 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aerobic oxidation of isosorbide and isomannide employing TEMPO/laccase†
Johannes
Gross
a,
Katharina
Tauber
a,
Michael
Fuchs
a,
Nina G.
Schmidt
a,
Aashrita
Rajagopalan
a,
Kurt
Faber
a,
Walter M. F.
Fabian
a,
Jan
Pfeffer
b,
Thomas
Haas
b and
Wolfgang
Kroutil
*a
aDepartment of Chemistry, Organic and Bioorganic Chemistry, University of Graz, Heinrichstrasse 28, A-8010 Graz, Austria. E-mail: Wolfgang.Kroutil@uni-graz.at; Fax: +43 316 380 9840; Tel: +43 316 380 5350
bEvonik Industries AG, CREAVIS Technologies & Innovation, Paul-Baumann-Strasse 1, 45772 Marl, Germany
First published on 30th January 2014
Abstract
The oxidation of the renewable diols isosorbide and isomannide was successfully achieved using a TEMPO/laccase system. Furthermore, various TEMPO-derivatives were tested leading to conversions of up to >99% for the oxidation of isosorbide, isomannide, indanol and a halohydrin to the corresponding ketone.
The diols isosorbide 1 and isomannide 2 (Scheme 1) are both gained from renewable resources like cellulose1 and starch.2 They represent platform chemicals for the potential replacement of oil-based products.3 Consequently, they are in focus of interest as starting materials for pharmaceutical applications as well as for organic solvents or fuels and for biopolymers.3,4 For instance, recently, 1 was derivatised to the corresponding diamine as a building block for polymers,5 or oxidised by chemical means.6 Amination of 1 by a ADH/transaminase bio-cascade led to only a low amount of mono-amination.7 Especially the transformation of isosorbide to valuable derivatives by functionalization or substitution of both hydroxyl groups is difficult due to steric hindrance and different reactivities of the two hydroxyl groups.
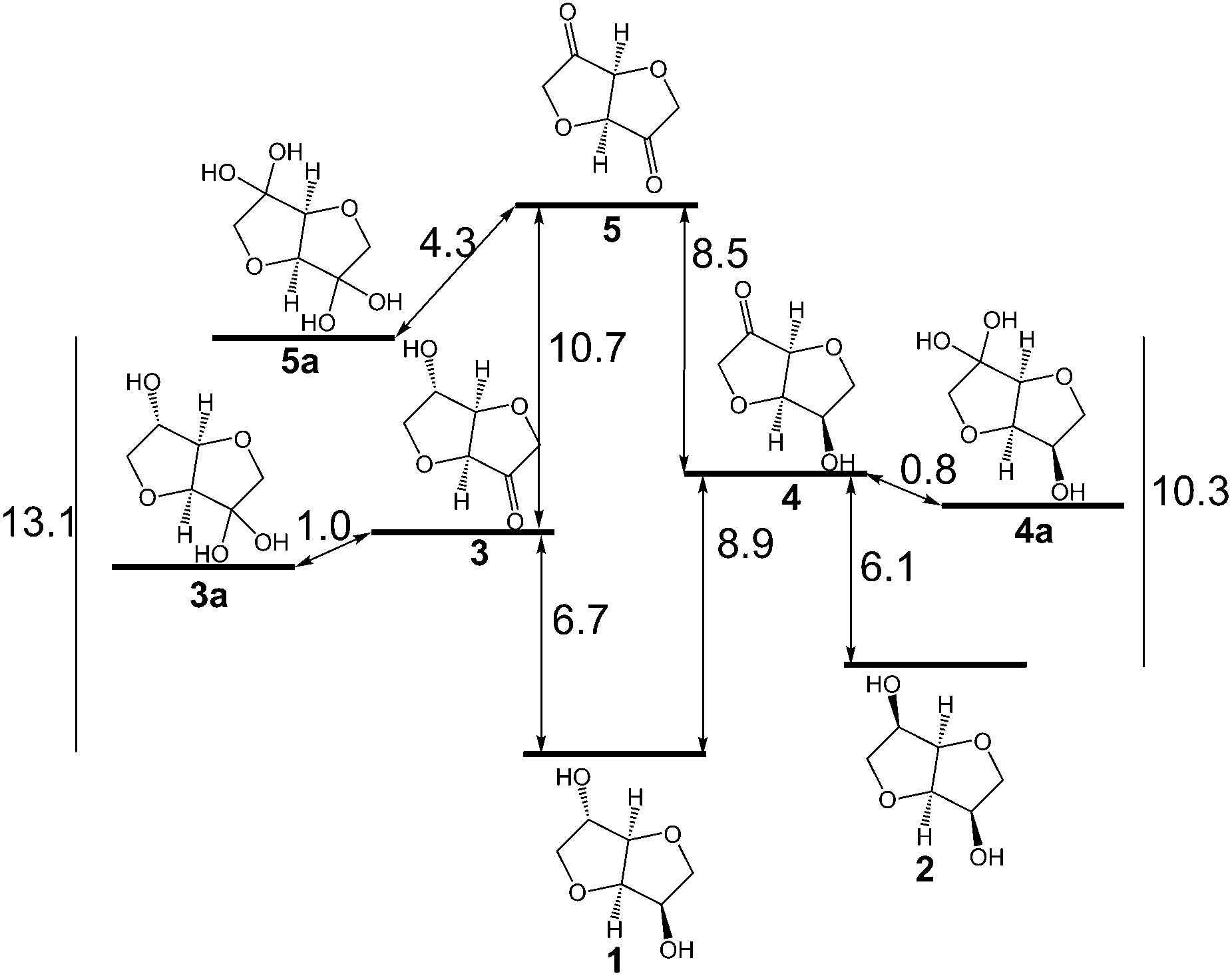 | ||
| Scheme 1 Relative Gibbs free energies (kcal mol−1) for the oxidation and hydration of isosorbide 1 and isomannide 2. Energies for oxidations were calculated for the reaction of the alcohol with acetone yielding the ketone plus 2-propanol. The calculations were performed using the MP2/cc-pVTZ//MP2/cc-pVDZ procedure in aqueous solution (IEFPCM solvation model). | ||
Results and discussion
