An effective, cost-efficient extraction method of biomass from wet microalgae with a functional polymeric membrane†
Gursong
Yoo‡
a,
Youngmin
Yoo‡
a,
Jong-Hee
Kwon
a,
Cornelius
Darpito
a,
Sanjiv K.
Mishra
b,
Kwanyong
Pak
a,
Min S.
Park
ab,
Sung Gap
Im
*ac and
Ji-Won
Yang
*ab
aDepartment of Chemical & Biomolecular Engineering, Korea Advanced Institute of Science & Technology, 291 Daehak-ro, Yuseong-gu, Daejeon 305-701, Republic of Korea. E-mail: sgim@kaist.ac.kr; jiwonyang@kaist.ac.kr; Fax: +82 42-350-3910; Tel: +82 42-350-3924
bAdvanced Biomass R&D Center, 291 Daehak-ro, Yuseong-gu, Daejeon 305-701, Republic of Korea
cKI for NanoCentury, 291 Daehak-ro, Yuseong-gu, Daejeon 305-701, Republic of Korea
First published on 2nd October 2013
Abstract
For energy-efficient extraction of biomass from microalgae, it is essential to extract the intracellular lipid directly from wet microalgae without drying the microalgal biomass. In this work, a novel, highly efficient cell disruption process was devised using a functional membrane coated with a cationic polymer. The proposed mechanism of cell disruption involves the perturbation of the local electrostatic equilibrium of the amphiphilic microalgal cell membrane caused by the direct contact with the tertiary-amine cations on the surface of the membrane. A tert-amine-containing polymer, poly-dimethylaminomethylstyrene (pDMAMS) film was conformally deposited on a nylon membrane by a vapor-phase polymerization process, termed as initiated chemical vapor deposition (iCVD). For the wet extraction with this membrane, the pDMAMS-coated membrane was immersed in a microalgal culture of Aurantiochytrium sp. KRS101. The microalgal culture was simply shaken together with the membrane to prompt the contact with the pDMAMS-coated membrane. With this ultimately simple procedure, the bursting of cells was clearly observed. Surprisingly, by this simple, energy-efficient process, a significantly high disruption yield of 25.6 ± 2.18% was achieved. The membrane-based extraction process is highly desirable in that (1) the process does not require an energy-consuming drying procedure, and (2) the proposed cell disruption method with a functional membrane is extremely simple and highly efficient.
Introduction
For the last decade, microalgae have received much attention as a potential feedstock for alternative renewable energy sources and high-valued natural products.1–3 The microalgae-based biodiesel production is expected to cause minimal impact on the environment, with higher productivity and greater energy return on investment.4 The produced microalgal biodiesel is also thought to be compatible directly with the existing energy infrastructure.5 Microalgae grow rapidly with high oil contents (up to 75%).4 They are capable of producing 2–10 times more biomass per unit land area than any terrestrial crop system.6Despite the numerous advantages of microalgae as a bio-product resource, the technology of biodiesel production from microalgae is still suffering from high production costs.7 At present, microalgal biofuel is not yet cost competitive with the fuels derived from petroleum. The production process of biodiesel from microalgae consists of four major steps, i.e. microalgal biomass culture, harvest, lipid extraction from the harvested microalgae, and the conversion of extracted lipids to fatty acid methyl ester (FAME).8,9 Among the downstream processes in entire microalgae-based biodiesel production, harvesting10–12 and lipid extraction13 are highly energy-consuming, which is one of the huge challenges in cost-efficient biodiesel production.14,15 Especially, the current lipid extraction method involves removing huge amounts of water from the wet microalgae culture, which requires heating and pressurizing the microalgal biomass. Hence, enormous energy consumption is inevitable for the extraction process. As a result, the lipid separation process reportedly accounts for approximately 50% of the total energy consumption for the production of microalgal biodiesel.16 Since the lipid of microalgae is enveloped by the multiple membrane layers of the cell wall,17,18 an intensive energy input is required to disrupt the microalgal cell.19,20 Hence, a cost-efficient disruption of the microalgal cell to access intracellular lipids without drying the microalgae is one of the core challenges for making microalgal biodiesel production economically feasible.16,21,22 Wet extraction by the treatment of microalgae with organic solvents had been suggested for this purpose. However, the solvent extraction methods showed low efficiency yield due to the limited contact of microalgal cells in the water-phase with non-polar organic solvents.23–32 Furthermore, a significant amount of organic solvent is required for this wet extraction process. The used organic solvent potentially causes environmental issues to demand additional process of solvent recovery, which will also result in the increase of the production cost. Mechanical stresses such as cell homogenizers, bead mills, ultrasounds, autoclave and spray drying or chemical stresses including osmotic shock and enzyme reactions have also been tested to improve the extraction yield of the targeted metabolites from microalgae.13,33 Most of the above-mentioned processes are at least either energy-intensive or less effective, or both. For the cost-efficient biodiesel production, the desired properties of cell disruption process will be (1) demanding minimal energy consumption, (2) space- and time-efficient, (3) minimizing the load of dewatering, and (4) compatible with continuous mode.
