Rapid Wolff–Kishner reductions in a silicon carbide microreactor†
Stephen G.
Newman
a,
Lei
Gu
a,
Christoph
Lesniak
b,
Georg
Victor
b,
Frank
Meschke
b,
Lahbib
Abahmane
b and
Klavs F.
Jensen
*a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA 02139, USA. E-mail: kfjensen@mit.edu; Fax: (+1) 617-258-8992
bTechnology & Innovation Department at ESK, ESK Ceramics GmbH & Co. KG, Max-Schaidhauf-Straße 25, 87437 Kempten, Germany
First published on 12th November 2013
Abstract
Wolff–Kishner reductions are performed in a novel silicon carbide microreactor. Greatly reduced reaction times and safer operation are achieved, giving high yields without requiring a large excess of hydrazine. The corrosion resistance of silicon carbide avoids the problematic reactor compatibility issues that arise when Wolff–Kishner reductions are done in glass or stainless steel reactors. With only nitrogen gas and water as by-products, this opens the possibility of performing selective, large scale ketone reductions without the generation of hazardous waste streams.
Introduction
The Wolff–Kishner reaction is a classical method for reducing ketones and aldehydes to the corresponding alkanes.1 In general, the transformation involves mixing the substrate with a stoichiometric amount of hydrazine to form water and an intermediate hydrazone that, in the presence of a strong base, liberates nitrogen gas to give the final product. While many procedures for performing Wolff–Kishner reductions exist, the most common is the Huang-Minlon modification, where a high boiling solvent is used and the temperature of the reactor is gradually elevated over the course of several hours.2 The volatile water and, subsequently, hydrazine are thus removed from the reaction, driving the equilibrium towards the intermediate hydrazone over the course of the temperature ramp. The final loss of N2 occurs only at the elevated temperature. As such, the reaction can be considered to take place over three steps – equilibrium formation of the hydrazone under reflux, temperature ramp and distillation to remove water and hydrazine and to drive the equilibrium towards the hydrazone, and irreversible thermal reduction (Scheme 1). This procedure has been reported on mole scale, for instance in the preparation of docosanedioic acid,3 or towards the synthesis of an imidazole API precursor from Merck & Co.4 Both processes required several hours of operation over three stages and a large excess of hydrazine.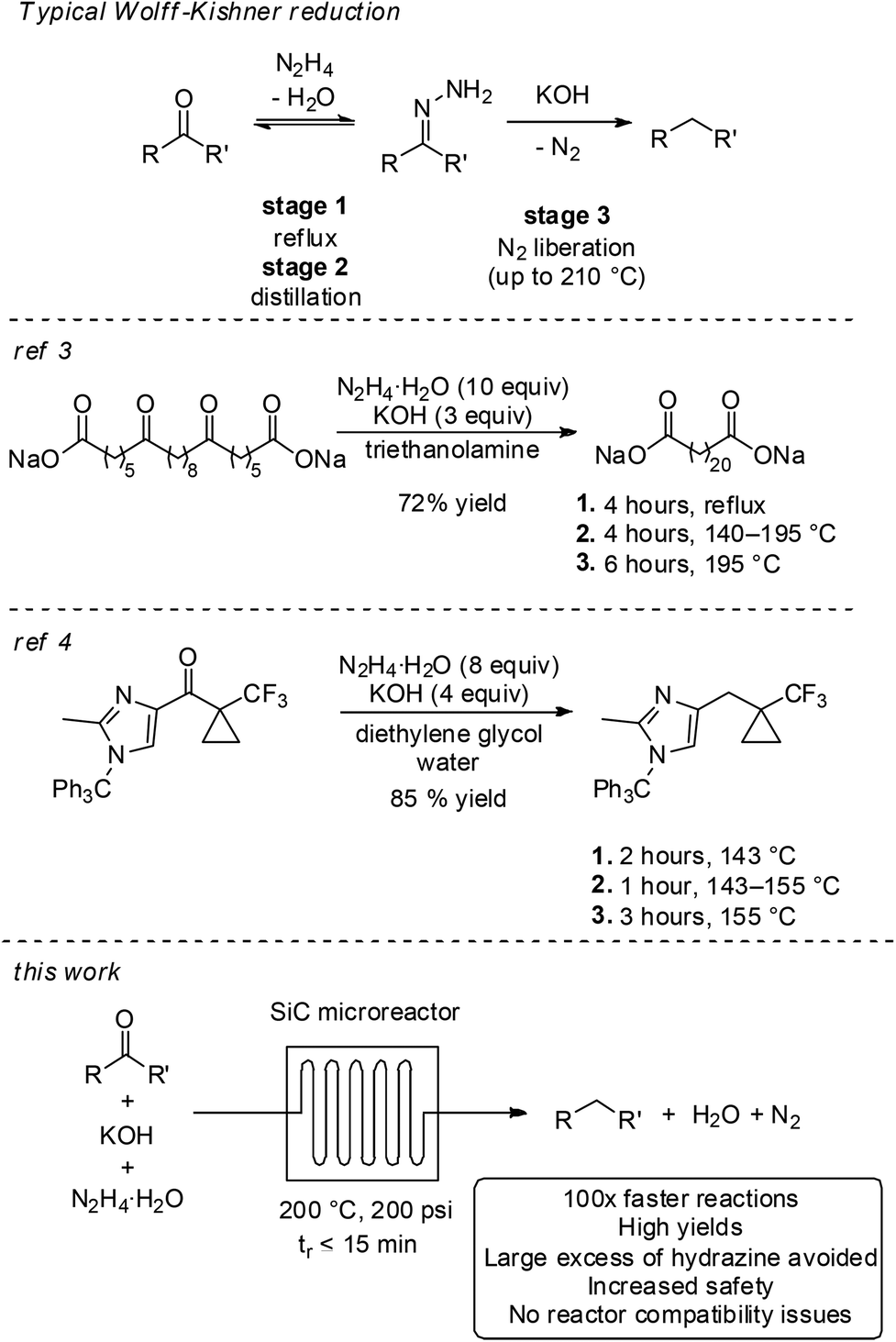 | ||
| Scheme 1 Wolff–Kishner reductions in batch and flow. | ||