Alcohol oxidation employing alcohol dehydrogenases (ADHs)7,8 represents one possible common biocatalytic method to prepare ketones or aldehydes from the corresponding alcohols.9 Consequently, various ADHs were investigated to oxidise isosorbide 1 as well as isomannide 2 to the corresponding diketone 5. However, the levels of conversion were unsatisfactory. A maximum of 4% conversion was reached at 50 mM substrate concentration over 24 hours. Calculating the required Gibbs free energy for the oxidation revealed that the energy needed for a mono-oxidation to the hydroxy ketone followed by hydration to the favoured hydrate (3a, 4a) is rather high (Scheme 1), even higher than for the disfavoured biocatalytic oxidation of halohydrins by ADHs (4–6 kcal mol−1).10 For instance, for the oxidative hydrogen transfer of isosorbide 1 to the exo-hydroxy ketone 3 6.7 kcal mol−1 are required while 8.9 kcal mol−1 are needed to get the endo-hydroxy ketone 4. Considering the hydrated forms (3a, 4a), the energy decreases by approximately 1 kcal mol−1, still representing a significant burden for the hydrogen transfer to yield high amounts of ketone. The higher energy needed to get the endo-hydroxy ketone 4 can be explained by the higher steric hindrance of the hydride to be removed at the exo-hydroxy of 1. Also for the second oxidation (transformation of 3 and 4, respectively, to diketone 5 and its hydrate 5a) a significant energy barrier has to be overcome. To address the problem of steric hindrance for the oxidation of the exo-hydroxy moiety as well as to provide sufficient energy, an oxidation method was needed involving a small mediator as an oxidizing agent and a high energy terminal oxidant like molecular oxygen. In case of success, the same approach could be applied for the oxidation of compounds 6–8, which are also difficult susbtrates for ADHs.Consequently, the combination of laccase and TEMPO M1 as a mediator was tested employing O2 as an oxidant (Scheme 2).11 Laccases12 can be produced in large quantities,13 and the oxidation system can be employed at ambient temperature (20–30 °C) and in aqueous medium. Although TEMPO has been employed for the oxidation of various sec-alcohols using various chemical reoxidation reagents,14sec-alcohols have only been sparsely oxidised with the TEMPO/laccase system.15 TEMPO enables an aerobic green oxidation.16
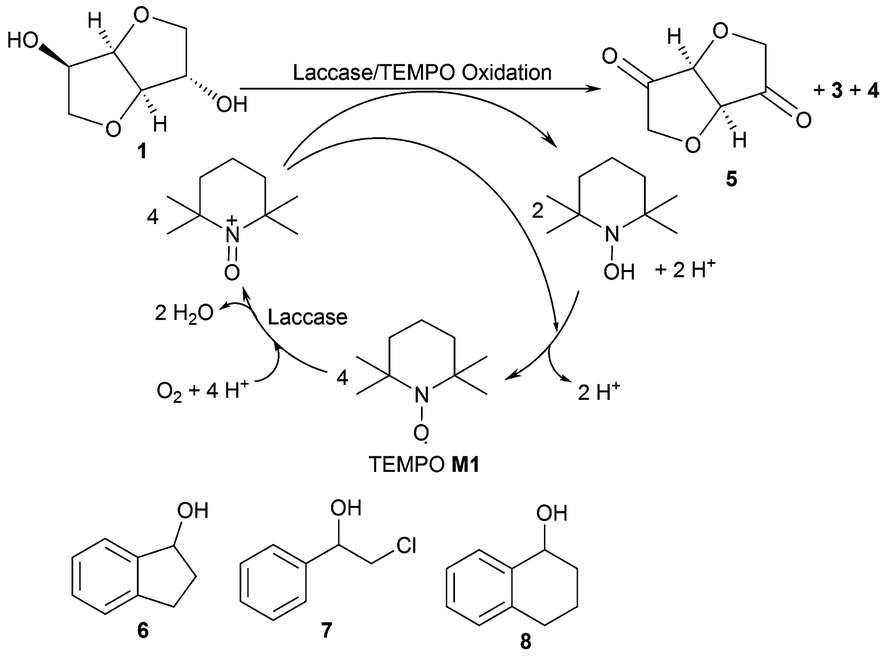 | ||
| Scheme 2 Oxidation of isosorbide 1 employing TEMPO/laccase and further substrates investigated. | ||