For this purpose, a new efficient method was developed to disrupt a microalgal cell membrane by use of a functional membrane coated with a tertiary-amine-containing polymer. The proposed mechanism for cell rupture activity is that the polycations in the tertiary-amine-containing polymers perturb the local electrostatic equilibrium of the cell membrane composed of the self-assembly of amphiphilic phospholipid bilayers, as described in Fig. 1. This mechanism of cell rupture by cationic coating had been well reported previously for the sterilization of bacteria and fungi.34–37 Here, we applied this anti-microbial coating to disrupt the microalgae for an efficient extraction of the intracellular lipids. The hydrophilic part of phospholipid bilayers is negatively charged, and exposed to external water environment. The contact of the negatively charged phospholipid bilayer membrane toward the positively charged polymer film on the functional membrane makes them electrostatically attract each other to cause local disorder in the self-assembled cell membranes in order to minimize the electrostatic charge interaction. The resulting local rearrangement of lipid bilayers of microalgae will induce a morphologic change in membrane layers, which will result in a rupture of the cell and consequently lead to the release of intracellular lipids in microalgae.38–41 To the best of our knowledge, positively charged polymer membranes have not yet been applied to the extraction of intracellular lipids from microalgal biomass. Particularly, the process sounds ideal in that it does not require any additional mechanical force or chemical stress to disrupt the cell. Moreover, the process is directly applicable under wet culture conditions and the coated membrane is reusable. Hence the operational cost of extraction can be significantly reduced.
 | ||
| Fig. 1 Schematic illustration of the proposed mechanism for cell disruption used in this work. Intact microalgal cells were enveloped with the multiple membrane layers of cell wall (left). The microalgae cells with negatively charged phospholipid bilayers are attracted to a functional membrane coated with positively charged tert-amine-containing polymer by electrostatic interaction. The perturbation of the local electrostatic equilibrium of the amphiphilic microalgal cell membrane is induced by the direct contact with the tertiary-amine cations (middle). The resulting local rearrangement of lipid bilayers of microalgae results in a rupture of the cell membrane and consequently leads to the release of intracellular lipid in microalgae (right). Inset image in the middle figure shows the chemical structure of tert-amine-containing polymer, pDMAMS. | ||
In this work, a tertiary-amine-containing polymer, poly-dimethylaminomethylstyrene (pDMAMS), was coated onto a commercially available nylon membrane by a vapor-phase polymerization process, termed as initiated chemical vapor deposition (iCVD).42Aurantiochytrium sp. KRS101 (hereafter Aurantiochytrium) was treated by the newly developed method. Aurantiochytrium is reported to produce a significant amount of lipids up to 80% of its total mass. More than half the amount of lipid is polyunsaturated fatty acid (PUFA) such as docosahexaenoic acid (DHA).43,44 Using fed-batch process45 and low-cost nutrients such as brewery wastewater,46 it can produce high amounts of biodiesel and DHA efficiently. Therefore, cost-efficient separation of fatty acids from Aurantiochytrium has been of great interest in terms of biodiesel production as well as high valued products. Here, we showed that the cell disruption by the pDMAMS-coated membrane is highly promising for the cost-efficient production of microalgal bio-products.
Experimental
iCVD coating of tertiary-amine-containing polymers on a nylon membrane
pDMAMS was deposited onto a nylon membrane using a custom built iCVD reactor (Daeki Hi-Tech Co., Ltd). For the polymerization, a monomer, N-4-vinylbenzyl-N,N-dimethylamine (DMAMS, Acros, 90%), and an initiator, tert-butyl peroxide (TBPO, Aldrich, 98%), were used without further purification. To achieve a designed flow rate of vaporized monomer, DMAMS was heated at 50 °C and introduced into the iCVD reactor at a flow rate of 1.2 sccm. TBPO was volatile enough and entered the iCVD reactor at a flow rate of 0.6 sccm without additional heating. The reactor pressure was maintained at 200 mTorr and the substrate temperature was held at 30 °C. By setting the filament temperature at 200 °C, iCVD polymerization was initiated. The thickness of the deposited pDMAMS was monitored by a He–Ne laser (JDS Uniphase) interferometer. All films were deposited with 1 μm thickness on both sides of the nylon membrane.The Fourier-transform infrared spectroscopy (FT-IR) was performed with an Alpha FT-IR spectrometer (BRUKER). The structure of the coated fabric was verified using scanning electron microscopy (SEM; Nova230, FEI) and optical microscopy.
Zeta potential analysis
Zeta potential of the nylon membrane with/without pDMAMS coating and Aurantiochytrium cell was measured using an ELSZ-1000ZS zeta potential analyzer (Otsuka Electronics, Japan) equipped with a flat surface cell unit. While zeta potential of Aurantiochytrium cell was normally measured without using the flat surface cell unit, nylon membrane samples were cut into a size of 10 mm × 30 mm and installed onto the flat surface cell unit for measurement. The sample was installed at a flat zeta cell unit. Monitor particle (zeta potential 0 mV) was dispersed in 10 mM NaCl (aq). After 10 mM NaCl solution was injected into the cell, zeta potential was measured by flowing the dispersed monitor particle into the flat zeta cell unit.Microalgal cultivation
Aurantiochytrium strain was gifted by Korea Research Institute of Bioscience & Bioengineering (KRIBB). The strain was inoculated to 100 ml complex glucose medium which comprised 10 g l−1 yeast extract (Duchefa Biochemie), 60 g l−1D-(+)-glucose, 9 g l−1 potassium phosphate monobasic, 15 g l−1 sea salts, and 10 mg l−1 tetracycline (Sigma-Aldrich) in a 500 ml volume culture flask. The cultures were heterotrophically cultivated in an incubator (SI-900R, Jeio Tech) with 100 RPM at 25 °C for 4 days. Cellular growth was observed by measuring the optical density at 750 nm using a UV spectrophotometer (UV-1800, Shimadzu, Japan). The cells from the culture broth were harvested by centrifugation at 4000 rpm for 10 min; the cell concentration was adjusted to reach a sufficient biomass for GC analysis by adding the discharged culture medium.Cell disruption by treatment of pDMAMS-coated membranes and direct extraction by hexane
