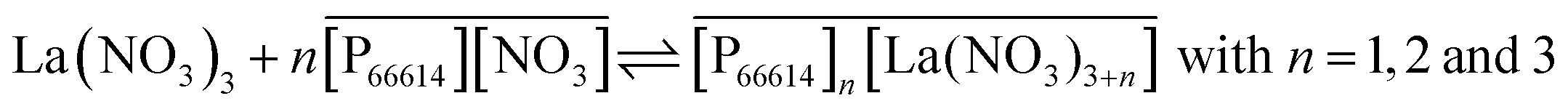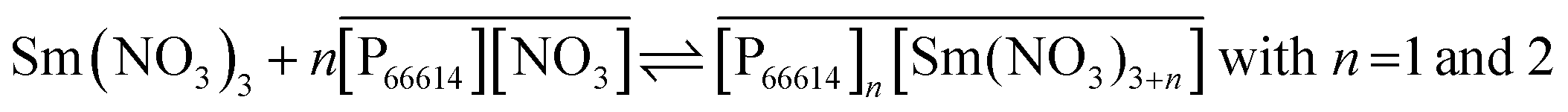DOI:
10.1039/C3GC41577E
(Paper)
Green Chem., 2014,
16, 1594-1606
Highly efficient separation of rare earths from nickel and cobalt by solvent extraction with the ionic liquid trihexyl(tetradecyl)phosphonium nitrate: a process relevant to the recycling of rare earths from permanent magnets and nickel metal hydride batteries
Received
2nd August 2013
, Accepted 6th January 2014
First published on 7th January 2014
Abstract
A solvent extraction process with the ionic liquid trihexyl(tetradecyl)phosphonium nitrate has been developed to extract rare earths and separate them from nickel or cobalt. The process is environmentally friendlier than traditional solvent extraction processes, since no volatile and flammable diluents have to be used. Compared to conventional ionic liquid metal extraction systems, the advantage of using the new ionic liquid is that expensive and persistent fluorinated ionic liquids can be avoided. The ionic liquid can be prepared by a simple metathesis reaction from the commercially available ionic liquid trihexyl(tetradecyl)phosphonium chloride (Cyphos IL 101). The extraction is facilitated by an inner salting-out effect of a highly concentrated metal nitrate aqueous phase. Feed solutions containing 164 g L−1 cobalt(II) and 84 g L−1 samarium(III), or 251 g L−1 nickel(II) and 61 g L−1 lanthanum(III) were tested. Percentage extractions of more than 99% were obtained for the rare earths and after a subsequent scrubbing step, the purity of the rare earth in the loaded ionic liquid phase was 99.9%. Complete stripping and regeneration of the ionic liquid could be performed using no chemicals other than pure water. Special attention was paid to the viscosity of the loaded ionic liquid phase and the kinetics of the extraction process, because the high viscosity and the slow mass transfer are the reasons why non-fluorinated ionic liquids have always been diluted in the past with conventional hydrophobic organic solvents such as kerosene, toluene or chloroform. The extraction mechanism of the rare earths samarium and lanthanum was studied and it was shown that different anionic complexes are formed. Lanthanum(III) is extracted at maximal loading via the hexakis anionic complex [La(NO3)6]3−, whereas samarium(III) is extracted at maximal loading via the pentakis anionic complex [Sm(NO3)5]2−. The difference in electrical charge of the anions has a pronounced effect on the viscosity of the ionic liquid phases. The separation of lanthanum and samarium from nickel or cobalt, out of highly concentrated metal salt solutions by solvent extraction, is of importance for the recycling samarium–cobalt permanent magnets or nickel metal hydride (NiMH) batteries.
Introduction
Liquid–liquid extraction or solvent extraction is the most often used technique for the separation and purification of metals.1 A mixture of metals, dissolved in the aqueous phase, is brought into contact with an organic phase, containing a specific extractant and a diluent such as toluene, dodecane, kerosene, chloroform, etc. The separation is based on differences in chemical interaction between the metal ion and the extractant and differences in the solubility of the respective complex in both the organic and aqueous phases. The efficiency of the separation process can often be tuned by changing extraction parameters such as the pH, temperature, the concentration of the metal ions in the aqueous feed and the concentration of the extractant in the organic phase.
Ionic liquids (ILs) are a relatively new class of solvents. These solvents consist entirely of ions and they are typically organic salts with a melting point below 100 °C.2–4 The application of ionic liquids for the extraction of metal ions was first reported in 1998.5,6 Ionic liquids have advantages compared to traditional organic solvents, which turn them into interesting solvents for extraction studies. They have a negligible vapor pressure at room temperature, so that they are non-volatile.7 They are also less flammable, have a higher thermal stability and a broader liquidus range than conventional organic solvents.8,9 Due to the growing concern about the environmental impact and safety issues related to volatile organic solvents, there is a strong drive for replacing these organic solvents by less noxious alternatives. Ionic liquids are often called designer solvents, due to a large variety of cations and anions, among which a selection can be made to get ionic liquids with suitable properties for a given application.10 In the case of solvent extraction, the ionic liquids can be designed to be immiscible with water (hydrophobic ionic liquids) and at the same time to be able to solubilise the extracted metal complexes. Different examples of hydrophobic ionic liquids have been tested for solvent extraction studies, but often fluorinated anions such as hexafluorophosphate (PF6−) or bis(trifluoromethylsulfonyl)imide (Tf2N−) anions were selected, because these anions give not only hydrophobic, but also low viscous ionic liquids.11–14 Unfortunately, these ionic liquids have several disadvantages as well: the price of fluorinated compounds is often high, they are persistent and in some cases (e.g. the hexafluorophosphate ionic liquids), they also show hydrolysis with formation of dangerous hydrofluoric acid.15 When extracting positively charged metal ions, extraction often occurs via an ion exchange mechanism in which the hydrophilic cation of the ionic liquid diffuses to the water phase and forms a water soluble ionic liquid in combination with the anion of the metal. In this way, part of the ionic liquid is inevitably lost in the water phase.16–18 Another approach is to make the cation more hydrophobic by introducing long alkyl chains.19–23 In this way, simple, stable and cheap anions can be chosen and loss of the cation to the water phase is avoided by the hydrophobicity of the cation. Several ionic liquids of this type have already been reported, although they are often diluted in molecular solvents to reduce the viscosity of the ionic liquid.24–42
Recently, we have reported the separation of some main transition metals (Co, Fe, Zn, Cu and Mn) from rare earths with the non-fluorinated and undiluted ionic liquid trihexyl(tetradecyl)phosphonium chloride (Cyphos® IL 101).20 Although the elements could be separated very efficiently, this separation process has also some disadvantages. Low metal loadings or concentrations require the use of huge solvent volumes and large solvent extraction equipment. From an economical point of view, this is not beneficial for industrial processes. Due to saturation effects, the percentage extraction of cobalt by trihexyl(tetradecyl)phosphonium chloride and the purity of the aqueous phase decreased significantly when going to higher metal loadings in the ionic liquid phase, so the concentration of the metal in the aqueous feed had to be kept low.20,21 Another disadvantage of this ionic liquid extraction system is the difficult stripping of iron from the loaded ionic liquid phase.20 The extraction system also did not allow the separate rare earths from nickel.
In this paper, we show how an efficient solvent extraction system for rare earths can be designed by replacing the chloride ionic liquid by the corresponding nitrate one, i.e. replacing trihexyl(tetradecyl)phosphonium chloride by trihexyl(tetradecyl)phosphonium nitrate. An advantage of trivalent rare-earth ions is that they can form anionic complexes with bidentate nitrate ligands, whereas most other elements cannot. Therefore, the rare earths can be extracted selectively from a concentrated aqueous nitrate solution, leaving behind the transition metal ions. The trihexyl(tetradecyl)phosphonium nitrate ionic liquid system is used for samarium–cobalt and lanthanum–nickel separations. These separations are highly relevant for the recycling of metal values from nickel metal hydride (NiMH) batteries or samarium–cobalt (SmCo) permanent magnets.43–46
Experimental
Chemicals
Trihexyl(tetradecyl)phosphonium chloride (>97%, Cyphos® IL 101) was purchased from IoLiTec (Heilbronn, Germany). Sm(NO3)3·6H2O (99.9%), Co(NO3)2·6H2O (99%) and Ni(NO3)2·6H2O (99%) were purchased from Acros Organics (Geel, Belgium). Nd(NO3)2·6H2O (99.9%) was obtained from Alfa Aeser (Karlsruhe Germany), KNO3 (99%+), NH4NO3 (99%+) and La(NO3)3·6H2O from Chempur (Karlsruhe, Germany) and Fe(NO3)3·6H2O (99%) from J.T. Baker. The silicone solution in isopropanol was obtained from SERVA Electrophoresis GmbH (Heidelberg, Germany) and the gallium (1000 g L−1) standard from Merck (Overijse, Belgium). All chemicals were used as received, without further purification.
Synthesis of trihexyl(tetradecyl)phosphonium nitrate, [P66614][NO3]
Trihexyl(tetradecyl)phosphonium chloride (91.03 g, 0.176 mol) was mixed three times with 100 mL of a 2 M KNO3 solution and stirred for 1 hour. Afterwards it was left for several hours until the organic layer was well separated from the aqueous one. After the metathesis reaction, the ionic liquid was washed further with 100 mL of water to remove chloride and KNO3 impurities. The presence of potassium and chloride impurities after reaction was checked by TXRF. The ionic liquid was obtained as a colorless liquid.
Yield: 111.21 gram, 0.160 mol, 99%. 1H NMR (300 MHz, CDCl3, δ/ppm): 2.27 (m, 8H, 4× CH2), 1.49 (m, 18H, 9× CH2), 1.30 (s, 12H, 6× CH3), 1.26 (m, 18H, 9× CH2), 0.89 (s, 12H, 6× CH3). 13C NMR (75.47 MHz, CDCl3, δ/ppm): 31.9, 31.0, 30.9, 30.7, 30.6, 30.4, 29.6, 29.5, 29.4, 28.9, 22.7, 22.4, 21.8, 21.7, 19.1, 18.5. 31P NMR (161.96 MHz, CDCl3, δ/ppm): 49.2 (impurity starting product, phosphine oxide), 33.6. FTIR (ATR, cm−1): 2955, 2923, 2853, 1464, 1412, 1334, 1213, 1110, 987, 829, 720. Elemental analysis calculated for C32H68NO3P·½H2O (M = 554.87 g mol−1) (%): C 69.27, H 12.53, N 2.52; found (%): C 68.93, H 13.72, N: 2.68. Melting point: 6 °C. Chloride content: 400 mg L−1. Dry ionic liquid: water content: 0.04 wt%, density: 0.914 g cm−3 (22 °C), viscosity: 1440 cP (22 °C). Water saturated ionic liquid: water content: 3.92 wt%, density: 0.914 g cm−3 (22 °C), viscosity: 265 cP (22 °C).
Instrumentation and analysis methods
Extraction experiments were performed with a Nemus Life Thermo Shaker TMS-200. After each extraction, the mixtures were centrifuged with a Heraeus Megafuge 1.0 centrifuge for 5 minutes at 5300 rpm. Metal concentrations were determined with a benchtop total reflection X-ray fluorescence (TXRF) spectrometer (Picofox S2, Bruker). After each experiment, the aqueous and ionic liquid phases were measured for their metal content. A gallium internal standard was added to a small fraction of the aqueous phase in order to obtain a volume of 1 mL. The quartz glass sample carriers were first treated with 20 μL of silicone solution in isopropanol, dried for 5 minutes in a hot air oven at 60 °C, followed by the addition of 5 μL of the sample and again a drying process of 20 minutes at the same temperature. The metal concentrations in the aqueous phase were measured for 200 seconds. For the ionic liquid phase, the gallium internal standard was added to a small amount of the ionic liquid phase (10–20 mg) and was further diluted with ethanol until 1 mL. Pretreatment of the sample carrier, sampling volume, drying procedure and measuring time have been performed in the same way for the ionic liquid phase as described for the aqueous phase. After separation, the metal content of one of the metals is sometimes very low in the presence of a large excess of the other metal. In these cases, the concentration was measured for 2000 seconds to obtain a higher signal to noise ratio. All the samples were diluted with MilliQ water, if necessary. The viscosity of the ionic liquid phase was measured using an automatic Brookfield plate cone viscometer, Model LVDV-II+P CP (Brookfield Engineering Laboratories, USA). Densities were measured with a pycnometer (volume: 5 mL). The melting point of the ionic liquid was determined with a Mettler-Toledo DSC 882 apparatus. A Mettler-Toledo DL39 coulometric Karl Fischer titrator was used with Hydranal® AG reagent to determine the water content of the ionic liquid. The water contents of the ionic liquid phase loaded with metals could not be measured due to interference of redox active metal ions with the Karl Fischer reagent. CHN-analysis was performed on a CE Instruments EA-1110 element analyser; FTIR spectra were recorded on a Bruker Vertex 70 spectrometer and analysed with OPUS software. 1H and 13C NMR have been recorded on a Bruker Avance 300 MHz NMR spectrometer and 31P NMR with H3PO4 as an external reference with a Bruker Avance 600 MHz spectrometer and analysed with the SPINWORKS software package. The NMR spectra of trihexyl(tetradecyl)phosphonium nitrate were recorded in deuterochloroform.