Despite these large scale examples, there are many challenges encountered in the Wolff–Kishner reduction. For instance, dangerous anhydrous hydrazine may be required.5 Pre-formation and isolation of the hydrazone intermediate is sometimes necessary, making formation of azine by-products a significant issue.6 The presence of large quantities of explosive, gaseous hydrazine in the batch reactor head space presents a major safety concern in scale-up. Moreover, reactor compatibility can be problematic. Hydrazine decomposition can be catalyzed by metals, so contact with stainless steel should generally be avoided.7 Glass is also undesirable due to the ability of strong base to etch the surface at elevated temperatures.8
Many other methods for performing similar ketone and aldehyde reductions exist. The Clemmensen reduction, using stoichiometric zinc, is a complimentary acidic reaction.9 The use of silanes or borohydrides along with a Lewis acid is also common.10 These and most other methods all have large waste streams, making them particularly environmentally unfriendly and costly on large scale. For example, researchers at Eli Lilly recently performed a TFA-promoted triethylsilane reduction of a diaryl ketone to the corresponding methylene on 100 mol scale; however an alternative route was sought due to challenges associated with the disposal of the fluoride and silane waste.11 Catalytic reduction with hydrogen gas is the greenest method, but suffers from limited scope and functional group compatibility.12 While the Wolff–Kishner reduction uses inexpensive hydrazine as a reducing agent and generates only water and nitrogen gas as waste products, alternative reduction methods are often chosen due to issues with high temperatures, high pH, reactor incompatibility, the need for multiple stages, and potential decomposition of hydrazine.
Flow chemistry can overcome many limitations of typical batch operation.13 From a safety standpoint, smaller reactors can often be used with improved heat transfer, reducing the risk of potential thermal runaway and explosions. Due to the ease with which temperature and pressure can be manipulated, a novel process window is introduced, giving access to a much wider range of reaction conditions. The enhanced mass and heat transfer, faster rates from the novel process window, continuous collection of product, and option to operate multiple reactors simultaneously all contribute to the improved scalability of reactions done in flow. Lastly, the absence of head space in flow reactors mitigates the risk associated with the accumulation of explosive gases. We proposed many of the limitations of Wolff–Kishner reactions could be overcome by operating in a continuous manner. However, most flow reactors are prepared out of base-sensitive silicon or glass,8 thermally sensitive polymer tubing, or steel that may react with hydrazine.14 To overcome these materials limitations, we designed a microreactor prepared from sintered silicon carbide (SiC).