The nylon membranes coated with pDMAMS were cut to the dimension of 5 cm by 5 cm and placed at the bottom of a 100 ml conical flask. 15 ml of Aurantiochytrium culture (14.6 g l−1) was poured into each flask. Flasks containing only microalgal culture were also prepared as controls. The flasks were placed on a tabular shaker (VS-202D, Vision Scientific) and horizontally shaken at 100 rpm for increasing contact time, for example 1, 3, 6, 9 12, 18 and 24 h. After the treatment, the whole flasks were subjected to direct extraction by hexane in order to collect the released lipid that is attached to the surface of the membrane. Hexane (LC grade, Merck) with the same volume of the applied Aurantiochytrium cell culture was added to each flask after each membrane contact. The mixture of the treated culture and hexane was gently mixed with 100 rpm at 25 °C for 1 h. Subsequently, the mixture was transferred into a 50 ml centrifuge tube and centrifuged at 4000 rpm for 10 minutes. After centrifugal separation, the organic phase of the sample tubes was carefully separated using a micropipette and collected in Teflon-sealed screw-capped Pyrex tubes. Hexane was evaporated completely under nitrogen gas stream; after the complete evaporation of hexane, a yellowish lipid at the bottom of the Pyrex tubes was quantified by GC analysis. For the analysis of total non-polar lipid content, same amount as the applied culture was completely dried and ground into flour. 2 ml of hexane was added to 10 mg of the ground Aurantiochytrium cells to extract the non-polar lipid.Gas chromatography (GC) analysis
The isolated lipid was converted to FAME for gas chromatography (GC) analysis. All the residual lipid samples from the above experiments were re-dissolved in 2 ml of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of chloroform and methanol in Teflon-sealed screw-capped Pyrex tubes. 1 ml of chloroform containing 0.5 mg of nonadecanoic acid (C19:0) purchased from Sigma was added to each tube as an internal standard, and 1 ml of methanol (Merck) and 300 μl of 98% sulfuric acid (Sigma-Aldrich) were added for the transesterification reaction under 100 °C temperature using a heating block for 20 min. After the reaction, the samples were cooled to room temperature, and 1 ml of deionized water was added to wash out the residual methanol and sulfuric acid. The samples were centrifuged for phase separation, and lower chloroform phase was separated and filtered through a 0.2 μm PVDF syringe filter (Whatman) prior to GC analysis. GC analysis for all the samples was carried out using Agilent 6890 gas chromatography equipped with a flame ionized detector and HP19091N-213 HP-INNOWax polyethylene glycol column (Agilent). Each FAME peak was identified and quantified referring a 37 component FAME standard mix (Supelco), and the total FAME content was calculated by summing up all peaks except solvent peaks.47
:1 mixture of chloroform and methanol in Teflon-sealed screw-capped Pyrex tubes. 1 ml of chloroform containing 0.5 mg of nonadecanoic acid (C19:0) purchased from Sigma was added to each tube as an internal standard, and 1 ml of methanol (Merck) and 300 μl of 98% sulfuric acid (Sigma-Aldrich) were added for the transesterification reaction under 100 °C temperature using a heating block for 20 min. After the reaction, the samples were cooled to room temperature, and 1 ml of deionized water was added to wash out the residual methanol and sulfuric acid. The samples were centrifuged for phase separation, and lower chloroform phase was separated and filtered through a 0.2 μm PVDF syringe filter (Whatman) prior to GC analysis. GC analysis for all the samples was carried out using Agilent 6890 gas chromatography equipped with a flame ionized detector and HP19091N-213 HP-INNOWax polyethylene glycol column (Agilent). Each FAME peak was identified and quantified referring a 37 component FAME standard mix (Supelco), and the total FAME content was calculated by summing up all peaks except solvent peaks.47
BODIPY staining
Invitrogen BODIPY® 505/515 (Life Technologies) was dissolved in anhydrous dimethyl sulfoxide (DMSO; Sigma-Aldrich). It was diluted in phosphate buffered saline (PBS) solution (Sigma-Aldrich), and the final concentrations of BODIPY and DMSO were 10 μM and 0.2 wt%.48 This solution was mixed with the microalgal culture in 1:9 (v/v) ratios, respectively. This mixture was incubated in the dark at 25 °C for 10 min prior to confocal microscopic observation.
Microscopic observation
Optical microscopic images of the ruptured cells were obtained using a DM2500 microscope equipped with a DFC425C camera (Leica Microsystems). Samples stained with BODIPY were observed with a Eclipse Ti confocal microscope equipped with a D-Eclipse C1 si (Nikon Instruments Inc). The wavelength of the excitation laser and the detection wavelength were set at 488 nm and at 515 nm, respectively. The bright-field image was merged with the fluorescence image in order to show lipid bodies and cell wall at the same time–space.Results and discussion
iCVD coating of pDMAMS on a nylon membrane
In this work, an effective disruption of a microalgal cell was induced by the electrostatic charge interaction of tertiary-amine groups in a pDMAMS-coated membrane with microalgal cells. The highly effective rupture of the cell membrane of bacteria and fungi by a cationic polymer film based on the electrostatic charge interaction has been reported previously.35–37 Here, a vapor-phase polymerization process iCVD was applied to deposit the positively charged pDMAMS polymer film conformally onto a nylon membrane. A radical polymerization reaction occurred in the vapor phase by flowing vaporized initiators and monomers into the vacuum reactor.42,49,50 The temperature of the substrate in the iCVD process was mostly in the range of 15–40 °C, which made the iCVD process applicable to various non-conventional substrates such as paper,51 fabric,52 and membranes53 without damaging the substrate materials. The fabrication process of a cationic polymer membrane using the iCVD process is also quite cost-competitive. Typical iCVD process pressures (50 to 1000 mTorr) do not require the use of a high-vacuum apparatus; a basic rotary pump is sufficient to operate the iCVD reactor. The solvent-free iCVD process dramatically reduces the cost for the use of a solvent and the removal of the solvent after the coating process. The iCVD process requires only a heated filament of 200–300 °C which is lower even than that of the temperature of the candle light. Moreover, the iCVD process has proven to be scalable to large batch and semicontinuous roll-to-roll reactors.The polymer film could be coated onto various complex surface morphologies of the substrates in a conformal manner, which is extremely desirable for the modification of membrane surfaces.54
Fig. 2(a) shows FT-IR spectra of the DMAMS monomer and iCVD pDMAMS film. In the FT-IR spectrum of the DMAMS monomer, the peak for the unsaturated carbon C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C was observed at 1630 cm−1, and the stretching and bending vibrational peaks of C–H bonds in the vinyl group (CH and CH2) were detected at 3085 and 900 cm−1, respectively. The peak at 860 cm−1 corresponds to the C–H bond in disubstituted benzene. The peak at 2770 cm−1 corresponding to tertiary amino–methyl C–H moieties in the amide group was also found in both spectra. Exclusively, in the spectrum of iCVD pDMAMS, the peaks corresponding to the CC double bond of the monomer were significantly reduced, as marked by asterisks in Fig. 2(a). The maintenance of the tertiary amine peak and the disappearance of this vinyl group in the spectrum of iCVD pDMAMS film clearly indicate the successful retention of tertiary amine functionalities during the polymerization of the DMAMS monomer in the iCVD process.
