 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A DFT study of furan hydrogenation and ring opening on Pd(111)
Shengguang
Wang
,
Vassili
Vorotnikov
and
Dionisios G.
Vlachos
*
Department of Chemical and Biomolecular Engineering, Catalysis Center for Energy Innovation, and Center for Catalytic Science and Technology, University of Delaware, Newark, Delaware 19716, USA. E-mail: vlachos@udel.edu
First published on 20th August 2013
Abstract
The reaction energies and the corresponding energy barriers of hydrogenation and ring opening of furan on Pd(111) for the formation of tetrahydrofuran (THF), 1-butanol and small hydrocarbons were studied using density functional theory (DFT). THF forms via sequential hydrogenation of carbon atoms of the furan ring in the order of α-carbon, adjacent β-carbon, second β-carbon, and the remaining α-carbon. Upon hydrogenation of the α-carbon of furan, ring opening becomes facile. Thus, hydrofuran (HF) is a reactive intermediate in both hydrogenation and ring opening. The fate of HF determines the selectivity of the overall reaction. A simple kinetic analysis indicates that coverage effects are important and the hydrogen partial pressure is a key factor in controlling selectivity. Dihydrofuran (DHF) was found to be a stable intermediate, consistent with experimental findings. Once DHF is formed, ring opening is not favored due to the high energy barriers of ring opening of DHF, trihydrofuran (TriHF) and THF. 1-Butanol is a thermodynamically favored product, while THF is kinetically preferred. Our theoretical work agrees well with experimental observations that 1-butanol is a major product at high temperatures whereas THF is a major product at low temperatures. Insights gained into selectivity toward ring hydrogenation and ring opening can assist future studies in catalyst selection.
Introduction
With increasing interest in renewable fuels, fuel additives, and chemicals, considerable attention has recently shifted toward furan derivatives, which can be obtained from sugars. The hydrogenation and hydrogenolysis of the furan derivatives can modify or remove side functional groups, saturate the furan ring with hydrogen, and open the furan ring to form alcohols and hydrocarbons. Hydrogenation products of the furan compounds include 2-methylfuran (MF), 2,5-dimethylfuran (DMF), tetrahydrofuran (THF), 2-methyltetrahydrofuran (MTHF), dihydroxymethyl furan (DHMF), and furfuryl alcohol (FA).Furan is a five-member aromatic ring system in which one of the two lone pairs of electrons of the oxygen is delocalized over the π-system of the ring. The hydrogenation of furan on metal catalysts has been studied experimentally and theoretically. The catalytic hydrogenation of furan was first carried out in 1949 by Smith and Fuzek1 over Adams platinum (platinum dioxide reduced in hydrogen). It was argued that the reaction proceeded along one of two pathways. The furan ring could hydrogenate to yield dihydrofuran (DHF) and then THF or open to form butanol. The conversion of DHF or THF to butanol was not observed. Recently, experiments have shown that furan hydrogenation on Pd/ZrO2 produces little THF at a low H2/furan ratio at 373 K, but solely THF at a high H2/furan ratio.2 Kliewer et al.3 researched furan hydrogenation experimentally over Pt(111), Pt(100) and Pt nanoparticles and found that both THF and butanol were produced. The hydrogenation of furfural, a singly substituted furan compound, can produce different distributions of furfuryl alcohol, furan, and ring-opening product on SiO2-supported Cu, Pd, and Ni catalysts, and the selectivity to decarbonylation and ring opening products increases at the expense of furfuryl alcohol with increasing temperature.4
Xu performed DFT calculations for furan ring opening and dehydrogenation on Pd(111) up to CO formation.5 It was found that the decomposition of furan begins with ring opening at the C–O bond, giving a C4H4O aldehyde species that rapidly loses the α-H to form C4H3O (see Fig. 1 for labeling of C atoms). C4H3O further dehydrogenates at the δ position to form C4H2O before the αC–βC bond dissociates to form CO. This research successfully explained the mechanism of furan decarbonylation. However, the conclusions of that work do not extend to the reaction of furan in the presence of hydrogen. Vorotnikov et al. have carried out dispersion-corrected DFT calculations on the mechanism of furfural conversion to furan, furfuryl alcohol and 2-methylfuran on Pd(111). It has been found that thermodynamics favors the production of furan and CO, while the activation energy for formation of furfuryl alcohol is lower than that for decarbonylation to furan.6 That work provided a detailed mechanism of the hydrogenolysis of furfural and its derivatives but did not consider the important problem of ring opening and hydrogenation.
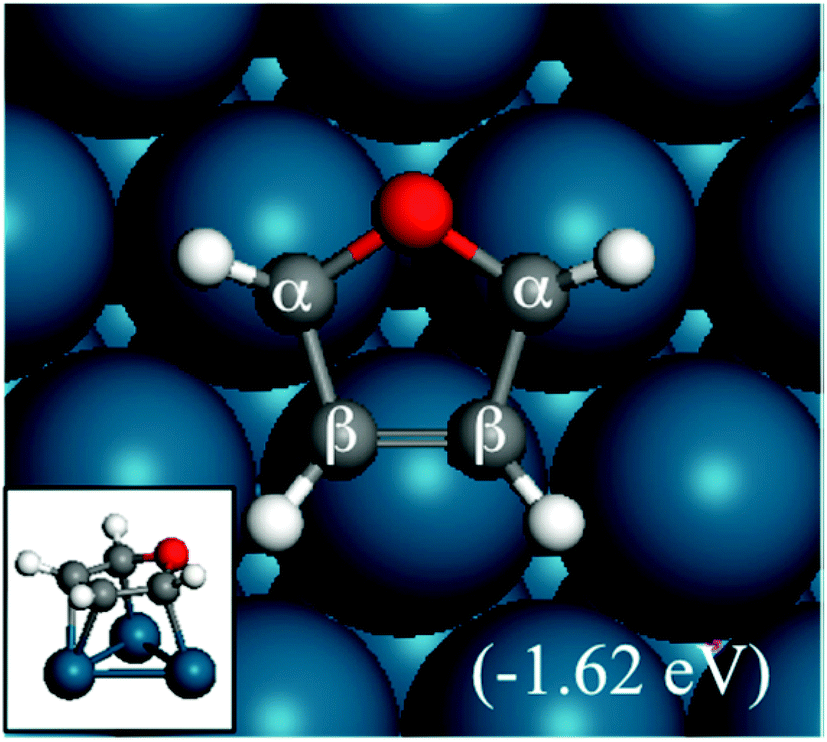 | ||
| Fig. 1 Structure and adsorption energy of furan adsorbed on a Pd(111) surface. The inset shows a side view. | ||
Ring opening of furans is important in producing open structure products, such as butanol, diols, etc. In contrast, ring opening and hydrogenation of the furan family (e.g., furfural, 5-hydroxymethyl furan) can lead to selectivity loss in experiments targeting alkylated furans, e.g., in converting furfural to methyl furan. In order to understand ring hydrogenation and ring opening, in this paper, we perform DFT calculations for the first time on the mechanism of furan hydrogenation to THF and ring opening followed by decarbonylation to C3 species and CO or hydrogenation to butanol-1. Our focus is on Pd(111). Yet, our calculations provide significant insights into the principles of selectivity control that can guide catalyst selection.
Methods and models
We carried out plane-wave DFT calculations using the Vienna Ab Initio Simulation Package (VASP), version 5.2.12.7,8 The electron–electron exchange and correlation energies were computed using the Perdew, Burke, and Ernzerhof functional with the latest dispersion correction, PBE-D3, which self-consistently calculates the total energy and forces.9,10 The projector augmented-wave method was used for the electron–ion interactions.11,12 We used a plane-wave basis set with an energy cutoff of 400 eV. For bulk calculations, a tetrahedron method with Blochl corrections and a 15 × 15 × 15 Monkhorst–Pack k-point mesh was used.13,14 The bulk lattice constant was obtained using the Birch–Murnaghan equation of state.15,16 The Pd fcc lattice constant was calculated to be 3.95 Å using the PBE and 3.90 Å using the PBE-D3, both of which are in good agreement with the experimental value of 3.89 Å.17 The metal slab was modeled with a 4 × 4 unit cell composed of four atomic layers. The bottom two layers were frozen. The vacuum between the slabs was set at 20 Å to minimize the effect of the interaction between them. The Brillouin zone was sampled with a 3 × 3 × 1 k-point grid. For accurate total energies, we used the Methfessel–Paxton method with a smearing parameter of 0.1. Surface relaxation was performed until all forces were smaller than 0.05 eV Å−1. The adsorption energy was computed as Eads = Eslab+i − Eslab − Ei, where Eslab+i is the total electronic energy of the metal slab–adsorbate system, Eslab is the total electronic energy of a clean slab, and Ei is the total electronic energy of the adsorbate in the gas-phase. The supercell for all gas-phase calculations was chosen to be 20 × 20 × 20 Å. The transition states were located using the climbing nudged elastic band (cNEB) method.18Furan hydrogenation intermediates
In this subsection, we discuss the adsorption structures and energies of the intermediates during the furan hydrogenation reaction. We tested various possible adsorption structures of furan on Pd(111) and found the structure shown in Fig. 1 to be the most stable. It has been reported that furan prefers to adsorb with its molecular plane lying parallel to the Pd(111) surface so that either the α–β or the β–β C–C bond is located on the top of a Pd atom, and the center of the ring is located either over a threefold site or a bridge site. The difference in adsorption energy of furan at different sites is small (0.01 eV using PBE and 0.05 eV using PBE-D3), and this indicates that the furan molecule is not sensitive to the adsorption site as long as the molecule stays parallel to the surface. Both PBE5 and PBE-D3 calculations gave the same conclusion regarding the most stable structure of furan on the Pd(111) surface. The adsorption structure of furan on Pd(111) was investigated by Knight et al.19 using NEXAFS and scanned-energy mode photoelectron diffraction (PhD), and they found that the furan molecule is adsorbed nearly parallel (within 10°) to the Pd(111) surface. This is in agreement also with STM images taken by Loui and Chiang.20The calculated adsorption energy of furan is −1.62 eV. Xu calculated the adsorption structures of furan using PBE and a four-layer slab with a (3 × 3) surface unit cell, and the adsorption energy of furan was reported to be −1.00 eV at the fcc site, which is lower than ours using the PEB-D3/(4 × 4) surface unit cell. The difference mainly comes from the selection of different functionals. The PBE-D3 selected here includes vdW interactions, resulting in stronger interaction compared with the PBE functional. Our bigger unit cell, which means a lower surface coverage, could be another reason for stronger binding. A TPD study by Ormerod et al. has found that the maximum molecular desorption temperature observed on Pd(111) is 360 K at doses higher than 0.25 L, which means that the desorption barrier for furan is ∼0.9 eV.21 However, when furan is dosed below 0.25 L, only CO and H2 desorb. This indicates that the binding of furan is stronger than its decomposition at low coverage.