Extraction experiments
All experiments were performed by shaking the undiluted trihexyl(tetradecyl)phosphonium nitrate ionic liquid with a water phase for 1 hour at 60 °C at 1800 rpm in vials of 4 mL and in a horizontal position. Due to the high viscosity and slower kinetics, scrubbing was done at higher temperature (80 °C) for 2 hours at 1900 rpm. Unless otherwise reported, 1 mL of the ionic liquid phase (0.914 gram) was mixed with 1 mL of the aqueous phase. Volume changes by the uptake or loss of large amounts of metals were ignored to simplify the calculations. Ammonium nitrate or sodium nitrate was used as a nitrate source. Data points were measured only once in all the experiments showing trends. The main errors are originating from sample preparation and the measurements with TXRF. A sixfold sample preparation procedure and measurement for all the used metals had relative standard deviations smaller than 4%.
Separation studies.
The stock solution of a samarium–cobalt mixture mimics the molar ratio of the main metals found in a SmCo5 magnet. In the first instance, the extraction percentage of samarium was determined by stepwise increasing the feed concentration from 6.6 g L−1 cobalt (20.4 g L−1 Co(NO3)2 and 3.3 g L−1 samarium (7.5 g L−1 Sm(NO3)3) to 202.5 g L−1 cobalt (629 g L−1 Co(NO3)2) and 104.8 g L−1 of samarium (234 g L−1 Sm(NO3)3). The initial nitrate concentration of the aqueous phase therefore increased from 0.3 to 9.1 M. Whereas most data points were obtained by shaking for 1 hour at 60 °C, the data point with the highest loading was obtained by shaking for 15 hours at 80 °C to avoid that equilibrium is not reached due to the very high viscosity of the ionic liquid phase under these conditions. The other extraction experiments were most often performed with a concentration of around 84.0 g L−1 samarium and 164.0 g L−1 cobalt to avoid problems with high viscosity, too long heating and shaking times. Secondly, we tried to improve the separation by salting out with NH4NO3 or by increasing the HNO3 concentration during the extraction. Therefore, 30 grams of NH4NO3 was dissolved in 50 mL of water to obtain a solution of 7.5 M of NH4NO3. Due to the hygroscopic behaviour of NH4NO3, the real nitrate concentration was probably slightly lower. A constant volume of the stock solution was mixed with different amounts of the NH4NO3 solution and further diluted with MilliQ water to obtain 1 mL. The metal concentrations were 80.2 g L−1 cobalt and 42.0 g L−1 samarium and were kept lower in order to obtain similar nitrate concentrations as in the initial extraction study without the addition of an extra nitrate source. In the case of the addition of nitric acid, the feed solution was made by mixing the stock solution with different amounts of nitric acid and further dilution with MilliQ water to 1 mL. The metal concentration here was 161.7 g L−1 cobalt and 84.5 g L−1 samarium. In all cases, the pH of the aqueous phase was lying between 3 and 4, except for the experiments performed with addition of HNO3. The influence of ionic liquid metal saturation effects and the purity of samarium in the ionic liquid phase were studied by mixing 1 mL of an aqueous phase containing 202.5 g L−1 cobalt and 104.8 g L−1 samarium with volumes of the ionic liquid phase ranging from 0.66 to 1.52 mL.
Scrubbing, stripping and further processing of the scrub and feed.
First, extractions were performed from a feed solution containing 162.3 g L−1 cobalt and 83.9 g L−1 samarium. Afterwards, 1 mL (1.057 gram) of the ionic liquid phase was scrubbed with different volumes of a 7.5 M NH4NO3 solution. The scrubbing was performed at 80 °C for 2 hours at 1900 rpm due to the slower kinetics of the highly viscous loaded ionic liquid. Stripping experiments were performed by first extracting 162.4 g L−1 cobalt and 85.0 g L−1 samarium from an aqueous solution and a sub-sequential scrubbing step with 7.5 M NH4NO3. Afterwards, the ionic liquid phase was stripped with different concentrations of HNO3 or three times with water. Further processing of the scrub and feed solution was performed to avoid losses of samarium in the cobalt streams and to purify the aqueous cobalt phase. First, 162.4 g L−1 cobalt and 85.0 g L−1 samarium were extracted from the aqueous phase, followed by a scrubbing step with 7.5 M NH4NO3. The feed solution containing now mainly cobalt was brought in contact with a fresh ionic liquid phase to remove the remaining samarium from the aqueous phase and to transfer it to the ionic liquid phase. The ionic liquid phase as well was then brought into contact with the used 7.5 M NH4NO3 scrub solution to remove the remaining samarium from the scrub phase.
Lanthanum–nickel and samarium–cobalt separations.
The samarium–cobalt separation was performed again under optimal conditions but the nitrate source was changed from NH4NO3 to the less hygroscopic NaNO3 in order to obtain correct nitrate values and to make it possible to separate cobalt from the nitrate source by precipitating it with NaOH and obtaining again NaNO3, which can be brought again into the system. Starting concentrations were 162.0 g L−1 cobalt and 83.8 g L−1 samarium. A similar process has been tested as well on the separation of lanthanum from nickel which could be interesting for the recycling of NiMH batteries. In this case 250.8 g L−1 nickel and 65.5 g L−1 lanthanum were mixed; other elements were not included. The lanthanum concentration was chosen in such a way that it mimics the total rare-earth concentration in a recycling scheme described by Larsson et al.46 The nickel concentration has been chosen in such a way that it represents approximately all the other transition metals and their nitrate anions. Extraction, scrubbing and stripping were performed in the same way as in the case of a samarium cobalt separation. Due to the high purity of both the aqueous nickel stream as well as the ionic liquid phase after scrubbing, further processing of the phases was not performed.
Kinetics, viscosity and extraction mechanism.
The reaction kinetics were studied by shaking 1 mL of 83.9 g L−1 samarium and 162.2 g L−1 cobalt at 60 °C with 1 mL of the ionic liquid phase at different times. The viscosity of the ionic liquid phase as a function of the temperature was studied by performing first an extraction on a 1 mL sample of the aqueous phase containing 83.8 g L−1 samarium and 162.0 g L−1 cobalt and obtaining an ionic liquid phase containing 83.2 g L−1 samarium and 3.2 g L−1 cobalt. The data points for the viscosity as a function of the loading were obtained from the separation experiment of samarium from cobalt without extra addition of nitrate ions. Extractions for the slope analysis were performed with 1 mL of an aqueous solution containing 10 M of NH4NO3. The metal concentrations in the aqueous phases were 4 g L−1 samarium or 4.8 g L−1 lanthanum. The organic phase was made by increasing the amount of ionic liquid from 0.01 gram to 0.09 gram and by diluting the ionic liquid further with toluene until a total volume of 0.1 mL was obtained. Extractions were performed for 2 h at 70 °C and 2000 rpm. The ionic liquid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :rare earth ratios at maximal loading of the ionic liquid were determined by the extraction of the rare earths from aqueous solutions containing 37.0 g L−1 lanthanum and 163.0 g L−1 nickel or 104.8 g L−1 samarium and 202.5 g L−1 cobalt, respectively, and by decreasing the volumes of the ionic liquid phase.
:rare earth ratios at maximal loading of the ionic liquid were determined by the extraction of the rare earths from aqueous solutions containing 37.0 g L−1 lanthanum and 163.0 g L−1 nickel or 104.8 g L−1 samarium and 202.5 g L−1 cobalt, respectively, and by decreasing the volumes of the ionic liquid phase.
Percentage extraction
The percentage extraction (%E) is defined as the amount of metal extracted to the ionic liquid phase over the total amount of metal in both phases and is given by the following expression:| | 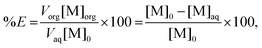 | (1) |
where Vorg and Vaq are the volumes of the organic and aqueous phases, respectively, and [M]0, [M]aq, and [M]org are the metal concentrations in the initial water phase, in the aqueous phase after extraction and in the ionic liquid phase after extraction, respectively. For the separation experiments, the first part of eqn (1) has been used to calculate the percentage extraction of cobalt, whereas for samarium, the remaining concentration in the aqueous phase was used to calculate the percentage extraction. For the scrubbing experiments, the following equation has been used to define the percentage stripping (%S) of the metals:| | 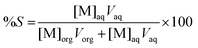 | (2) |
After extraction, metals are removed from the ionic liquid phase with nitric acid or water. To calculate the percentage stripping (%S) in the stripping phase, the following equation has been used:
| | 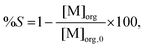 | (3) |
where [M]
org,0 is the metal concentration in the ionic liquid phase after the scrubbing, calculated by subtracting the losses of samarium in the aqueous phase after extraction and after the scrubbing from the initial metal concentration. For the experiments where further processing of the feed and scrub phase were performed, only the purity of cobalt and the samarium concentration in the ionic liquid phase have been reported due to small, but inevitable, losses of the aqueous and organic phases, when one of the phases has to be removed and another one has to be added.
Results and discussion
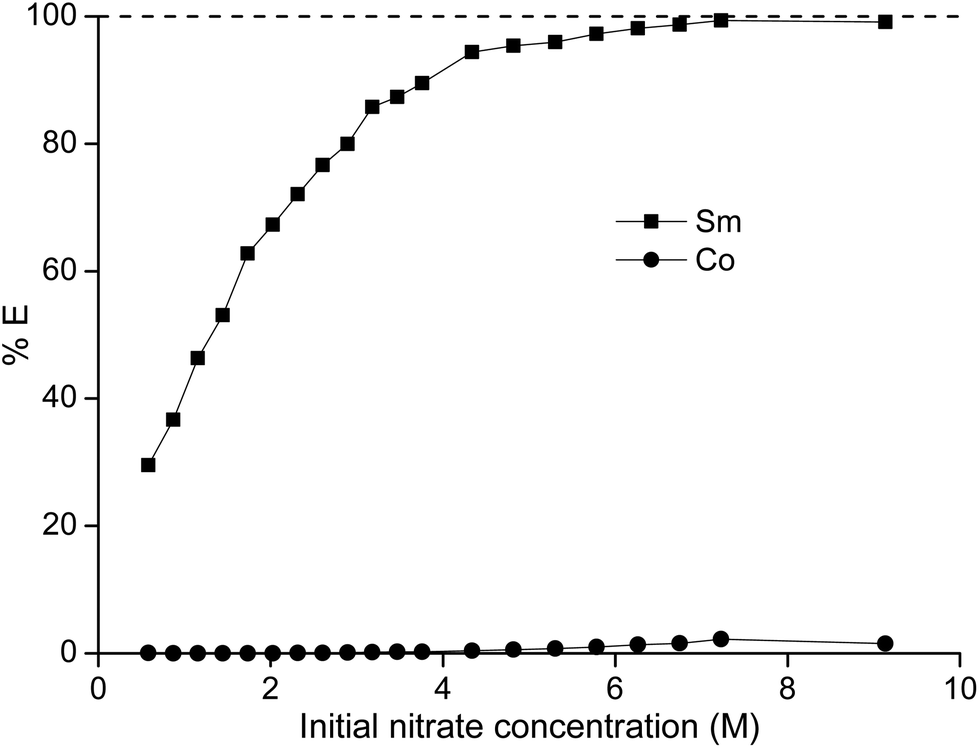
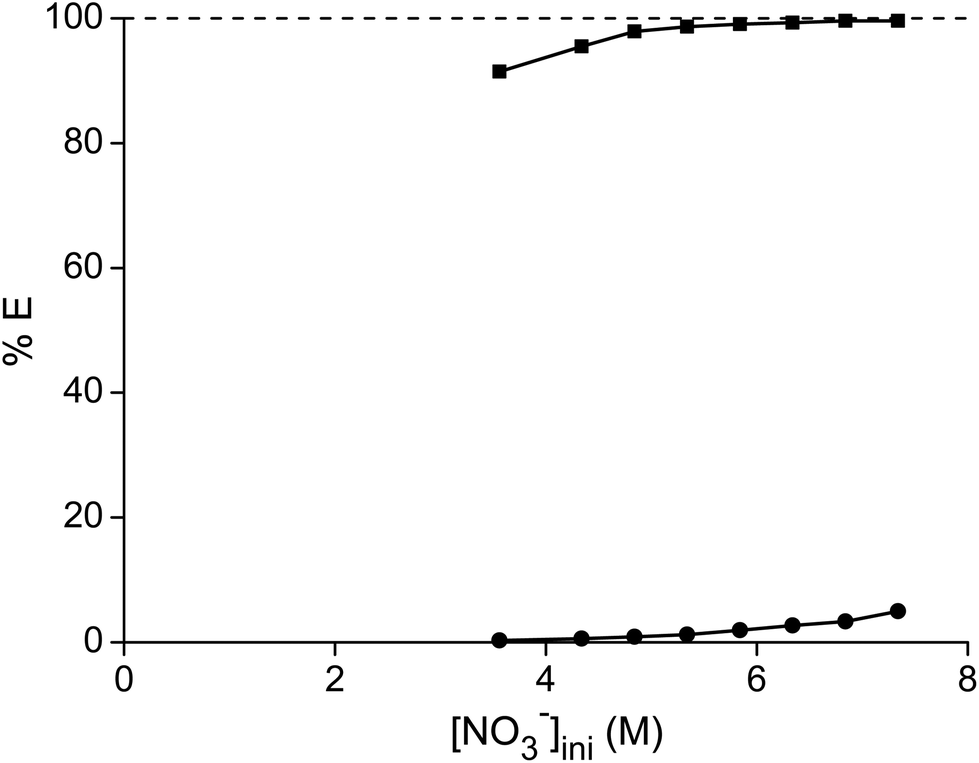
In a first series of experiments, where we tried to separate samarium from cobalt, the dependence of the separation on the nitrate concentration and acidity was studied. The concentration ratio of cobalt over samarium was chosen in a way that it mimicked the metal ratios found in a SmCo5 magnet. First, the total metal concentration and thus the nitrate concentration were stepwise increased without the addition of another salting-out agent (Fig. 1). It can be concluded that the higher the nitrate concentration, the higher the percentage extraction of samarium with a maximum of 99% at an initial nitrate concentration of 9.1 M. Cobalt is virtually not extracted, although at the highest nitrate concentration, the percentage extraction is not negligible any longer and %E values close to 2% were found. Whereas the purity for samarium in the ionic liquid phase was about 100% for the first data points at the lowest NO3− concentrations, the purity of the extracted samarium for the highest NO3− concentration was only 90% due to the increasing extraction of cobalt.