C was observed at 1630 cm−1, and the stretching and bending vibrational peaks of C–H bonds in the vinyl group (CH and CH2) were detected at 3085 and 900 cm−1, respectively. The peak at 860 cm−1 corresponds to the C–H bond in disubstituted benzene. The peak at 2770 cm−1 corresponding to tertiary amino–methyl C–H moieties in the amide group was also found in both spectra. Exclusively, in the spectrum of iCVD pDMAMS, the peaks corresponding to the CC double bond of the monomer were significantly reduced, as marked by asterisks in Fig. 2(a). The maintenance of the tertiary amine peak and the disappearance of this vinyl group in the spectrum of iCVD pDMAMS film clearly indicate the successful retention of tertiary amine functionalities during the polymerization of the DMAMS monomer in the iCVD process.
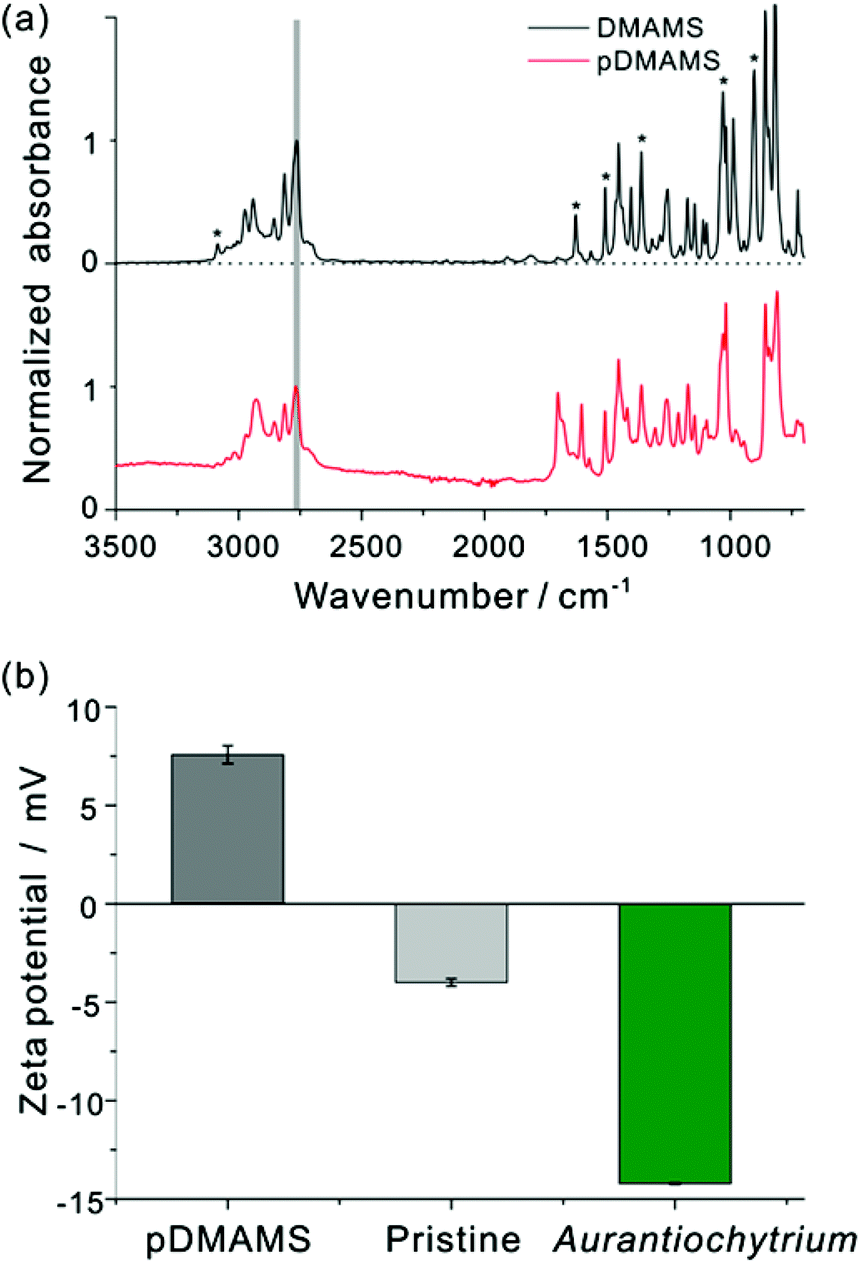 | ||
| Fig. 2 (a) FT-IR spectra of DMAMS monomer (top) and iCVD deposited pDMAMS polymer (bottom). The dark gray bar located at 2770 cm−1 represents the tertiary amino-methyl C–H moieties in DMAMS and pDMAMS. Asterisks illustrate peaks (900, 1030, 1360, 1510, 1630, 3090 cm−1) corresponding to the vinyl group in DMAMS monomer. (b) Zeta potential of Aurantiochytrium, pristine nylon membrane and pDMAMS-coated nylon membrane. The surface potentials of Aurantiochytrium and pristine bare nylon membrane are shown negatively charged: −14.22 ± 0.05 and −4.00 ± 0.19 mV, respectively. On the other hand, the pDMAMS-coated membrane was positively charged with the surface potential of 7.69 ± 2.37 mV. | ||
Since the proposed mechanism of cell rupture activity involves in the electrostatic charge interaction between the positively charged membrane and the negatively charged cell membrane of microalgae (see ESI Fig. S1†), zeta potential was measured to investigate a surface electrical interaction with the pDMAMS-coated nylon membrane and the Aurantiochytrium cell (Fig. 2(b)). Aurantiochytrium showed −14.22 ± 0.05 mV of zeta potential while the pDMAMS-coated nylon membrane has a positive value of 7.69 ± 2.37 mV; the pristine nylon membrane showed a negative zeta potential of −4.00 ± 0.19 mV. Therefore, the negatively charged Aurantiochytrium culture would electrostatically attract the nylon membrane with polycations in the pDMAMS, which was hypothesized to induce the local perturbation of the electrostatic assembly of microalgal cell membrane bilayers. The observation of zeta potential and water contact angle results clearly demonstrate that the membrane surface was modified with a positively charged tertiary-amine group of pDMAMS.