The adsorption structures of HF are shown in Fig. 2. HF is more stable with the extra hydrogen atom located on the α- (Fig. 1b) or the β-carbon (Fig. 1c), and these structures are nearly isoenergetic. Therefore, both intermediates would exist and should be considered in subsequent transition state searches. The hydrogenation of the oxygen atom has a very high energy, as expected, since the oxygen is saturated with two C–O bonds.
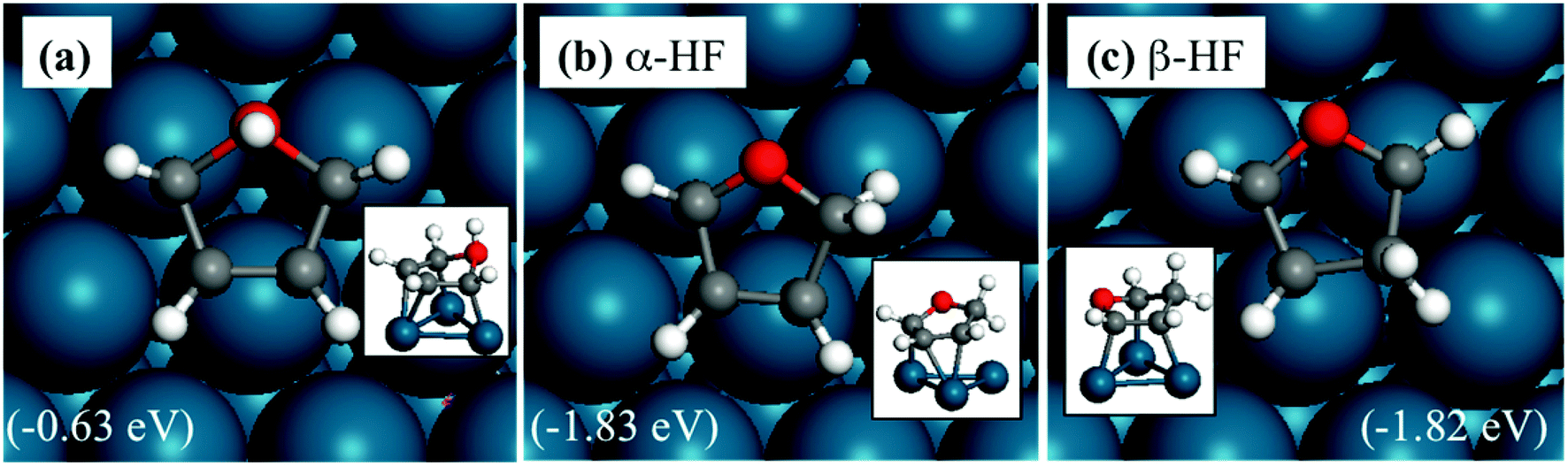 | ||
| Fig. 2 Structures of HF adsorbed on a Pd(111) surface. The energies in brackets are relative to furan(g), 0.5H2(g) and a clean Pd(111) slab. Panels (a, b and c) are hydrogenated at oxygen, α-carbon and β-carbon, respectively. The insets show side views. | ||
The side view in Fig. 2 shows that all oxygen and carbon atoms on the ring are almost in the same plane for α-HF, whereas β-HF has two atoms high or farther from (O and beta C) and the rest three atoms low or closer compared to the metal surface. This is because in β-HF, the oxygen atom and the β-carbon are saturated, and have repulsive interactions with the Pd(111) surface, while the α-carbon in between still binds with the surface. In contrast, in α-HF, the saturated oxygen and α-carbon are adjacent and the repulsive interaction does not largely distort the ring. This helps explain the trends in reaction barriers described later.
The optimized structures of DHF are shown in Fig. 3. As shown in Fig. 3(a), hydrogenation of the oxygen atom is also difficult. The most stable structure is α,β-DHF, with the other two carbon atoms binding with the Pd surface (Fig. 3(b)). The saturated oxygen and α- and β-carbon atoms move away from the surface. Fig. 3(e) shows the structure where both α-carbon atoms are hydrogenated and the molecule binds to the surface with two β-carbon atoms. This structure, with the saturated atoms adjacent to each other, has a low energy although this is not the most stable one. The structure shown in Fig. 3(d) is similar to the above structure. The only difference is that this structure has β-carbons binding to the same surface Pd atom, while the structure in Fig. 3(e) binds to two individual Pd atoms. The structure in Fig. 3(c) also has a relatively high energy, since the ring is largely distorted.
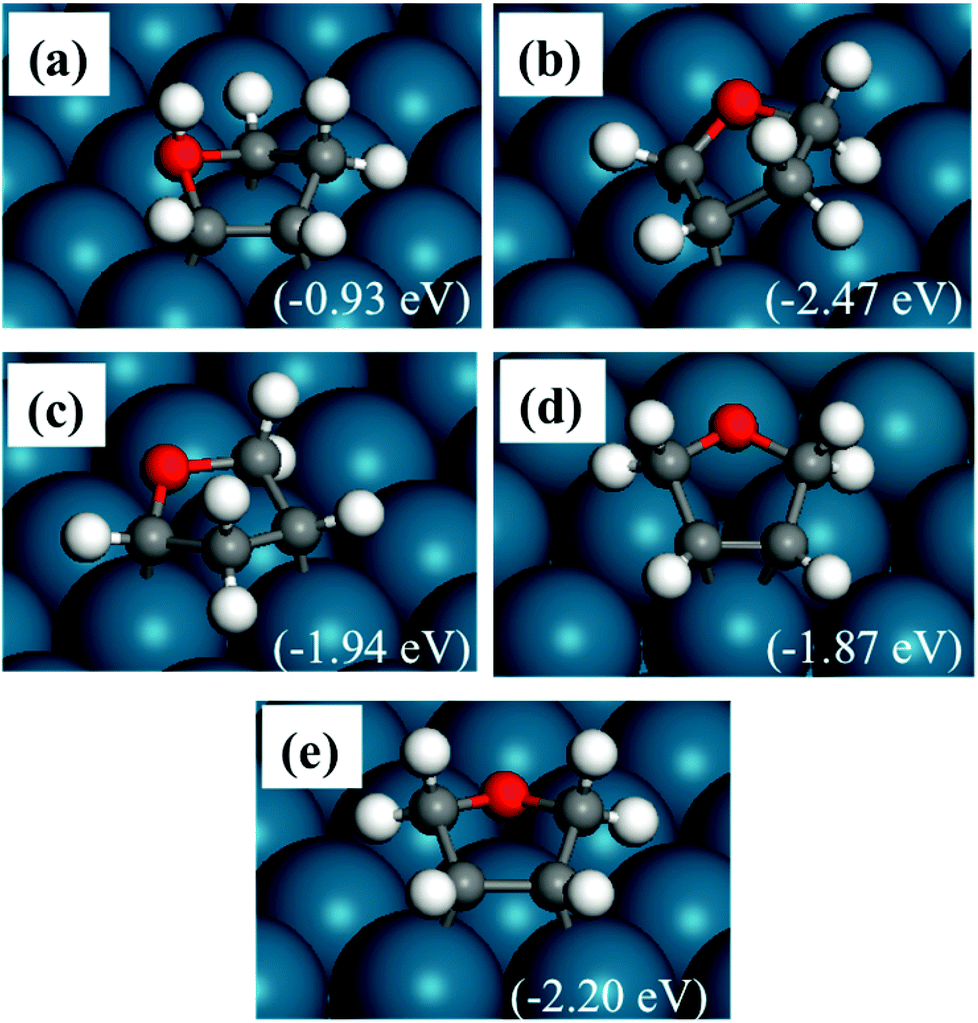 | ||
| Fig. 3 Structures of DHF adsorbed on a Pd(111) surface. The energies in brackets are relative to furan(g), H2(g) and a clean Pd(111) slab. Panel (a) is oxygen, β-DHF, panels (b and c) are α,β-DHF with different relevant positions, and panels (d and e) are α,α-DHF at different sites. | ||
The above analysis indicates that the energy of adsorbed DHF is affected by three factors. The hydrogenation on carbon is favored over the hydrogenation on oxygen. More C–Pd bonds and less distortion of the five-member ring also lower the energy. A similar trend is seen in the binding strength of furan and HF.