Results and discussion
SiC is a ceramic material with exceptional chemical compatibility, temperature stability, and thermal conductivity. These properties make it a useful component for demanding applications such as in automobile brake discs and high temperature semiconductors. In chemical synthesis, Kappe and co-workers used a microwave vial made out of SiC to perform high temperature chemical reactions which would corrode typical Pyrex microwave vials.15 Industrial flow reactors constructed of SiC have recently been commercialized by Boostec16 and ESK17 allowing access to otherwise ‘forbidden’ chemistries on large scale. Microreactors prepared out of robust ceramic materials are known;18 however, to our knowledge, none have been generally applicable for fine chemical synthesis. As such, we wanted to design a universally corrosion resistant microreactor constructed of SiC for the purpose of small scale reaction development. Such reactors would facilitate development of chemistries that may otherwise be impossible in traditional glass or metal reactors, without needing to commit the large amounts of material necessary to operate industrial scale SiC reactors.The design we chose was based on a silicon chip previously developed in our lab.19 To ensure general applicability to a range of transformations, two inlets, a quench line, and an outlet were included (Fig. 1a). This cooled inlet and outlet zone is thermally separated from a spiral heated zone by a halo etch. The reactor was produced by assembling a stack of two EKasic® SiC plates with identical external dimensions and individual internal designs. The bottom plate has the manifold channels which were manufactured in the SiC green body using Nd:YAG scanning laser erosion machining. The trapezoid-shaped channels (Fig. 1b) are in the hundreds of microns-range, giving approximately 470 μL total internal volume. The trapezoid cross-section of the channels is a result of the laser etching process; the edge shades the laser light making it difficult to produce completely rectangular cross-sections. The thermal insulation holes and all other features were milled in the sintered material post firing of each layer. Before being joined to a single-body, the top and bottom plates were precisely aligned to define the fluidic structures, i.e., inlet and outlet holes were aligned with channels. For diffusion bonding, the stacked plates were subjected to high temperature sintering at ∼1700 °C and 15 bar isostatic pressure under inert atmosphere to give the monolithic reactor (Fig. 1c). During this process the plates fused to form a strong bond. The outside of the reactor shows a small seam due to the low pressure applied on the edges during bonding (Fig. 1d). However, a cross-sectional image of the inside of the fully bonded reactor shows no interface is evident where the two plates were joined (Fig. 1e). Thus, the original stack of plates has become a monolithic piece of ceramic with homogenous mechanical properties. The microreactor channels are gas-tight and separated which is particularly important when hazardous substances are handled. The final assembled reactor was successfully tested at 48 bar and 300 °C without failure.
 | ||
| Fig. 1 (a) An image of the bottom plate before diffusion bonding. (b) An illustration of the trapezoid-shaped channels. (c) The finished reactor. (d) A bonding seam can be seen between the two plates. (e) A cross-sectional close-up of the interface after bonding and treatment with iron cyanide reveals the SiC crystals have grown from one plate to the other during diffusion, giving a seamless union in the interior. The dotted line illustrates where the two plates contacted each other prior to bonding. | ||
With the new SiC microreactor in hand, we used benzophenone as a simple substrate for optimization of the Wolff–Kishner reduction. Parameters studied included pressure, temperature, residence time, concentration in substrate, equivalents of hydrazine, equivalents of KOH, and solvent. While many examples in the literature use a large excess of hydrazine, we proposed that the absence of reactor headspace and operation under pressure would allow efficient reactions to take place with just 1.5 equivalents. With this limitation in mind, we found optimal conditions to be reaction at 200 °C and 200 psi backpressure with 3 equivalents of KOH at 0.8 M concentration in diethylene glycol monoethylether (carbitol) as the solvent. Under these conditions, an 83% yield of diphenylmethane could be obtained with a residence time of just 5 minutes,20 corresponding to a production rate of 630 mg h−1 or 15.12 g per day with a excellent space-time yield of 61.9 kg L−1 h−1 (Table 1, entry 1). Operation at lower pressure, temperature, residence time, equivalence of hydrazine, or concentration were all found to decrease the yield of the reaction (entries 2–6). Use of lower temperatures or fewer equivalents of KOH at 0.8 M concentration led to clogging of the reactor due to buildup of insoluble azine. The effect of lowered base concentration on yield is apparent at 0.4 M (entry 7). The more common Wolff–Kishner solvent, diethylene glycol, was less effective at dissolving the reaction mixture, and could only be tested at 0.4 M, where it was found to give a lower yield than carbitol (entry 8). Other solvents such as ethylene glycol and triethanolamine are highly viscosity and did not completely dissolve the reagents, making them poor choices for flow experiments.