In a first attempt, commercial laccases from Trametes versicolor (TvLac) and Agaricus bisporus (AbLac) were applied using sodium acetate buffer at varied pHs or in deionised water (Table 1). Best results were achieved with TvLac while AbLac led to very low conversions. Based on the published sequences of laccases from these fungi, they are similar in size (TvLac: 519aa, AbLac: 520aa), but have only 47.5% sequence identity.17 A redox potential is only published for TvLac (E0′ = 785 mV vs. normal hydrogen electrode).18 TEMPO/TvLac oxidised the two equal hydroxy groups of isomannide 2 as well as the two energetically different hydroxy groups of isosorbide 1 to the diketone 5 at pH 5.5 and 6.5. For 1 both possible intermediates – the endo- and the exo-hydroxyyketone – were observed at pH 4.5, indicating that in the first oxidation step no absolute discrimination between the two hydroxy moieties occurred; for 2 obviously only the endo-hydroxyketone can be formed due to the stereochemistry of the substrate. Blank experiments showed that in the absence of an enzyme or a mediator no product formation was observed. Interestingly, just using water without buffer salts (last entry, pH 6.5) led to perfect conversion, offering another opportunity to minimize waste. Isomannide 2 was in general transformed faster than 1. Although these first tests gave promising results, optimisation studies were required to decrease the concentration of the mediator and the enzyme.
| Substr. | Buffer | pH | Laccaseb | endo-OH-ketone 4 [%] | exo-OH-ketone 3 [%] | Diketone 5 [%] |
|---|---|---|---|---|---|---|
| a Relative amounts as peak areas measured by GC-FID. Substrate (50 mM), laccase (10 U), TEMPO (30 mol%), 24 h at 30 °C, 120 rpm. b TvLac = Laccase from Trametes versicolor; AbLac = Agaricus bisporus. c In the absence of TEMPO. | ||||||
| 1 | NaOAc | 5.5 | None | <0.1 | <0.1 | <0.1 |
| 1 | NaOAc | 4.5 | AbLac | <0.1 | <0.1 | <0.1 |
| 1 | NaOAc | 5.5 | AbLac | 1 | <0.1 | <0.1 |
| 1 | H2O dist. | 6.5 | AbLac | 2 | <0.1 | <0.1 |
| 1 | NaOAc | 4.5 | TvLac | 8 | 13 | 79 |
| 1 | NaOAc | 5.5 | TvLac | <0.1 | <0.1 | >99 |
| 1 | NaOAc | 6.5 | TvLac | <0.1 | <0.1 | >99 |
| 1 | NaOAc | 5.5 | TvLacc | <0.1 | <0.1 | <0.1 |
| 2 | NaOAc | 4.5 | AbLac | 4 | <0.1 | <0.1 |
| 2 | NaOAc | 4.5 | TvLac | 23 | <0.1 | 77 |
| 2 | NaOAc | 5.5 | TvLac | <0.1 | <0.1 | >99 |
| 2 | H2O dist. | 6.5 | TvLac | <0.1 | <0.1 | >99 |
In the next experiments the TEMPO concentration was lowered to 5 mol%. In two buffer systems (acetate and phosphate) the highest conversion was achieved at low buffer concentration (Fig. 1). In general, best results were obtained at pH 6 and low buffer concentrations (5–10 mM).