The relatively stable structures of trihydrofuran (TriHF) are shown in Fig. 4(a) and (b). The product of hydrogenation on the oxygen atom still has high energies. The structure with an α-carbon not hydrogenated (a) has lower energy compared to the structure with the β-carbon not hydrogenated (b). The energy difference between them is 0.18 eV. The TriHF molecule binds to the surface with only one C–Pd bond. Fig. 4(c–e) show that when the oxygen atom is hydrogenated the energies are high.
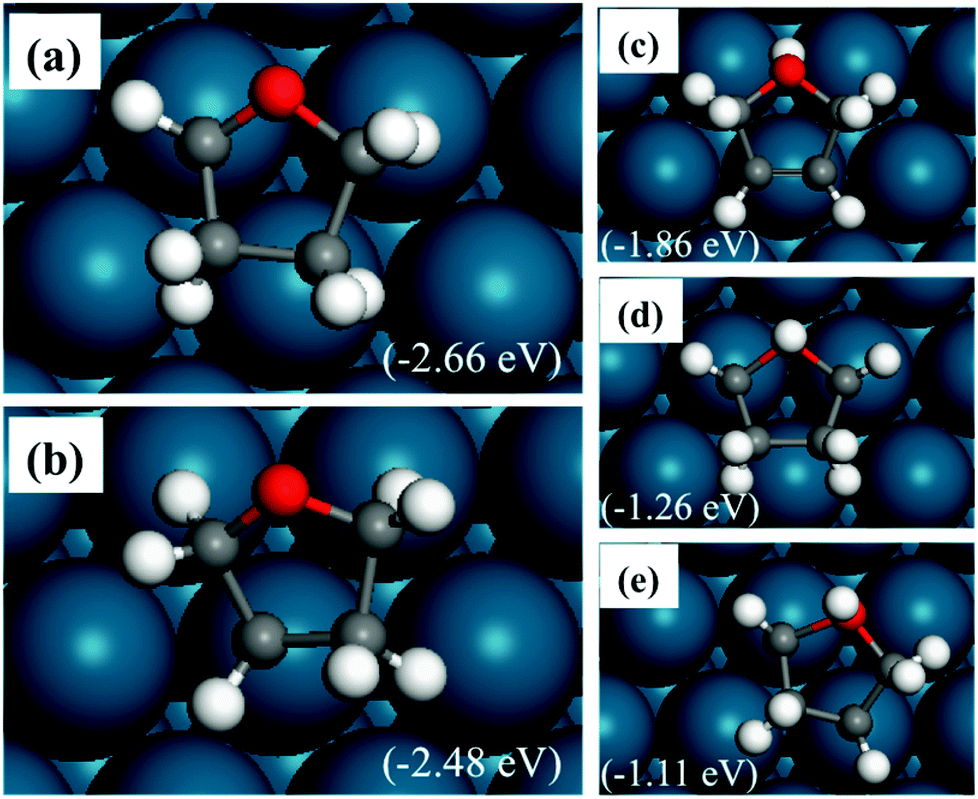 | ||
| Fig. 4 Structures of TriHF adsorbed on a Pd(111) surface. The energies in brackets are relative to furan(g), 1.5H2(g) and a clean Pd(111) slab. Panels (a) and (b) have an oxygen atom un-hydrogenated and panels (c–e) have an oxygen atom hydrogenated. | ||
The structures of THF are shown in Fig. 5. The most stable structure is shown in Fig. 5(a) with the THF ring bending down to the surface. It is noted that the stable structure in the gas-phase is similar to the conformation of THF with a distorted ring, shown in Fig. 5(b) and (d). In Fig. 5(b), the ring also bends downward to the surface; however, the distorted ring is not favored compared to the interaction with the flat surface. Fig. 5(c) and 5(d) show that the vertical adsorbed THF on the surface is not energetically favored compared to the bent structures. The adsorption energy of the most stable structure of THF is −1.13 eV. The binding strength is weaker than that of furan.
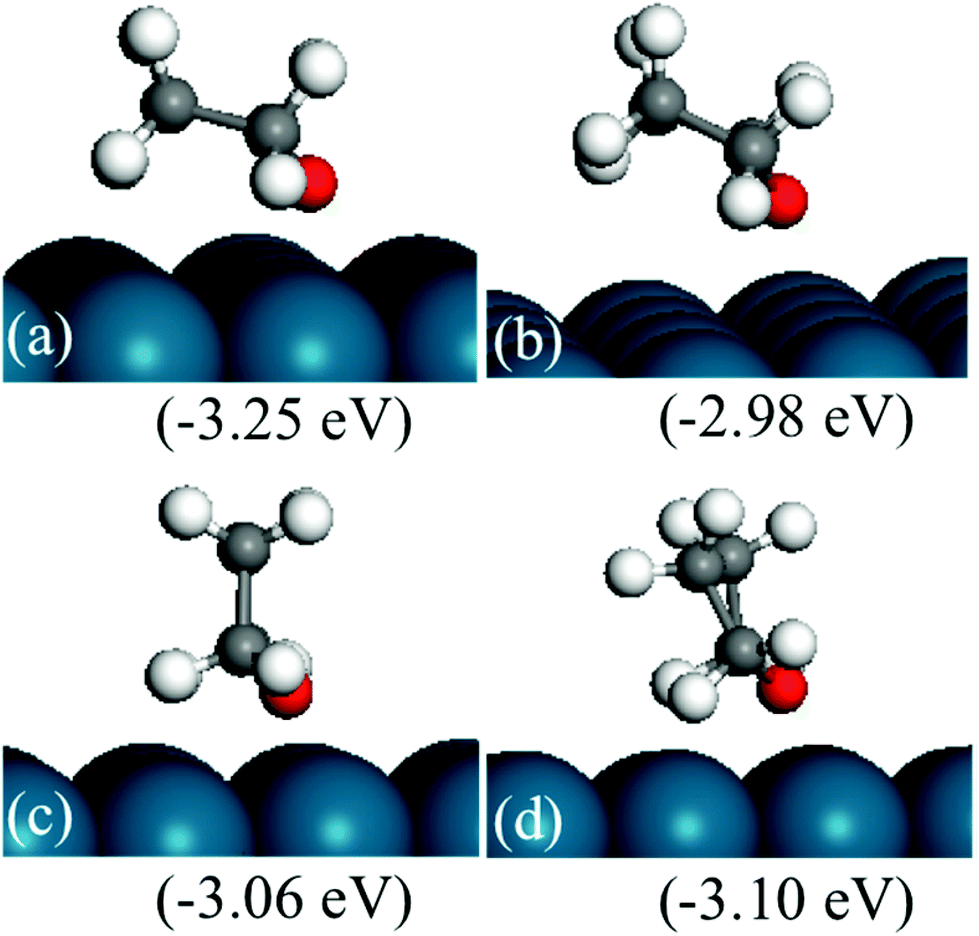 | ||
| Fig. 5 Structures of THF adsorbed on a Pd(111) surface. The energies in brackets refer to furan(g), 2H2(g) and a clean Pd(111) slab. The strongest (panel (a)) adsorption energy of THF is −1.13 eV. Structures in panels (a) and (b) bend down to the surface, while structures in panels (c) and (d) are adsorbed in a vertical configuration. | ||
There are four C–Pd bonds in adsorbed furan, and three, two, one and zero for the most stable HF, DHF, TriHF and THF adsorbed structures, respectively. With increasing degree of hydrogenation, the binding strength of the closed shell intermediate decreases. THF is expected to desorb faster than DHF and furan. The relative energies with respect to the reactants (gas-phase furan and H2) decrease with increasing degree of hydrogenation, i.e., the hydrogenation reaction is thermodynamically favorable.