|
|
||
|---|---|---|
| Entry | Change from standard conditions | Yielda |
| a Yield determined by GC with dodecane as internal standard. b Temperature below 185 °C lead to clogging of the reactor. | ||
| 1 | None | 83% |
| 2 | 100 psi | 44% |
| 3 | 185 °Cb | 73% |
| 4 | t res = 3 min | 71% |
| 5 | 1.1 equiv. N2H4·H2O | 65% |
| 6 | 0.4 M | 65% |
| 7 | 0.4 M, 1.0 equiv. KOH | 33% |
| 8 | 0.4 M, diethylene glycol instead of carbitol | 36% |
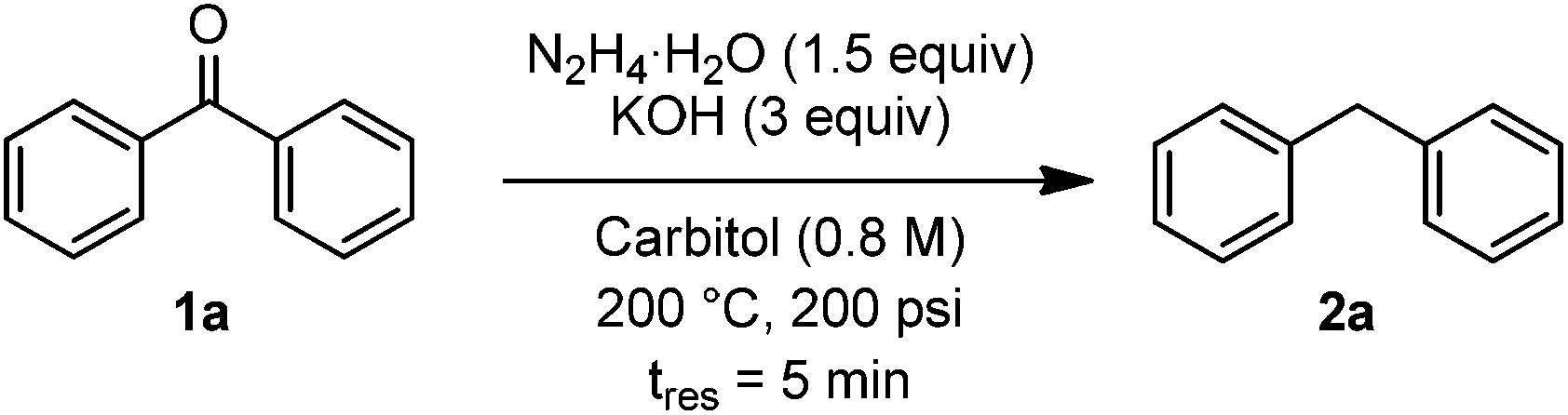
Two remarkable aspects of the optimized results are apparent. Firstly, relatively few equivalents of hydrazine are required. A typical batch experiment uses between 2 and 8 equivalents, adding additional costs and hazards, and complicating waste disposal. Secondly, the reaction time (5 min) is two orders of magnitude lower than in batch. The faster reaction is presumably due to the ability to operate safely in a closed system under pressure which prevents hydrazine from boiling out of the reactor, and thus avoiding the need to perform the reaction in a series of stages. This likely has a further benefit of keeping all reaction components prior to the N2 liberation in equilibrium. In batch after the excess hydrazine has been removed at elevated temperature, irreversible azine formation or reversion to starting material could occur. Azine formation was also evident in the flow experiment at 100 psi (Table 1, entry 2), presumably due to the relatively low concentrations of hydrazine in solution to drive the equilibrium towards the hydrazone.
To investigate the scope of the transformation, the reactivity of a range of aldehydes and ketones was explored (Table 2). Diphenylmethane could be isolated in good yield (entry 1). Further diluting hydrazine with water gives added safety from violent decomposition.21 Towards this end, an extra 3 equivalents of water relative to hydrazine (i.e. 31% aqueous hydrazine) were added, and a similar yield was obtained provided the residence time was lengthened. Electron-rich (entry 2) and electron-poor (entry 3) diaryl ketones could be reduced in high yield. 