The SEM images of the pristine membrane (left) and pDMAMS-coated membrane (right) showed no significant change in morphology of the surface of the nylon membrane following the iCVD process, which illustrates the conformal coverage of functional pDMAMS polymer films onto the tortuous nylon membrane (Fig. 3). The hydrophobic nylon membrane showed the water contact angle (WCA) of 111°, which was lowered to 92° on the pDMAMS-coated membrane. The WCA observation indicates that the hydrophilic cationic functionality was added conformally to the nylon membrane.
 | ||
| Fig. 3 SEM images of the bare nylon membrane (left) and the pDMAMS-coated nylon membrane (right). No noticeable difference in the morphology of nylon membrane observed from the SEM result indicates the conformal coverage of the iCVD pDMAMS coating. Each inset image shows the water contact angle image of each nylon membrane. The hydrophobic bare nylon membrane surface with the water contact angle of 111° (inset, left) became hydrophilic with the 200 nm of pDMAMS coating with the water contact angle of 92° (inset, right). | ||
Cell disruption caused by iCVD pDMAMS coating
The prepared pDMAMS-coated membrane was applied to the extraction of intracellular lipids of Aurantiochytrium. One of the greatest advantages of this process is that the extraction process is extremely simple; no further energy such as heat or electric shock was necessary for the extraction process. A square sheet of pDMAMS-modified membrane (5 cm by 5 cm) was immersed into the Aurantiochytrium culture (cell density: 14.6 g l−1). The cell culture was gently shaken with the immersed cationic membrane up to 24 h. From this ultimately simple extraction process, amazingly, the bursting of cells and the release of intercellular organelle was clearly observed in optical and confocal microscopic images (Fig. 4), whereas the control experiment without the membrane treatment did not show any noticeable bursting of cells except that it could even be caused by autolysis or by shear stress from shaking for 24 h. No significant cell disruption was observed under treatment of the pristine membrane. Only 0.47% yield increased from 0.77% initial cell disruption yield, which was similar to results shown in the control experiment (Fig. S4 in ESI†). This result clearly shows that the nylon-meshed membrane has no meaningful effect on cell disruption. The change of cellular morphology became dominant with the increase of treatment time (see Fig. 4 and ESI Fig. S2†); the aggregation of microalgal cells was clearly observed with the increased treatment time due to the increased amount of released lipid which is highly viscous, sticky, and immiscible with the culture medium (Fig. S4 in ESI†). Thus, the released lipid “wets” the neighboring microalgae to form a cluster of cell aggregates. The release of intracellular lipids was also confirmed by an observation in confocal microscopic images showing the green fluorescence of BODIPY-binding lipids (Fig. 4(c–e)). Fig. 4(d) clearly shows that the intercellular components of microalgae were released from a single ruptured hole on the surface of the microalgal cell. The image of the disrupted Aurantiochytrium cells after the treatment with the pDMAMS-coated membrane just seemed like a deflated balloon poked by a pin, which strongly supports the hypothesis that cell disruption occurred at a local point where the polycationic group of pDMAMS contacted and penetrated into the cell. | ||
| Fig. 4 Optical microscope (top) and confocal microscope (bottom) images of Aurantiochytrium cells with the treatment of the pDMAMS-coated nylon membrane for 0 h (a, c), 9 h (d) and 24 h (b, e). The release of the intracellular lipid of Aurantiochytrium was clearly observed. Ruptured cells in the microscope images are indicated with arrows in (b). Lipid was stained to green colour using BODIPY in the confocal microscope images. | ||
Cell disruption yield by FAME analysis
Cell disruption yield could be evaluated by the ratio of the amount of lipids released from the disrupted cell to total cellular lipids.24,27,29–31,47,55 However, the released lipids were highly viscous and form agglomerates of lipid, cell debris, and intact cells, which made it extremely challenging to quantify the exact amount of released lipids from disrupted cells exclusively. In this work, the exact evaluation of the cell disruption yield was achieved by using an organic extractant, hexane. Hexane is a well-known solvent for extraction of non-polar lipids from dried biomass.13,22 The released lipid from Aurantiochytrium by this membrane extraction process was readily soluble in hexane,56–60 whereas