Activation energies in furan hydrogenation
For furan hydrogenation to HF, we calculated the transition states of the hydrogenation of α- and β-carbons. The hydrogenations of α- and β-carbon are endothermic with reaction energies of ∼0.5 eV. For α-carbon hydrogenation, we tested other reaction patterns. We chose the one with lowest transition state energy (TS-fh1 in Fig. 6) for further discussion. The hydrogenation of α-carbon has an energy barrier of 1.22 eV relative to the separately adsorbed furan and H. For β-carbon hydrogenation, we tested various reaction patterns and discuss the lower transition state energy (TS-fh2 in Fig. 6). The hydrogenation of β-carbon has an energy barrier of 1.18 eV, which is very close to that of α-carbon hydrogenation.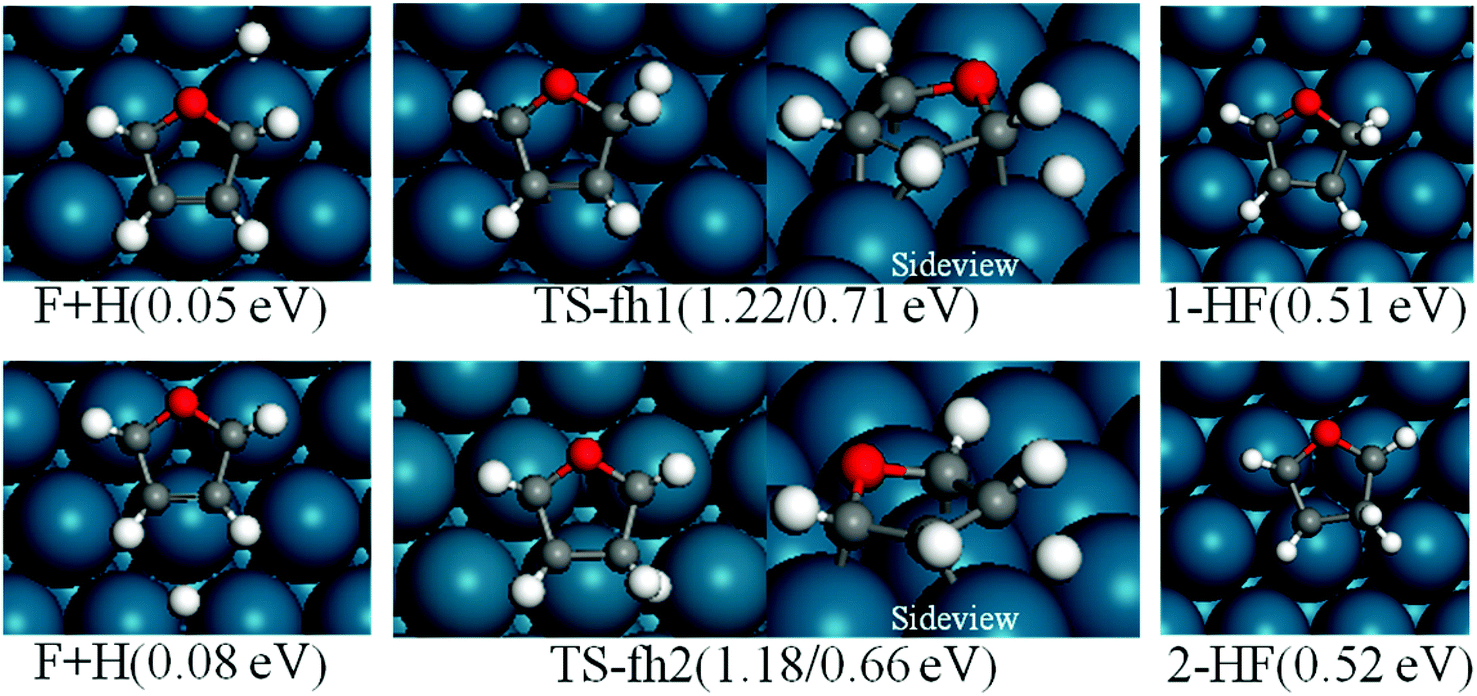 | ||
| Fig. 6 Structures of reactants, transition states and products of the first furan (F) hydrogenation. The energies in brackets for F + H refer to separately adsorbed furan and H, and the energies in brackets for the transition states are forward and backward energy barriers. | ||
The reaction energy (∼1.7 eV) of oxygen atom hydrogenation is even higher than the energy barrier (∼1.2 eV) of carbon atom hydrogenations. We expect that the energy barrier of the oxygen atom hydrogenation will be even higher. For this reason we did not search for the transition state for oxygen atom hydrogenation. In summary, the first hydrogenation step of furan produces either α-HF or β-HF. The reaction energies and barriers of the two pathways are very close.
Fig. 7 shows the structures and energies of HF, DHF and TriHF hydrogenation reactions. Since α-HF and β-HF are isoenergetic and can form via furan hydrogenation, we calculated the hydrogenation of both of them and found that the energy barriers are 0.67 and 1.33 eV, respectively. Therefore, the hydrogenation of the β-carbon of α-HF is more favorable for the formation of DHF. So far we have seen the pathway of furan hydrogenation to DHF, which is a stable molecule found in experiments. The reaction energies of α-HF and β-HF hydrogenation are nearly energy neutral (both 0.08 eV).
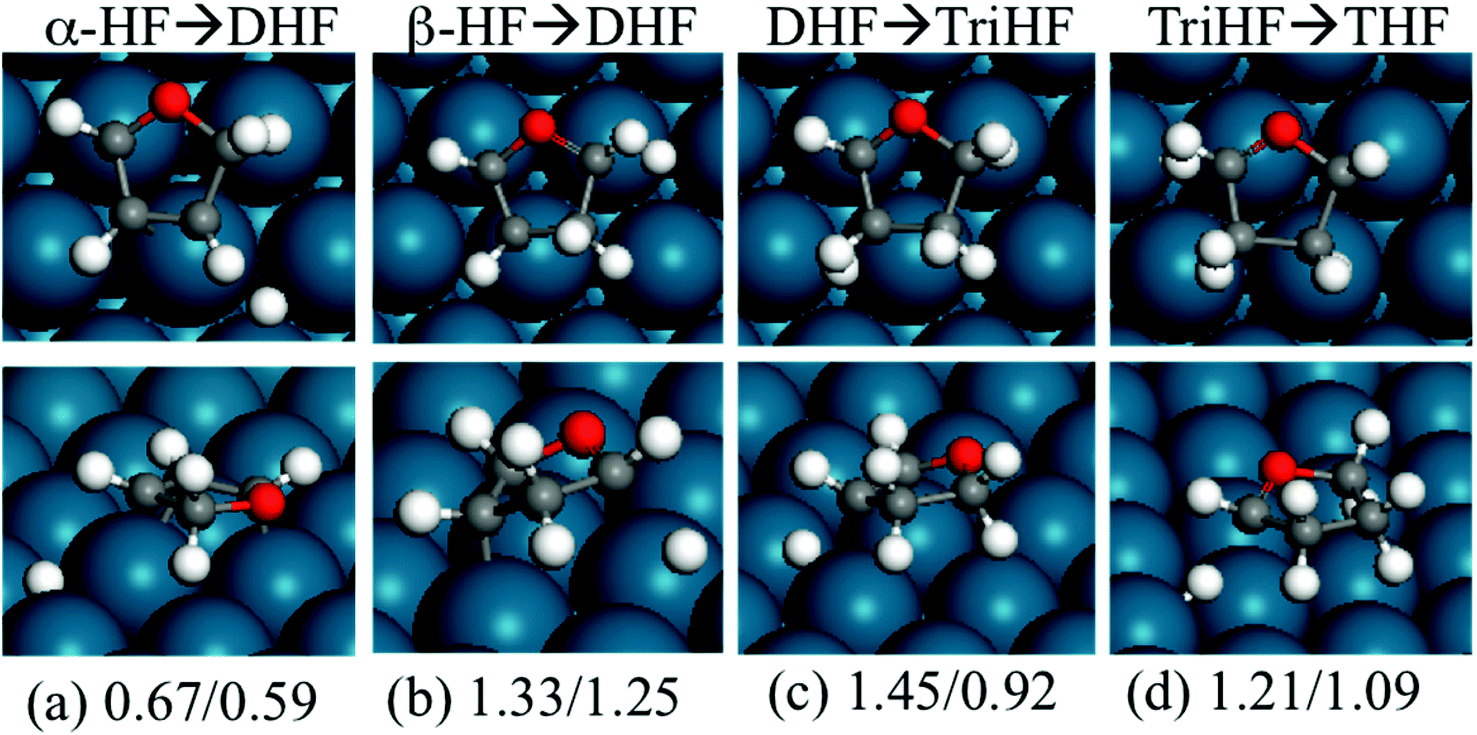 | ||
| Fig. 7 Top view (top row) and side view (bottom row) of the transition states of α-HF (a), β-HF (b), DHF (c) and TriHF (d) hydrogenation. The energies are forward and backward energy barriers (in eV) relative to the separately adsorbed HF + H, DHF + H and TriHF + H, respectively. | ||
It is noted that β-HF hydrogenation has a higher barrier than α-HF hydrogenation, although they have similar reaction energies. As mentioned above (Fig. 2(b) and 2(c)), α-HF has all atoms on the ring in the same plane while β-HF does not. For α-HF, during the bonding of hydrogen to the β carbon to form α,β-DHF, the β carbon slightly tilts up while the ring still remains planar. However, in the hydrogenation of β-HF, during the bonding of the hydrogen atom to the α carbon, the α carbon tilts up and the αC–O–βC–γC dihedral changes dramatically (from a partially zigzag ring to a planar ring). Although this reaction finally forms the same product as α-HF hydrogenation, there is a significant skeleton change in β-HF hydrogenation. This is the reason for the high energy of the transition state in β-HF hydrogenation.
For DHF hydrogenation to TriHF, the energy barrier is 1.45 eV, which is higher than the formation of DHF from HF. DHF hydrogenation has a higher reaction energy than HF hydrogenation (0.53 vs. 0.08 eV). The neutral reaction energy of HF hydrogenation and the high reaction energy of DHF hydrogenation rationalize the high stability of DHF. For TriHF hydrogenation to THF, the calculated forward and backward energy barriers are 1.21 and 1.09 eV, respectively.
In summary, most hydrogenation barriers are about 1.2 eV or slightly higher, except the HF hydrogenation to DHF, where the forward energy barrier of HF hydrogenation is only 0.67 eV. This small barrier indicates that HF is a reactive intermediate.