3-Chloro and -bromo substituents were also tolerated (entries 4 and 5). Aryl alkyl (entries 6 and 7) and alkyl alkyl ketones (entry 8), as well as several aldehydes (entries 9–11) could also be reduced efficiently. 4-Bromobenzophenone was also attempted, but significant reduction of the C–Br bond occurred and an accurate isolated yield could not be obtained. Due to the differing solubilities of the starting materials and intermediates, concentration was optimized separately for each example, and ranged from 0.1 to 0.8 M. The reactor could be run continuously for several hours without issue, and no corrosion was evident over the course of our studies. The potential to perform such chemistries continuously on larger scale is thus very promising.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | t res (min) | Yieldb [%] |
| a Typical reaction conditions: substrate 1 (4 mmol), N2H4·H2O (6 mmol), KOH (12 mmol), 0.8 M in carbitol, 200 °C. Concentration varied per entry. See ESI. b Yield of isolated product. c 3 Equiv. additional H2O added. tres = 10 min. d 220 °C. | ||||
| 1 |
|
|
5 | 82% |
| 84%c | ||||
| 2 |
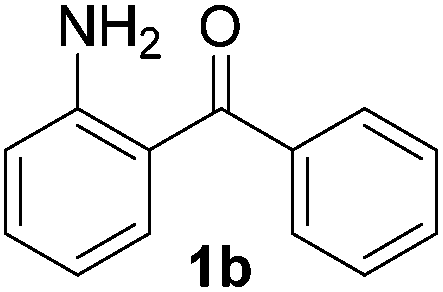
|
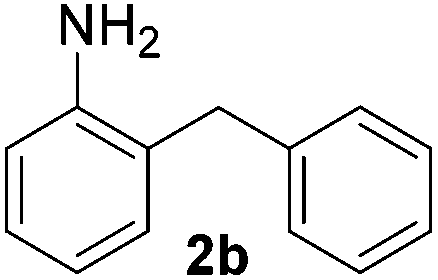
|
15 | 82 |
| 3 |
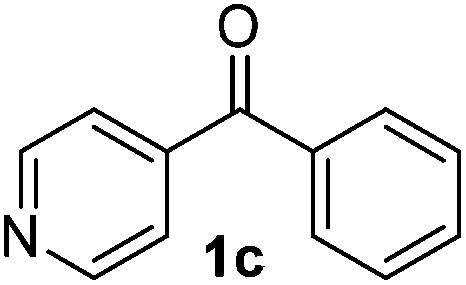
|
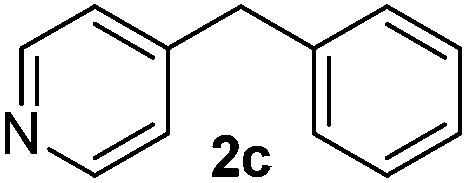
|
3 | 94 |
| 4 |
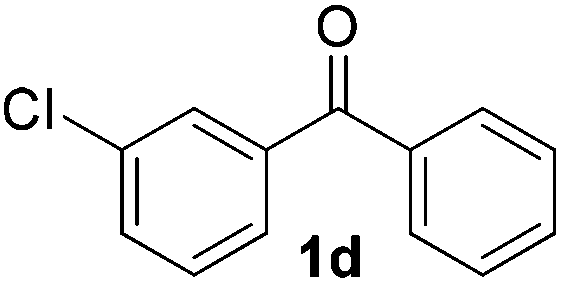
|
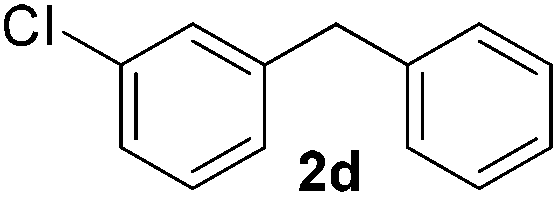
|
3 | 84 |
| 5 |
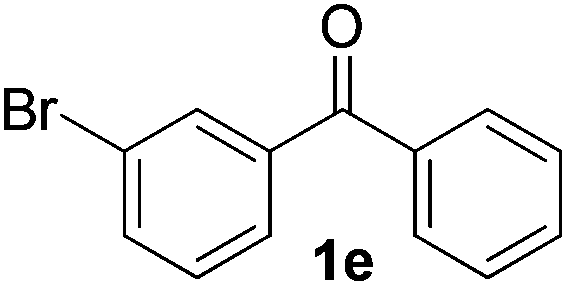
|
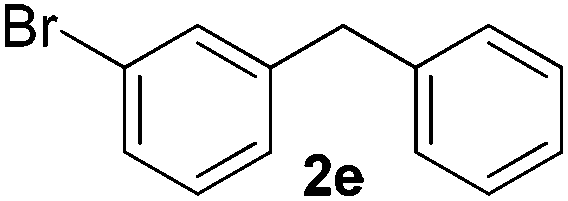
|
10 | 76 |
| 6 |
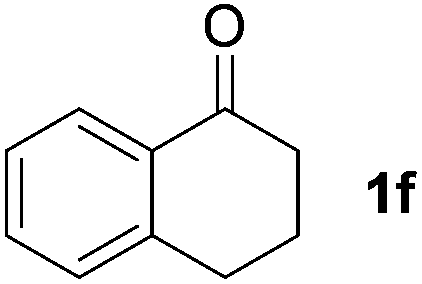
|
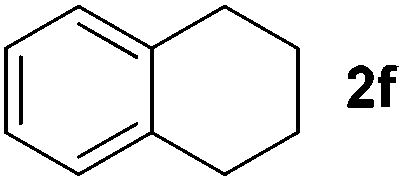
|
15 | 78 |
| 7 |
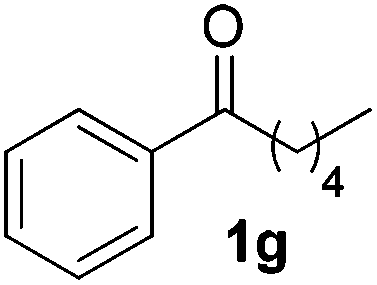
|
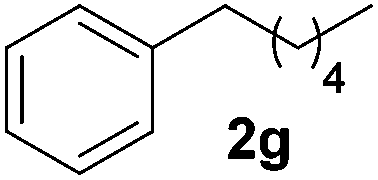
|
10 | 96d |
| 8 |
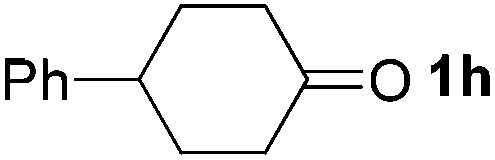
|
|
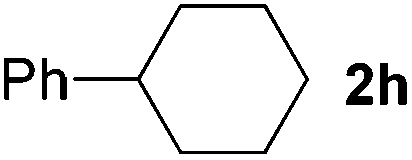
2 | 83 |
| 9 |
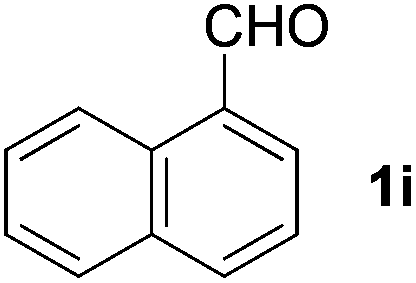
|
|
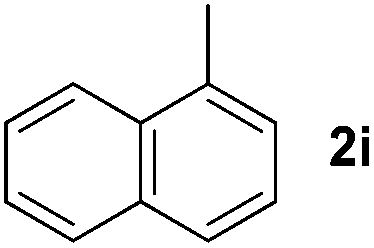
3 | 91 |
| 10 |
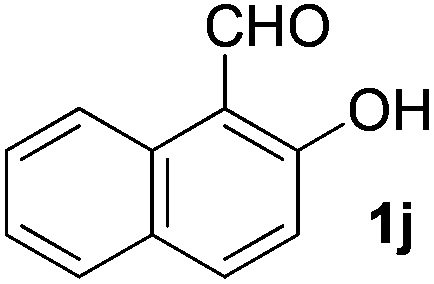
|
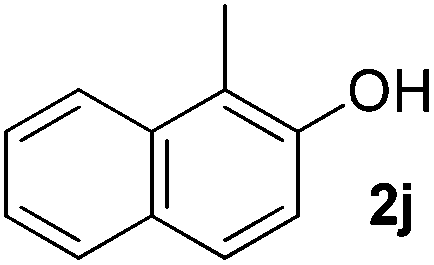
|
5 | 90 |
| 11 |
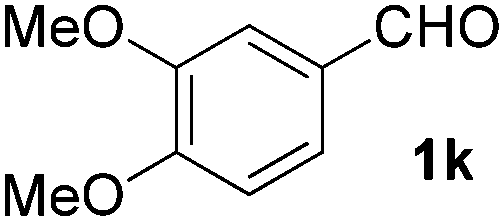
|
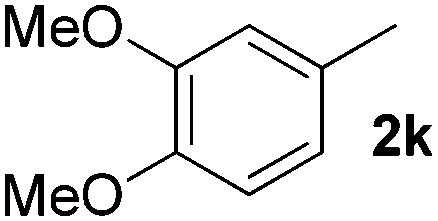
|
5 | 87 |
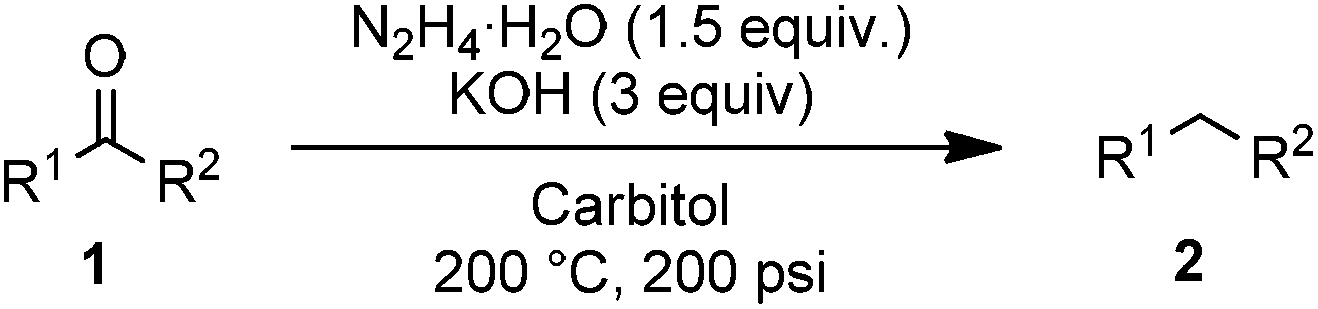
Conclusions
