatures. The sample was maintained at each temperature for 1 hour before NMR analysis. ▼ denotes the peaks of β-anomer; ◆ denotes the peaks assigned to the boron complex; ■ denotes the peaks assigned to the dimer; △ denotes the peaks for α-anomer. Note: the peaks at around 23 and 170 ppm are split upon the addition of boric acid, which may not be obviously seen in the figure. | ||
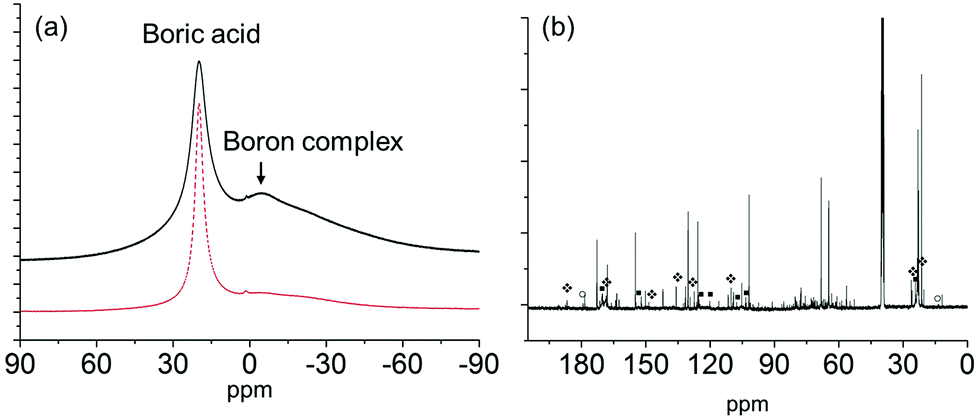 | ||
Fig. 8 (a) 11B NMR of boric acid (dash line) and boric acid with NAG (solid line) at room temperature; (b) 13C NMR of the sample in DMSO-d6 after heating at 160 °C for 1 h.  denotes the peaks for the major product 3A5AF; ■ possibly denotes 4-(acetylamino)-1,3-benzenediol; ○ denotes the peaks for acetic acid. denotes the peaks for the major product 3A5AF; ■ possibly denotes 4-(acetylamino)-1,3-benzenediol; ○ denotes the peaks for acetic acid. | ||
After heating at 50, 80 and 110 °C, the peaks assigned to the boron complex keep decreasing and new peaks intensify concurrently (Fig. 7c–e), indicating that the boron complex is intermediate for further conversion. The new peaks can be categorized into two groups. By referring to the standard 13C NMR spectra, one group of the rising peaks can be unambiguously assigned to the β-anomer of NAG. Meanwhile, another group is likely to be assigned to the dimer of NAG. The formation of β-anomer suggests that boric acid facilitates the ring-opening reaction of the α-anomer, since the linear form of NAG is required for β-anomer formation. At the same time, this provides a plausible explanation for the promotional effect of boric acid, as the formation of 3A5AF starts with the ring opening of NAG. After heating at 160 °C, the color of the sample became dark black and a variety of peaks appeared in the 13C NMR spectrum (see Fig. 8b). The peaks belonging to 3A5AF, 4-(acetylamino)-1,3-benzenediol and acetic acid have been assigned. Levoglucosenone is not found possibly due to its low concentration. There are many other peaks in the carbon–carbon double bond (100–150 ppm) and unsaturated alcoholic alkane range (50–80 ppm), which might be assigned to precursors for chitin–humins and other side products.
4. Conclusions
For the first time, direct conversion of chitin into a nitrogen-containing furan derivative (3A5AF) was systematically investigated, opening up a new avenue for generating value-added, renewable chemicals bearing nitrogen. Boric acid is identified as the best additive and NMP as the best solvent. Under optimized conditions, 3A5AF reaches a yield of 7.5% from chitin in two hours. NMR studies and poison tests provided mechanistic insights by confirming the formation of a six-membered ring boron complex intermediate. Water is detrimental to the reaction and its content should be kept below 1%. The entire reaction network from chitin to 3A5AF and side products has been established by a combination of instrumental techniques. Kinetic studies suggest that hydrolysis of the crystalline region of chitin is likely to be rate-determining, and the product is not stable at high temperatures or for elongated reaction times.The yield of 3A5AF from chitin is low in the current work. Two strategies to further increase the one-pot conversion of chitin into nitrogen containing chemicals such as 3A5AF can be envisaged. Chitin pretreatment may be essential for chitin depolymerization as it can break down the robust structure and hydrogen bonding network effectively. Solvents that exhibit better solubility towards chitin dissolution, such as ionic liquids, could also be attempted. Investigations along these directions are currently underway in the lab.