our preliminary experiment showed that hexane could not extract intracellular lipids from the intact Aurantiochytrium culture (Fig. S5 in ESI†). In addition, hexane is highly volatile so as to be removed easily from the extracted lipid mixture for better quantification of the extraction yield. In order to extract the total released lipid from the disrupted microalgal cells only,33,61,62 the same volume of hexane was applied to the membrane-treated cell culture. The released lipid amount by the membrane treatment was quantified with respect to the FAME contents of non-polar lipids extracted by hexane. Total FAME contents from the maximum extractible neutral lipid contents were determined by the hexane extraction from a fully dried Aurantiochytrium powder biomass, where the total dry biomass was assumed to be completely ruptured by a thorough grinding procedure. To minimize the effect of cellular growth during polymer treatment on lipid contents, Aurantiochytrium in early stationary phase was sampled. The cell disruption yield according to FAME contents yield was plotted with respect to the treatment time (Fig. 5). Cell disruption yield was saturated after 18 h treatment of the pDMAMS membrane, while no noticeable release of lipid was detected in the control experiment. Surprisingly, the maximal extraction yield was as high as 25.6% and the standard deviation of the yield was ±2.18 by this novel simple extraction process. The highest degree of cell disruption rate appeared in the time period from 3 to 6 h treatment, which suggests that the meaningful disruption of microalgal cell leading to whole leak of intercellular components could not happen just by a temporary contact of the microalgae with the tertiary amine group on the pDMAMS-coated membrane. The delayed response of the cell is ascribed to the complexity and rigidity of the outer microalgal cell wall, which is hypothesized to be resistant against the penetration of the positively charged polymer. After 6 h of treatment, the cell disruption rate decreased gradually with the increased treatment time, which should be due to either the fouling of the functional membrane surface, or the aggregation of intact microalgae cells with the released lipids. These phenomena would decrease the contact between the microalgae and the polymer membrane, and accordingly, the cell-rupturing capacity of the pDMAMS membrane. To improve the space- and time-efficiency of cell disruption it is critical to develop a continuous module that can alleviate biofouling on the polymer-coated membrane and to provide reliable contact events between the pDMAMS membrane and the microalgal cells.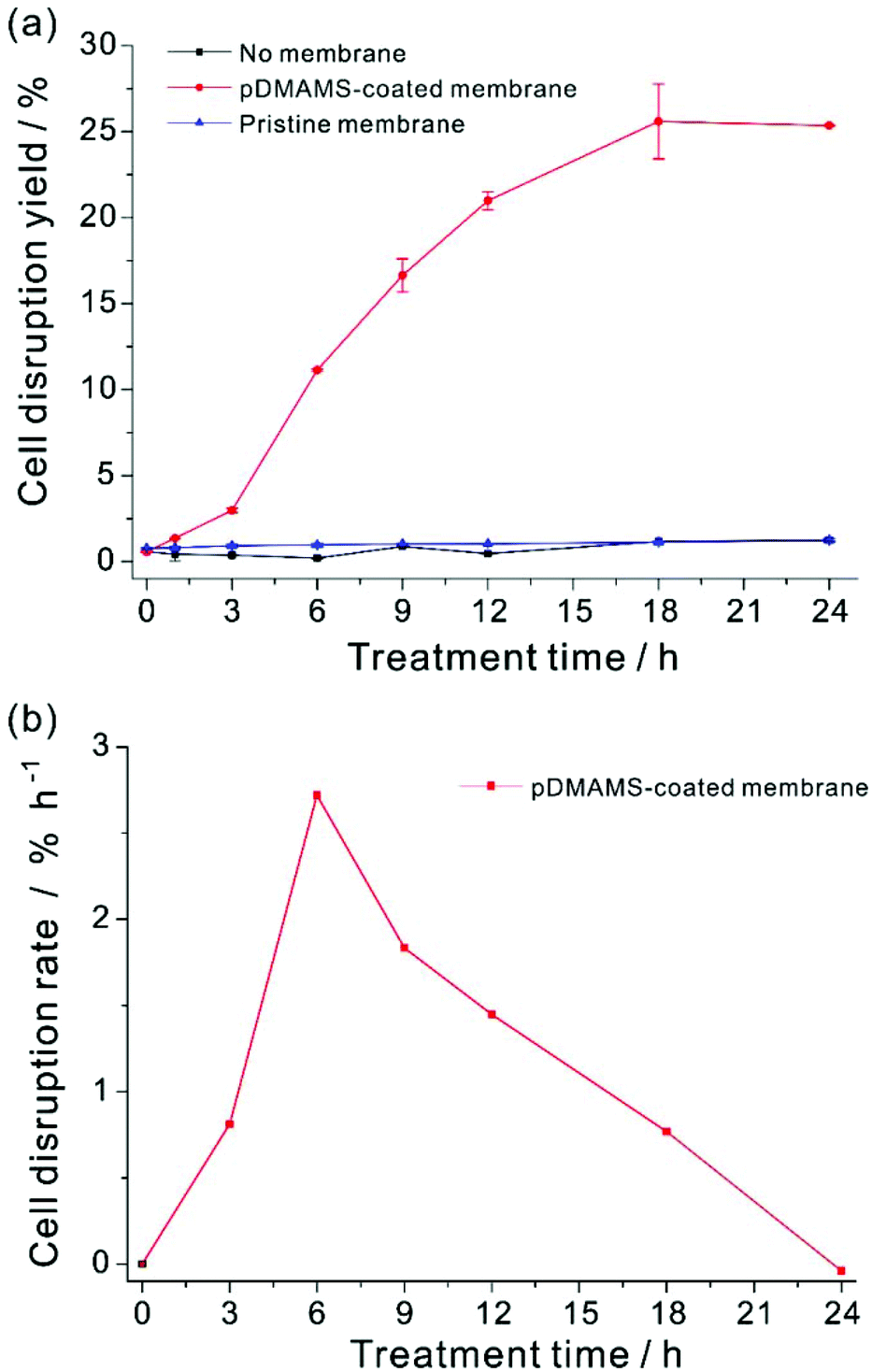 | ||
| Fig. 5 (a) Cell disruption yield and (b) cell disruption rate of the lipid extraction by the contact of Aurantiochytrium culture with pDMAMS-coated membrane. Cell disruption rate represents time-efficiency of cell disruption within each treatment period. Plotted values for cell disruption yield correspond to the average of three independent measurements. Some standard error bar was covered by symbol size. | ||
Conclusion
A simple, cost-effective, and highly efficient extraction method of microalgal intracellular biomass was introduced for the first time, based on a functional membrane with a tertiary-amine-containing polymer (pDMAMS) film, conformally deposited by the iCVD process. FT-IR analysis confirmed that the iCVD pDMAMS maintained the tertiary amine functionality after the vapor-phase polymerization procedure. The microscopic observation and the quantitative analysis on the released lipid contents from disrupted cells clearly demonstrate that the tertiary-amine group in pDMAMS played a significant role in the disruption of multiple membrane layers and extraction of intracellular lipids from the Aurantiochytrium microalgae. Our ultimately simple extraction process showed dramatically high cell disruption yield of up to 25.6 ± 2.18%. We believe that the simple and efficient membrane-based process is highly promising for cost-competitive extraction of valuable intercellular bio-products from direct fluid-culture. We strongly assure that time efficiency can be optimized further with increase of volume productivity, which will make the membrane-based wet microalgal extraction method a breakthrough for the application to commercial biomass production.Acknowledgements
This work was supported by the Advanced Biomass R&D Center (ABC) of Korea Grant funded by the Ministry of Education, Science and Technology (ABC-2010-0029728) and (ABC-2011-0031356).Notes and references
- Y. Chisti, Trends Biotechnol., 2008, 26, 126–131 CrossRef CAS PubMed.