Ring opening intermediates
The structures of ring-opening products of furan, HF, DHF, TriHF and THF were optimized, and the results are shown in Fig. 8. Ring-opening products are all five-membered chain molecules. There are many adsorption conformations for each molecule, considering the possible bending degrees of the skeleton and adsorption sites. The optimization of the entire set of possible conformations of all molecules is computationally infeasible. Because the intermediates are produced from opening of rings (furan, HF, DHF, TriHF and THF), the bent conformations at proximal adsorption sites to their respective rings are the most important. These conformations can be formed with one elementary step (ring opening).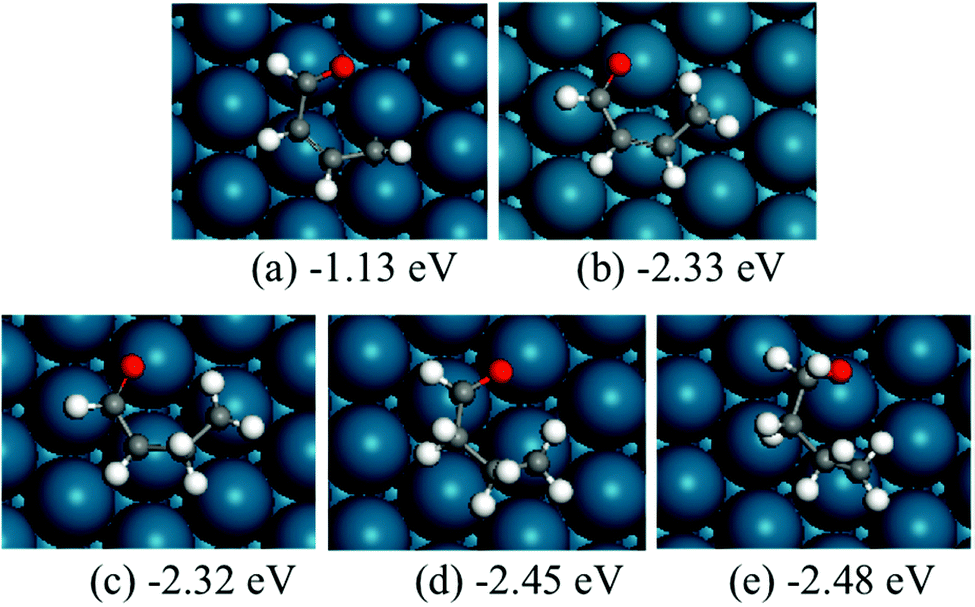 | ||
| Fig. 8 Structures and energies of the ring opening products of F (a), HF (b), DHF (c), TriHF (d) and THF (e). The energies refer to furan(g) and H2(g). | ||
The linear structures are stable for gas-phase molecules. The stabilities of the structures in Fig. 8 are also compared to straight linear conformations. The furan ring-opening product (CHCHCHCHO, Fig. 8a) is more stable than its straight-chain conformation. The linearization barriers and relative stabilities of the major product of ring-opening (ring-opened HF) are discussed in the ‘Reactions following ring opening’ subsection.
We tested various possible structures for C–C bond breaking. During the optimization, many of the initial structures form a ring. This is usually a sign that the ring-opened structures have very poor stabilities. Indeed, the optimized ring-opened furan (CHCHOCHCH) has high energy.
In summary, ring opening may start with the breaking of C–O bonds, according to thermodynamics. We further performed transition state searches to verify this finding.
Activation energies in ring opening
For ring opening of furan, there are three possible bond-breaking pathways, i.e., O–αC, αC–βC and βC–βC cleavage. We calculated the energy barrier of all the three possibilities (Fig. 9(a–c)). Both the C–C bond breaking have high energy barriers (2.37 and 1.84 eV) and are not competitive compared to C–O bond breaking (1.24 eV). | ||
| Fig. 9 Structures of the transition states of F (a–c), HF (d and e), DHF (f), TriHF (g) and THF (h) ring opening reactions. The numbers are forward and backward energy barriers in eV. | ||
For O–αC bond breaking, the initial state (Fig. 1) in this paper is adsorbed furan and the final state (Fig. 8a) has an α carbon moved to a neighboring bridge site, while the transition state has a structure in between. There is an apparent displacement of the reacting carbon atom, while other atoms stay at their adsorption sites. Xu reported a barrier of 0.82 eV using the PBE functional and the dimer method.5 Xu's initial state (furan) is the same as ours, but the final state is adsorbed at a totally different site compared to the initial state. In Xu's transition state structure, both the reacting carbon and oxygen at the two ends of the breaking C–O bond shift to the same direction and the whole skeleton slightly rotates counter clockwise, compared to the initial state. Due to these differences in the final and transition states, further comparison of the results is impossible.
For the ring opening of HF, we no longer considered the C–C bond breaking reactions, since they are expected to be slow compared to the C–O bond breaking. However, there are two types of C–O bonds in HF molecules, i.e., CH2–O and CH–O. We therefore calculated the CH2–O bond breaking to form CH2CHCHCHO (Fig. 9(d)) and the CH–O bond breaking to form CHCHCHCH2O (Fig. 9(e)). For the first reaction, the forward and backward energy barriers are 0.22 and 0.72 eV, respectively. The low energy barrier and the fact that this is an exothermic reaction indicate that the reaction is facile. Comparatively, the energy barrier and reaction energy of the second reaction are high (1.91 and 0.85 eV), which means that the CH–O bond breaking is not competitive compared to the CH2–O bond breaking.
Thus, hydrogenation has a large effect on ring opening. There are three additional effects of hydrogenation. First, the stability of the HF decreases compared to furan. Second, the –CH2– in adsorbed HF is saturated and cannot bind to the Pd(111) surface. Both of these effects increase the energy of the adsorbed HF, and in turn decrease the energy barrier. Third, CH2–O has a lower bond dissociation energy than CH–O, which means the C–O bond becomes less stable after hydrogenation of the α-carbon atom. This explains why the CH2–O breaking barrier is lower than that of CH–O breaking.
As mentioned before, DHF has α- and β-carbons hydrogenated. There are CH–O and CH2–O bonds in the molecule. We only calculated the bond breaking of the CH2–O bond based on the above discussion of the effect of hydrogenation on the activation of C–O bonds. The energy barrier of CH2–O breaking to form CH2CH2CHCHO is 2.11 eV (Fig. 9(f)), which is higher than that of CH2–O bond breaking in HF. This is not surprising, since DHF is a stable molecule without an unpaired electron. Experimentally, DHF is a side product in furan hydrogenation to THF.3
The reaction energy and energy barrier of TriHF ring opening are 0.21 and 1.77 eV, respectively. Similar to HF, TriHF is an instable intermediate.
The energy barrier of THF ring opening is high (2.32 eV), which is comparable to that of DHF. This indicates that once the THF has formed, it will not be easy to dissociate. The other reason for the high energy barrier of THF ring opening is the adsorbed pattern of the molecule. All carbon atoms in THF are saturated, resulting in weak adsorption of the molecule. Fig. 5 shows that the THF weakly binds to the surface, and the carbon atom is far away from the surface. This means that the Pd(111) surface can hardly dissociate the THF molecule.
In summary, the ring opening reaction happens via C–O bond cleavage, but not C–C bond cleavage (C–C bond cleavage energy barriers are 0.5–1.0 eV higher than those of C–O bond cleavage for furan). Hydrogenation of the CH–O to form CH2–O decreases the barrier of C–O cleavage, rendering α-HF more prone to ring-opening compared to furan or β-HF. However, with more hydrogenation of the ring, the intermediates lift up because of the weak interaction of saturated carbon with the Pd(111) surface. This weakens the catalytic effect of the Pd(111) surface. Therefore, only partial hydrogenation (furan to HF) promotes ring opening, while non-hydrogenated and over-hydrogenated rings are difficult to decompose.
Kinetic analysis
In order to obtain insight into the competition between hydrogenation and ring-opening reactions, we performed a simple kinetic analysis. As shown in Fig. 10(a), we first compared the energy barriers. The furan ring opening reaction has a slightly higher energy barrier than furan ring hydrogenation (0.06 eV). The energy barrier of HF ring opening is 0.45 eV lower than its hydrogenation. Based on energy barriers, one may draw the conclusion that HF does not get hydrogenated. However, experiments show THF as a product.3 For DHF and TriHF, the ring opening has a much higher energy barrier than hydrogenation. Therefore, once DHF forms, it will not decompose given sufficient hydrogen in the system.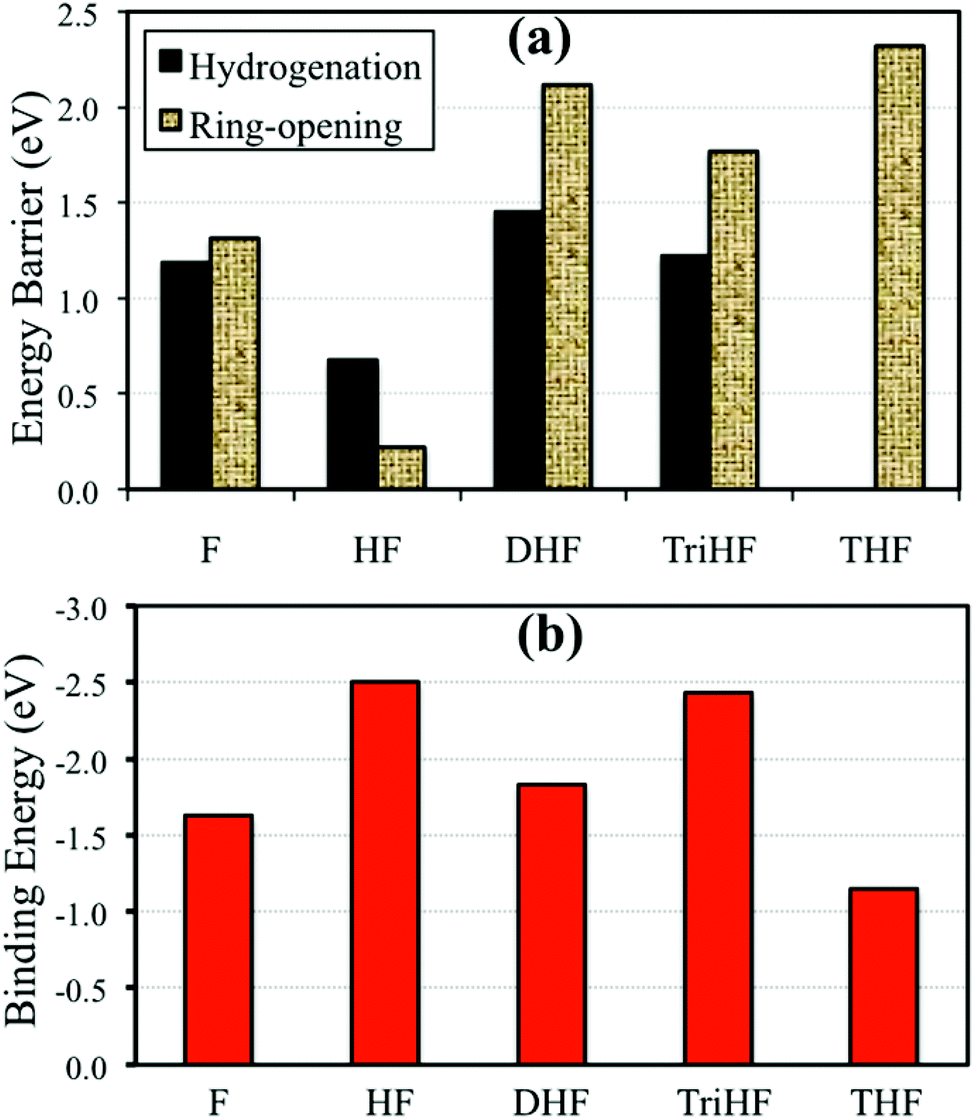 | ||
| Fig. 10 Forward energy barriers of hydrogenation and ring-opening reactions (a) and binding energies of hydrogenated derivatives of furan (b). | ||
Next we perform a further analysis by calculating kinetic rate constants and considering the surface coverages of the intermediates. For furan hydrogenation, as shown in eqn (1), the forward reaction rate is affected by the coverages of furan and hydrogen atoms. Ring opening is affected by the coverages of furan and empty sites (*), as shown in eqn (2). The reaction rates are then written as eqn (3) and (4), respectively. From eqn (5), we can calculate the ratio between the reaction rates of the hydrogenation and ring opening reactions, which reflects the competition between these reactions.