In conclusion, we have designed a new microreactor constructed of SiC, capable of withstanding exceptionally harsh reaction conditions. This was employed in performing continuous Wolff–Kishner reductions, which could be completed quickly and safely. Compared with typical batch conditions, reaction times are decreased by up to two orders of magnitude. We believe our microreactor will provide an important bridge between existing industrial scale flow reactors and small scale academic reaction development. The results obtained herein may facilitate Wolff–Kishner reductions on previously inaccessible large scales due to the significant rate increase and the use of robust ceramic reactor materials. Since no hazardous by-products are generated, this can provide an environmentally friendly alternative to more frequently employed hydride-, silane-, and metal-based methods.Experimental
General procedure: the substrate, KOH (3 equiv.), and 4 mL of diethylene glycol monoethyl ether (carbitol) were combined in a 5 mL volumetric flask. The contents were placed in an ultrasonic cleaning bath and sonicated until the mixture became homogeneous. Hydrazine monohydrate (1.5 equiv.) was added and the flask was brought to 5 mL total volume by dropwise addition of carbitol. The reactor was connected to a 200 psi backpressure regulator at the outlet and brought to 200 °C. The reacting solution was pumped through at 94 μL min−1 (for tres = 5 min) using a Syrris Asia syringe pump with 500/250 μL glass syringes. The second inlet and quench line were plugged. After waiting for the reactor to reach steady state (∼3 residence times), a sample vial was placed at the outlet and the product was collected for ∼2 residence times. The sample was worked up and purified by flash column chromatography. Further details on the apparatus, variations in specific examples, and workup procedure can be found in the ESI.†Acknowledgements
We thank the Novartis-MIT Center for Continuous Manufacturing for support of this work. S. G. N. thanks the Natural Sciences and Engineering Research Council of Canada (NSERC) for financial support in the form of a postdoctoral fellowship.Notes and references
- (a) D. Todd, Org. React., 1948, 4, 378 CAS; (b) R. O. Hutchins and M. K. Hutchins, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, New York, 1991, vol. 8, p. 327 Search PubMed; (c) N. Kishner, J. Russ. Chem. Soc., 1911, 43, 582 Search PubMed; (d) L. Wolff, Justus Liebigs Ann. Chem., 1912, 394, 86 CrossRef.
- Huang-Minlon, J. Am. Chem. Soc., 1946, 68, 2487 CrossRef.
- S. Hünig, E. Lücke, W. Brenniger, F. E. Mumford, E. A. LaLancette, W. J. Middleton and B. C. McKusick, Org. Syn., 1963, 43, 34 Search PubMed.
- J. T. Kuethe, K. G. Childers, Z. Peng, M. Journet, G. R. Humphrey, T. Vickery, D. Bachert and T. T. Lam, Org. Process Res. Dev., 2009, 13, 576 CrossRef CAS.
- D. H. R. Barton, D. A. J. Ives and B. R. Thomas, J. Chem. Soc., 1955, 2056 CAS.
- (a) D. J. Cram, M. R. V. Sahyun and G. R. Knox, J. Am. Chem. Soc., 1962, 84, 1734 CrossRef CAS; (b) M. F. Grundon, H. B. Henbest and M. D. Scott, J. Chem. Soc., 1963, 1855 RSC.
- J.-P. Schirmann and P. Bourdauducq, Hydrazine, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002 Search PubMed.
- For example, silicon 100 is etched at a rate of 246 μm h−1 in 20% KOH at 100 °C. While the presence of alcohol solvent slows this rate down, the etch rate rapidly increases with temperature with an approximate activation energy of E/R = 7000 K. At 200 °C the etch rate is ∼10 mm h−1. Given the channel sizes of a typical silicon microreactor are several hundred μm, the chemical environment will rapidly destroy the channel walls. See: H. Seidel, L. Csepregi, A. Heuberger and H. Baumgärtel, J. Electrochem. Soc., 1990, 137, 3612 CrossRef CAS PubMed.
- E. Clemmensen, Chem. Ber., 1914, 47, 681 CrossRef CAS.
- (a) D. Mitchell, K. P. Cole, P. M. Pollock, D. M. Coppert, T. P. Burkholder and J. R. Clayton, Org. Process Res. Dev., 2012, 16, 70 CrossRef CAS; (b) S. Chandrasekhar, C. R. Reddy and B. N. Babu, J. Org. Chem., 2002, 67, 9080 CrossRef CAS PubMed; (c) D. M. Ketcha, B. A. Lieurance, D. F. J. Homan and G. W. Gribble, J. Org. Chem., 1989, 54, 4350 CrossRef CAS; (d) I. Smonou, Tetrahedron Lett., 1994, 35, 2071 CrossRef CAS.