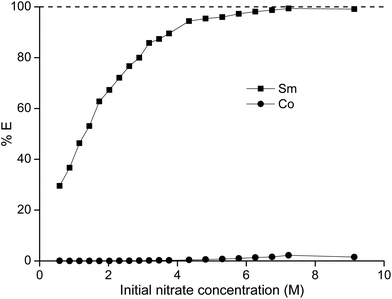 |
| | Fig. 1 Percentage extraction (%E) of samarium (■) and cobalt (●) as a function of the initial nitrate concentration in the aqueous phase. | |
Secondly, we tried to improve the separation by the addition of NH4NO3 as an extra salting-out agent (Fig. 2). The nitrate concentration on the X-axis is the sum of counter ions of the metals and the nitrate anions from NH4NO3. At similar total aqueous nitrate concentrations, the percentage extraction of both metals was slightly higher for both samarium and cobalt. This is due to the lower metal concentrations in the aqueous phase, and thus a lower loading of the ionic liquid, which has an influence on the extraction equilibrium. The purity of samarium in the loaded ionic liquid phase drops to 80% for the last data point with the highest nitrate concentrations, because of the higher percentage extraction of cobalt. Besides the lower concentrations, also the lower purity of samarium is a drawback in comparison to the NH4NO3-free system.
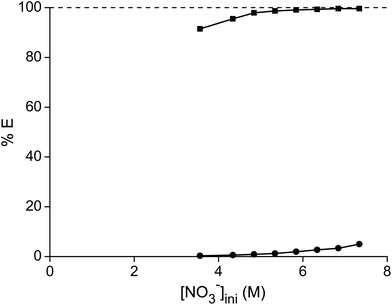 |
| | Fig. 2 Percentage extraction (%E) of samarium (■) and cobalt (●) as a function of the initial nitrate concentration in the aqueous phase. The nitrate concentration is the sum of the nitrate concentrations of the metals and of NH4NO3. The metal concentration was kept constant at 80.2 g L−1 cobalt and 42.0 g L−1 samarium. | |
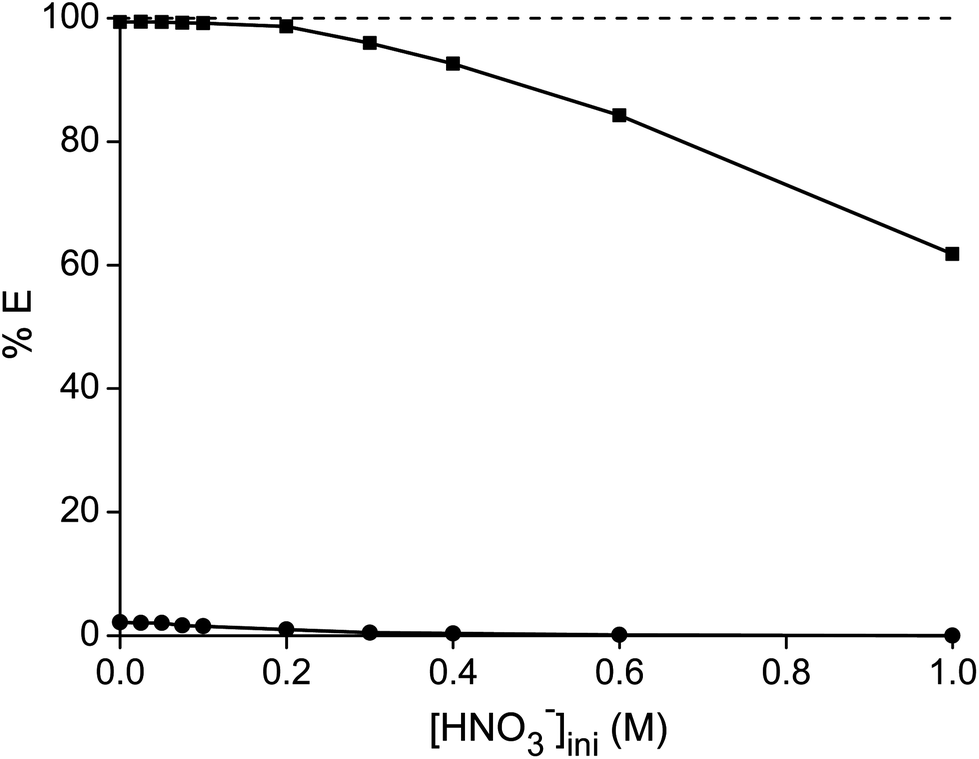
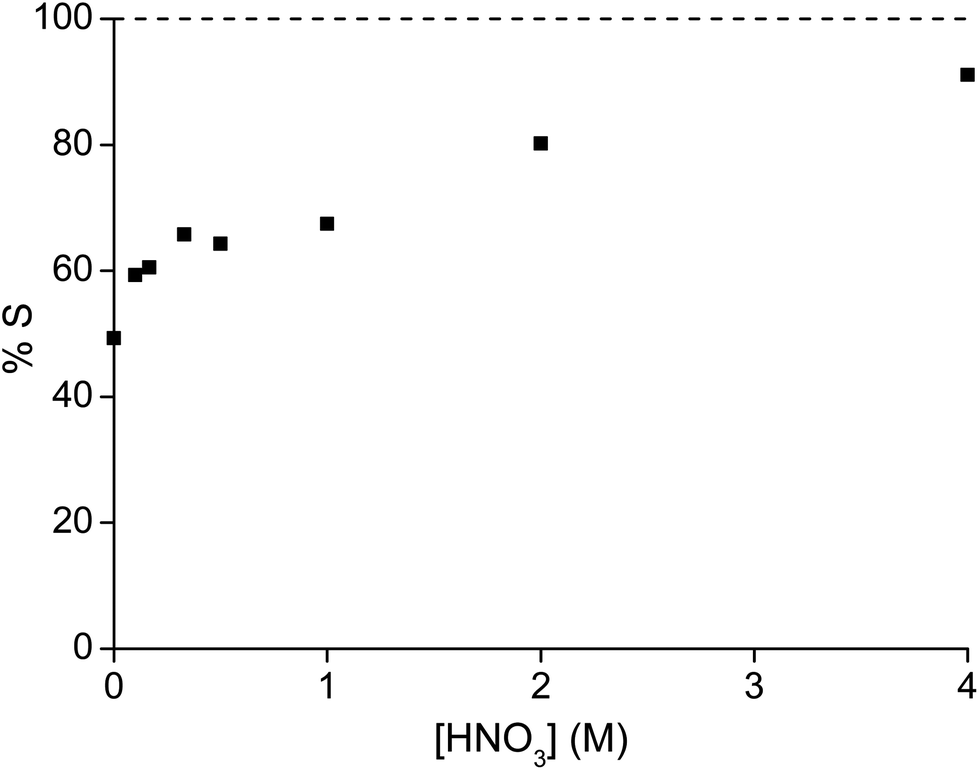
Thirdly, different concentrations of nitric acid were added to a highly concentrated metal solution. A higher nitric acid concentration decreases the percentage extraction of both metals, due to the competitive extraction of nitric acid (Fig. 3).47 All experiments showed an increase in pH after the extraction, which also supports the hypothesis that competitive nitric acid extraction occurs. The purity of samarium in the ionic liquid phase increased with increasing nitric acid concentration, but the significantly lower extraction capacity of the ionic liquid is a drawback. Moreover, the ionic liquid phase has been loaded with high concentrations of nitric acid which had to be removed before the maximal loading of the ionic liquid can be obtained in a new extraction experiment.
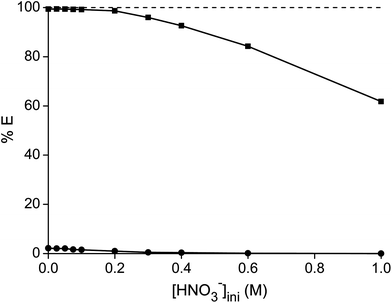 |
| | Fig. 3 Percentage extraction (%E) of samarium (■) and cobalt (●) as a function of the initial HNO3 concentration in the aqueous phase. The metal concentration was kept constant at 161.7 g L−1 cobalt and 84.5 g L−1 of samarium. | |
Dried trihexyl(tetradecyl)phosphonium nitrate has a viscosity of 1440 cP at room temperature (22 °C). After saturation with water, the viscosity decreases to 265 cP, but significantly increases again when it has been loaded with more samarium metal. The density of the ionic liquid at room temperature, loaded with 83.0 g L−1 samarium and 3.2 g L−1 cobalt, increases from 0.914 g mL−1 to 1.057 g mL−1 (Table 1). Although the density of the ionic liquid loaded with metals is now higher than that of pure water, no phase inversion was observed during the extraction or scrubbing steps. This is because the density of the water phase, containing 161 g L−1 cobalt after the extraction and 600 g L−1 NH4NO3 (7.5 M) after the scrub, is still significantly higher than the density of the ionic liquid phase.
Table 1 Physical properties of trihexyl(tetradecyl)phosphonium nitrate at 22 °C in the dry, water saturated and metal-loaded states
| |
[M]a (g L−1) |
H2Oa (wt%) |
ρ
(g mL−1) |
η
(cP) |
m.p.a (°C) |
|
[M] = metal concentration; H2O = water content; ρ = density; η = viscosity; m.p. = melting point.
n.d., not determined.
|
| [P66614][NO3], dry |
0 |
0.04 |
0.914 |
1440 |
6 |
| [P66614][NO3], H2O |
0 |
3.92 |
0.914 |
265 |
n.d.b |
| [P66614][NO3], loaded |
Sm: 83.0 Co: 3.2 |
n.d.b |
1.057 |
3730 |
n.d.b |
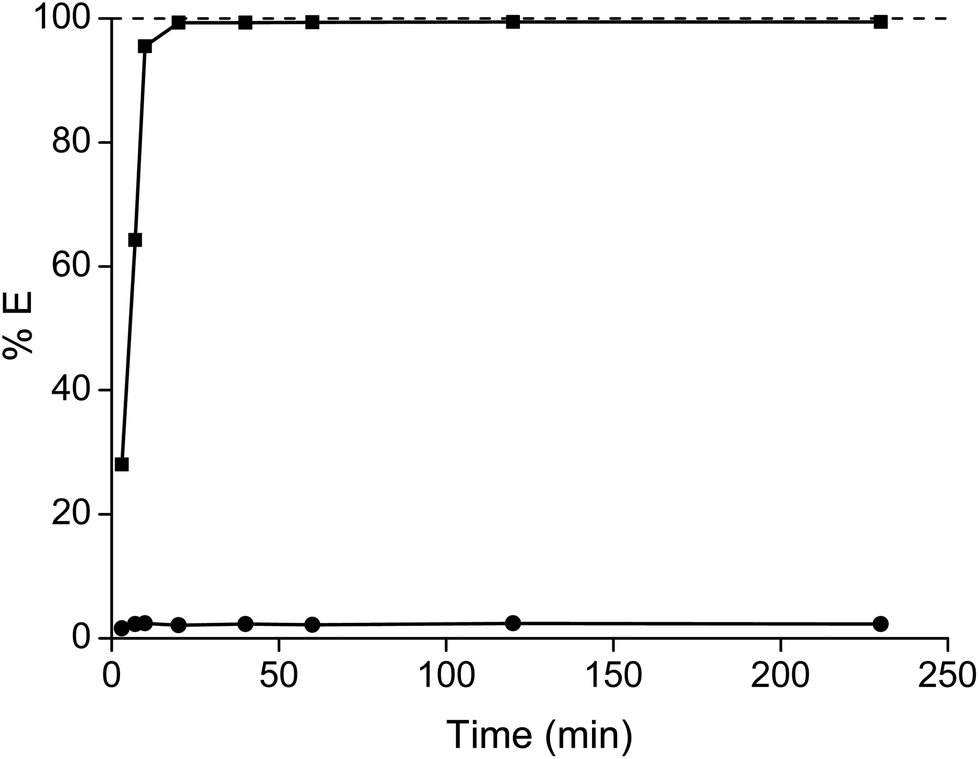
The high viscosity has a negative effect on the mass transfer and thus on the kinetics. That is why in the past non-fluorinated ionic liquids were diluted in organic solvents. It was observed that equilibrium was approached after 20 minutes, although the percentage extraction of samarium slightly further increased over time (Fig. 4). After 4 hours, the percentage extraction of samarium had increased further by only 0.12%, which is only a marginal improvement taking into account the required extra shaking and heating efforts (and thus additional energy input). Therefore, it was decided to perform all the extraction for 1 hour at 60 °C.
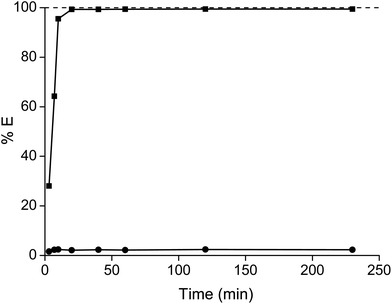 |
| | Fig. 4 Percentage extraction (%E) of samarium (■) and cobalt (●) as a function of the shaking time at 1800 rpm and 60 °C. The metal concentration was kept constant at 162.2 g L−1 cobalt and 83.9 g L−1 samarium. | |
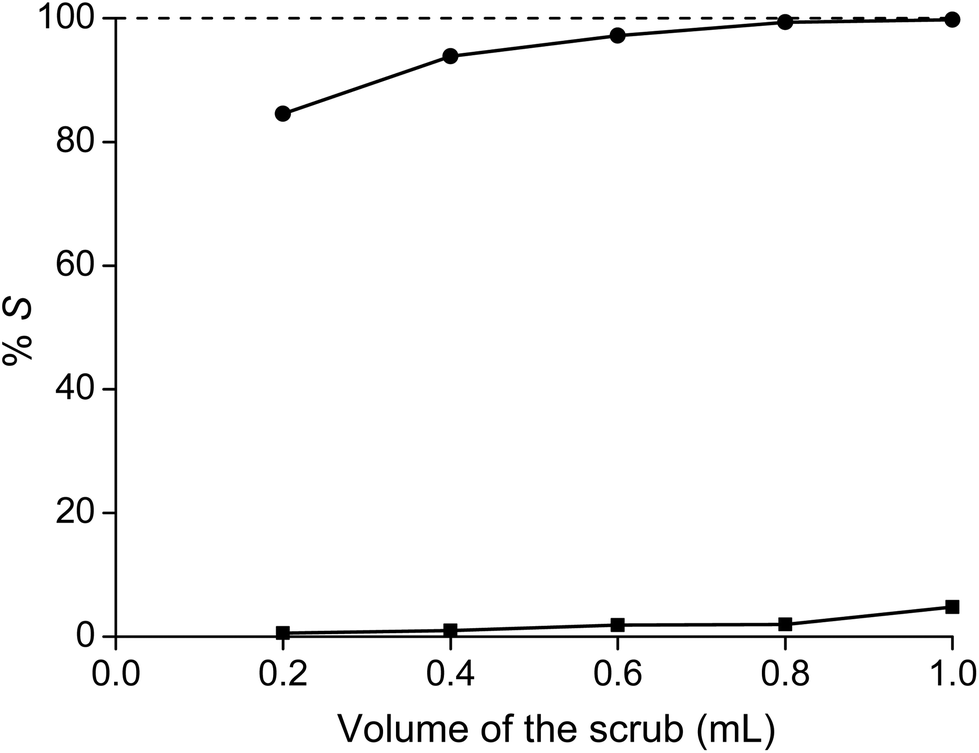
Because the addition of HNO3 or NH4NO3 did not improve the separation of samarium from cobalt, further separation was performed with a scrub step. Therefore, a solution of 7.5 M of NH4NO3 was brought into contact with the ionic liquid phase loaded with mainly samarium. The volume of the scrub was decreased in order to obtain a scrub phase which has higher concentrations of cobalt and could perhaps be added back to the feed solution after use. At a volume ratio of the aqueous over the ionic liquid phase of 1:1, more than 99.8% of cobalt and only about 4.8% of samarium was scrubbed from the ionic liquid phase (Fig. 5). An extraction percentage of 95.2% for samarium at a 7.5 M NH4NO3 concentration is slightly higher than the value reported for the extraction of europium by 30% tricaprylmethylammonium nitrate in xylene with 8.2 M NH4NO3 (%E = 94%).33,37 After the scrub step, the ionic liquid contained samarium with a purity of 99.9%. Smaller volumes also decreased the scrubbing of both metals, but had the disadvantage that the purity of samarium significantly decreased to below 99% for a scrub step with 0.2 mL of 7.5 M NH4NO3. Therefore, scrubbing was in the subsequent experiments always performed with 1 mL of 7.5 M NH4NO3 solution. The removal of cobalt from the ionic liquid phase can be observed easily by the disappearance of the pink cobalt colour of the ionic liquid phase (Fig. 6). The mass transfer rate in the scrub step is significantly slower than for extraction, because the viscosity of the ionic liquid loaded with samarium is very high from the start, whereas the viscosity of the ionic liquid in the case of extraction is only very high at the end of the extraction when maximal loading and equilibrium are almost reached. Therefore, the scrubbing step was performed at higher temperature (80 °C) for 2 hours, with shaking at 1900 rpm.
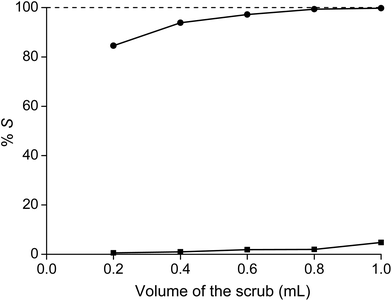 |
| | Fig. 5 Percentage stripping (%S) of samarium (■) and cobalt (●) from a 1 mL ionic liquid phase sample as a function of the volume of the aqueous scrub phase. | |
 |
| | Fig. 6 Lanthanum extraction with traces of nickel in the ionic liquid phase (A); scrubbing of the ionic liquid phase from (A) to obtain 99.9% pure lanthanum in the ionic liquid phase (B); extraction experiment with a neodymium iron mixture where we obtained an iron precipitate (C); samarium extraction with traces of cobalt in the ionic liquid phase (D); scrubbing of the ionic liquid phase from (D) to obtain 99.9% pure samarium in the ionic liquid phase (E). | |
After the extraction and scrubbing step, samarium has to be removed from the ionic liquid phase. This could be achieved in two different ways: (1) by washing the ionic liquid phase with a HNO3 solution or (2) by reducing the nitrate concentration by washing with pure water. Stripping increased with increasing nitric acid concentrations (Fig. 7). 91% of samarium could be removed in one strip step when the aqueous phase contained 4 M of nitric acid and already 50% of samarium was removed when using pure, neutral water. In the second and third stripping steps with pure water, another 41% and 5% of samarium was removed and only 0.3 g L−1 or 0.3% of the initial samarium concentration remained in the ionic liquid phase (Table 2). The use of water as a stripping agent is not only environmentally friendlier, but also avoids loading the ionic liquid with nitric acid and the requirement to remove it before the ionic liquid can be reused.
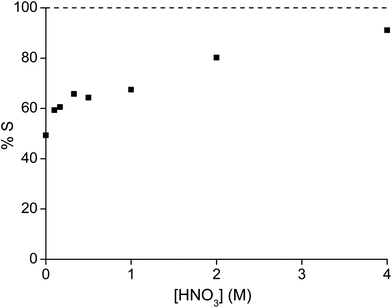 |
| | Fig. 7 Stripping of samarium as a function of the nitric acid concentration in the aqueous phase. | |
Table 2 Percentage stripping (%S) of samarium from the ionic liquid phase in each stripping step with 1 mL of water when NH4NO3 was used during scrubbing
| Stripping step |
%S of samarium |
| 1 |
53.6 |
| 2 |
40.6 |
| 3 |
5.4 |
| Total |
99.7 |
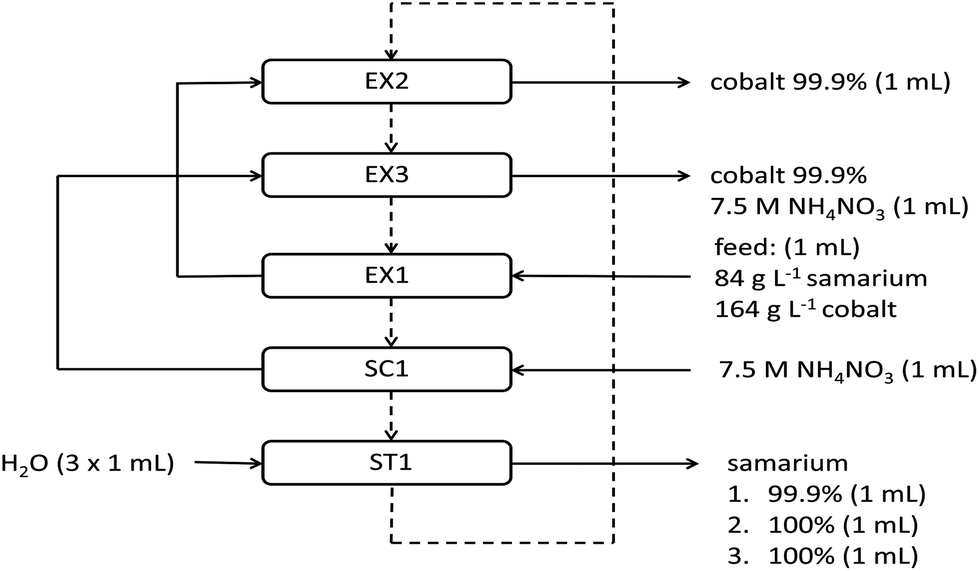
After the extraction and removal of samarium from the feed, the water phase is a highly concentrated solution of cobalt with a purity of 99.8%. This phase could be brought back into contact with fresh or stripped ionic liquid wherein the remaining samarium left the aqueous phase after the first extraction step can be removed from the water phase. Therefore, the purity of cobalt further increased to more than 99.9%. Afterwards, the NH4NO3 scrub solution, containing some samarium as well, was brought into contact with the ionic liquid phase. In this way, a second less concentrated cobalt stream was generated, which was also 99.9% pure in cobalt. NH4NO3 can be separated from cobalt and reused by going to alkaline pH values where cobalt precipitates as a pure hydroxide and where NH4NO3 remains in solution. Moreover, samarium lost during extraction and scrubbing could be brought back into the system by loading it in the ionic liquid. The ionic liquid contained at that time 3.2 g L−1 cobalt and 7.5 g L−1 samarium and can be brought again into contact with feed solution. A full flow chart of the ionic liquid solvent extraction system for the separation of samarium and cobalt is shown in Fig. 8. The whole extraction system was built up in such a way that we tried to minimize the volume of waste streams as much as possible. The ionic liquid can be reused and losses to the aqueous phase are minimal because of its high hydrophobicity. 1 mL of water is used three times to strip samarium from the organic phase without other metals or salts present in the stripping solution. Cobalt(II) nitrate is the only metal salt left in the feed solution after removal of samarium. The NH4NO3 scrub solution can be reused by slightly changing the pH to alkaline values in order to precipitate cobalt as a hydroxide salt and to obtain again a pure NH4NO3 scrub.
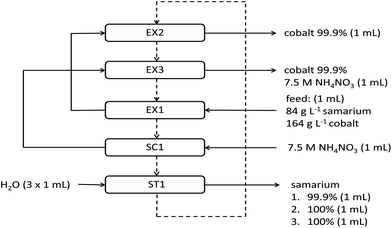 |
| | Fig. 8 Flow chart of a samarium cobalt separation. EX = extraction, SC = scrubbing, ST = stripping. The dashed line is the ionic liquid stream and the full line is the water stream. Purities of the final water streams are also given. | |
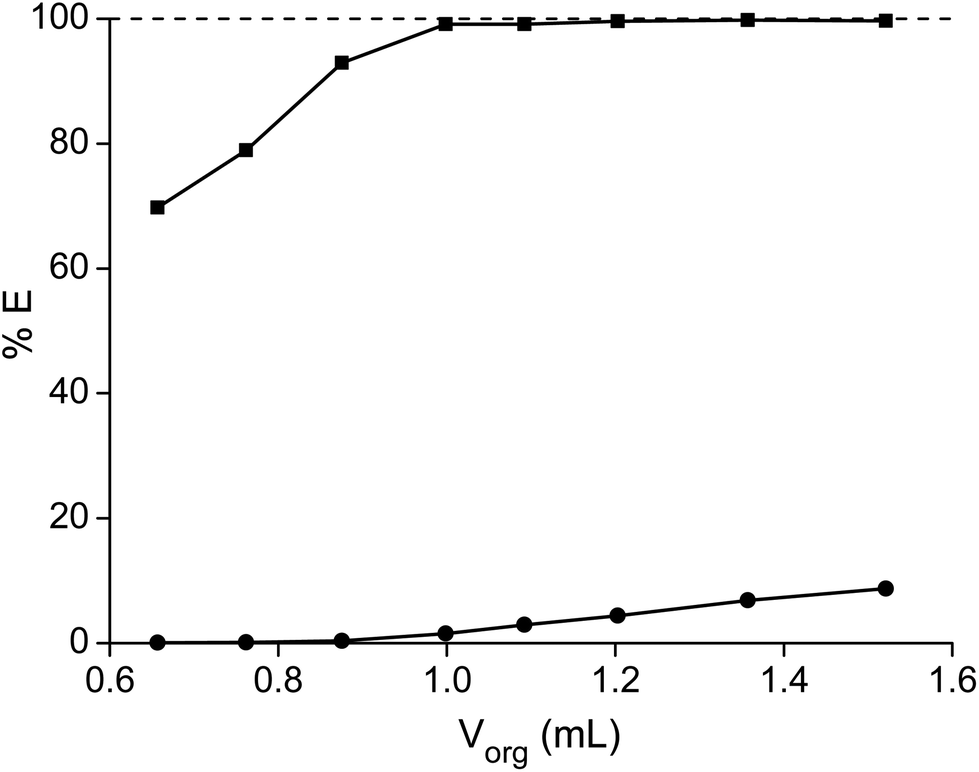
Finally, the extraction of samarium with different volumes of the ionic liquid phase was tested (Fig. 9). Therefore 1 mL of feed solution with a cobalt concentration of 202.5 g L−1 and a samarium concentration of 104 g L−1 was mixed for 15 hours at 80 °C with different volumes of the ionic liquid phase. At a phase volume ratio of 1:1, the percentage extraction of samarium was 99.1% and 1.5% for cobalt and the purity of samarium in the ionic liquid phase was 86.3%. The percentage extraction increased both for samarium and cobalt when using higher volumes of ionic liquid. In addition, when using smaller amounts of ionic liquid, the percentage extraction decreased for both elements. When the ionic liquid was saturated with samarium, the percentage extraction of samarium significantly dropped, but the purity of samarium in the ionic liquid phase considerably increased, because of this saturation effect (Fig. 10). For instance, when the ionic liquid phase was slightly saturated with samarium by decreasing the volume of the ionic liquid phase and the percentage extraction of samarium was dropped therefore to 92.9%, the purity of samarium in the ionic liquid phase increased from 86.3% to 97.9%, due to a cobalt extraction percentage of only 0.4%. In this case, the ionic liquid phase contained 111.0 g L−1 samarium and only 1 g L−1 cobalt. The purity could be increased even further by decreasing the volume of the ionic liquid phase, but this also resulted in a significant decrease in samarium extraction and thus to a lower purity of the aqueous phase. This system has not been studied further, due to the higher viscosity and the lower purity of the aqueous phase.
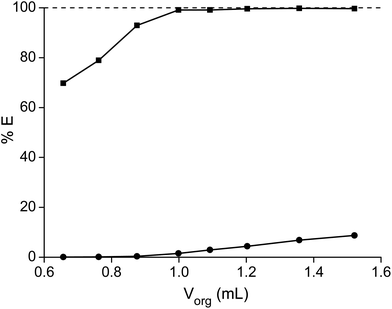 |
| | Fig. 9 Percentage extraction (%E) of a 104.8 g L−1 samarium solution (■) and a 202.5 g L−1 cobalt solution (●) as a function of the volume of the ionic liquid phase at a constant volume of the water phase (1 mL). | |
 |
| | Fig. 10 Visual representation of the ionic liquid phase when the volume of ionic liquid was decreased and less cobalt was extracted due to a saturation effect. | |
A similar process as described in Fig. 8 has been tested as well on the separation of lanthanum from nickel, which could be interesting for the recycling of NiMH batteries. In this case 250.8 g L−1 nickel and 65.5 g L−1 lanthanum were mixed, and other elements were not included. The specific choice for these concentrations was described in the Experimental section. The results of these experiments are given in Tables 3 and 4. First of all, it was observed that for lanthanum–nickel separation no further purification of the feed solution or scrub was necessary, because of the quantitative extraction of lanthanum in a single extraction step. This is due to a higher nitrate concentration in comparison with the samarium–cobalt system, but is also due to stronger complex formation formed between the ionic liquid nitrate ions and the lighter lanthanides. It is known from the literature that the percentage extraction of lanthanide ions decreases with an increase in atomic number.48,49 The remaining nickel is removed during the scrub step to obtain lanthanum in the ionic liquid phase with a purity of 99.9%. By comparing the samarium–cobalt system described in Fig. 8 with the results obtained in Table 3, some conclusions can be drawn about the influence of the scrub type. The samarium in the ionic liquid phase had a purity of 99.9%, after scrubbing with 7.5 M NH4NO3 solution, but a purity of only 99.4% in the case of scrubbing with 7.5 M NaNO3 solution. The difference in percentage stripping of the remaining nickel from the ionic liquid phase in the scrub step with a NH4NO3 solution (99.7%) or a NaNO3 solution (only 94.1%) is the underlying reason for the difference in purity. The ionic liquid phase contained only 0.04 g L−1 samarium after three stripping steps with water. This value is also lower than the value of 0.3 g L−1 found in the case when stripping was performed after a scrub with a NH4NO3 solution. The purity of cobalt in the aqueous phase after further purification of the scrub and feed solution (EX1 and EX2) was over 99.9%. The ionic liquid phase, ready to be brought back into contact with new feed solution contained 16.6 g L−1 cobalt and 2.4 g L−1 samarium.
Table 3 Percentage extraction (%E), percentage stripping (%S) from the ionic liquid phase and purity of nickel and lanthanum after an extraction and a scrub step with 7.5 M NaNO3 from a solution containing 162.0 g L−1 cobalt and 83.8 g L−1 samarium and with an initial nitrate concentration of 7.2 M. Purities of the cobalt streams after EX2 and EX3 are also given
| |
|
|
Purity (%) |
| |
Sm |
Co |
Sm,org |
Co,aq |
| EX1 |
%E = 99.3% |
%E = 2.2% |
90.0 |
99.8 |
| SC1 |
%S = 1.0% |
%S = 94.1% |
99.4 |
— |
| EX2 |
— |
— |
— |
99.9 |
| EX3 |
— |
— |
— |
99.9 |
Table 4 Percentage extraction (%E), percentage stripping (%S) and purity of nickel and lanthanum after an extraction and a scrub step with 7.5 M NaNO3 from a solution containing 250.8 g L−1 nickel and 65.5 g L−1 lanthanum, with an initial nitrate concentration of 10.0 M
| |
|
|
Purity (%) |
| |
La |
Ni |
La,org |
Ni,aq |
| EX1 |
%E = 99.4% |
%E = 2.3% |
83.4 |
99.9 |
| SC1 |
%S = 2.0% |
%S = 99.6% |
99.9 |
— |
Also the separation of iron from neodymium was tried by mixing both metals in a molar ratio of 6.7, which is close to the elemental molar ratio found in Nd2Fe14B permanent magnets. The pH of the stock solution was −1.5, which is due to the partial hydrolysis of iron(III) and the resulting formation of nitric acid. When mixing that aqueous solution with the ionic liquid, HNO3 was extracted strongly to the ionic liquid phase, and the removal of HNO3 significantly increased the pH of the aqueous phase. The extraction of nitric acid by tricaprylmethylammonium nitrate in different molecular solvents has been studied by Cerná et al.47,49 The high concentrations of nitric acid in the ionic liquid phase have not only a large decreasing influence on the percentage extraction of the metals to the ionic liquid phase, as shown already above. However, due to the higher pH values, iron(III) hydroxide precipitated in the aqueous during the extraction process (Fig. 6). For this reason, the separation of neodymium from iron was not further investigated.
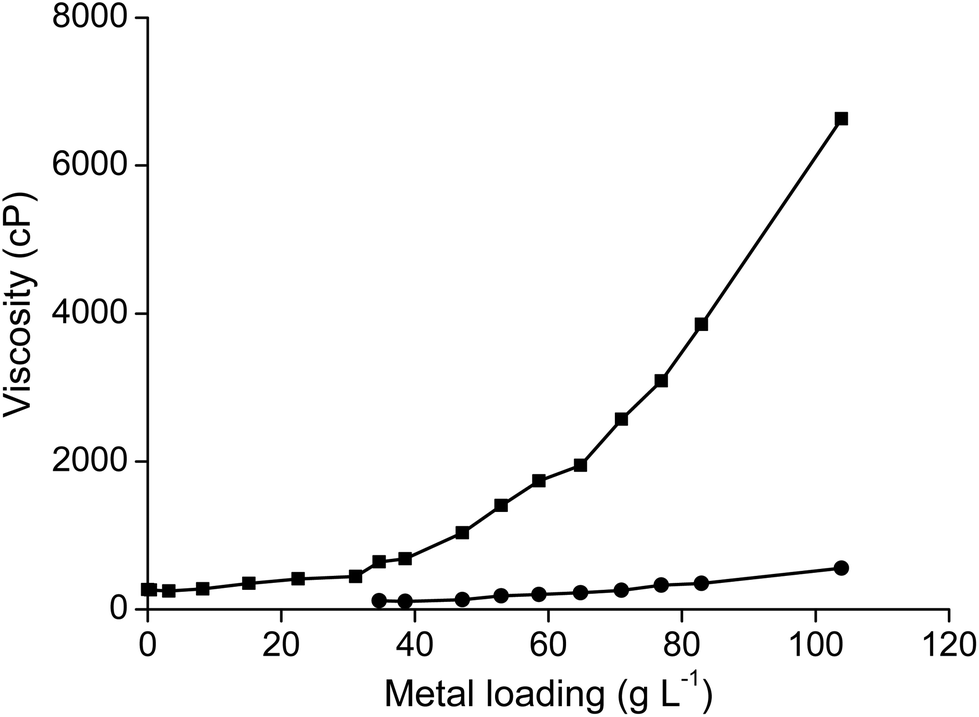
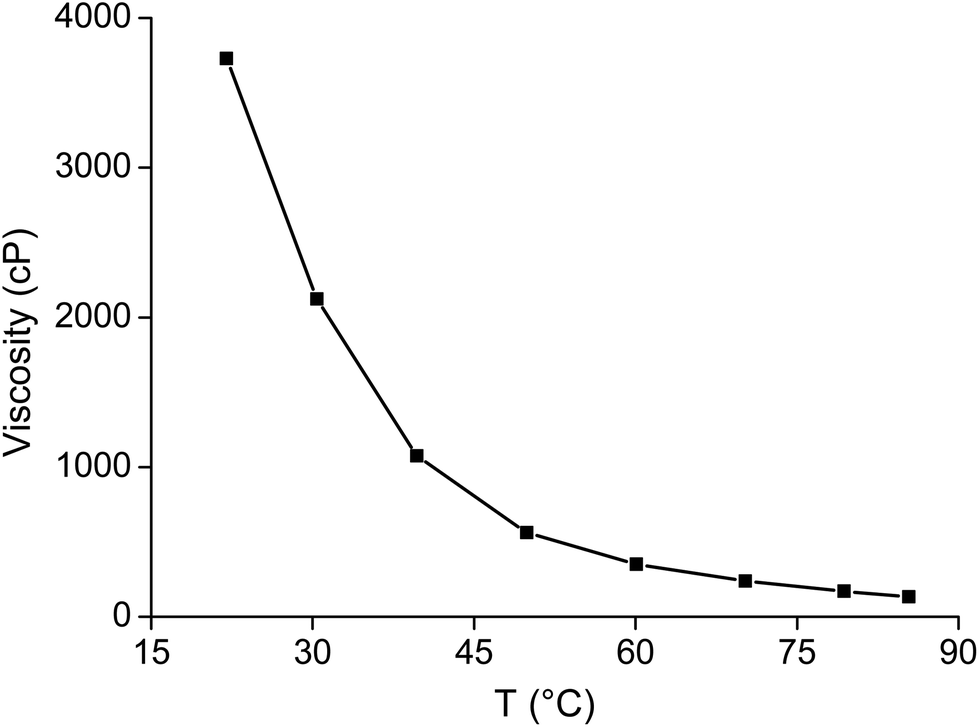
As mentioned above, the viscosity of dried trihexyl(tetradecyl)phosphonium nitrate is 1440 cP and that of the water-saturated ionic liquid is 265 cP (at 22 °C). The viscosity of the ionic liquids increased when it was loaded with samarium (Fig. 11). The viscosity of the ionic liquid phase was found to be 6630 cP at room temperature when it was loaded with 103.9 g L−1 samarium and 5.9 g L−1 cobalt. The viscosity could be decreased by an increase in temperature to 350 cP at 60 °C or even 170 cP and 80 °C (Fig. 12). A similar increase in viscosity was observed when loading the ionic liquid trihexyl(tetradecyl)phosphonium chloride with cobalt(II) in highly concentrated chloride media, due to the change in charge of the anion.20 The −1 charged chloride ion is changed for a [CoCl4]2− anion with a −2 charge, which increases the electrostatic interactions between the cation and the anion of the ionic liquid and thus leads to an increase in viscosity. In contrast, no increasing viscosity was observed for iron(III), which forms in strong chloride media the −1 charged [FeCl4]− anion. The significant increase in viscosity of the ionic liquid with increasing samarium concentrations is an indication that the anion of the metal complex in the ionic liquid has maybe a charge more negative than −1. A significant difference in viscosity was observed between the ionic liquid loaded with samarium and that loaded with lanthanum. The viscosity of the water-saturated ionic liquid phase loaded with 65.5 g L−1 lanthanum was 8970 cP at 22 °C, whereas the ionic liquid loaded with 103.9 g L−1 samarium and 6.5 g L−1 cobalt had still a lower viscosity (6630 cP) under the same experimental conditions, although the metal salt concentration in the ionic liquid phase was higher than in the case of samarium extraction. These differences in viscosity are an indication that the stoichiometry of the extracted species is different for lanthanum and samarium.
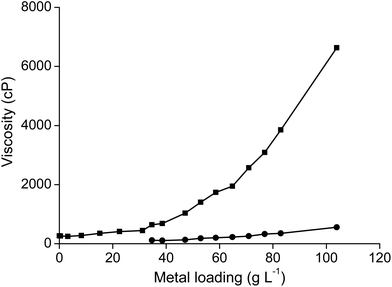 |
| | Fig. 11 Viscosity of the ionic liquid as a function of the metal loading at 22 °C and at 60 °C. | |
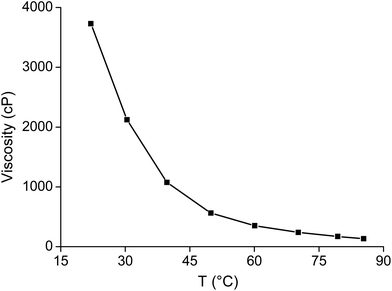 |
| | Fig. 12 Viscosity of the ionic liquid loaded with 83.2 g L−1 samarium and 3.6 g L−1 cobalt as a function of the temperature. | |
Slope analysis is the most used method for the determination of the number of extractant molecules involved in the extraction process. The general extraction mechanism for lanthanide extraction can be written as:
| | | Ln3+ + 3NO3− + n[P66614][NO3] ⇌ [Ln(NO3)3+n][P66614]n | (4) |
The stability constant βn of the extracted complex is defined as
| | 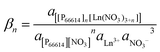 | (5) |
where
a is the activity of each species involved in the extraction and where
n is the number of ionic liquid molecules surrounding one rare-earth ion. Activities rather than concentrations have to be used if the aqueous salt concentrations are very high. The activity for a compound
i can be written as
with
γi being the activity coefficient and
Ci the concentration of the corresponding species. The dielectric constants for ionic liquids and toluene are moderate and low, respectively, and is decreasing with temperature.
50–52 Therefore, dissociation of the ionic liquid is negligible and can be considered to be non-electrolyte.
50,53,54 This also means that the activity coefficient
γi is close to one and the activity of the species in the organic phase (the ionic liquid and the complex) can be approached by the concentration (
ai,org ≈
Ci,org).
| | 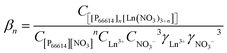 | (7) |
Eqn (7) can be rewritten by taking the logarithm and by introducing the distribution ratio D, which is defined as the concentration of the rare earth in the organic phase ([P66614]n[Ln(NO3)3+n]) over the concentration in the aqueous phase (Ln3+).
| | 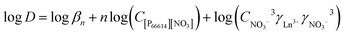 | (8) |
The activity coefficients γ can be rewritten by introducing the Debye–Hückel equation: 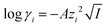 , where A is a constant, z is the charge of the ion and I is the ionic strength. The value of A in water at extraction temperature (60 °C) is 0.5495 L1/2 mol−1/2.57Eqn (8) is therefore equal to:
, where A is a constant, z is the charge of the ion and I is the ionic strength. The value of A in water at extraction temperature (60 °C) is 0.5495 L1/2 mol−1/2.57Eqn (8) is therefore equal to:
| | 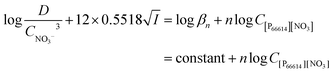 | (9) |
The distribution ratio D and the changing nitrate concentration in the aqueous phase CNO3− can be calculated from the measured and changing rare-earth concentration in the aqueous phase. The ionic strength is changing during extraction and can be calculated by the following expression:
| | 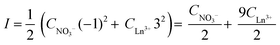 | (10) |
Therefore, the final equation becomes:
| | 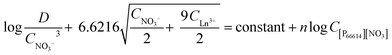 | (11) |
A slope analysis was performed by extracting 4 g L−1 samarium or 4.8 g L−1 lanthanum in 1 mL of aqueous phase containing 10 M NH4NO3. Toluene was chosen as the organic phase but the total volume of the organic phase was kept small and the weight percentage of the ionic liquid went up to 87 vol% to come as close as possible to the conditions where the ionic liquid is undiluted. No pure ionic liquid was measured as problems could occur related to the higher viscosity and slower kinetics. CLn3+ was measured after extraction. D and CNO3− are calculated from the changing lanthanide concentration CLn3+. Therefore, the left part of eqn (11) can be calculated and the concentration of [P66614][NO3] was varied and is known. A straight line with a slope of n, the number of ionic liquid molecules involved in the extraction, should be obtained when the left part of eqn (11) is plotted as a function of the changing ionic liquid concentration.
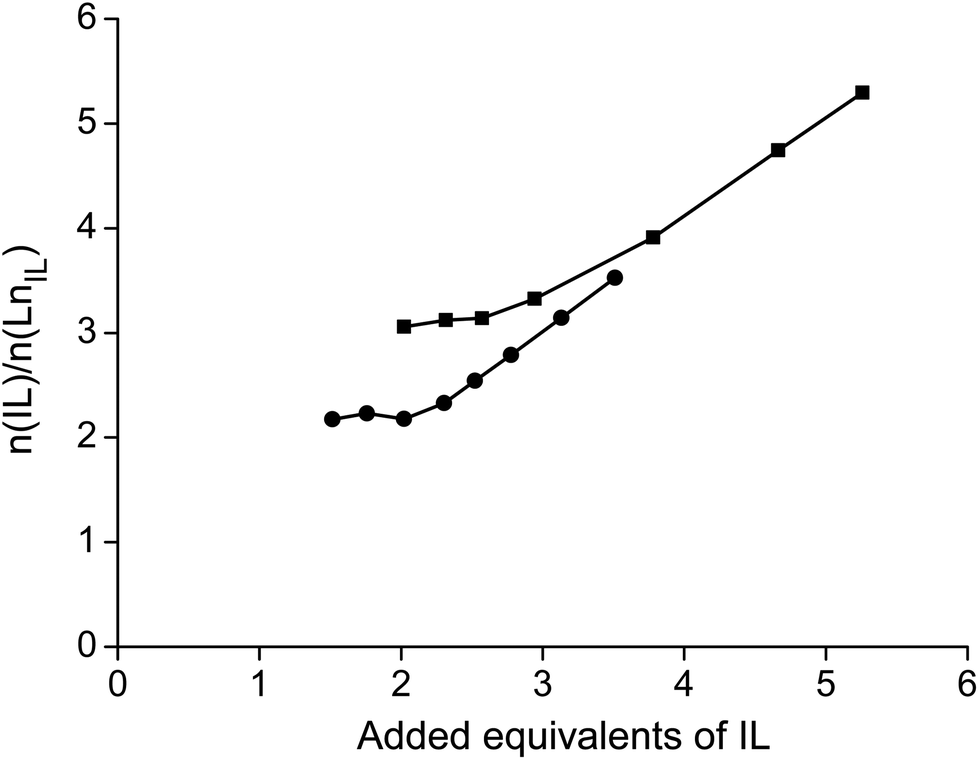
No straight lines were obtained in our slope analysis experiments, meaning that the extraction does not occur via a single complex (Fig. 13). Therefore, an extra experiment was performed where the maximal loading of the ionic liquid phase was tested. Samples (1 mL) of the aqueous phase containing 104.8 g L−1 samarium and 202.5 g L−1 cobalt were brought into contact with decreasing volumes of ionic liquid, until the point was reached where the ionic liquid was saturated with metal ions and no free extractant or ionic liquid was left. The excess of metal ions remained in the aqueous phase. The mathematical expression for the complexation constant was already given in eqn (5). The concentration of the ionic liquid is constant and about 1.7 M if it is present in undiluted form. Percentage extractions were all above 99% if the ionic liquid is present in an excess and βn is under the same conditions higher than 100. High percentage extraction in combination with high stability constants means that no free ionic liquid will be present once there is a shortage of ionic liquid. In Fig. 14, it is shown that when the number of ionic liquid equivalents added to the system was decreased below two, the molar ratio of the ionic liquid over the number of moles of samarium extracted in the ionic liquid phase remained almost constant at a value of two, or n(IL)/n(SmIL) = 2. This means that each samarium(III) ion is extracted by two molecules of the ionic liquid trihexyl(tetradecyl)phosphonium nitrate, [P66614][NO3] at maximal loading of the ionic liquid phase. A similar experiment was performed with lanthanum(III), but here, the initial aqueous metal concentrations were 37.0 g L−1 lanthanum and 163.0 g L−1 nickel. For lanthanum, a constant value n(IL)/n(LaIL) is already obtained in the region lower than three equivalents of ionic liquid. This means that in the case of saturation, three ionic liquid molecules are necessary for extracting one lanthanum(III) ion.
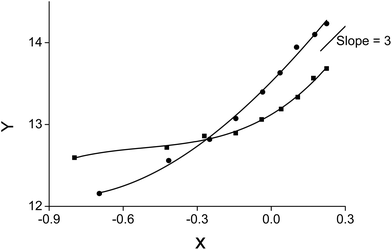 |
| | Fig. 13 Slope analysis of 4.8 g L−1 lanthanum (■) or 4 g L−1 samarium (●) from an aqueous solution containing 10 M NH4NO3 to a 0.1 mL of the organic phase containing the ionic liquid in toluene. X = log C[P66614][NO3] and 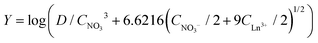 . A straight line with a slope of 3 is given for comparison. . A straight line with a slope of 3 is given for comparison. | |
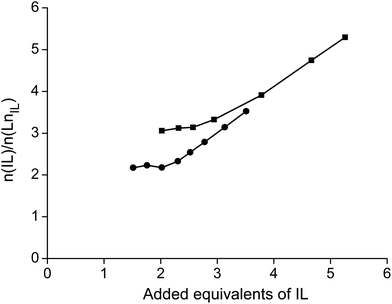 |
| | Fig. 14 The number of moles of IL over the number of moles of extracted lanthanide (n(IL)/n(Lnorg)) as a function of the number of ionic liquid equivalents added, at constant initial metal concentrations of lanthanum (■) or samarium (●). | |
From slope analysis and from the loading experiment, it can be concluded that probably different extraction mechanisms can occur depending on the kind of rare earth and the concentration of the metal, ionic liquid or nitrate anions in the aqueous or organic phase:
| | 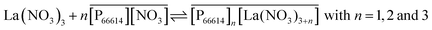 | (12) |
| | 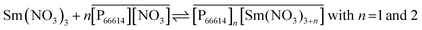 | (13) |
The formation of a −3 charged anion with lanthanum(III) and a −2 charged anion with samarium(III) in the ionic liquid has a significant effect on the interactions between the cation and the anion of the ionic liquid and this is the main reason why the viscosity of an ionic liquid phase loaded with lanthanum is much higher than that of one loaded with samarium. Several papers on rare-earth nitrate systems in the presence of bulky cations reported on the formation of hexakis complexes [Ln(NO3)6]3− for the lighter lanthanides (La, Ce, Pr, and Nd), while pentakis complexes [Ln(NO3)5]2− were formed during extraction with the heavier lanthanides.49,55–57 A non-polar solvent such as toluene, xylene or hexane was used in the case of solvent extraction experiments. The pentakis or hexakis nitrato complexes formed during extraction experiments in the organic phase are different from those obtained in a pure water phase where mainly mono- and dinitrate complexes exist.58 Besides the radius of the lanthanide ion, the formation of hexakis [Ln(NO3)6]3− complexes in an organic phase depends also on the nature of the cation.57 The crystal structure of lanthanum(III) forming inner sphere complexes with six nitrate anions, giving the high coordination number of 12, was described already in several crystallographic papers.59–63 Although there are six nitrates surrounding lanthanum(III) at high loadings, it is not certain whether this rare-earth ion has a coordination number of 12 in solution as well.
Conclusions
The undiluted ionic liquid trihexyl(tetradecyl)phosphonium nitrate, [P66614][NO3], was successfully used to extract samarium(III) or lanthanum(III) from highly concentrated metal solutions containing cobalt or nickel, respectively. The highly efficient extraction is based on an inner salting-out effect. After one extraction and a subsequent scrub step, purities better than 99.9% could be obtained both for the transition metals and the rare earths. Stripping could be performed with a nitric acid solution, but also in an environmentally friendlier way by three washing steps with pure water. Apart from the starting salts, the ionic liquid and a nitrate source such as ammonium nitrate or sodium nitrate, no other chemicals were necessary to separate the samarium–cobalt and the lanthanum–nickel mixtures and to regenerate the ionic liquid. It must be stressed that no volatile or flammable molecular organic solvents are used in our solvent extraction process. The viscosity of the ionic liquid increased considerably when loading it with rare-earth metals. By increasing the temperature, viscosity could be significantly reduced and equilibrium times of about 20 minutes could be obtained. The extraction mechanism occurs probably via two or three different complexes. The extraction at high lanthanum(III) loadings occurs mainly via a hexanitrato complex whereas the extraction of samarium(III) at high loadings occurred mainly via a pentakis nitrato complex. The extraction processes presented in this paper serve as model systems for the recovery of the main metals from samarium–cobalt permanent magnets and nickel metal hydride batteries. However, both these magnets and battery alloys contain small admixtures of other elements than samarium and cobalt, or lanthanum and nickel, respectively. These minor components can influence the solvent extraction process. The simplified model system presented in this paper will be used in the near future as the basis for the development of environmentally friendly recycling processes of real SmCo magnets and NiMH batteries.
Acknowledgements
The authors thank the KU Leuven for financial support (GOA/13/008 and IOF-KP RARE3). Support by IoLiTec (Heilbronn, Germany) and Cytec Industries (Niagara Falls, Ontario, Canada) is also gratefully acknowledged.
References
-
J. Rydberg, M. Cox, C. Musikas and G. R. Choppin, Solvent Extraction: Principles and Practice, Marcel Dekker, Inc., New York, 2004 Search PubMed
 .
.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed .
- K. R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68, 351–356 CrossRef CAS .
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS .
- J. G. Huddleston, H. D. Willauer, R. P. Swatloski, A. E. Visser and R. D. Rogers, Chem. Commun., 1998, 1765–1766 RSC .
- S. Dai, H. Ju and E. Barnes, J. Chem. Soc., Dalton Trans., 1999, 1201–1202 RSC .
- M. J. Earle, J. M. S. S. Esperanca, M. A. Gilea, J. N. Canongia Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831–834 CrossRef CAS PubMed .
- M. Smiglak, W. M. Reichert, J. D. Holbrey, J. S. Wilkes, L. Sun, J. S. Thrasher, K. Kirichenko, S. Singh, A. R. Katritzky and R. D. Rogers, Chem. Commun., 2006, 2554–2556 RSC .
- D. M. Fox, W. H. Awad, J. W. Gilman, P. H. Maupin, H. C. De Long and P. C. Trulove, Green Chem., 2003, 5, 724–727 RSC .
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed .
- I. Billard, A. Ouadi and C. Gaillard, Anal. Bioanal. Chem., 2011, 400, 1555–1566 CrossRef CAS PubMed .
- P. R. Vasudeva Rao, K. A. Venkatesan, A. Rout, T. G. Srinivasan and K. Nagarajan, Sep. Sci. Technol., 2012, 47, 204–222 CrossRef CAS .
- Z. Kolarik, Solvent Extr. Ion Exch., 2013, 31, 24–60 CrossRef CAS .
- G. C. Tian, J. Li and Y. X. Hua, Trans. Nonferrous Met. Soc. China, 2010, 20, 513–520 CrossRef CAS .
- R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361–363 RSC .
- M. L. Dietz and J. A. Dzielawa, Chem. Commun., 2001, 2124–2125 RSC .
- M. P. Jensen, J. Neuefeind, J. V. Beitz, S. Skanthakumar and L. Soderholm, J. Am. Chem. Soc., 2003, 125, 15466–15473 CrossRef CAS PubMed .
- M. L. Dietz, J. A. Dzielawa, I. Laszak, B. A. Young and M. P. Jensen, Green Chem., 2003, 5, 682–685 RSC .
- D. Parmentier, S. J. Metz and M. C. Kroon, Green Chem., 2013, 15, 205–209 RSC .
- T. Vander Hoogerstraete, S. Wellens, K. Verachtert and K. Binnemans, Green Chem., 2013, 15, 919–927 RSC .
- S. Wellens, B. Thijs and K. Binnemans, Green Chem., 2012, 14, 1657–1665 RSC .
- S. Wellens, B. Thijs, C. Möller and K. Binnemans, Phys. Chem. Chem. Phys., 2013, 15, 9663–9669 RSC .
- W. Wang, H. Yang, H. Cui, D. Zhang, Y. Liu and J. Chen, Ind. Eng. Chem. Res., 2011, 50, 7534–7541 CrossRef CAS .
- R. Mishra, P. C. Rout, K. Sarangi and K. C. Nathsarma, Hydrometallurgy, 2011, 108, 93–99 CrossRef CAS PubMed .
- A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 13, 471–481 RSC .
- A. Rout, K. A. Venkatesan, T. G. Srinivasan and P. R. Vasudeva Rao, Sep. Purif. Technol., 2012, 95, 26–31 CrossRef CAS PubMed .
- A. Cieszynska and M. Wisniewski, Sep. Purif. Technol., 2011, 80, 385–389 CrossRef CAS PubMed .
- H. F. Aly, M. El-Garhy and S. El-Reefy, Microchem. J., 1972, 17, 431–435 CrossRef CAS .
- T. Sato, T. Shimomura, S. Murakami, T. Maeda and T. Nakamura, Hydrometallurgy, 1984, 12, 245–254 CrossRef CAS .
- F. Fabrega and M. B. Mansur, Hydrometallurgy, 2007, 87, 83–90 CrossRef CAS PubMed .
- M. Regel-Rosocka, Sep. Purif. Technol., 2009, 66, 19–24 CrossRef CAS PubMed .
- D. Kogelnig, A. Stojanovic, F. Jirsa, W. Körner, R. Krachler and B. K. Keppler, Sep. Purif. Technol., 2010, 72, 56–60 CrossRef CAS PubMed .
- F. L. Moore, Anal. Chem., 1966, 38, 510–512 CrossRef CAS .
-
D. J. Bauer and R. E. Lindstrom, Report: Differential Extraction of Rare-earth Elements in Quaternary Ammonium Compound-Chelating Agent Systems, U.S. Dept. of Interior, Bureau of Mines, Washington, D.C., 1971 Search PubMed .
- I. Komasawa, K. Hisada and M. Miyamura, J. Chem. Eng. Jpn., 1990, 23, 308–315 CrossRef CAS .
- D. Lu, J. S. Horng and Y. C. Hoh, J. Less-Common Met., 1989, 149, 219–224 CrossRef .
-
F. L. Moore, United States Patent, US/1966/3294494, 1966 Search PubMed .
-
D. J. Bauer and R. E. Lindstrom, United States Patent, US/1967/3323857, 1965 Search PubMed .
- A. Landgren and J. O. Liljenzin, Solvent Extr. Ion Exch., 1999, 17, 1387–1401 CrossRef CAS .
- P. Giridhar, K. A. Venkatesan, T. G. Srinivasan and P. R. Vasudeva Rao, Hydrometallurgy, 2006, 81, 30–39 CrossRef CAS PubMed .
- N. S. Egorova, V. V. Belova, A. A. Voshkin, A. I. Khol'kin, A. K. Pyartman and V. A. Keskinov, Theor. Found. Chem. Eng., 2008, 42, 708–713 CrossRef CAS .
- M. Regel-Rosocka, L. Nowak and M. Wisniewski, Sep. Purif. Technol., 2012, 97, 158–163 CrossRef CAS PubMed .
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Clean. Prod., 2013, 51, 1–22 CrossRef CAS PubMed .
-
European Commission, Report: Critical raw materials for the EU, Report of the Ad-hoc Working Group on defining critical raw materials, 2010 .
-
U. S. Department of Energy, Report: 2011 Critical Materials Strategy, 2011 .
- K. Larsson, C. Ekberg and A. Oÿdegaard-Jensen, Hydrometallurgy, 2012, 129–130, 35–42 CrossRef CAS PubMed .
- M. Cerná, V. Bízek, J. Št'astová and V. Rod, Chem. Eng. Sci., 1993, 48, 99–103 CrossRef .
- Y. Marcus and I. Abrahamer, J. Inorg. Nucl. Chem., 1961, 22, 141–150 CrossRef CAS .
- M. Cerná, E. Volaufov and V. Rod, Hydrometallurgy, 1992, 28, 339–352 CrossRef .
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed .
- J. Hunger, A. Stoppa, S. Schrödle, G. Hefter and R. Buchner, ChemPhysChem, 2009, 10, 723–733 CrossRef CAS PubMed .
- M. M. Huang, Y. Jiang, P. Sasisanker, G. W. Driver and H. Weingärtner, J. Chem. Eng. Data, 2011, 56, 1494–1499 CrossRef CAS .
-
W. J. Hamer, Theoretical mean activity coefficients of strong electrolytes in aqueous solutions from 0 to 100 °C, U.S. Govt. Print. Off., Washington, 1968 Search PubMed .
-
V. S. Kislik, Solvent Extraction Classical and Novel Approaches, Elsevier, Oxford, 2012 Search PubMed .
- V. V. Bagreev and S. O. Popov, Polyhedron, 1985, 4, 929–932 CrossRef CAS .
- J. C. G. Bünzli, B. Klein, G. O. Pradervand and P. Porcher, Inorg. Chem., 1983, 22, 3763–3768 CrossRef .
- I. M. Walker and D. H. Weeden, Inorg. Chem., 1973, 12, 772–777 CrossRef CAS .
- S. Andersson, K. Eberhardt, Ch. Ekberg, J.-O. Liljenzin, M. Nilsson and G. Skarnemark, Radiochim. Acta, 2006, 94, 469–474 CAS .
- A. S. R. Chesman, D. R. Turner, G. B. Deacon and S. R. Batten, J. Coord. Chem., 2007, 60, 2191–2196 CrossRef CAS .
- F. A. Hart, M. B. Hursthouse, K. M. A. Malik and S. Moorhouse, J. Chem. Soc., Chem. Commun., 1978, 549–550 RSC .
- Y. Tang, Y. Feng, X. Gan, M. Tan and K. Yu, Polyhedron, 1996, 15, 3219–3223 CrossRef CAS .
- G. H. Tao, Y. Huang, J. A. Boatz and J. M. Shreeve, Chem.–Eur. J., 2008, 14, 11167–11173 CrossRef CAS PubMed .
- S. P. Ji, M. Tang, L. He and G. H. Tao, Chem.–Eur. J., 2013, 19, 4452–4461 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence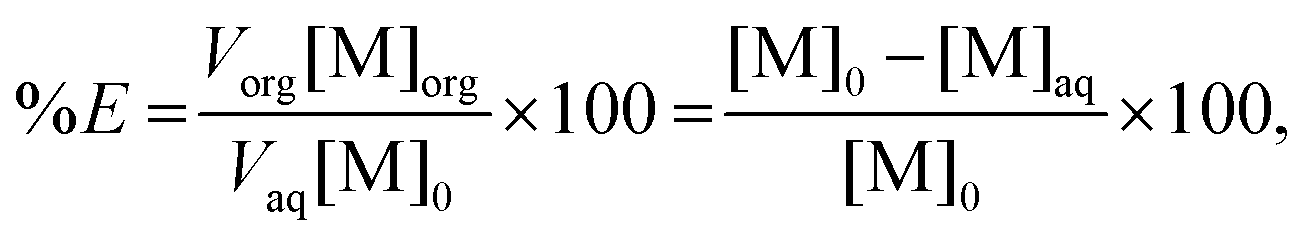
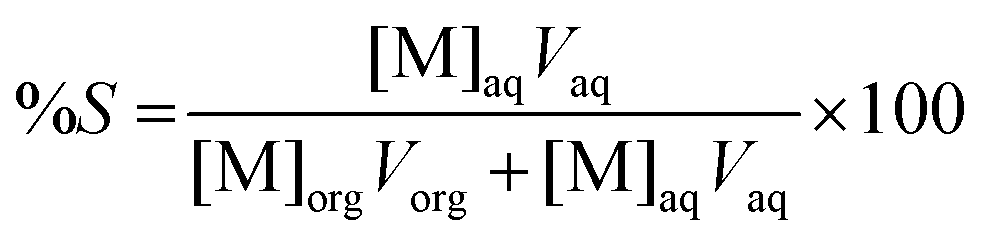
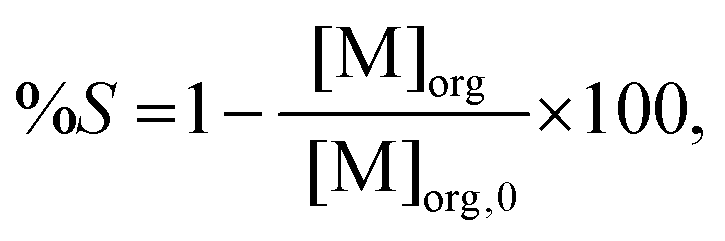
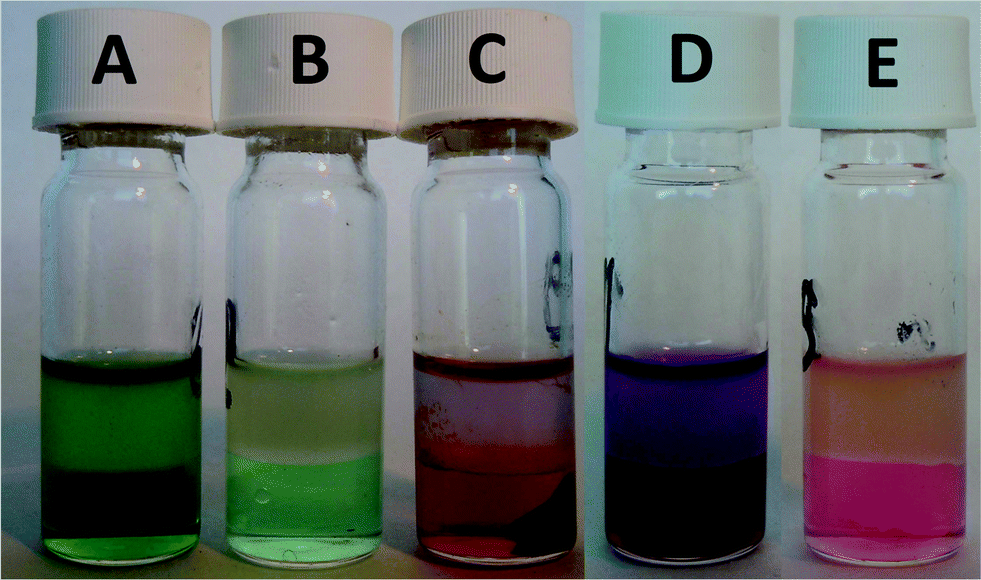

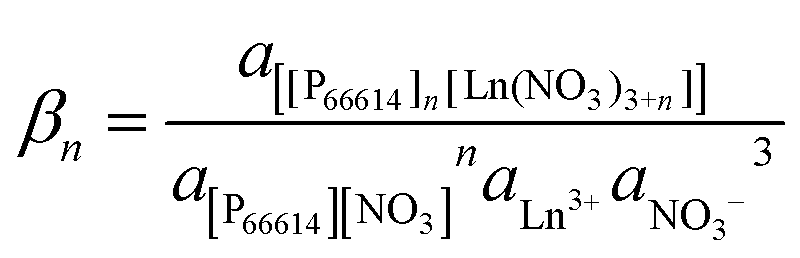
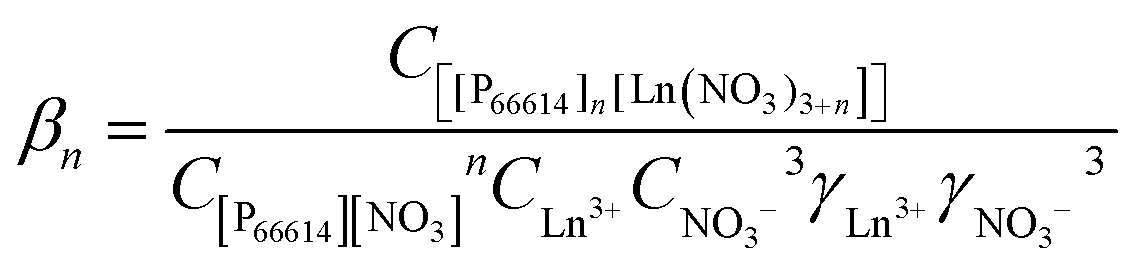
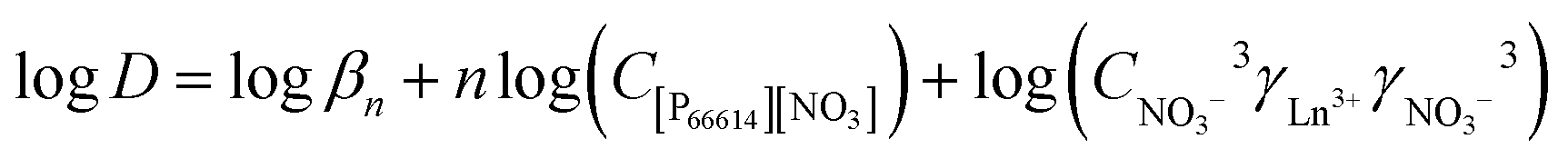
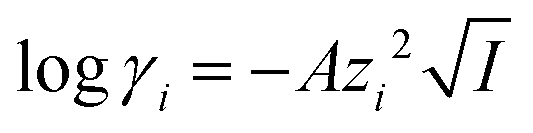 , where A is a constant, z is the charge of the ion and I is the ionic strength. The value of A in water at extraction temperature (60 °C) is 0.5495 L1/2 mol−1/2.57Eqn (8) is therefore equal to:
, where A is a constant, z is the charge of the ion and I is the ionic strength. The value of A in water at extraction temperature (60 °C) is 0.5495 L1/2 mol−1/2.57Eqn (8) is therefore equal to: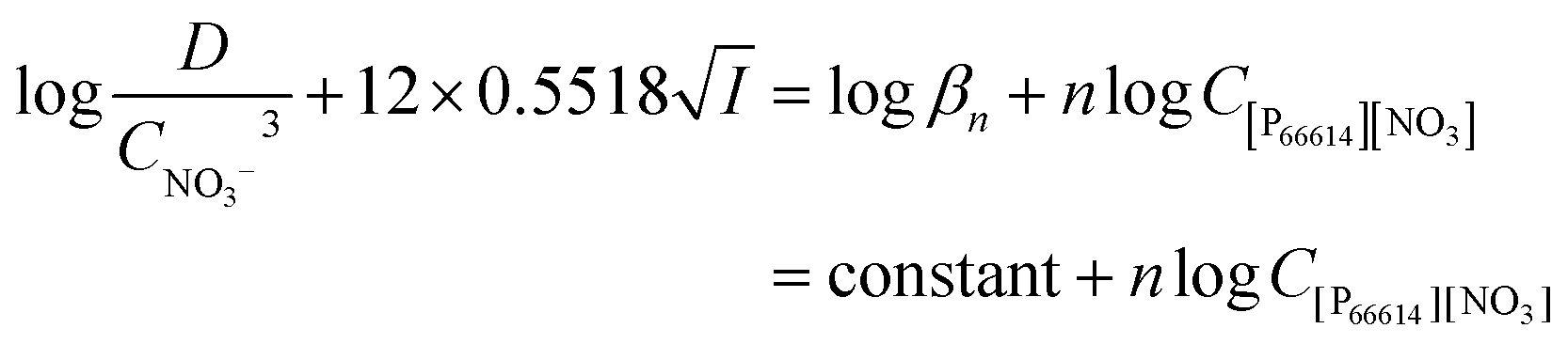
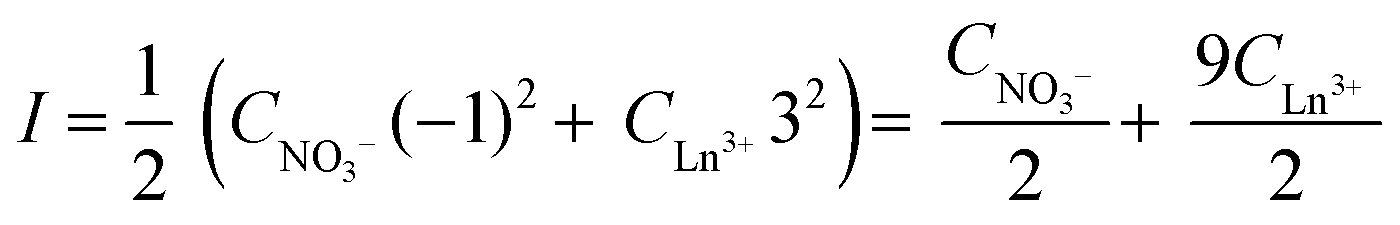
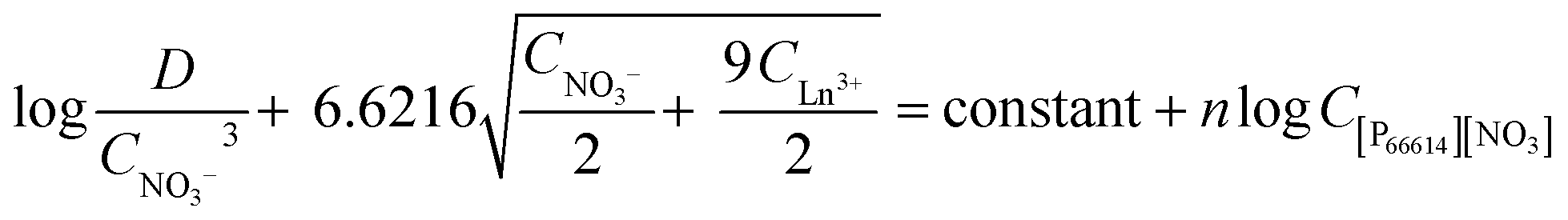
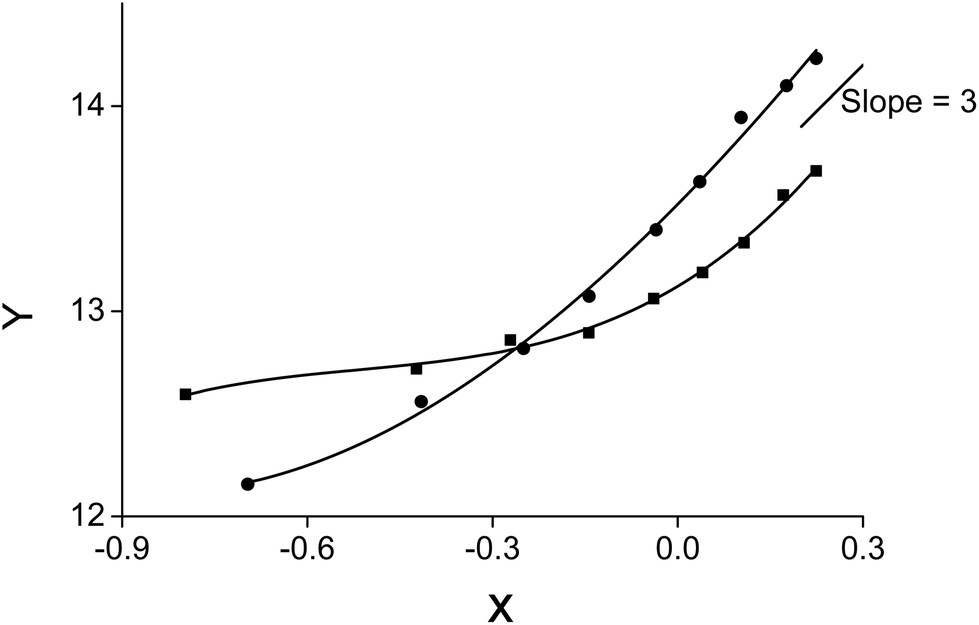
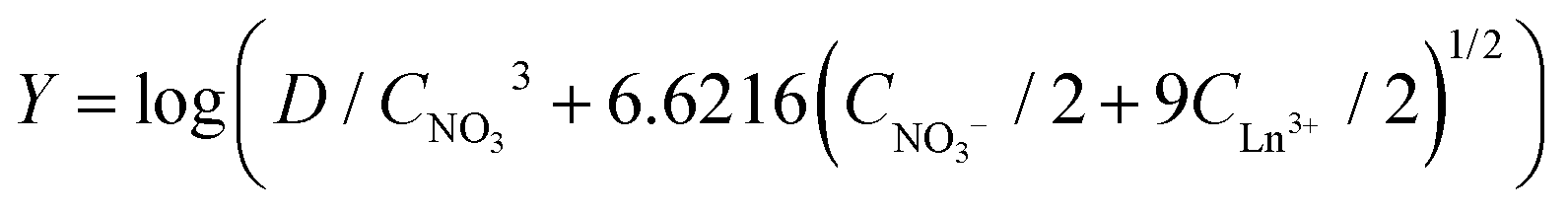 . A straight line with a slope of 3 is given for comparison.
. A straight line with a slope of 3 is given for comparison.