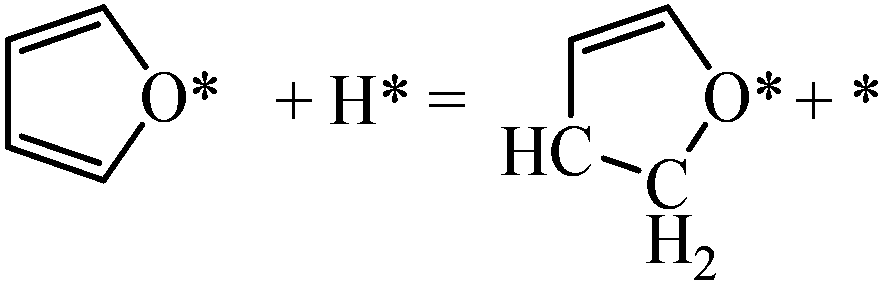 | (1) |
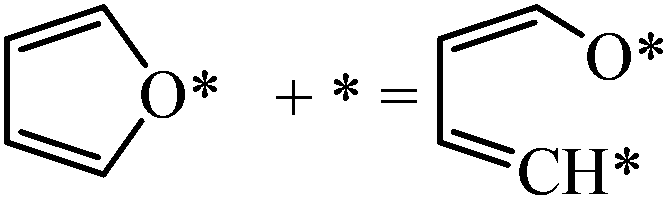 | (2) |
| rh = khθFθH | (3) |
| rro = kroθFθ* | (4) |
 | (5) |
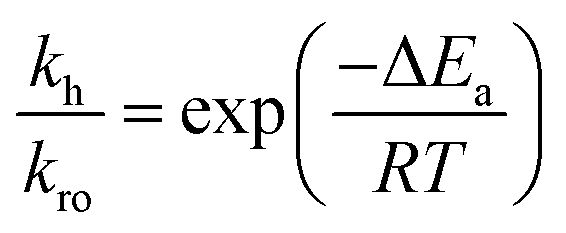 | (6) |
At a test temperature of 100 °C, the ratio between the rate constants (kh/kro) of furan hydrogenation and ring opening reactions is ∼6 when the difference between the energy barriers is only 0.06 eV. We also calculated the ratio between the coverages of adsorbed hydrogen atoms and empty sites via the dissociative adsorption Gibbs free energy of hydrogen. The coverage ratio was calculated based on the equilibrium of H2 adsorption on Pd(111). The energies of H2(g), H/slab and slab were calculated using the PBE-D3. The Gibbs free energy and its variation with the temperature of H2(g) were calculated with the Shomate equation where the parameters were fitted to experimental results.22 The contribution of solid states (slab and H/slab) to the Gibbs free energy was ignored. It was found that the coverage of empty sites is very low compared to that of hydrogen. The ratio between the coverage of H and empty sites is in the range of 5 × 106–1 × 108 for a H2 partial pressure of 1–10 bar (Fig. 11). Thus, the reaction rate of furan hydrogenation is much faster than ring opening based on eqn (5). Therefore, furan will not decompose with sufficient hydrogen in the system.
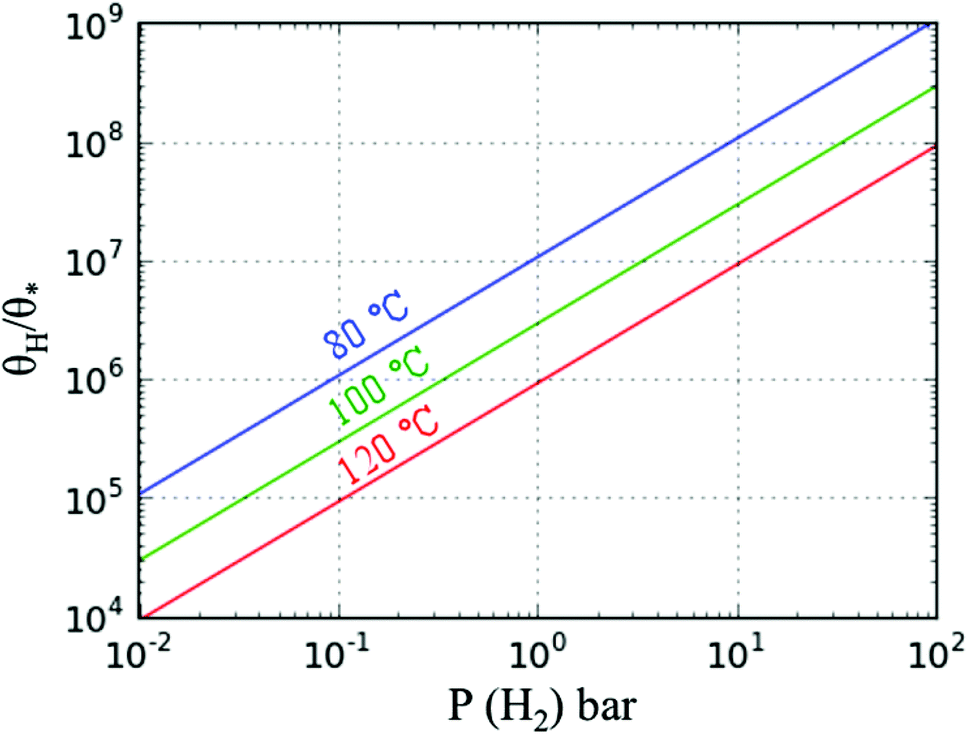 | ||
| Fig. 11 Ratio of the surface coverages of adsorbed H and empty sites* vs. partial pressure of H2 at the three temperatures shown. | ||
A similar analysis was also carried out for HF hydrogenation and ring opening reactions at 100 °C. The ratio between the rate constants of hydrogenation and ring opening is ∼8 × 10−7, based on the difference in energy barriers of 0.45 eV. At low H2 pressure, the reaction rate of hydrogenation is comparable to that of ring opening, based on eqn (5), i.e., ring opening is possible. When the H2 pressure is high, the ring-opening reaction is not favorable. Apart from the H2 pressure, according to experimental results, the ring opening reaction is more competitive at high temperatures. Since the theoretical rates of HF hydrogenation and ring opening reactions are close to each other, the selectivity is then sensitive to reaction conditions. However, for furan, DHF and TriHF, the selectivity is not sensitive to operating conditions, due to the big difference in energy barriers. Obviously, a more complete analysis requires a full microkinetic model along with the effect of the reversibility of all reactions.
In summary, the ring opening reaction becomes feasible when HF is formed via hydrogenation of furan, while hydrogenation is favorable for all intermediates because of the high H* surface coverage. The ring opening reaction is not competitive for other intermediates than HF.
Fig. 10(b) shows binding energies of various furan derivatives with increasing degree of hydrogenation. THF is the weakest adsorbed species among them (adsorption energy of −1.14 eV), which means that THF is easy to desorb from Pd(111). Desorption of DHF is difficult when compared to THF. This agrees with experiments showing that THF has a higher selectivity than DHF.3 HF and TriHF are not stable gaseous species and have high adsorption energies.
No direct correlation between the binding energies and the energy barriers was found (compare Fig. 10(a) and (b)). It was found that both HF and TriHF bind strongly to Pd(111). However, HF is much more reactive than TriHF and other intermediates; see energy barriers in Fig. 10(a). The low reactivity of TriHF may be due to its binding with Pd(111) via only one α-carbon. Taking Fig. 7(d) as an example, the structure of the transition state of TriHF hydrogenation to form THF has no Pd–C bond. This indicates that Pd(111) does not stabilize the transition state structure, which results in a high transition state energy. For comparison, Pd(111) stabilizes the transition state of α-HF hydrogenation (Fig. 7(a)) via two Pd–C bonds, and results in a low transition state energy. The reactivity of an intermediate cannot simply be identified by its own adsorption structure, as it depends on the type of transition state.