- A. Demirbas and M. Fatih Demirbas, Energ. Convers. Manage., 2011, 52, 163–170 CrossRef PubMed.
- R. H. Wijffels and M. J. Barbosa, Science, 2010, 329, 796–799 CrossRef CAS PubMed.
- Y. Chisti, Biotechnol. Adv., 2007, 25, 294–306 CrossRef CAS PubMed.
- M. Yoshida, Y. Tanabe, N. Yonezawa and M. M. Watanabe, Biofuels, 2012, 3, 761–781 CrossRef CAS.
- R. Sayre, Using Microalgae To Produce Biomass, Mitigate Carbon Emissions, and Recycle Nutrients, Boston, USA, 2013 Search PubMed.
- M. K. Lam and K. T. Lee, Biotechnol. Adv., 2012, 30, 673–690 CrossRef CAS PubMed.
- L. Gouveia and A. C. Oliveira, J. Ind. Microbiol. Biotechnol., 2009, 36, 269–274 CrossRef CAS PubMed.
- P. D. Patil, H. Reddy, T. Muppaneni, A. Mannarswamy, T. Schuab, F. O. Holguin, P. Lammers, N. Nirmalakhandan, P. Cooke and S. Deng, Green Chem., 2012, 14, 809 RSC.
- J. Kim, B. G. Ryu, K. Kim, B. K. Kim, J. I. Han and J. W. Yang, Bioresour. Technol., 2012, 123, 164–170 CrossRef CAS PubMed.
- J. Kim, B. G. Ryu, B. K. Kim, J. I. Han and J. W. Yang, Bioresour. Technol., 2012, 111, 268–275 CrossRef CAS PubMed.
- W. Farooq, Y.-C. Lee, J.-I. Han, C. H. Darpito, M. Choi and J.-W. Yang, Green Chem., 2013, 15, 749–755 RSC.
- P. Mercer and R. E. Armenta, Eur. J. Lipid Sci. Technol., 2011, 113, 539–547 CrossRef CAS.
- J. Kim, G. Yoo, H. Lee, J. Lim, K. Kim, C. W. Kim, M. S. Park and J. W. Yang, Biotechnol. Adv., 2013, 31, 862–876 CrossRef CAS PubMed.
- K. Boer, N. R. Moheimani, M. A. Borowitzka and P. A. Bahri, J. Appl. Phycol., 2012, 24, 1681–1698 CrossRef CAS.
- L. Lardon, A. Helias, B. Sialve, J. P. Steyer and O. Bernard, Environ. Sci. Technol., 2009, 43, 6475–6481 CrossRef CAS.
- K. Roberts, M. Gurney-Smith and G. J. Hills, J. Ultra. Mol. Struct. R., 1972, 40, 599–613 CrossRef CAS.
- E. Braun and H. G. Aach, Planta, 1975, 126, 181–185 CrossRef.
- S. S. Save, A. B. Pandit and J. B. Joshi, Chem. Eng. J. Biochem. Eng. J., 1994, 55, B67–B72 CrossRef.
- V. D. Farkade, S. T. L. Harrison and A. B. Pandit, Biochem. Eng. J., 2006, 31, 25–30 CrossRef CAS PubMed.
- L. Xu, D. W. Wim Brilman, J. A. Withag, G. Brem and S. Kersten, Bioresour. Technol., 2011, 102, 5113–5122 CrossRef CAS PubMed.
- R. Halim, M. K. Danquah and P. A. Webley, Biotechnol. Adv., 2012, 30, 709–732 CrossRef CAS PubMed.
- B. D. Wahlen, R. M. Willis and L. C. Seefeldt, Bioresour. Technol., 2011, 102, 2724–2730 CrossRef CAS PubMed.
- C.-Z. Liu, S. Zheng, L. Xu, F. Wang and C. Guo, Appl. Energ., 2013, 102, 971–974 CrossRef CAS PubMed.
- Y. Shen, Z. Pei, W. Yuan and E. Mao, Int. J. Agri. Biol. Eng., 2009, 2, 51–57 CAS.
- G. Young, F. Nippgen, S. Titterbrandt and M. J. Cooney, Sep. Purif. Technol., 2010, 72, 118–121 CrossRef CAS PubMed.
- S. Balasubramanian, J. D. Allen, A. Kanitkar and D. Boldor, Bioresour. Technol., 2011, 102, 3396–3403 CrossRef CAS PubMed.