Acknowledgements
This work was financially supported by the Start-up Grant from NUS (WBS: R-279-000-368-133) and a MOE Tier-1 project (WBS: R-279-000-387-112).Notes and references
- J. R. Rostrup-Nielsen, Science, 2005, 308, 1421–1422 CrossRef CAS PubMed.
- C. Somerville, H. Youngs, C. Taylor, S. C. Davis and S. P. Long, Science, 2010, 329, 790–792 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- C. S. K. Lin, R. Luque, J. H. Clark, C. Webb, C. Du, C. S. K. Lin, R. Luque, J. H. Clark, C. Webb and C. Du, High Energy Dens. Phys., 2011, 4, 1471–1479 CAS.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 118, 5285–5287 CrossRef.
- N. Yan, C. Zhao, C. Luo, P. J. Dyson, H. Liu and Y. Kou, J. Am. Chem. Soc., 2006, 128, 8714–8715 CrossRef CAS PubMed.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed.
- N. Yan, Y. Yuan, R. Dykeman, Y. Kou and P. J. Dyson, Angew. Chem., Int. Ed., 2010, 49, 5549–5553 CrossRef CAS PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- N. Yan and P. J. Dyson, Curr. Opin. Chem. Eng., 2013, 2, 178–183 CrossRef PubMed.
- B. R. Caes, M. J. Palte and R. T. Raines, Chem. Sci., 2013, 4, 196–199 RSC.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 119, 7780–7783 CrossRef.
- T. Deng and H. Liu, Green Chem., 2013, 15, 116–124 RSC.
- H. Kobayashi and A. Fukuoka, Green Chem., 2013, 15, 1740–1763 RSC.
- F. M. Kerton, Y. Liu, K. W. Omari and K. Hawboldt, Green Chem., 2013, 15, 860–871 RSC.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS PubMed.
- H. Sashiwa and S.-i. Aiba, Prog. Polym. Sci., 2004, 29, 887–908 CrossRef CAS PubMed.
- K. Gopalan Nair, A. Dufresne, A. Gandini and M. N. Belgacem, Biomacromolecules, 2003, 4, 1835–1842 CrossRef PubMed.
- H. Sashiwa and Y. Shigemasa, Carbohydr. Polym., 1999, 39, 127–138 CrossRef CAS.
- A. K. Metreveli, P. K. Metreveli, I. E. Makarov and A. V. Ponomarev, High Energy Chem., 2013, 47, 35–40 CrossRef CAS.
- L. T. Zeng, C. Q. Qin, L. S. Wang and W. Li, Carbohydr. Polym., 2011, 83, 1553–1557 CrossRef CAS PubMed.
- J. Zawadzki and H. Kaczmarek, Carbohydr. Polym., 2010, 80, 394–400 CrossRef CAS PubMed.
- A. Einbu and K. M. Varum, Biomacromolecules, 2008, 9, 1870–1875 CrossRef CAS PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sørlie and V. G. H. Eijsink, Science, 2010, 330, 219–222 CrossRef CAS PubMed.
- M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 859–861 CrossRef CAS PubMed.
- K. W. Omari, J. E. Besaw and F. M. Kerton, Green Chem., 2012, 14, 1480–1487 RSC.
- Y. Wang, C. M. Pedersen, T. Deng, Y. Qiao and X. Hou, Bioresour. Technol., 2013, 143, 384–390 CrossRef CAS PubMed.
- M. W. Drover, K. W. Omari, J. N. Murphy and F. M. Kerton, RSC Adv., 2012, 2, 4642–4644 RSC.
- K. W. Omari, L. Dodot and F. M. Kerton, ChemSusChem, 2012, 5, 1767–1772 CrossRef CAS PubMed.
- X. Hu, Y. Du, Y. Tang, Q. Wang, T. Feng, J. Yang and J. F. Kennedy, Carbohydr. Polym., 2007, 70, 451–458 CrossRef CAS PubMed.
- Y.-B. Huang and Y. Fu, Green Chem., 2013, 15, 1095–1111 RSC.
- B. Lindman, G. Karlström and L. Stigsson, J. Mol. Liq., 2010, 156, 76–81 CrossRef CAS PubMed.
- C. K. S. Pillai, W. Paul and C. P. Sharma, Prog. Polym. Sci., 2009, 34, 641–678 CrossRef CAS PubMed.
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. L. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326–333 RSC.
- J. Fan, M. De Bruyn, V. L. Budarin, M. J. Gronnow, S. Breeden, D. J. Macquarrie, J. H. Clark, P. S. Shuttleworth, J. Fan, M. De Bruyn, V. L. Budarin, M. J. Gronnow, S. Breeden, D. J. Macquarrie, J. H. Clark and P. S. Shuttleworth, J. Am. Chem. Soc., 2013, 135, 12728–12731 Search PubMed.