Reactions following ring opening
Since the ring opening reaction of HF has very low forward and backward energy barriers, one may expect that this step will be equilibrated, and reactions after it may be rate-determining steps for the production of ring-opened products. Three reactions were considered after the HF ring opening reaction, namely, the formation of linear conformation, the formation of butanol via further hydrogenation, and decarbonylation.Once the HF ring opens, it will not be easy to close back the ring, since the intermediate is believed to easily become linear. We explored this hypothesis by calculating the stability of linear conformation of CH2CHCHCHO, which is the ring-opened product of HF. The most stable linear conformation is 0.04 eV higher in energy than the curved conformation. The energy barrier for the formation of the linear conformation is 2.23 eV (Fig. 13). The high energy barrier can be rationalized because during the reaction, the hydrogen atom on β-carbon needs to rotate to the other side of the carbon atom, and also all carbon and oxygen atoms bind to the surface since one of them is saturated with hydrogen. Based on the above results, we propose that the ring-opened HF does not easily switch to a linear conformation. It is possible that the intermediate may still form due to the low (0.7 eV) backward reaction energy barrier and the high linear conformation energy barrier. Upon further hydrogenation, the intermediates may become linear more easily due to weaker binding with the surface. Fig. 14 indicates that hydrogenated furan derivatives, ring open products and hydrogenated ring open products tend to uplift from Pd(111). For intermediates of hydrogenation after ring opening, optimization calculations were started with conformations similar to the furan ring. The optimized 1-butanol tilts up into a spiral local minimum energy structure, which shows a clear tendency of linearization.
 | ||
| Fig. 12 Structures and energies in eV of ring-opened HF and the most stable intermediates for each hydrogenation step. The energies are relative to gas-phase furan and H2. For comparison, the adsorption energy of butanol is −0.82 eV. | ||
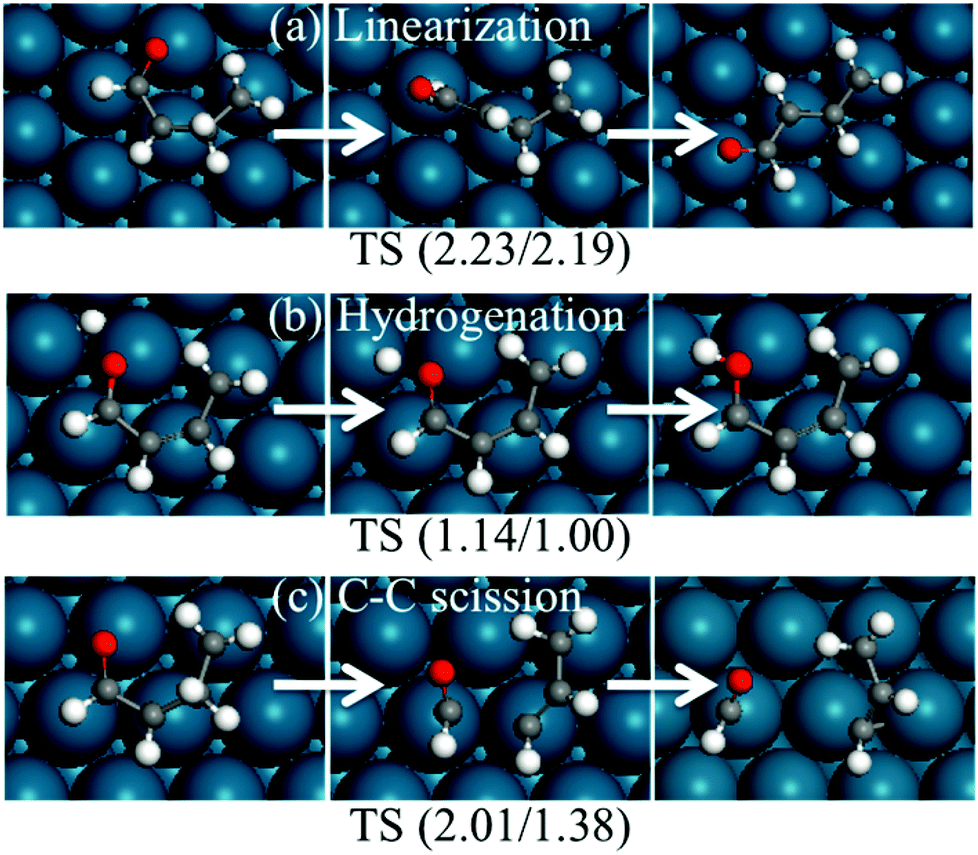 | ||
| Fig. 13 Structures of reactants, transition states and products of possible reactions after ring opening. The energies in parentheses are forward and backward energy barriers in eV. | ||
 | ||
| Fig. 14 Structures of intermediates along the pathways of furan hydrogenation (blue), ring opening (red) and hydrogenation (black) after ring opening. The figure shows the intermediates uplifting with increasing degree of hydrogenation. | ||
The second reaction considered after ring opening of HF is the hydrogenation of the intermediate to butanol, which has been reported as the byproduct of furan hydrogenation to THF on the Pt surface.3 The relative energies and structures of the possible products of the first hydrogenation of ring-opened HF are shown in Fig. 12. The energy barriers of the forward and backward reactions are 1.14 and 1.00 eV, respectively (Fig. 13). This indicates that the hydrogenation of ring-opened HF is kinetically feasible. The energy barriers of further hydrogenation after ring opening are comparable with those of the hydrogenation reactions of the ring to THF.