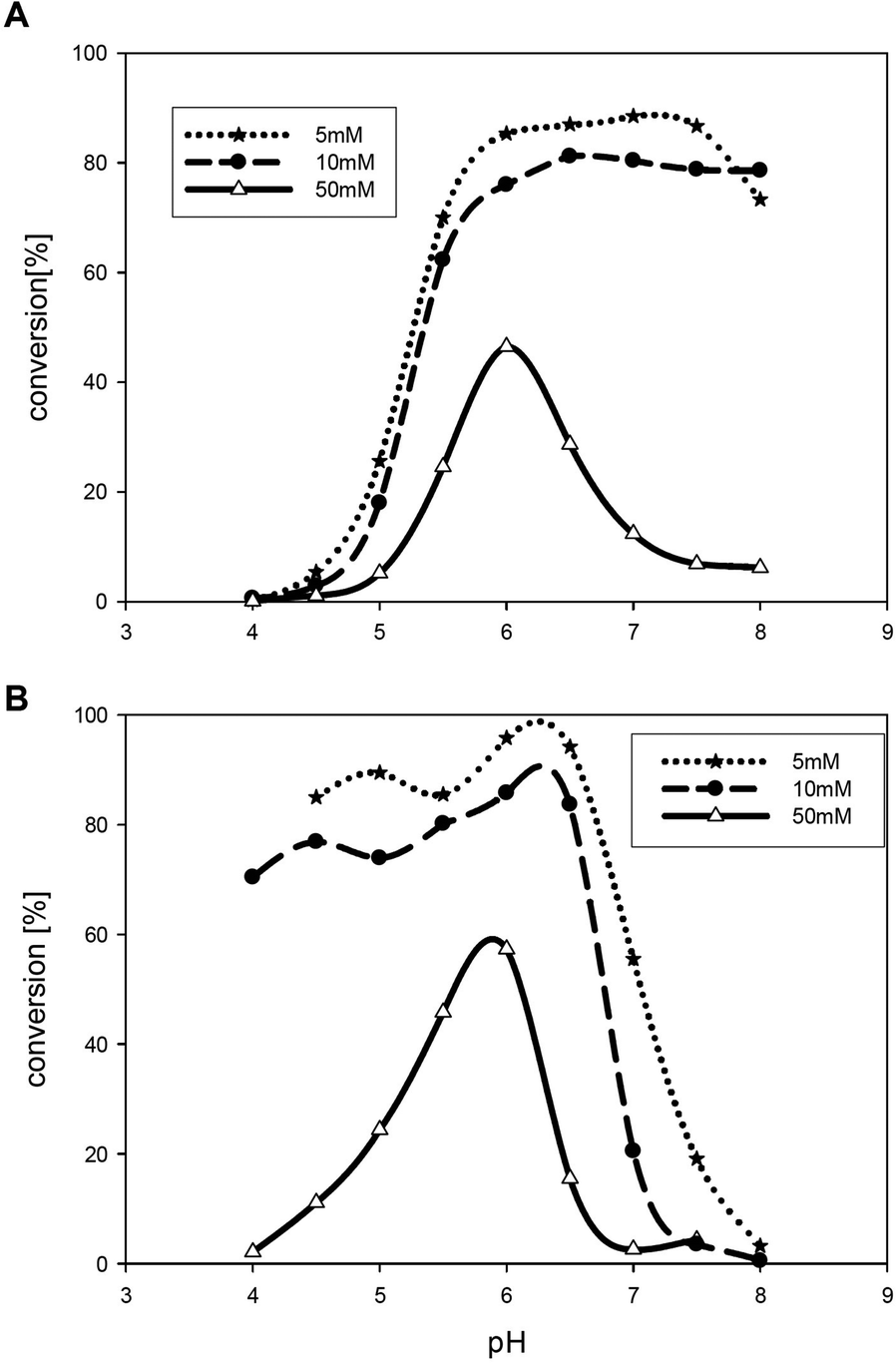 | ||
| Fig. 1 Conversions at varied pH in acetate buffer (A) and phosphate buffer (B) (5 mM, 10 mM and 50 mM) using 1 as a substrate. Conversions were measured by GC-FID. Reaction conditions: TEMPO (5 mol%), isosorbide (50 mM), TvLac (0.5 mg ml−1); 24 h at 30 °C at 120 rpm. Lines between the measured points are only drawn as a guide for the eye to ease reading and not to interpolate values. | ||
Interestingly, for bis-tris buffer and sodium citrate buffer conversion did not exceed 3%, although oxidation of glycols and various alcohols is reported using sodium citrate buffer at a pH of 4.5 and high buffer concentrations.15c Obviously these conditions are not favourable for isosorbide or isomannide as a substrate. Further decrease of the buffer concentration to 0.5–1.5 mM improved the conversion and led to full diketone formation as was observed in the deionised water-system.
To minimize the concentration of the mediator, the TEMPO concentration was decreased successively. Reducing the TEMPO concentration from 30 mol% to 4 mol% (mol mediator/mol substrate) did not lead to a significant decrease of the diketone formation employing isosorbide 1 as the substrate over 24 hours under the conditions employed. However, as soon as the mediator concentration was below 4 mol%, conversion decreased rapidly (Fig. 2).
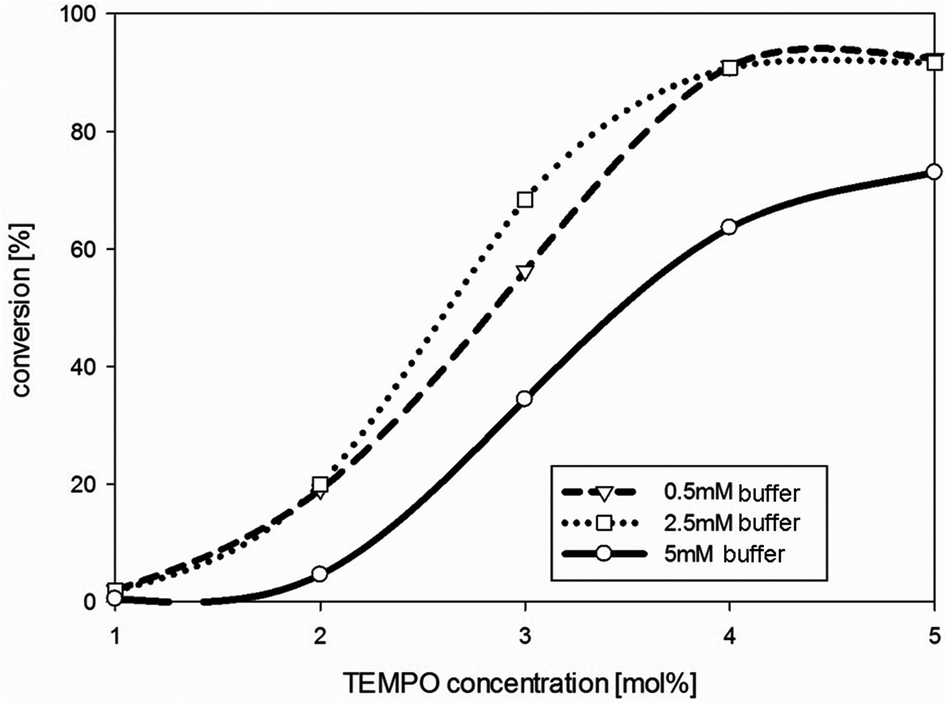 | ||
| Fig. 2 Variation of TEMPO concentration in phosphate buffer (0.5, 2.5 and 5 mM, pH 6) using isosorbide 1 as a substrate. Conversions measured by GC-FID. Reaction conditions: substrate (50 mM); TvLac (0.5 mg ml−1), 24 h at 30 °C, 120 rpm. Lines between the measured points are only drawn as a guide for the eye to ease reading and not to interpolate values. | ||
To identify the most suitable mediator,15a,19 a selection of different mediators M2–M15 were tested (Fig. 3).
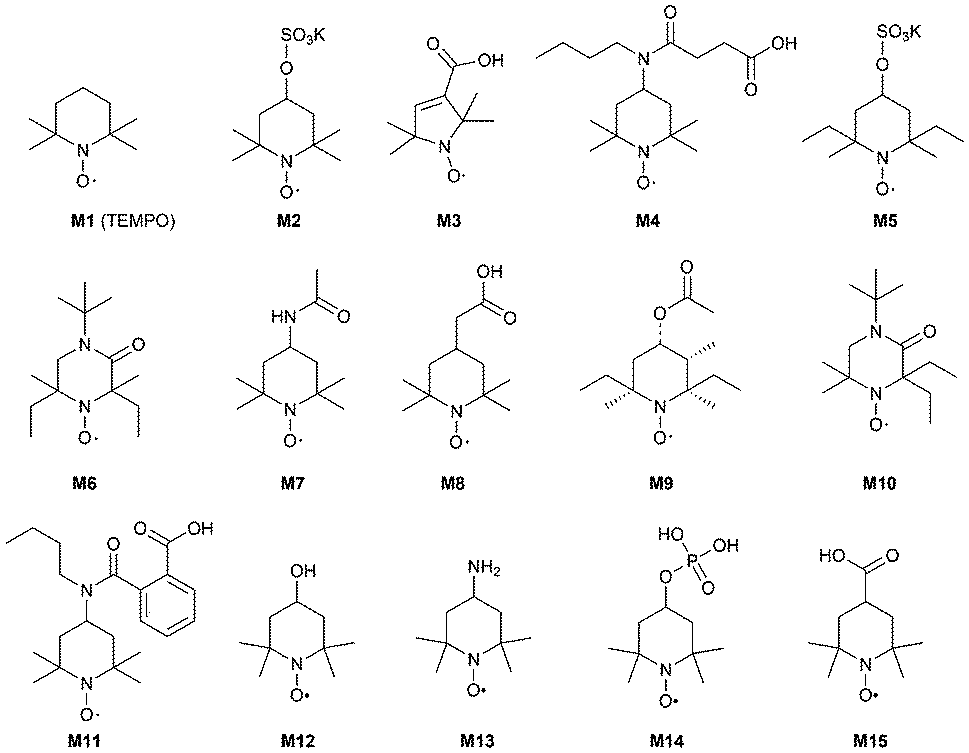 | ||
| Fig. 3 Selection of mediators: TEMPO and derivatives. | ||
However, for isosorbide 1 as a substrate, TEMPO M1 was superior to all other TEMPO-derivatives M2–M15.
Investigating the substrate scope of the oxidation method, also 1-indanol 6 was tested. Using 6 as a substrate, TEMPO M1 led to 45% conversion at 50 mM substrate concentration within 4 hours. For this substrate, a number of mediators proved to be superior compared to M1 (Fig. 4): for instance, M3 led to 99% conversion, M4 gave 65% conversion, M8 76%, and mediator M15 75%. It is noticeable that all these mediators possess a carboxylic acid moiety. Mediator M2 (41%) and mediator M14 (42%) led to results comparable to TEMPO M1.
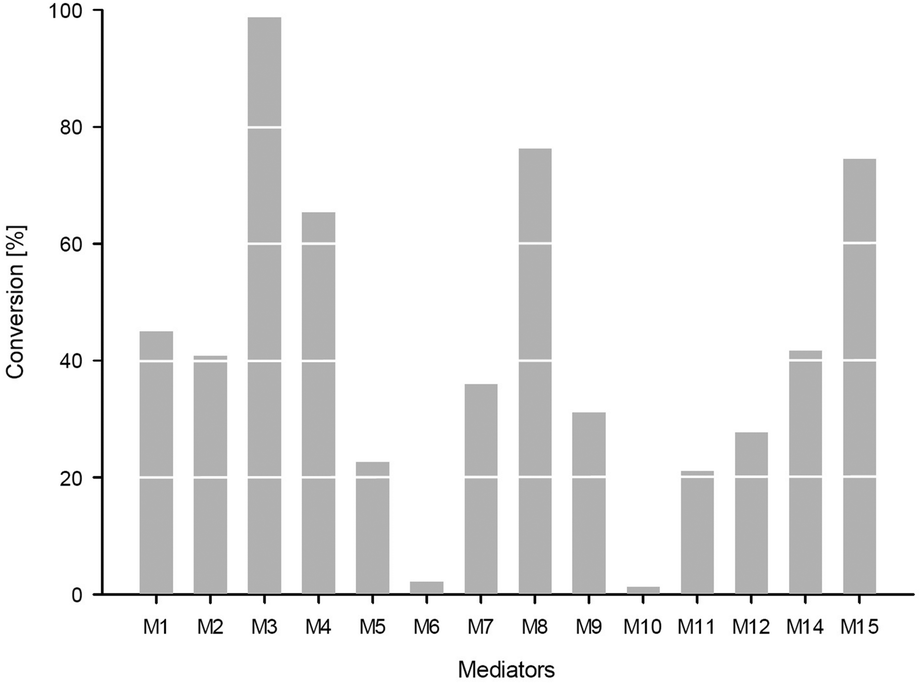 | ||
| Fig. 4 Oxidation of 1-indanol 6 (50 mM) employing different mediators (5 mol%) in phosphate buffer (pH 6, 5 mM) employing TvLac (0.5 mg ml−1); 4 h at 30 °C, 120 rpm. Conversions were measured by GC-FID. | ||
Increasing the mediator concentration of TEMPO M1 to 10 mol%, 1-indanol 6 was quantitatively oxidised within 24 hours (Fig. 5). In contrast, the halohydrin 2-chloro-1-phenyl ethanol 7 was oxidised only efficiently in the presence of elevated TEMPO concentration (45 mol%) giving 87% conversion after 24 hours.
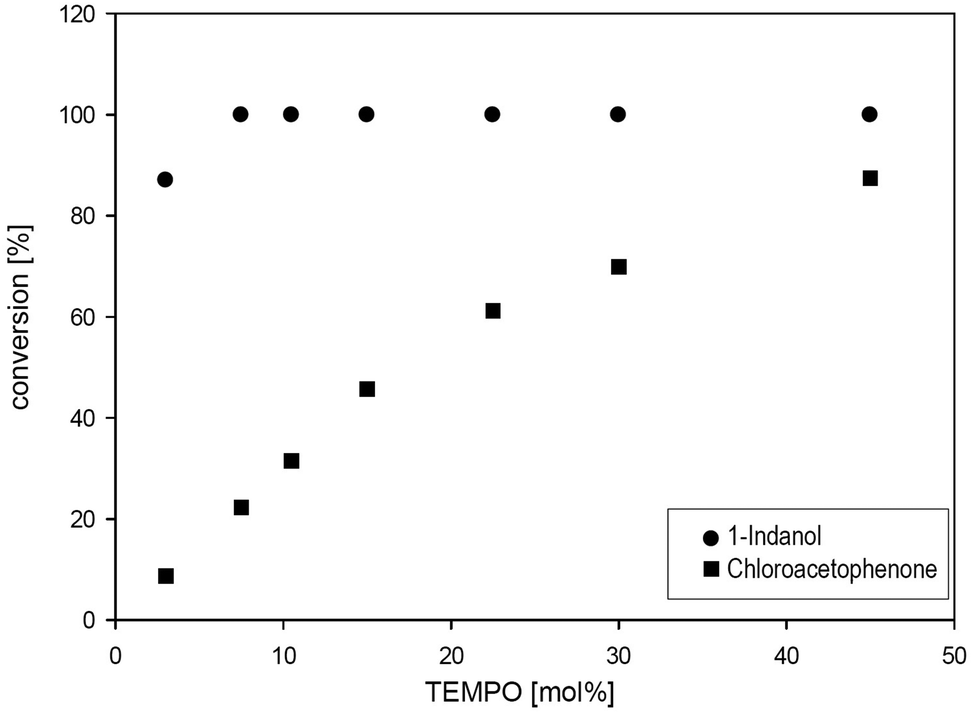 | ||
| Fig. 5 Variation of the concentration of M1 in phosphate buffer (pH 6, 5 mM) for the oxidation of 1-indanol 6 and chloroacetophenone 7 (50 mM). Conversions were measured by GC-FID. Conditions: TvLac (0.5 mg ml−1), 24 h at 30 °C, 120 rpm. | ||
While indanol (50 mM) could be oxidised using M3 as a mediator at only 2.5 mol% with full conversion to the corresponding ketone within 24 hours, substrate 7 led with M3 only to 4% conversion under the same conditions. On the other hand, 1,2,3,4-tetrahydronaphthalen-1-ol 8 was oxidised with 88% with 2.5 mol% M3. The results leading to highest conversions are summarized in Table 2.
Conclusion
Full conversions were achieved for the di-oxidation of the renewable platform compounds isosorbide 1 and isomannide 2 to the corresponding diketone 5 using a TEMPO/laccase system. Best results were obtained for instance in phosphate buffer (2.5 mM, pH 6.0) with 5 mol% of TEMPO. Alternative mediators were not as efficient as TEMPO for diols 1 and 2, but mediator M3 proved to be superior to TEMPO for the oxidation of indanol 6 and 1,2,3,4-tetrahydronaphthalen-1-ol 8. Additionally, employing a buffer with a very low salt content (2.5 mM) minimizes the amount of resources required.Experimental
Reaction conditions for isosorbide 1/isomannide 2
TEMPO/acetone-solution (15 μl, mediator concentration in the reaction: 2.5 mM) was transferred into Sarstedt tubes (15 ml) and acetone was evaporated. Then phosphate buffer (980 μl, 2.5 mM, pH 6 containing 50 mM isosorbide 1 or isomannide 2) was added and the oxidation was started by addition of the enzyme solution (20 μl, end concentration 0.5 mg ml−1). Reactions took place at 30 °C and 120 rpm on a rotatory shaker. For work-up the samples were lyophilised, extracted with acetone (1 ml) and centrifuged (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 3 min at room temperature). The organic layer was dried (Na2SO4) and analysed on GC-FID. Products were identified by comparison with authentic reference material.
000 rpm, 3 min at room temperature). The organic layer was dried (Na2SO4) and analysed on GC-FID. Products were identified by comparison with authentic reference material.
Reaction conditions for alcohols 6–8
TEMPO/acetone-solution (15 μl, mediator concentration in the reaction: 2.5 mM) was transferred into Sarstedt tubes (15 ml) and acetone was evaporated. Then phosphate buffer (980 μl, 2.5 mM, pH 6) as well as the substrate (50 mM) was added and the oxidation was started by addition of the enzyme solution (20 μl, end concentration 0.5 mg ml−1). Reactions took place at 30 °C and 120 rpm on a rotatory shaker. For work-up the samples were extracted with ethyl acetate (2 × 500 μl) and centrifuged (13000 rpm, 3 min at room temperature). The organic layer was dried (Na2SO4) and analysed on GC-MS. Products were identified by comparison with authentic reference material.
Acknowledgements
The research leading to these results has received funding from the German Federal Ministry of Education and Research (BMBF) within the project “Bioxamine” (grant no. 0316044B) and Evonik Industries AG. Some TEMPO derivatives were provided by BASF Schweiz AG.Notes and references
- (a) S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS; (b) P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- (a) Top Value Added Chemicals from Biomass Volume I-Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Principal Investigators: T. Werpy, G. Petersen, Contributing authors: A. Aden, J. Bozell, J. Holladay, J. White and A. Manheim, Pacific Northwest National Laboratory and the National Renewable Energy Laboratory, 2004, pp. 58–60 Search PubMed; (b) G. Flèche and M. Huchette, Starch, 1986, 38, 26–30 CrossRef.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- (a) J. Wu, P. Eduard, S. Thiyagarajan, L. Jasinska-Walc, A. Rozanski, C. F. Guerra, B. A. J. Noordover, J. van Haveren, D. S. van Es and C. E. Koning, Macromolecules, 2012, 45, 5069–5080 CrossRef CAS; (b) J. Wu, L. Jasinska-Walc, D. Dudenko, A. Rozanski, M. R. Hansen, D. van Es and C. E. Koning, Macromolecules, 2012, 45, 9333–9356 CrossRef CAS; (c) B. A. J. Noordover, V. G. van Staalduinen, R. Duchateau, C. E. Koning, R. A. T. M. van Benthem, M. Mak, A. Heise, A. E. Frissen and J. van Haveren, Biomacromolecules, 2006, 7, 3406–3416 CrossRef CAS PubMed.
- S. Imm, S. Bähn, M. Zhang, L. Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599–7603 CrossRef CAS PubMed.
- (a) U. Dingerdissen, J. Pfeffer, T. Tacke, T. Haas, H. Schmidt, F. Klasovsky, R. Sheldon, M. Janssen, M. Volland, M. Rimbach and S. Rinker, Evonik Degussa GMBH, 2010, WO2010089213A1 Search PubMed; (b) K. Heyns, W.-P. Trautwein and H. Paulsen, Chem. Ber., 1963, 96, 3195–3199 CrossRef CAS.
- A. Lerchner, S. Achatz, C. Rausch, T. Haas and A. Skerra, ChemCatChem, 2013, 5, 3374–3383 CrossRef CAS.
- (a) Y. Ni and J.-H. Xu, Biotechnol. Adv., 2012, 30, 1279–1288 CrossRef CAS PubMed; (b) M. M. Musa and R. S. Phillips, Catal. Sci. Technol., 2011, 1, 1311–1323 RSC; (c) F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285–2313 RSC; (d) T. Matsuda, R. Yamanaka and K. Nakamura, Tetrahedron: Asymmetry, 2009, 20, 513–557 CrossRef CAS PubMed; (e) G. W. Huisman, J. Linag and A. Krebber, Curr. Opin. Chem. Biol., 2009, 14, 1–8 Search PubMed; (f) W. Kroutil, H. Mang, K. Edegger and K. Faber, Curr. Opin. Chem. Biol., 2004, 8, 120–126 CrossRef CAS PubMed.
- (a) G. de Gonzalo, A. A. Orden and F. R. Bisogno, Curr. Org. Chem., 2012, 16, 2598–2612 CrossRef CAS; (b) C. Rodríguez, I. Lavandera and V. Gotor, Curr. Org. Chem., 2012, 16, 2525–2541 CrossRef; (c) F. Hollmann, I. W. C. E. Arends, K. Buehler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226–265 RSC; (d) K. Edegger, H. Mang, K. Faber, J. Gross and W. Kroutil, J. Mol. Catal. A: Chem., 2006, 251, 66–70 CrossRef CAS PubMed; (e) W. Kroutil, H. Mang, K. Edegger and K. Faber, Adv. Synth. Catal., 2004, 346, 125–142 CrossRef CAS.
- (a) F. R. Bisogno, E. Garcia-Urdiales, H Valdes, I. Lavandera, W. Kroutil, D. Suarez and V. Gotor, Chem.–Eur. J., 2010, 16, 11012–11019 CrossRef CAS PubMed; (b) F. G. Mutti, A. Orthaber, J. H. Schrittwieser, J. G. de Vries, R. Pietschnig and W. Kroutil, Chem. Commun., 2010, 46, 8046–8048 RSC; (c) I. Lavandera, A. Kern, V. Resch, B. Ferreira-Silva, A. Glieder, W. M. F. Fabian, S. de Wildeman and W. Kroutil, Org. Lett., 2008, 10, 2155–2158 CrossRef CAS PubMed.
- (a) A. Diaz-Rodriguez, I. Lavandera, S. Kanbak-Aksu, R. A. Sheldon, V. Gotor and V. Gotor-Fernandeza, Adv. Synth. Catal., 2012, 354, 3405–3408 CrossRef CAS; (b) S. A. Tromp, I. Matijosyte, R. A. Sheldon, I. W. C. E. Arends, G. Mul, M. T. Kreutzer, J. A. Moulijn and S. de Vries, ChemCatChem, 2010, 2, 827–833 CrossRef CAS; (c) S. Mathew and P. Adlercreutz, Bioresour. Technol., 2009, 100, 3576–3584 CrossRef CAS PubMed; (d) S. R. Couto and J. L. T. Herrera, Biotechnol. Adv., 2006, 24, 500–513 CrossRef PubMed.
- (a) V. Madhavi and S. S. Lele, BioResources, 2009, 4, 1694–1717 Search PubMed; (b) S. Riva, Trends Biotechnol., 2006, 24, 219–226 CrossRef CAS PubMed.
- L. Gianfreda, F. Xu and J. M. Bollag, Biorem. J., 1999, 3, 1–26 CrossRef CAS.
- (a) M. Turks, V. Rodins, E. Rolava, P. Ostrovskis and S. Belyakov, Carbohydr. Res., 2013, 375, 5–15 CrossRef CAS PubMed; (b) M. Shibuya, M. Tomizawa and Y. Iwabuchi, J. Org. Chem., 2008, 73, 4751–4752 CrossRef PubMed; (c) I. A. Ansari and R. Gree, Org. Lett., 2002, 4, 1507–1509 CrossRef CAS PubMed; (d) A. Dijksman, A. Marino-Gonzalez, A. M. Payeras, I. W. C. E. Arends and R. A. Sheldon, J. Am. Chem. Soc., 2001, 123, 6826–6833 CrossRef CAS PubMed.
- (a) I. W. C. E. Arends, Y. Li, R. Ausan and R. A. Sheldon, Tetrahedron, 2006, 62, 6659–6665 CrossRef CAS PubMed; (b) A. M. Barreca, M. Fabbrini, C. Galli, P. Gentili and S. Ljunggren, J. Mol. Catal. B: Enzym., 2003, 26, 105–110 CrossRef CAS PubMed; (c) M. Fabbrini, C. Galli, P. Gentili and D. Macchitella, Tetrahedron Lett., 2001, 42, 7551–7553 CrossRef CAS.
- S. Wertz and A. Studer, Green Chem., 2013, 15, 3116–3134 RSC.
- The comparison is based on the sequences with the numbers GI: 166334 and GI: 1172163. For the alignment of the sequences see ESI.†.
- B. Reinhammar, Laccase, in Copper Proteins and Copper Enzymes, ed. R. Lontie, CRC Press, Boca Raton, FL, 1984, pp. 1–35 Search PubMed.
- M. Fabbrini, C. Galli and P. Gentili, J. Mol. Catal. B: Enzym., 2002, 16, 231–240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analytics, sources of chemical and enzymes, computational details. See DOI: 10.1039/c3gc41855c |
| This journal is © The Royal Society of Chemistry 2014 |