- O. A. El Seoud, H. Nawaz and E. P. Arêas, Molecules, 2013, 18, 1270–1313 CrossRef CAS PubMed.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- M. Ohara, A. Takagaki, S. Nishimura and K. Ebitani, Appl. Catal., A, 2010, 383, 149–155 CrossRef CAS PubMed.
- J. Wang, J. Ren, X. Liu, G. Lu and Y. Wang, AIChE J., 2013, 59, 2558–2566 CrossRef CAS.
- J. N. Chheda and J. A. Dumesic, Catal. Today, 2007, 123, 59–70 CrossRef CAS PubMed.
- A. S. Amarasekara, L. D. Williams and C. C. Ebede, Carbohydr. Res., 2008, 343, 3021–3024 CrossRef CAS PubMed.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed.
- B. F. M. Kuster and H. S. van der Baan, Carbohydr. Res., 1977, 54, 165–176 CrossRef CAS.
- T. S. Hansen, J. M. Woodley and A. Riisager, Carbohydr. Res., 2009, 344, 2568–2572 CrossRef CAS PubMed.
- T. Ståhlberg, S. Rodriguez-Rodriguez, P. Fristrup and A. Riisager, Chem.–Eur. J., 2011, 17, 1456–1464 CrossRef PubMed.
- E. A. Khokhlova, V. V. Kachala and V. P. Ananikov, ChemSusChem, 2012, 5, 783–789 CrossRef CAS PubMed.
- T. S. Hansen, J. Mielby and A. Riisager, Green Chem., 2011, 13, 109–114 RSC.
- S. Zhao, M. Cheng, J. Li, J. Tian and X. Wang, Chem. Commun., 2011, 47, 2176–2178 RSC.
- Z. Sun, M. Cheng, H. Li, T. Shi, M. Yuan, X. Wang and Z. Jiang, RSC Adv., 2012, 2, 9058–9065 RSC.
- Y. Zhang, V. Degirmenci, C. Li and E. J. M. Hensen, ChemSusChem, 2011, 4, 59–64 CrossRef CAS PubMed.
- E. A. Pidko, V. Degirmenci and E. J. M. Hensen, ChemCatChem, 2012, 4, 1263–1271 CrossRef CAS.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- S. Hu, Z. Zhang, J. Song, Y. Zhou and B. Han, Green Chem., 2009, 11, 1746–1749 RSC.
- Z. Zhang, Q. Wang, H. Xie, W. Liu and Z. Zhao, ChemSusChem, 2011, 4, 131–138 CrossRef CAS PubMed.
- Y. Su, H. M. Brown, X. Huang, X.-d. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117–122 CrossRef CAS PubMed.
- J. Potvin, E. Sorlien, J. Hegner, B. DeBoef and B. L. Lucht, Tetrahedron Lett., 2011, 52, 5891–5893 CrossRef CAS PubMed.
- R. C. Remsing, R. P. Swatloski, R. D. Rogers and G. Moyna, Chem. Commun., 2006, 1271–1273 RSC.
- Y. Wang, Y. Chang, L. Yu, C. Zhang, X. Xu, Y. Xue, Z. Li and C. Xue, Carbohydr. Polym., 2013, 92, 90–97 CrossRef CAS PubMed.
- M. Osada, C. Miura, Y. S. Nakagawa, M. Kaihara, M. Nikaido and K. Totani, Carbohydr. Polym., 2013, 92, 1573–1578 CrossRef CAS PubMed.
- F. G. Pearson, R. H. Marchessault and C. Y. Liang, J. Polym. Sci., 1960, 43, 101–116 CrossRef.
- G. Cárdenas, G. Cabrera, E. Taboada and S. P. Miranda, J. Appl. Polym. Sci., 2004, 93, 1876–1885 CrossRef.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- I. van Zandvoort, Y. Wang, C. B. Rasrendra, E. R. H. van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CAS.
- Z. Srokol, A.-G. Bouche, A. van Estrik, R. C. J. Strik, T. Maschmeyer and J. A. Peters, Carbohydr. Res., 2004, 339, 1717–1726 CrossRef CAS PubMed.
- A. Kruse and A. Gawlik, Ind. Eng. Chem. Res., 2002, 42, 267–279 CrossRef.
- T. M. Aida, Y. Sato, M. Watanabe, K. Tajima, T. Nonaka, H. Hattori and K. Arai, J. Supercrit. Fluids, 2007, 40, 381–388 CrossRef CAS PubMed.
- R. van den Berg, J. A. Peters and H. van Bekkum, Carbohydr. Res., 1994, 253, 1–12 CrossRef CAS.
- M. Vincendon, Die Makromol. Chem., 1985, 186, 1787–1795 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of solvent/additive screening and optimization, EA/GPC characterization and additional mechanistic investigation. See DOI: 10.1039/c3gc42436g |
| This journal is © The Royal Society of Chemistry 2014 |