- (a) D. Mitchell, K. P. Cole, P. M. Pollock, D. M. Coppert, T. P. Burkholder and J. R. Clayton, Org. Process Res. Dev., 2012, 16, 70 CrossRef CAS; (b) A. N. Campbell, K. P. Cole, J. R. Martinelli, S. A. May, D. Mitchell, P. M. Pollock and K. A. Sullivan, Org. Process Res. Dev., 2013, 17, 273 CrossRef CAS.
- For recent examples, see: (a) M. A. Alotaibi, E. F. Kozhevnikova and I. V. Kozhevnikov, Chem. Commun., 2012, 48, 7194 RSC; (b) X. Xu, Y. Gong, P. Zhang, H. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987 CrossRef CAS PubMed; (c) J. G. Stevens, R. A. Bourne, M. V. Twigg and M. Poliakoff, Angew. Chem., Int. Ed., 2010, 49, 8856 CrossRef CAS PubMed; (d) T. S. Hansen, K. Barta, P. T. Anastas, P. C. Ford and A. Riisager, Green Chem., 2012, 14, 2457 RSC.
- For recent overviews, see: (a) J.-i. Yoshida, in Flash Chemistry. Fast Organic Synthesis in Microsystems, Wiley-Blackwell, 2008 Search PubMed; (b) C. Wiles and P. Watts, in Micro Reaction Technology in Organic Synthesis, CRC Press, 2011 Search PubMed; (c) S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456 RSC; (d) B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300 CrossRef CAS PubMed; (e) C. Wiles and P. Watts, Green Chem., 2012, 14, 38 RSC; (f) J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17 CrossRef CAS.
- For examples on the safe use of hydrazine in flow, see (a) A. DeAngelis, D.-H. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 3434 CrossRef CAS PubMed; (b) T.-H. Yoon, S.-H. Park, K.-I. Min, X. Zhang, S. J. Haswell and D.-P. Kim, Lab Chip, 2008, 8, 1454 RSC; (c) C. Wiles, P. Watts and S. J. Haswell, Org. Process Res. Dev., 2004, 8, 28 CrossRef CAS; (d) E. Garcia-Egido, V. Spikmans, S. Y. F. Wong and B. H. Warrington, Lab Chip, 2003, 3, 73 RSC; (e) R. Eluri and B. Paul, J. Nanopart. Res., 2012, 14, 800 CrossRef; (f) D. Cantillo, M. Baghbanzadeh and C. O. Kappe, Angew. Chem., Int. Ed., 2012, 51, 10190 CrossRef CAS PubMed.
- B. Gutmann, D. Obermayer, B. Reichart, B. Prekodravac, M. Irfan, J. M. Kremsner and C. O. Kappe, Chem.–Eur. J., 2010, 16, 12182 CrossRef CAS PubMed.
- S. Elgue, A. Conte, C. Gourdon and Y. Bastard, Chim. Oggi, 2012, 30, 18 CAS.
- L. Abahmane, EKasic® modular flow reactor systems for chemical production under severe conditions. 4th Symposium on Continuous Flow Reactor Technology for Industrial Applications, Lisbon (Portugal), September 26–27, 2012.
- (a) Christian, M. Mitchell, D. P. Kim and P. J. A. Kenis, J. Catal., 2006, 241, 235 CrossRef CAS PubMed; (b) R. Knitter, D. Gohring, P. Risthaus and J. Hausselt, Microsyst. Technol., 2001, 7, 85 CrossRef; (c) F. Meschke, G. Riebler, V. Hessel, J. Schürer and T. Baier, Chem. Eng. Technol., 2005, 28, 465 CrossRef CAS; (d) K. Jain, C. Wu, S. V. Atre, G. Jovanovic, V. Narayanan, S. Kimura, V. Sprenkle, N. Canfield and S. Roy, Int. J. Appl. Ceram. Technol., 2009, 6, 410 CrossRef CAS.
- R. L. Hartman, J. R. Naber, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Org. Process Res. Dev., 2010, 14, 1347 CrossRef CAS.
- The stated residence time is defined as volume of the reactor divided by the feed rate of reactants. Since nitrogen gas is generated and some thermal expansion of the heated solution is likely, the actual time that the reactants spend in the reactor will be lower.
- J. K. Niemeier and D. P. Kjell, Org. Process Res. Dev., 2013 DOI:10.1021/op400120g.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and NMR spectra. See DOI: 10.1039/c3gc41942h |
| This journal is © The Royal Society of Chemistry 2014 |