- C. Dejoye, M. A. Vian, G. Lumia, C. Bouscarle, F. Charton and F. Chemat, Int. J. Mol. Sci., 2011, 12, 9332–9341 CrossRef CAS PubMed.
- R. B. Levine, T. Pinnarat and P. E. Savage, Energy Fuels, 2010, 24, 5235–5243 CAS.
- G. Jin, F. Yang, C. Hu, H. Shen and Z. K. Zhao, Bioresour. Technol., 2012, 111, 378–382 CrossRef CAS PubMed.
- J. Y. You, C. Peng, X. Liu, X. J. Ji, J. Lu, Q. Tong, P. Wei, L. Cong, Z. Li and H. Huang, Bioresour. Technol., 2011, 102, 6088–6094 CrossRef CAS PubMed.
- J. Sheng, R. Vannela and B. E. Rittmann, Environ. Sci. Technol., 2011, 45, 3795–3802 CrossRef CAS PubMed.
- M. Cooney, G. Young and N. Nagle, Sep. Purif. Rev., 2009, 38, 291–325 CrossRef CAS.
- A. M. Bieser and J. C. Tiller, Macromol. Biosci., 2011, 11, 526–534 CrossRef CAS PubMed.
- F. Siedenbiedel and J. C. Tiller, Polymers, 2012, 4, 46–71 CrossRef CAS.
- A. M. Carmona-Ribeiro and L. D. D. Carrasco, Int. J. Mol. Sci., 2013, 14, 9906–9946 CrossRef CAS PubMed.
- T. P. Martin, S. E. Kooi, S. H. Chang, K. L. Sedransk and K. K. Gleason, Biomaterials, 2007, 28, 909–915 CrossRef CAS PubMed.
- J. C. Tiller, C. J. Liao, K. Lewis and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5981–5985 CrossRef CAS PubMed.
- M. A. Gelman, B. Weisblum, D. M. Lynn and S. H. Gellman, Org. Lett., 2004, 6, 557–560 CrossRef CAS PubMed.
- E. R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- J. Lin, S. Y. Qiu, K. Lewis and A. M. Klibanov, Biotechnol. Bioeng., 2003, 83, 168–172 CrossRef CAS PubMed.
- S. G. Im and K. K. Gleason, AIChE J., 2011, 57, 276–285 CrossRef CAS.
- W. K. Hong, C. H. Kim, D. Rairakhwada, S. Kim, B. K. Hur, A. Kondo and J. W. Seo, Bioprocess Biosyst. Eng., 2012, 35, 129–133 CrossRef CAS PubMed.
- W. K. Hong, D. Rairakhwada, P. S. Seo, S. Y. Park, B. K. Hur, C. H. Kim and J. W. Seo, Appl. Biochem. Biotechnol., 2011, 164, 1468–1480 CrossRef CAS PubMed.
- K. Kim, E. J. Kim, B. G. Ryu, S. Park, Y. E. Choi and J. W. Yang, Bioresour. Technol., 2013, 135, 269–274 CrossRef CAS PubMed.
- B. G. Ryu, K. Kim, J. Kim, J. I. Han and J. W. Yang, Bioresour. Technol., 2013, 129, 351–359 CrossRef CAS PubMed.
- G. Yoo, W. K. Park, C. W. Kim, Y. E. Choi and J. W. Yang, Bioresour. Technol., 2012, 123, 717–722 CrossRef CAS PubMed.
- M. S. Cooper, W. R. Hardin, T. W. Petersen and R. A. Cattolico, J. Biosci. Bioeng., 2010, 109, 198–201 CrossRef CAS PubMed.
- J. B. You, K. I. Min, B. Lee, D. P. Kim and S. G. Im, Lab Chip, 2013, 13, 1266–1272 RSC.
- J. M. Jeong, M. S. Oh, B. J. Kim, C. H. Choi, B. Lee, C. S. Lee and S. G. Im, Langmuir, 2013, 29, 3474–3481 CrossRef CAS PubMed.
- W. E. Tenhaeff and K. K. Gleason, Adv. Funct. Mater., 2008, 18, 979–992 CrossRef CAS.
- Y. Yoo, J. B. You, W. Choi and S. G. Im, Polym. Chem., 2013, 4, 1664–1671 RSC.
- A. Asatekin and K. K. Gleason, Nano Lett., 2011, 11, 677–686 CrossRef CAS PubMed.
- S. H. Baxamusa, S. G. Im and K. K. Gleason, Phys. Chem. Chem. Phys., 2009, 11, 5227–5240 RSC.
- F. Adam, M. Abert-Vian, G. Peltier and F. Chemat, Bioresour. Technol., 2012, 114, 457–465 CrossRef CAS PubMed.
- R. Halim, R. Harun, M. K. Danquah and P. A. Webley, Appl. Energ., 2012, 91, 116–121 CrossRef CAS PubMed.
- C. Gudin and D. Chaumont, Bioresour. Technol., 1991, 38, 145–151 CrossRef.
- J. Sheng, R. Vannela and B. E. Rittmann, Water Sci. Technol., 2012, 65, 567–573 CrossRef CAS PubMed.
- M. M. Mendes-Pinto, M. F. J. Raposo, J. Bowen, A. J. Young and R. Morais, J. Appl. Phycol., 2001, 13, 19–24 CrossRef.
- H. Zheng, J. Yin, Z. Gao, H. Huang, X. Ji and C. Dou, Appl. Biochem. Biotechnol., 2011, 164, 1215–1224 CrossRef CAS PubMed.
- A. Carrero, G. Vicente, R. Rodríguez, M. Linares and G. L. del Peso, Catal. Today, 2011, 167, 148–153 CrossRef CAS PubMed.
- E. Ryckebosch, K. Muylaert and I. Foubert, J. Am. Oil Chem. Soc., 2011, 89, 189–198 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3gc41695j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |