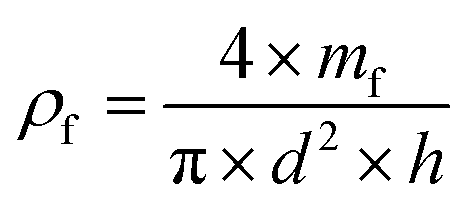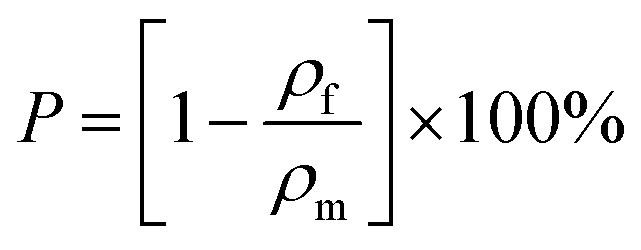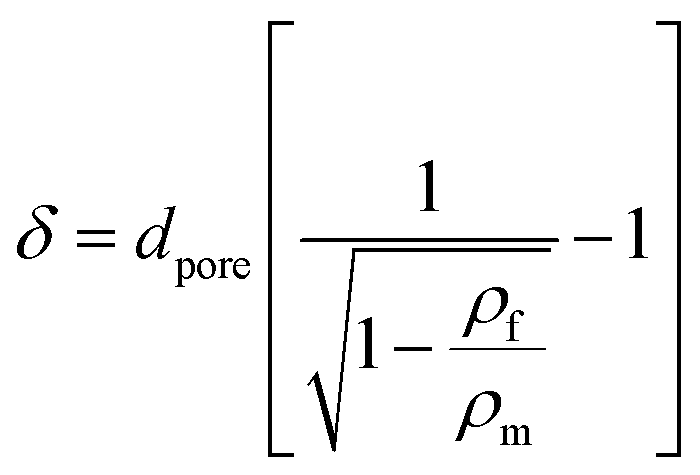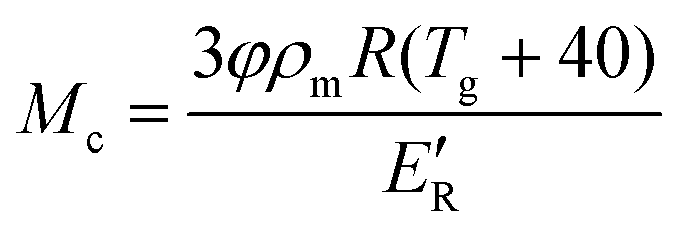DOI:
10.1039/C3GC41807C
(Paper)
Green Chem., 2014,
16, 1931-1940
Tailored for simplicity: creating high porosity, high performance bio-based macroporous polymers from foam templates†
Received
1st September 2013
, Accepted 19th November 2013
First published on 20th November 2013
Abstract
Mechanical frothing can be used to create gas–liquid monomer foams, which can then be subsequently polymerised to produce macroporous polymers. Until recently, this technique was limited to producing low porosity macroporous polymers with poor pore morphology and compression properties. In this study, we show that high porosity (75–80%) biobased macroporous polymers with excellent mechanical compression properties (E = 163 MPa, σ = 4.9 MPa) can be produced by curing of epoxy resin foams made by mechanical frothing. The key to this is to utilise the very viscous nature and very short working time of a biobased epoxy resin. It was found that increasing the frothing time of the biobased epoxy resin reduces the pore diameter of the resulting macroporous polymers. These macroporous polymers were also found to be partially interconnected. The compression properties of the macroporous polymers with smaller average pore diameter were found to be higher than those of foams with larger pore diameters. Unlike emulsion templating, which uses high internal phase emulsions to produce macroporous polymers, called polyHIPEs, the mechanical frothing technique has the advantage of creating macroporous polymers from monomers which cannot be easily emulsified.
Introduction
High porosity, low-density polymer foams or macroporous polymers are ubiquitous materials. Most polymer foams are made from petrochemicals. Nonetheless, plant oil-derived monomers have also been used as building blocks for polyurethane foams since the 1980s.1–3 Macroporous polymers are ideal for applications where weight saving is critical, such as sandwich panels,4 insulation and packaging.5 The global market value for macroporous polymers was estimated to be approximately US$ 82.6 billion in 2012 and is projected to reach approximately US$ 130 billion by 2018.6 This exponential growth of the polymer foam industry is mainly driven by the growth of the Asian market, predominantly in the automotive, packaging, building and construction industries. In addition to this, macroporous polymers have also found many applications in tissue engineering7–9 and supports for catalyst10 if they are open-cell, and thermal insulation11 if they possess a closed-cell pore structure.
Numerous methods can be used to produce macroporous polymers. These include the use of physical and chemical blowing agents,12–14 thermally induced phase separation (TIPS)15 and polymerising the continuous phase of a suitable emulsion, otherwise known as emulsion templating.16–20 Emulsion templating has become a very active research area for the fabrication of high porosity macroporous polymers – otherwise known as polymerised high internal phase emulsions (polyHIPEs) with tailored porosity and pore structure. This is most commonly achieved by first creating water-in-monomer emulsions stabilised by either surfactants21 and/or particles,22 followed by the subsequent polymerisation of the monomer phase and drying to remove the (dispersed) water phase. The pores in emulsion templated macroporous polymers are created by removal of the dispersed water droplets, i.e. the template, from the polymerised emulsions. Whilst this technique is very versatile, the method of polymerisation of the continuous phase is almost exclusively, with a few exceptions, limited to free radical polymerisations. To the best of the authors’ knowledge, there has only been one study that uses an epoxy based (bisphenol-A diglycidyl ether) monomer.23 However, 4-methyl-2-pentanon was used as the solvent, presumably to reduce the viscosity of the monomer to aid emulsification. In addition to this, surfactant or particulate emulsifiers22,24–26 are typically needed to stabilise an emulsion template – sometimes up to 20 wt% surfactant is used.20,27,28 Not only does the need for large surfactant concentrations to create stable emulsion templates increase the cost of making emulsion templated macroporous polymers but it also poses problems during the purification steps, as surfactants cannot be easily removed from a closed-cell macroporous polymers. In addition to this, surfactants can also act as plasticisers for polymers.29 Moreover, the drying step to remove the water template to produce polyHIPEs is very energy intensive. As a first approximation,§ an energy of 929 kJ kg−1 of polymer is required to remove the water from a macroporous polymer with a porosity and foam density of 80% and 200 kg m−3, respectively.
In addition to emulsion templating, non-aqueous (air) foams can also be used as template to produce highly porous structures. Monolithic structures of air templated macroporous polymers were first produced by Murakami and Bismarck.30 The authors used oligomeric tetrafluoroethylene (OTFE) particles to stabilise air bubbles in a monomer, followed by UV-polymerisation of this non-aqueous foam, resulting in a closed-cell macroporous polymer. We have also previously shown that very viscous acrylated epoxidised soybean oil (AESO), which is industrially used as a co-monomer for solvent free, radiation curing coating ink systems31–33 and natural fibre reinforced biocomposites,34,35 can be mechanically frothed to create a monomer foam. This monomer foam can be cured by microwave irradiation to produce biobased macroporous polymers.36 However, the compression properties of these macroporous polyAESO were rather poor (E = 52–166 MPa and σ = 183–343 kPa)¶ due to their irregular pore structures (Fig. 1), which is a direct result of bubble coalescence and non-uniform air bubble expansion during polymerisation, and low cross-link density of polyAESO. Moreover, these foams also possess rather low porosity of less than 60%. Nevertheless, it can be anticipated that mechanical frothing, which is intrinsically scalable, provides new means for fabrication of environmental friendly macroporous polymers. Therefore, in this work, we present a solution to solving the challenges associated with the poor pore morphology and low porosity of biobased macroporous polymers produced by polymerisation of air-in-epoxy resin foams made by mechanical frothing.
 |
| | Fig. 1 Scanning electron micrograph of acrylated epoxidised soybean oil based macroporous polymers manufactured by mechanical frothing. Obtained from Lee et al.36 with kind permission from the Royal Society. | |
Results and discussion
Mechanical frothing of a biobased epoxy resin
Herein, we report the use of mechanical frothing to create epoxy foam templates which can be cured to fabricate high porosity, high performance biobased macroporous polymers. To achieve this, we address the aforementioned challenges in mechanical frothing by using very viscous (∼2500 mPa s) plant derived epoxy resins that have a short gel time (<1 h). Consequently, these biobased epoxy resins cannot be fully degassed to remove the air bubbles trapped during the mixing of the epoxy resin with hardener because of the short working time. This renders this new generation of biobased epoxy resins impractical for the manufacturing of high performance structural greener composites. However, the high viscosity and short working time of these biobased epoxy resins are very favourable for the production of foams via mechanical frothing, which can be cured into macroporous polymers.
We have successfully prepared epoxy foam templates via mechanical frothing of a very viscous plant-derived epoxy and hardener using a hand mixer operating at maximum power output of 100 W for 10, 20 and 30 min, respectively. Here, we term macroporous polymers produced from foam templates frothed for 10 min, 20 min and 30 min as macroporous polymers 1, 2 and 3, respectively. These foam templates were also cured under different curing conditions, namely (i) at room temperature for 24 h (termed 1-A, 2-A and 3-A, respectively), (ii) at room temperature for 3 h, followed by post curing at 70 °C for 16 h (termed 1-B, 2-B and 3-B, respectively) and (iii) at room temperature for 24 h followed by post curing at 70 °C for 16 h (termed 1-C, 2-C and 3-C, respectively).
Structure and morphology of the macroporous polymers
Fig. 2 shows the scanning electron micrographs of the mechanically frothed biobased macroporous polymers. Unlike the pore structure of the macroporous polymers observed in our previous study36 (Fig. 1), which was highly irregular, spherical pores can be seen in the macroporous polymers produced in this study. The air bubbles trapped in the frothed epoxy resin were always in equilibrium (spherical shape to minimise surface tension) throughout the curing step. This is a direct result of the nature of the biobased epoxy resin, which cures at room temperature. No extra energy input (i.e., heating) is required to initiate the curing step. Therefore, the viscosity of the resin does not decrease and allows for the non-uniform expansion of the air bubbles during curing at elevated temperatures.
 |
| | Fig. 2 Pore structure and morphology of the macroporous epoxy resins prepared. 1, 2 and 3 denote macroporous polymers produced curing of mechanical frothed epoxy resin for 10, 20 and 30 min, respectively. (A), (B) and (C) represent the different curing condition of the mechanically frothed epoxy resin. A: cured at room temperature for 24 h, B: cured at room temperature for 3 h followed by post curing for 16 h at 70 °C and C: cured at room temperature for 24 h followed by post curing for 16 h at 70 °C. | |
Pore throats can also be seen in macroporous polymers 2 and 3. However, pore throats were not observed in macroporous polymers 1. The pre-requisite for pore throat formation is the rupture of the lamella layer separating two bubbles. In the case of emulsion templating, the film separating two liquid droplets is hypothesised to rupture as a result of the decrease in the solubility of the surfactants within the crosslinked polymer as polymerisation proceeds.37,38 It was also postulated that pore throats are formed due to the volume contraction when the monomer converts into a polymer.20 Our foam templating method however, does not involve the use of surfactants. Therefore, the rupture of the lamella is thought to be due to incomplete bubble coalescence during the curing of the frothed biobased epoxy resin. It should be noted that the viscosity increases as a function of time during to the curing process. This implies that the likelihood of full bubble coalescence decreases with increasing degree of curing. Therefore, pore throats are not observed in macroporous polymers 1. Instead, ‘dimples’ or ‘golf ball-like’ structures on the pore wall can be observed, suggesting incomplete bubble coalescence and lamella layer rupture. The volume contraction of the epoxy resin upon polymerisation cannot explain the formation of pore throats in our macroporous polymers as the highly compressible nature of air in the foam template allows uniform contraction of the pores. The presence of pore throats suggests that the macroporous polymers could be open porous with interconnected pores but gas permeability measurements showed that in fact all fabricated macroporous polymers were impermeable. This implies that the pores are not fully interconnected throughout the full length of the macroporous polymer monolith.
Porosity of the macroporous polymers
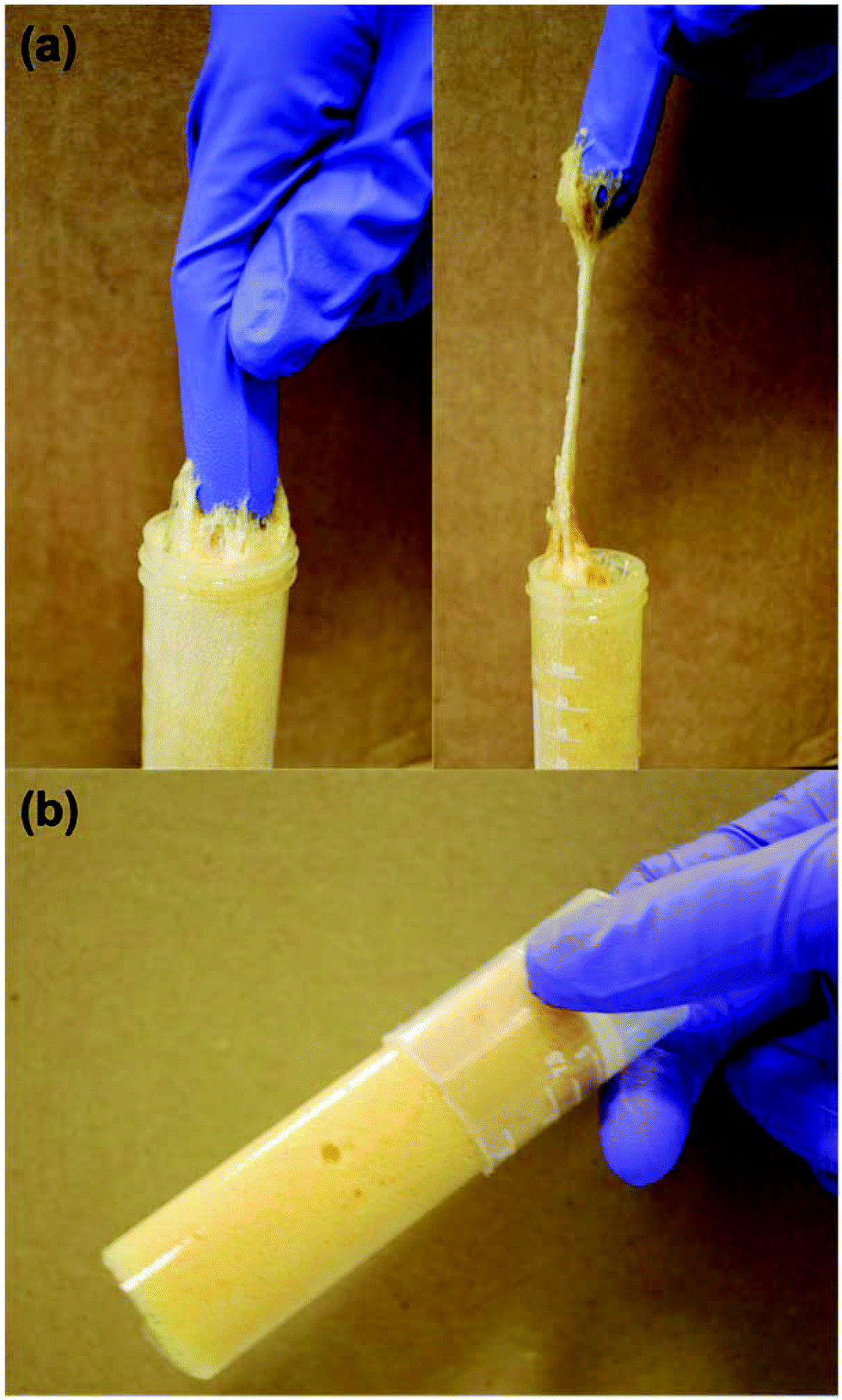
Both the measured true and foam densities of the macroporous polymers and the porosity are tabulated in Table 1. High porosity macroporous polymers with porosities ranging from 75 and 81% had been successfully produced. The highly porous nature of the macroporous polymers is a direct result of the high-energy frothing (mixing) process to introduce air bubbles into the biobased epoxy resin, estimated to be approximately 1 W g−1. It can also be seen from Fig. 2 that macroporous polymers 1 had the largest pore diameter compared to macroporous polymers 2 and 3 (see Table 1 for the average pore diameters). The larger pore diameter observed in macroporous polymers 1 is due to the phase separation of the liquid biobased epoxy foam, as liquid foams are inherently unstable.|| The phase separation of liquid foams starts with the gravitational drainage of the monomer between two adjacent bubbles into the Plateau border, resulting in a decrease of the lamella thickness.39 When two adjacent air bubbles are close enough, the capillary pressure in the lamella region will be larger than that of the Plateau border. At this point, capillary drainage becomes dominant and results in bubble coalescence.40 When the resin was only frothed for 10 min, the foam has more time for bubble coalescence to occur leading to the observed larger pore diameter prior to reaching the gel point compared to a foam frothed for 20 min and 30 min, respectively, as the gel point of the resin (approximately 1 h) is the same. This is also consistent with the observation that the macroporous polymers 2 and 3, which were frothed for 20 min and 30 min, respectively, possessed smaller average pore diameters, as the liquid resin in the foam templates, which starts curing already during frothing, after frothing are closer to the gel point. It can also be seen from Table 1 that macroporous polymers 1-B, 2-B and 3-B possess a slightly higher porosity and larger average pore diameters within the group of macroporous polymers 1, 2 and 3, respectively. It is worth recalling at this point that the macroporous polymers B differs from A and C in that the curing of these frothed biobased epoxy resin was conducted for 3 h at room temperature, followed by post curing at 70 °C for 16 h. The frothed resin is in a gel-like state after curing at room temperature for 3 h (Fig. 3). The heating of this gel-like foam during the post curing step to 70 °C resulted in the isotropic thermal expansion of air bubbles within the epoxy foam. This translates to the observed larger average pore diameter and the slight increase in the porosity of the porous polymers.
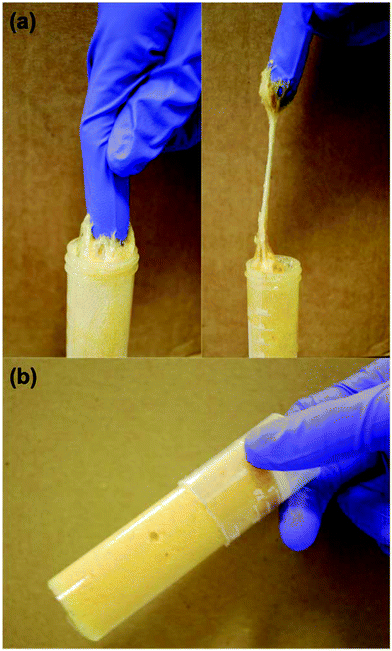 |
| | Fig. 3 Images showing (a) the gel-like state of the liquid foam 3 h after frothing and (b) macroporous polymer 24 h after mechanical frothing. The resin was frothed for 10 min. | |
Table 1 Morphological and thermal properties of the macroporous polymers. P, davg, twall and Td denote porosity, average pore diameter, pore wall thickness and onset thermal degradation temperature, respectively
| Sample |
ρ
m (g cm−3) |
ρ
f (g cm−3) |
P (%) |
d
avg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (μm) a (μm) |
t
wall (μm) |
T
d (°C) |
Residual mass (wt%) |
|
The error represents the standard error of the mean value.
|
|
1-A
|
1.151 ± 0.010 |
0.256 ± 0.004 |
78 ± 1 |
257 ± 30 |
34 ± 4 |
277 |
8.4 |
|
1-B
|
|
0.235 ± 0.010 |
80 ± 1 |
270 ± 42 |
32 ± 4 |
276 |
8.5 |
|
1-C
|
|
0.242 ± 0.011 |
79 ± 1 |
252 ± 35 |
32 ± 4 |
276 |
8.3 |
|
2-A
|
1.153 ± 0.010 |
0.283 ± 0.015 |
75 ± 1 |
238 ± 20 |
37 ± 3 |
277 |
8.1 |
|
2-B
|
|
0.224 ± 0.002 |
81 ± 1 |
273 ± 14 |
30 ± 2 |
276 |
8.0 |
|
2-C
|
|
0.266 ± 0.001 |
77 ± 1 |
222 ± 10 |
31 ± 1 |
281 |
8.4 |
|
3-A
|
1.150 ± 0.010 |
0.293 ± 0.001 |
75 ± 1 |
187 ± 7 |
29 ± 1 |
283 |
8.7 |
|
3-B
|
|
0.243 ± 0.002 |
79 ± 1 |
245 ± 9 |
31 ± 1 |
275 |
8.5 |
|
3-C
|
|
0.290 ± 0.002 |
75 ± 1 |
147 ± 7 |
23 ± 1 |
277 |
8.4 |
Thermal degradation behaviour of the macroporous polymers
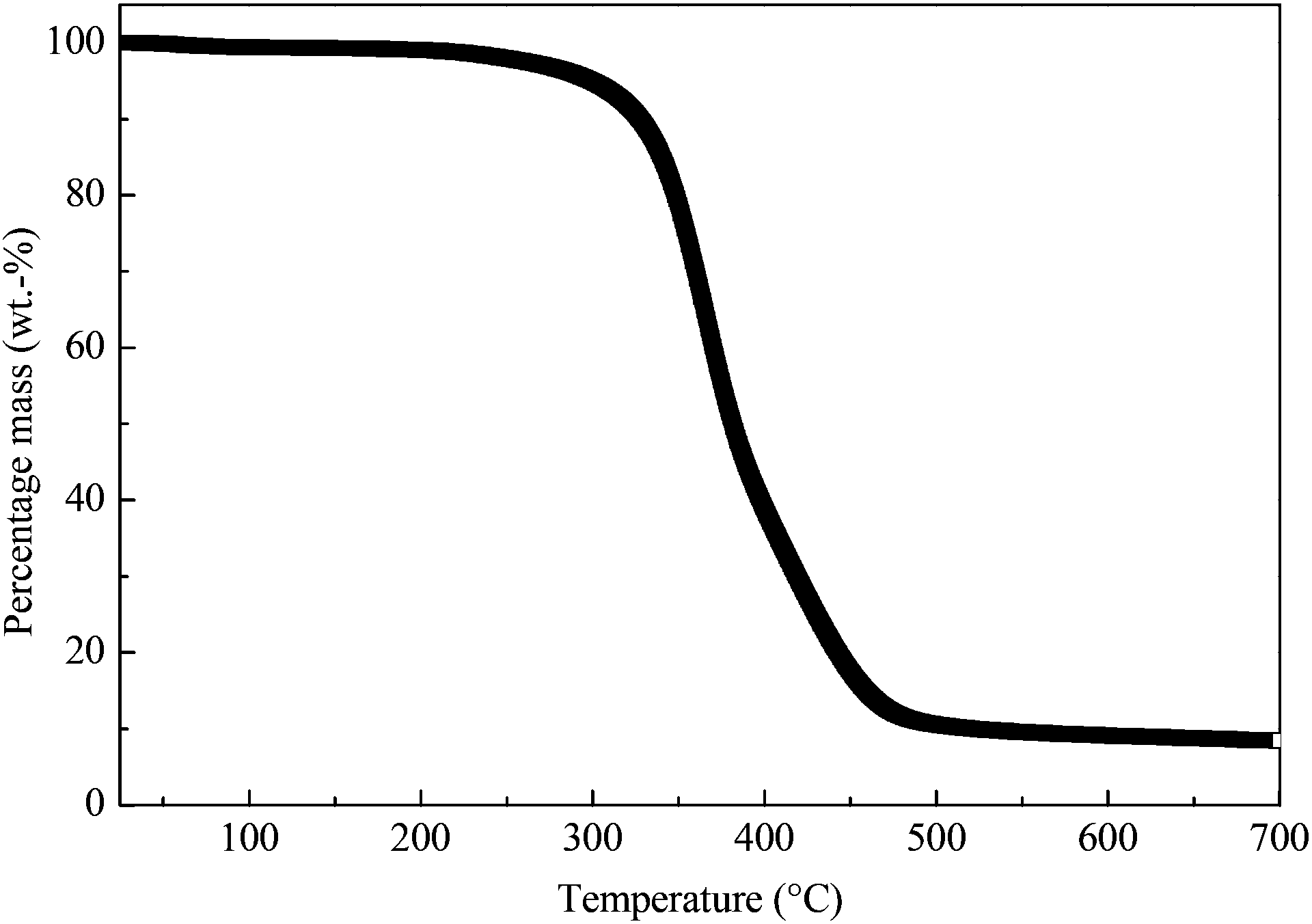
The representative thermal degradation behaviour of the macroporous polymers is shown in Fig. 4. The onset thermal degradation temperatures of these porous polymers are tabulated in Table 1. These macroporous polymers underwent a single step degradation in nitrogen atmosphere, with an onset thermal degradation temperature of approximately 280 °C. The thermal decomposition of an epoxy typically starts with the dehydration of the secondary alcohol leading to the formation of vinylene ethers.41 This is then followed by the chain scission of the allylic ethers formed. As the temperature increases, further decomposition of the epoxy resin produces light combustible gases and various hydrocarbons.42–44 The residual carbon content for all samples was found to be approximately 8.5 wt% (Table 1). This char formation is a result of the carbonisation of the epoxy resin in an inert atmosphere and is partially due to the Claisen rearrangement of allylic ethers/amides.45
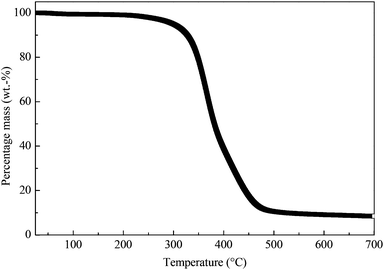 |
| | Fig. 4 Representative thermal degradation behaviour of the macroporous polymers. | |
Compression properties of the macroporous polymers
The mechanical performance of the resulting macroporous polymers determines their eventual real world applications. The compression properties of the macroporous polymers, along with their specific properties (the ratio between absolute compression properties and foam density) are tabulated in Table 2. It can be seen from this table that the macroporous polymers cured for 3 h at room temperature, followed by post-curing for 16 h at 70 °C (macroporous polymers 1-B, 2-B and 3-B) performed worse than the macroporous polymers polymerised at ambient conditions. At first glance, this could be attributed to the difference in porosities between the macroporous polymers. The effect of porosity on the compression performance of open- and closed-cell macroporous polymers is well established via the Ashby–Gibson models.46,47 However, the specific compression properties of macroporous polymers 1-B, 2-B and 3-B are still lower than that of macroporous polymers within the group of 1, 2 and 3, respectively. Therefore, the differences in compression performance of these macroporous polymers are not a result of differences in porosities as they hardly differ from each other.
Table 2 Compression properties of the macroporous polymers. E, σ, Especific and σspecific denote the compression modulus, compression strength, specific compression modulus and specific compression strength, respectively
| Sample |
E (MPa) |
σ (MPa) |
E
specific (MPa cm3 g−1) |
σ
specific (MPa cm3 g−1) |
|
1-A
|
113 ± 3 |
3.5 ± 0.1 |
435 ± 20 |
13.5 ± 0.6 |
|
1-B
|
88 ± 5 |
2.7 ± 0.3 |
383 ± 27 |
11.7 ± 1.4 |
|
1-C
|
114 ± 11 |
3.6 ± 0.4 |
475 ± 54 |
15.0 ± 1.8 |
|
2-A
|
124 ± 24 |
4.3 ± 0.3 |
443 ± 87 |
15.4 ± 1.2 |
|
2-B
|
97 ± 3 |
2.9 ± 0.1 |
441 ± 24 |
13.2 ± 0.8 |
|
2-C
|
126 ± 11 |
4.2 ± 0.1 |
467 ± 44 |
15.6 ± 0.7 |
|
3-A
|
148 ± 6 |
4.8 ± 0.1 |
510 ± 27 |
16.6 ± 0.7 |
|
3-B
|
114 ± 4 |
3.5 ± 0.1 |
475 ± 26 |
14.6 ± 0.7 |
|
3-C
|
163 ± 5 |
4.9 ± 0.1 |
562 ± 26 |
16.9 ± 0.7 |
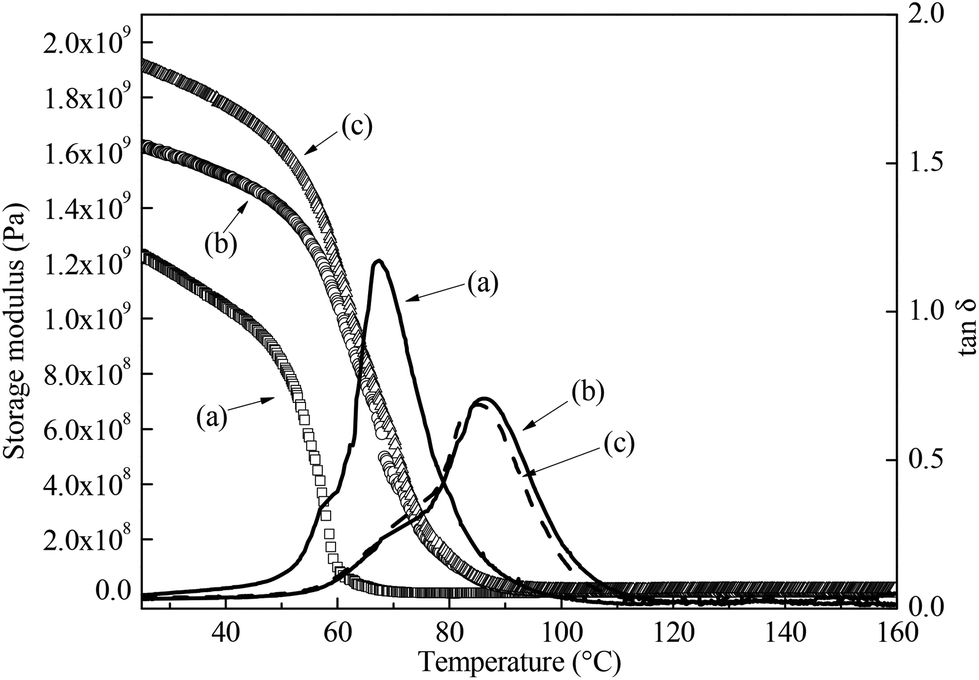
We then investigated the degree of crosslinking (q) of the resin polymerised using different conditions. q can be estimated by quantifying the average molecular weight between two cross-links Mc (eqn (5)) from the viscoelastic properties of the bulk polymers48,49 (Table 3). A higher q is characterised by a lower Mc. Fig. 5 shows the viscoelastic properties of these polymers as a function of temperature. As expected, bulk polymer A, which was cured for 24 h only, possess the lowest storage modulus, highest molecular weight between crosslinks and lowest mechanical Tg, defined as the peak of the tan δ curve, compared to bulk polymers B and C. This is attributed to the lack of a high temperature post-curing step, resulting in a higher Mc and hence lower storage modulus and Tg of the resulting macroporous polymer. Nevertheless, this result contradicts the compression properties of our macroporous polymers, which shows that macroporous polymers cured at room temperature for 24 h (macroporous polymers 1-A, 2-A and 3-A) and cured for 24 h at room temperature followed by post curing at 70 °C for 16 h (macroporous polymers 1-C, 2-C and 3-C) performed better in compression than macroporous polymers 1-B, 2-B and 3-B.
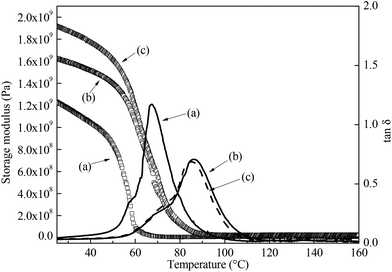 |
| | Fig. 5 Viscoelastic behaviour of the bulk epoxy resins cured using different conditions. (a) Room temperature for 24 h, (b) room temperature for 3 h, followed by 70 °C for 16 h and (c) room temperature for 24 h, followed by 70 °C for 16 h, respectively. The storage modulus is represented by the hollow icons and the tan δ is represented by the lines, respectively. | |
Table 3 Viscoelastic properties of the bulk polymers. E′, E′R, Tg denote the storage modulus at room temperature, the storage modulus of the rubbery plateau evaluated at Tg + 40 K and the mechanical glass transition temperature, respectively
| Sample |
E′ (GPa) |
E
′R (MPa) |
T
g (°C) |
M
c (g mol−1) |
|
A
|
1.22 ± 0.02 |
8.4 ± 0.8 |
67.7 ± 0.4 |
369 ± 34 |
|
B
|
1.68 ± 0.07 |
15.9 ± 3.1 |
86.0 ± 0.3 |
228 ± 45 |
|
C
|
1.83 ± 0.13 |
18.4 ± 2.4 |
85.3 ± 0.4 |
196 ± 26 |
Whilst the Ashby-Gibson model46,47 showed that the compression properties of polymer foams are independent of pore diameter, the effect of pore diameter has been shown to affect the compression properties of macroporous polymers.50–53 The compression properties of a macroporous polymer are determined by the bending properties of the materials making up the pore walls.1 Since the pore wall thickness and porosity are very similar for all samples (see Table 1), the number of pores per unit volume of the macroporous polymers must be larger for porous polymers with smaller pore diameter. This leads to the presence of more struts per unit cross-section of the porous polymers with smaller pore diameter. Therefore, the load required to compress the macroporous polymers possessing smaller pores per unit porous polymer area increases, leading to better compression properties compared to porous polymers with larger pore diameter (at constant pore wall thickness). This is consistent with our observation whereby macroporous polymers 1-B, 2-B and 3-B performed worse within the group of macroporous polymers 1, 2 and 3, respectively. The difference in compression properties between macroporous polymers 1, 2 and 3 is also consistent with the aforementioned hypothesis as the average pore diameter decreases in the order of macroporous polymers 1, followed by 2 and 3, respectively.
Discussion: comparing foam and emulsion templating techniques
Although polymer foams can be produced by a multitude of methods, here we are focusing our discussion on templating methods to produce foams, mainly emulsion and foam templating. Using liquid foams produced via mechanical frothing as templates to fabricate macroporous polymers offers several advantages over the more conventional emulsion templating technique; (i) purification and drying steps are not needed, (ii) the method is suitable for highly viscous (epoxy) monomers and (iii) no emulsifiers are needed. However, it should be noted that our foam templating technique does not yet allow for the fabrication of open-cell macroporous polymers. In addition to this, the porosity of the resulting macroporous polymers is independent of frothing time, as shown in this study. The fraction of gas entrained (ϕ) during mixing54 is expressed in the form of:| | 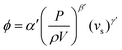 | (1) |
where α′, β′ and γ′ represent scale-independent constants. P, ρ, V and vs, denote the power input to the mixing, density of the mixing liquid, volume of the mixing vessel and superficial gas velocity, respectively. The combined term 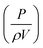 represents the average energy dissipated per unit mass in the mixing vessel. This equation shows that the amount of air bubbles entrained by the liquid foam templates during mechanical frothing (and, therefore, the porosity of the resulting macroporous polymers) can be controlled by the energy input during the frothing step. It is also worth mentioning at this point that in order to control the porosity of the resulting foam produced from a foam template the mechanically frothed foam must be stable during the curing process, i.e. creaming and bubble coalescence should not occur.
represents the average energy dissipated per unit mass in the mixing vessel. This equation shows that the amount of air bubbles entrained by the liquid foam templates during mechanical frothing (and, therefore, the porosity of the resulting macroporous polymers) can be controlled by the energy input during the frothing step. It is also worth mentioning at this point that in order to control the porosity of the resulting foam produced from a foam template the mechanically frothed foam must be stable during the curing process, i.e. creaming and bubble coalescence should not occur.
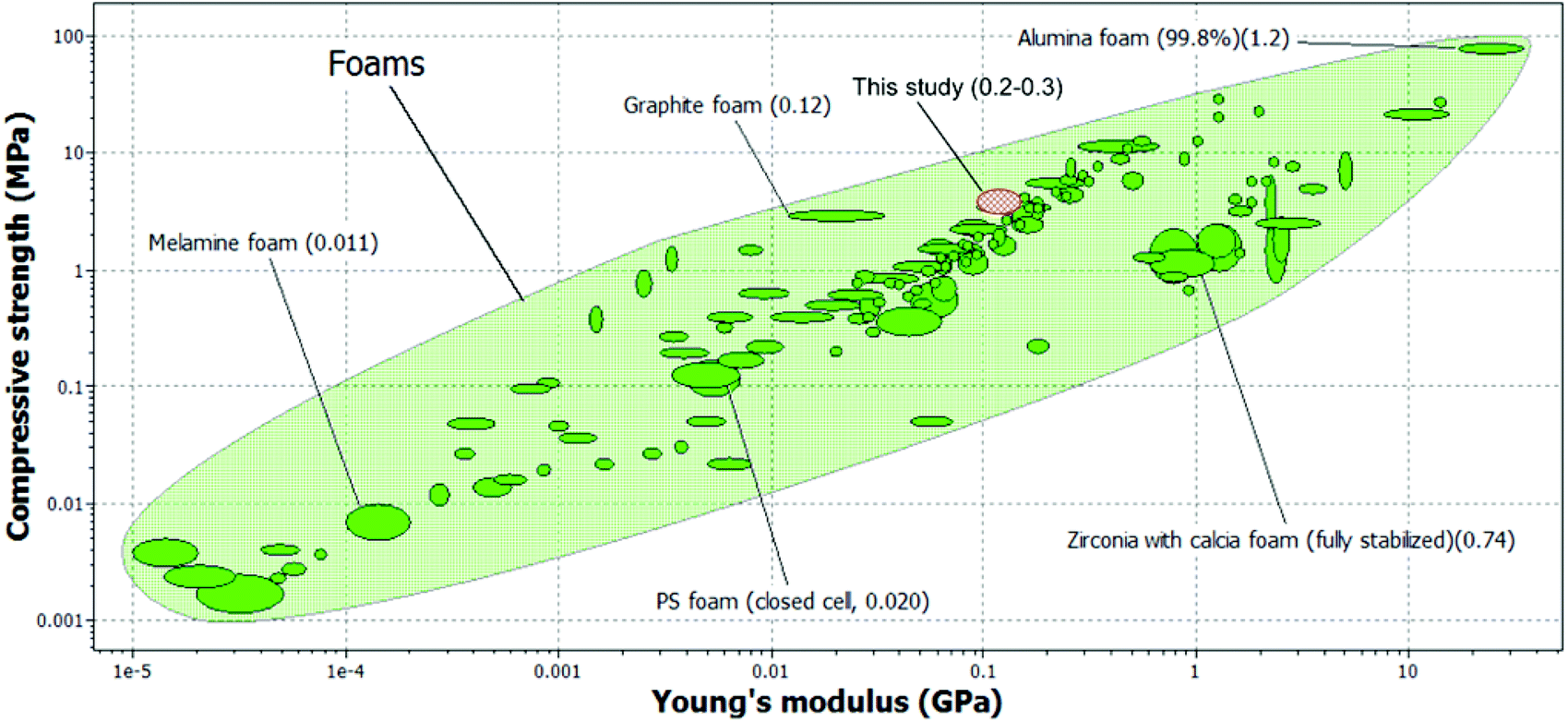
One of the biggest advantages of foam templating over emulsion templating is the possibility of using very viscous epoxy resin (or other monomers for that matter) as the monomer. This allows for the fabrication macroporous polymers with outstanding compression properties; the compression properties exceed those of typical polystyrene based macroporous polymers prepared by emulsion templating (E = 60 MPa and σ = 4.7 MPa) of similar foam density.27 Recently, it was shown that a compromise has to be made between the viscosity of the continuous minority water phase, determined by the concentration of dissolved monomer, so that a homogenous oil-in-water HIPE template can be obtained.55 The compression stiffness of our foam templated epoxy-based macroporous polymers is also comparable to the highest compression modulus (E = 130 MPa) reported in literature56 for emulsion templated macroporous poly(dicyclopentadiene)HIPEs. The compression strength, on the other hand, exceeds those of emulsion templated macroporous poly(dicyclopentadiene), which was found to be 2.5 MPa. However, it should be noted that these polyHIPEs are open porous whilst the biobased macroporous polymers manufactured in this study were closed-celled. Nonetheless, we have successfully fabricated truly high performance biobased macroporous polymers with compression properties that also exceed those of a supercritical carbon dioxide foamed thermosetting resin based on acrylated epoxidised soybean oil (E = 23 MPa and σ = 1.1 MPa) of similar foam density.57 An Ashby plot of absolute compression properties of engineering foams is shown in Fig. 6. The compression properties of engineering foams ranges from 0.1 MPa to 10 GPa and 1 kPa to 100 MPa in terms of compression stiffness and strength, respectively. The absolute compression properties of our manufactured macroporous polymers are also included in Fig. 6. It can be seen from this figure that our macroporous polymers perform much better than melamine, polystyrene and even graphite foams. Whilst it is true that our foam density is one order of magnitude higher than these foams (with graphite foams being the exception), it should be noted that the compression properties of these engineering foams are 2–3 orders of magnitude lower than the macroporous polymers manufactured in this study. Therefore, the specific compression properties of our manufactured macroporous polymers are still higher than that many of these engineering foams.
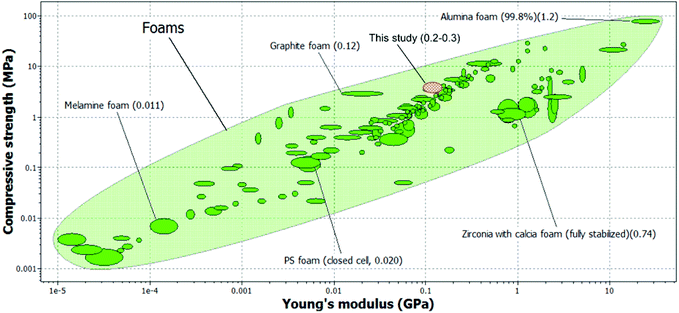 |
| | Fig. 6 A comparison between the compression properties of macroporous polymers manufactured in this study and various engineering foams. The values in the brackets indicate the foam density. Data obtained from Granta Design. | |
As with emulsion templating, foam templating also suffers from drawbacks. The major drawback of this technique is the inability to froth low viscosity monomers. The stability of liquid foams is governed by the drainage of liquid in the lamella region.40 If the viscosity of the monomer phase is low, the air bubbles will rise to the top surface of the monomer faster due to creaming. In addition to this, we have yet to produce open-cell macroporous polymers with foam templating technique. Unlike emulsion templated macroporous polymers, where the emulsifiers are postulated to aid the formation of pore throats,20,37,38 the mechanism for pore throat formations in foam templated macroporous polymers is not clear. There are some indications in this study that incomplete bubble coalescence could help creating pore throats. Creating highly interconnected foam templated macroporous polymers remains a challenge to be addressed.
Conclusions
In a previous study36 we showed that macroporous polymers could be produced by the polymerisation of mechanically frothed acrylated epoxidised soybean oil foams. However, the pore morphology and compression properties of the resulting macroporous polymers were poor. In this work, we successfully produced high porosity, high performance biobased macroporous epoxy resins by curing a biobased epoxy foam template. The key to this success was to utilise the highly viscous nature and fast gelation time of a plant-derived biobased epoxy resin. The foam templated macroporous polymers possess porosities of between 75 and 80%. The combination of different frothing times and curing conditions produced macroporous polymers with various pore structures and compression performance. It was found that increasing mechanical frothing time of the biobased epoxy resin leads to a decrease in the average pore diameter of the resulting porous polymers. This is due to the reduction of the standing time before gelation occurs, which significantly reduces the likelihood of air bubble coalescence. The pore diameter of these porous polymers is largest when the foamed biobased epoxy resin was cured for 3 h followed by a high temperature post-curing at 70 °C. This is attributed to the isotropic thermal expansion of the air bubbles induced by the heating of the gel-like foamed epoxy resin after 3 h curing. Pore throats were also observed in macroporous polymers that were produced by mechanical frothing of the biobased epoxy resin for 30 min. It is hypothesised that the presence of pore throats in these samples is due to the incomplete bubble coalescence as a result of reduction in time taken for gelation. These macroporous polymers possess compression moduli and strengths as high as ∼160 MPa and 4.9 MPa, respectively, which is the highest reported so far for biobased macroporous polymers. It was observed that the compression performance of these porous polymers increased with decreasing pore diameter. The average pore wall thickness was found to be constant irrespective of pore diameter and porosity of the porous polymers. Therefore, the increase in compression performance is due to the increase in the number pores, and hence struts, per unit volume of the macroporous polymers. This increases the load required per unit porous polymer area to compress the macroporous polymers.
Experimental
Materials
A high biomass carbon containing epoxy resin (Greenpoxy 55, biomass carbon percentage = 55 ± 2%, density = 1.15 ± 0.01 g cm−3, viscosity = 3000 mPa s @ 20 °C) and an amine-based hardener (GP505, biomass carbon percentage = 58 ± 3%, density = 0.99 ± 0.01 g cm−3, viscosity = 1700 mPa s @ 20 °C) were purchased from Matrix Composite Materials Company Ltd (Bristol, UK) and used as the resin for the preparation of macroporous polymers. Nitrogen gas (oxygen free) was used to study the gas permeability of the manufactured macroporous polymers and was purchased from BOC Industrial Gas (Morden, UK).
Biobased macroporous polymers preparation
The macroporous polymers were prepared via mechanical frothing following a protocol previously described.36 Briefly, 29.7 g of hardener was poured into a Pyrex glass bowl containing 74 g of epoxy resin. The epoxy and hardener were mixed using a hand mixer operating at a maximum power output of 100 W for 10 min (1) to introduce air bubbles into the mixture. The resulting air-resin foam was then shaped into cylindrical plastic centrifuge tubes (25 mm in diameter and 115 mm in height) using a spatula. Epoxy and hardener frothed for 20 min (2) and 30 min (3), respectively were also produced as previously described in this study. Three different curing conditions were investigated in this study; (i) cured at room temperature for 24 h (1-A, 2-A and 3-A), (ii) cured at room temperature for 3 h followed by a post curing step at 70 °C for 16 h (1-B, 2-B and 3-B) and (iii) cured at room temperature for 24 h followed by post curing at 70 °C for 16 h (1-C, 2-C and 3-C), respectively.
Preparation of biobased bulk polymers
In order to study the effect of curing condition on the mechanical performance of the previously described macroporous polymers, the resin was also cured into a dense polymer without air bubbles. 74 g of epoxy resin and 29.6 g of hardener were mixed using a spatula for 5 min. Gentle stirring was used during the mixing to minimise the entrapment of air bubbles. The mixed resin was then poured into a metal mould coated with a release agent (Frekote 770NC, Henkel, Düsseldorf, Germany**) to be polymerised into dimensions of 80 × 10 × 6 mm3. The curing conditions were the same as previously described. Prior to measurements, the rectangular bars were cut into dimensions of 80 × 10 × 3 mm3 using a diamond saw (Diadisc 5200, Mutronic, Rieden, Germany) to remove the top 3 mm section containing air bubbles.
Characterisations of the biobased macroporous and bulk polymers
Morphology of the macroporous polymers.
The morphology and internal structure of the produced macroporous polymers were investigated using variable pressure scanning electron microscopy (SEM) (JEOL 5610 LV, JEOL Ltd, Herts, UK). The accelerating voltage used was 20 kV. Prior to SEM, the macroporous polymers were cut using a band saw into approximately 10 × 10 × 10 mm3 cubes and stuck onto aluminium stubs using carbon tabs. These samples were coated with Au (K550 sputter coater, Emitech Ltd, Kent, UK) at 20 mA for 2 min prior to SEM. The average pore diameter (davg) was determined from SEM images, with a sample population of 50 pores.
Density and porosity of the macroporous polymers.
The (true) density of the polymer (ρm) was determined using He pyncnometry (Accupyc 1330, Micromeritrics Ltd, Dunstable, UK). Prior to the measurement, the macroporous polymers were crushed into powders using a mortar and a pestle. The foam density (ρf) was calculated using eqn (2):| | 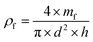 | (2) |
where mf, d and h denote the mass, the diameter and height of the macroporous polymer, respectively. With ρf and ρf known, the porosity (P) of the macroporous polymers is calculated from (3):| | 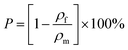 | (3) |
The mean pore wall thickness (δ) was calculated using the Aleksandrov's formula58 (eqn (4)):
| | 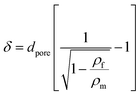 | (4) |
where
dpore is the average pore diameter.
Gas permeability of the macroporous polymers.
In order to avoid gas leakage via cross-flow, 15 mm diameter monoliths of the macroporous polymers were sealed with a non-permeable epoxy coating (Araldite 2020, Huntsman Advanced Materials, Cambridge, UK). The samples were secured in a 31 mm diameter cylindrical hollow PTFE mould and the resin was poured into the mould and left to cure for 24 h at room temperature. Once the resin has cured, the sample was removed from the mould and cut into 25 mm length. The ends were machined to reveal the faces of the sample. The N2 gas permeability of the macroporous polymers was measured using a homemade permeability apparatus using a pressure rise technique.59 Briefly, the previously coated and machined sample was sealed in the cell and a pressure differential was induced across the cell. The gas on the high-pressure side that flowed through the sample was collected in a vessel with known volume and the rate of pressure rise was determined. The viscous permeability and permeability coefficient can then be calculated as previously described.27
Thermal degradation of the macroporous polymers.
The thermal degradation behaviour of the macroporous polymers in nitrogen was characterised using thermal gravimetric analysis (TGA) (TGA Q500, TA Instruments, UK). A sample mass of 20 mg was heated from room temperature to 800 °C at a heating rate of 10 °C min−1 in a nitrogen flow rate of 60 mL min−1.
Compression properties of the macroporous polymers.
Compression tests were performed on the macroporous polymers using a Llyods EZ50 (Lloyds Instruments, Fareham, UK) in accordance to ASTM D1621-00. Cylindrical test specimens with the same height and diameter of 25 mm were compressed between two flat and parallel polished plates coated with Teflon. The load cell and crosshead speed used were 50 kN and 1 mm min−1, respectively. A total of 5 specimens were tested for each type of formulation. The compliance of the machine was found to be 3.5 × 10−5 mm N−1.
Degree of crosslinking of the bulk polymers.
The degree of crosslinking of the polymerised epoxy resin is estimated from the molecular weight between crosslinks (Mc) using eqn (5).48| | 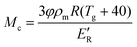 | (5) |
where φ, R, Tg and E′R are the front factor, which represents the mean square end-to-end chain distance in the polymer network over the chain distance in free space, universal gas constant, glass transition temperature (defined as the temperature at the peak of tan δ) and the storage modulus of the rubbery plateau, respectively. For a cured epoxy system, φ was found to be very close to unity.49E′R was determined using dynamic mechanical thermal analysis (DMTA) (Tritec 2000, Triton Technology Ltd, Keyworth, UK). DMTA was measured in 3 point bending mode from room temperature to 160 °C at a heating rate of 2 °C min−1 and a frequency of 1 Hz. The E′R used for the calculation of Mc is taken at Tg + 40 K.49
Acknowledgements
The authors greatly acknowledge the funding provided by the University of Vienna for LLC Wong and KYL. The authors would also like to thank Dr Ana G. Pereira-Medrano from Granta Design Ltd for generating the compression data for engineering foams shown in Fig. 6 in our manuscript.
References
-
K. Hill and R. Höfer, in Sustainable Solutions for Modern Economies, ed. R. Höfer, The Royal Society of Chemistry, Cambridge, UK, 2009 Search PubMed.
-
T. W. Abraham and R. Höfer, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, vol. 10, pp. 15–58 Search PubMed.
-
J. E. McGrath, M. A. Hickner and R. Höfer, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, vol. 10, pp. 1–3 Search PubMed.
-
L. J. Gibson and M. F. Ashby, Cellular solids: Structure and properties, Cambridge University Press, Cambridge, 1997 Search PubMed.
-
F. E. Bailey-Jr., in Handbook of Polymeric Foams and Foam Technology, ed. D. Klempner and K. C. Frisch, Oxford University Press, New York, 1991, pp. 47–72 Search PubMed.
- Polymer foam market by types (polyurethane, polystyrene, polyvinyl chloride, polyolefin, phenolic, melamine and others), applications (packaging, building & construction, furniture & bedding, automotive, wind energy and others) & geography – global trends & forecasts to 2018, TX Market Research Company and Consulting Firm, Dallas, 2013.
- R. C. Thomson, M. J. Yaszemski, J. M. Powers and A. G. Mikos, J. Biomater. Sci.,Polym. Ed., 1995, 7, 23–38 CrossRef CAS PubMed.
- J. M. Karp, P. D. Dalton and M. S. Shoichet, MRS Bull., 2003, 28, 301–306 CrossRef CAS.
- L. Safinia, K. Wilson, A. Mantalaris and A. Bismarck, Macromol. Biosci., 2007, 7, 315–327 CrossRef CAS PubMed.
- P. Ciambelli, V. Palma and E. Palo, Catal. Today, 2010, 155, 92–100 CrossRef CAS PubMed.
- J. W. Kang, J. M. Kim, M. S. Kim, Y. H. Kim, W. N. Kim, W. Jang and D. S. Shin, Macromol. Res., 2009, 17, 856–862 CrossRef CAS.
- W. T. Zhai, S. N. Leung, L. Wang, H. E. Naguib and C. B. Park, J. Appl. Polym. Sci., 2010, 116, 1994–2004 CAS.
- M. Mihai, M. A. Huneault, B. D. Favis and H. B. Li, Macromol. Biosci., 2007, 7, 907–920 CrossRef CAS PubMed.
- D. J. Kim, S. W. Kim, H. J. Kang and K. H. Seo, J. Appl. Polym. Sci., 2001, 81, 2443–2454 CrossRef CAS.
- J. J. Blaker, V. Maquet, R. Jerome, A. R. Boccaccini and S. N. Nazhat, Acta Biomater., 2005, 1, 643–652 CrossRef CAS PubMed.
- S. D. Kimmins and N. R. Cameron, Adv. Funct. Mater., 2011, 21, 211–225 CrossRef CAS.
- N. R. Cameron, Polymer, 2005, 46, 1439–1449 CrossRef CAS PubMed.
- I. Pulko and P. Krajnc, Macromol. Rapid Commun., 2012, 33, 1731–1746 CrossRef CAS PubMed.
- M. S. Silverstein, Prog. Polym. Sci., 2013 DOI:10.1016/j.progpolymsci.2013.1007.1003 , in press.
- N. R. Cameron, D. C. Sherrington, L. Albiston and D. P. Gregory, Colloid Polym. Sci., 1996, 274, 592–595 CAS.
- A. Menner, K. Haibach, R. Powell and A. Bismarck, Polymer, 2006, 47, 7628–7635 CrossRef CAS PubMed.
- A. Menner, V. Ikem, M. Salgueiro, M. S. P. Shaffer and A. Bismarck, Chem. Commun., 2007, 4274–4276, 10.1039/b708935j.
- J. Wang, C. Zhang, Z. Du, A. Xiang and H. Li, J. Colloid Interface Sci., 2008, 325, 453–458 CrossRef CAS PubMed.
- A. Menner, R. Verdejo, M. Shaffer and A. Bismarck, Langmuir, 2007, 23, 2398–2403 CrossRef CAS PubMed.
- V. O. Ikem, A. Menner and A. Bismarck, Angew. Chem., Int. Ed., 2008, 47, 8277–8279 CrossRef CAS PubMed.
- P. J. Colver and S. A. F. Bon, Chem. Mater., 2007, 19, 1537–1539 CrossRef CAS.
- S. S. Manley, N. Graeber, Z. Grof, A. Menner, G. F. Hewitt, F. Stepanek and A. Bismarck, Soft Matter, 2009, 5, 4780–4787 RSC.
- J. M. Williams and D. A. Wrobleski, Langmuir, 1988, 4, 656–662 CrossRef CAS.
- A. N. Ghebremeskel, C. Vernavarapu and M. Lodaya, Int. J. Pharm., 2007, 328, 119–129 CrossRef CAS PubMed.
- R. Murakami and A. Bismarck, Adv. Funct. Mater., 2010, 20, 732–737 CrossRef CAS.
-
G. W. Borden, O. W. Smith and D. J. Trecker, US3,979,270, 1976 Search PubMed.
-
D. Bartmann, A. Lott, U. Lübker, J. Metzger and W. Sack, DE10001476 B4, 2004 Search PubMed.
-
R. P. Balmer, M. P. Hazell and T. R. Mawby, WO2006042038, 2006 Search PubMed.
-
U. Riedel, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012 DOI:10.1016/B978-0-444-53349-4.00268-5, pp. 295–315.
- K.-Y. Lee, K. K. C. Ho, K. Schlufter and A. Bismarck, Compos. Sci. Technol., 2012, 72, 1479–1486 CrossRef CAS PubMed.
- K.-Y. Lee, L. L. C. Wong, J. J. Blaker, J. M. Hodgkinson and A. Bismarck, Green Chem., 2011, 13, 3117–3123 RSC.
- A. Menner and A. Bismarck, Macromol. Symp., 2006, 242, 19–24 CrossRef CAS.
- V. O. Ikem, A. Menner, T. S. Horozov and A. Bismarck, Adv. Mater., 2010, 22, 3588–3592 CrossRef CAS PubMed.
- D. K. Sarker, D. Bertrand, Y. Chtioui and Y. Popineau, J. Texture Stud., 1998, 29, 15–42 CrossRef.
-
P. Walstra, in Foams: Physics, chemistry and structure, ed. A. J. Wilson, Springer-Verlag, Berlin, York, 1989, pp. 1–16 Search PubMed.
- S. V. Levchik and E. D. Weil, Polym. Int., 2004, 53, 1901–1929 CrossRef CAS.
- J. C. Paterson-Jones, J. Appl. Polym. Sci., 1975, 19, 1539–1547 CrossRef CAS.
- J. C. Paterson-Jones, V. A. Percy, R. G. F. Giles and A. M. Stephen, J. Appl. Polym. Sci., 1973, 17, 1867–1876 CrossRef CAS.
- J. C. Paterson-Jones, V. A. Percy, R. G. F. Giles and A. M. Stephen, J. Appl. Polym. Sci., 1973, 17, 1877–1887 CrossRef CAS.
- S. C. Lin, B. J. Bulkin and E. M. Pearce, J. Polym. Sci., Part A: Polym. Chem., 1979, 17, 3121–3148 CAS.
- L. J. Gibson and M. F. Ashby, Proc. R. Soc. London, Ser. A, 1982, 382, 43–59 CrossRef CAS.
- L. J. Gibson, M. F. Ashby, G. S. Schajer and C. I. Robertson, Proc. R. Soc. London, Ser. A, 1982, 382, 25–42 CrossRef.
- A. V. Tobolsky, J. Polym. Sci., Part C: Polym. Symp., 1965, 9, 157–191 Search PubMed.
- T. Murayama and J. P. Bell, J. Polym. Sci., Part A2, 1970, 8, 437–445 CrossRef CAS.
- Y. P. Jeon, C. G. Kang and S. M. Lee, J. Mater. Process. Technol., 2009, 209, 435–444 CrossRef CAS PubMed.
- Z. G. Xu, J. W. Fu, T. J. Luo and Y. S. Yang, Mater. Des., 2012, 34, 40–44 CrossRef CAS PubMed.
- L. Maheo, P. Viot, D. Bernard, A. Chirazi, G. Ceglia, V. Schmitt and O. Mondain-Monval, Composites, Part B, 2013, 44, 172–183 CrossRef CAS PubMed.
- J. C. Lehuec, T. Schaeverbeke, D. Clement, J. Faber and A. Lerebeller, Biomaterials, 1995, 16, 113–118 CrossRef CAS.
-
J. C. Middleton and J. M. Smith, in Handbook of Industrial Mixing: Science and Practice, ed. E. L. Paul, V. A. Atiemo-Obeng and S. M. Kresta, John Wiley & Sons, Hoboken, New Jersey, 2004 Search PubMed.
- S. Zhou, A. Bismarck and J. H. G. Steinke, J. Mater. Chem. B, 2013, 1, 4736–4745 RSC.
- S. Kovacic, K. Jerabek, P. Krajnc and C. Slugovc, Polym. Chem., 2012, 3, 325–328 RSC.
- L. M. Bonnaillie and R. P. Wool, J. Appl. Polym. Sci., 2007, 105, 1042–1052 CrossRef CAS.
- F. A. Shutov, Adv. Polym. Sci., 1983, 51, 155–225 CrossRef CAS.
-
P. C. Carmen, Flow of gases through porous media, Academic Press, New York, 1956 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3gc41807c |
| ‡ Current address: Department of Chemical Engineering, University College London, Torrington Place, WC1E 7JE, London, UK. |
| § See ESI S1 for derivation of energy required to remove water from an emulsion templated macroporous polymer. |
| ¶ E and σ denote compression modulus and strength, respectively. |
| || Videos of the liquid foams frothed for 10, 20 and 30 min, respectively, undergoing phase separation can be found in ESI S2 (movies). |
| ** http://www.loctite.at/atd/content_data/111590_Frekote_Bro_E_0209.pdf |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 

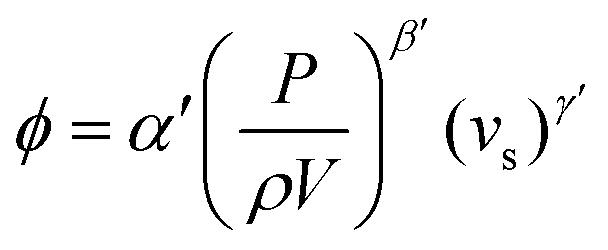
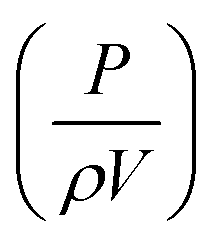 represents the average energy dissipated per unit mass in the mixing vessel. This equation shows that the amount of air bubbles entrained by the liquid foam templates during mechanical frothing (and, therefore, the porosity of the resulting macroporous polymers) can be controlled by the energy input during the frothing step. It is also worth mentioning at this point that in order to control the porosity of the resulting foam produced from a foam template the mechanically frothed foam must be stable during the curing process, i.e. creaming and bubble coalescence should not occur.
represents the average energy dissipated per unit mass in the mixing vessel. This equation shows that the amount of air bubbles entrained by the liquid foam templates during mechanical frothing (and, therefore, the porosity of the resulting macroporous polymers) can be controlled by the energy input during the frothing step. It is also worth mentioning at this point that in order to control the porosity of the resulting foam produced from a foam template the mechanically frothed foam must be stable during the curing process, i.e. creaming and bubble coalescence should not occur.