Finally, we considered the further decomposition (C–C bond breaking) for the formation of CO and small hydrocarbons. The transition state of the C3H4–CHO cleavage was calculated, and is shown in Fig. 13. The reaction energy and energy barrier of the C–C bond cleavage were found to be 0.62 and 2.01 eV, respectively. This indicates that the C3H4–CHO cleavage is not feasible. Xu reported that the reaction following the ring opening is dehydrogenation.
The hydrogenation of ring-opened HF has a moderate energy barrier. This reaction is feasible under mild reaction conditions. The barriers of the linearization and C–C scission of ring-opened HF are high, and they are not competitive compared to the hydrogenation.
Summary of reaction energetics and pathways
Fig. 15 shows the potential energy surface of the reaction network of furan hydrogenation. The reaction energies and energy barriers are listed in Table 1. In the potential energy surface, we have eliminated reaction steps with much lower reaction rates than their respective competitors. For example, furan direct ring opening has a comparable energy barrier to furan hydrogenation, but the estimated reaction rates from the simple kinetic analysis show that ring opening is very slow compared to hydrogenation.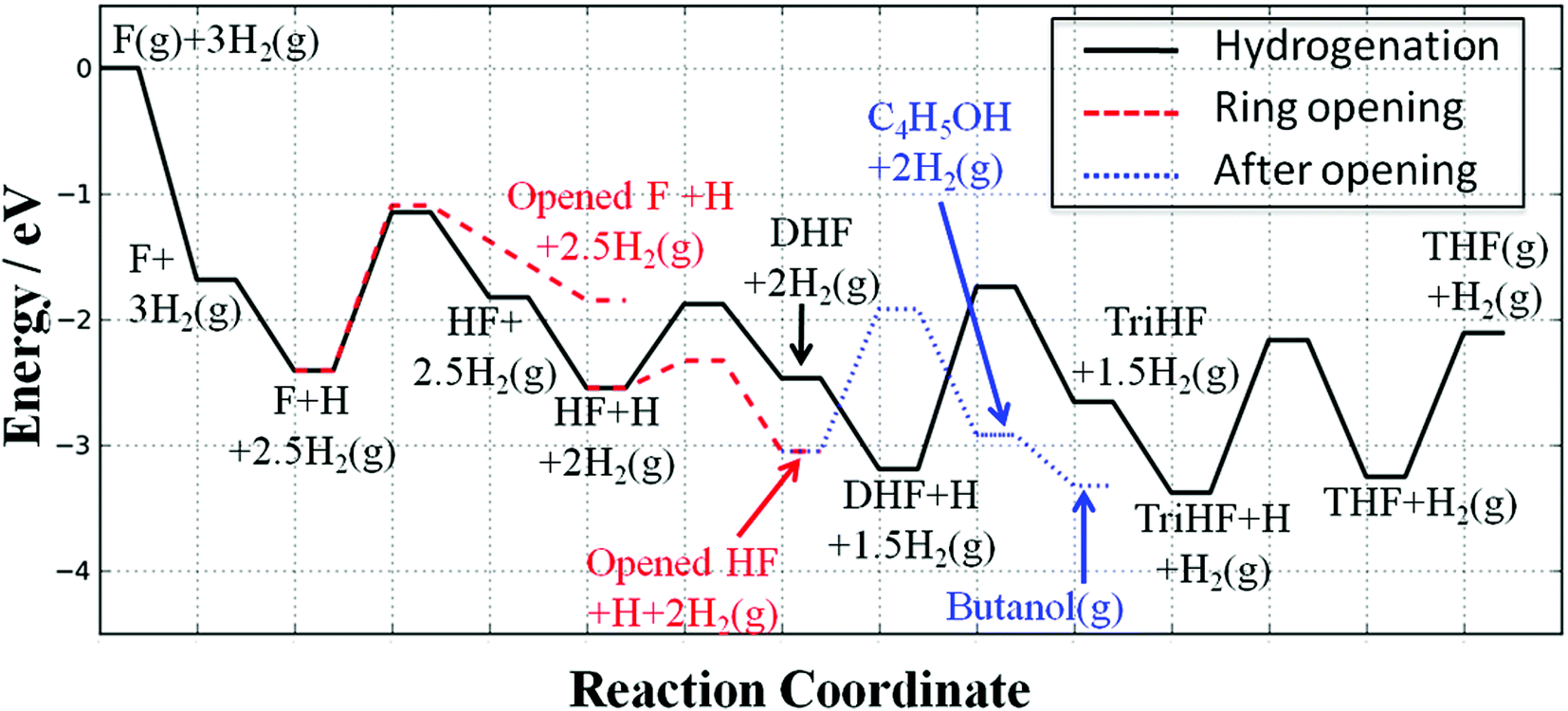 | ||
| Fig. 15 Potential energy surface of the furan hydrogenation and ring opening reactions. The energies are referenced to furan(g) + 3H2(g). For clarity, only major intermediates relevant to the respective elementary steps are shown in the labels. | ||
| Reaction | Chemical equation | E rxn/eV | E a(f/b)/eV |
|---|---|---|---|
| Gas phase reactions | |||
| Formation of THF |
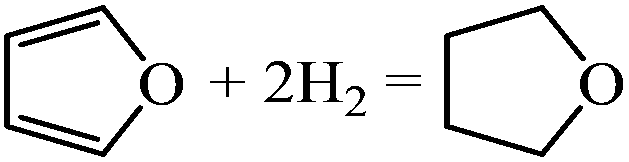
|
−2.12 | |
| Formation of DHF |
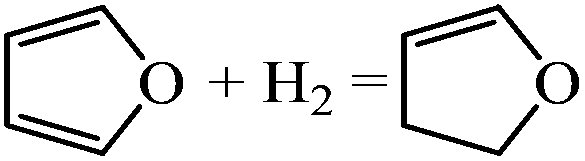
|
−0.66 | |
| Formation of 1-butanol |
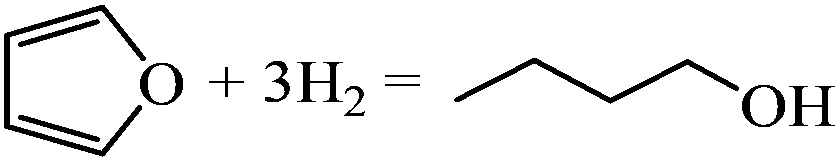
|
−3.34 | |
| Decarbonylation |
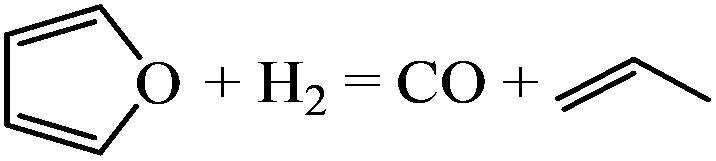
|
−0.11 | |
| Adsorptions/desorptions | |||
| Furan adsorption |
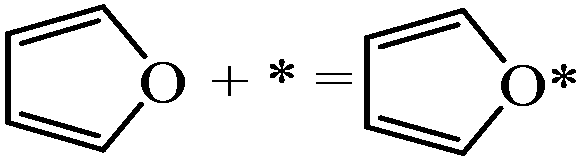
|
−1.62 | |
| Hydrogen adsorption | 0.5H2+* = H* | −0.72 | |
| DHF adsorption |
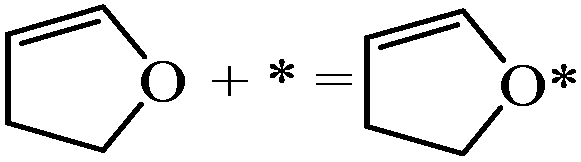
|
−1.82 | |
| THF adsorption |
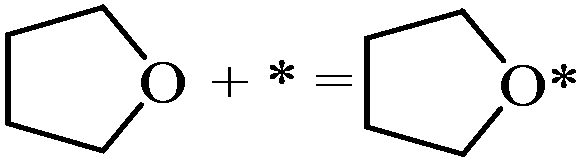
|
−1.14 | |
| 1-Butanol adsorption |
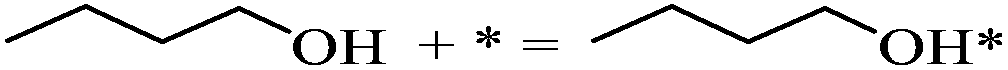
|
−0.83 | |
| Hydrogenations | |||
| F + H = α-HF |

|
0.51 | 1.22/0.71 |
| F + H = β-HF |
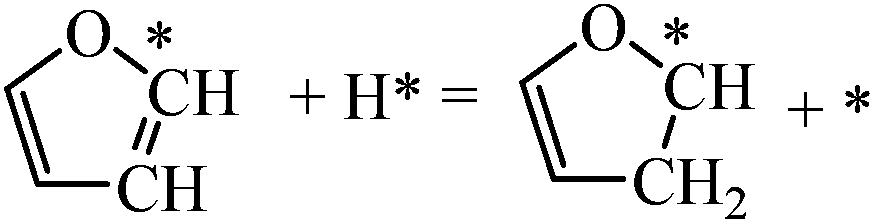
|
0.52 | 1.18/0.66 |
| α-HF + H = DHF |
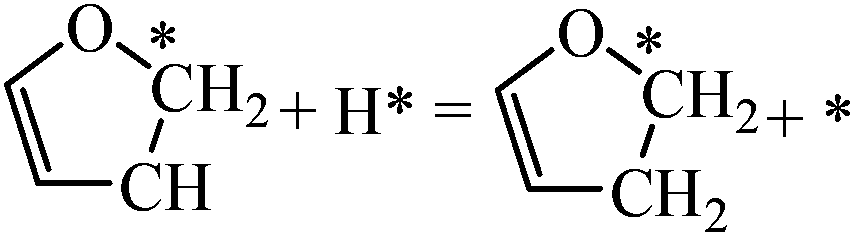
|
0.08 | 0.67/0.59 |
| β-HF + H = DHF |
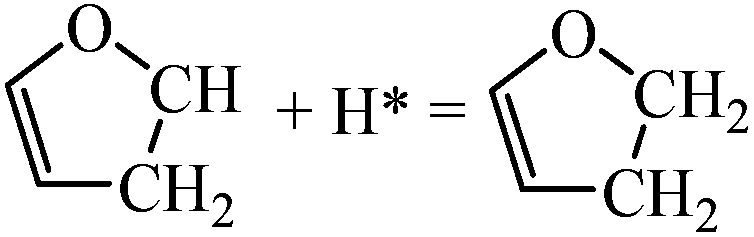
|
0.08 | 1.33/1.25 |
| DHF + H = TriHF |
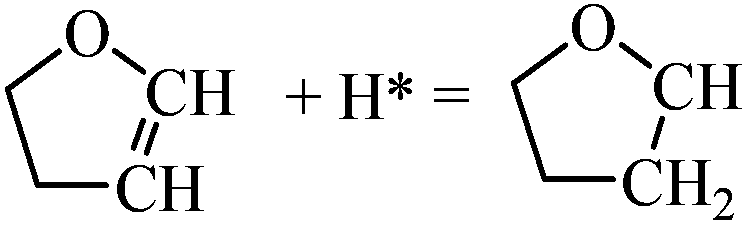
|
0.53 | 1.45/0.92 |
| TriHF + H = THF |

|
0.13 | 1.21/1.09 |
| Ring openings | |||
| Furan (CH–O scission) |
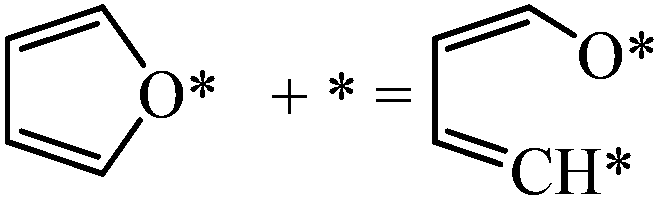
|
0.53 | 1.24/0.71 |
| HF (CH2–O scission) |
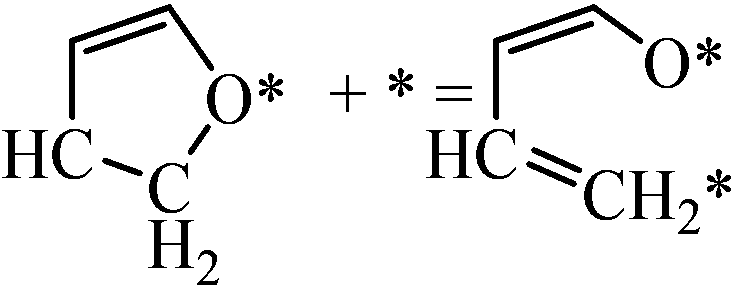
|
−0.50 | 0.22/0.72 |
| HF (CH–O scission) |
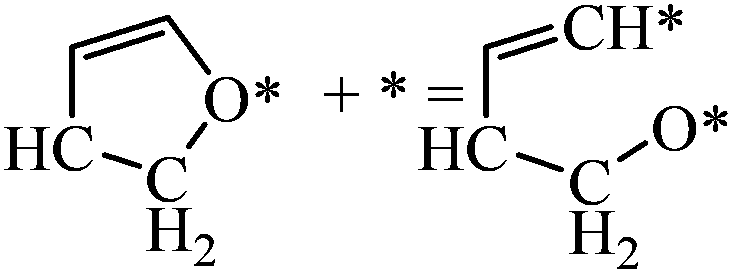
|
0.85 | 1.91/1.06 |
| DHF (CH2–O scission) |
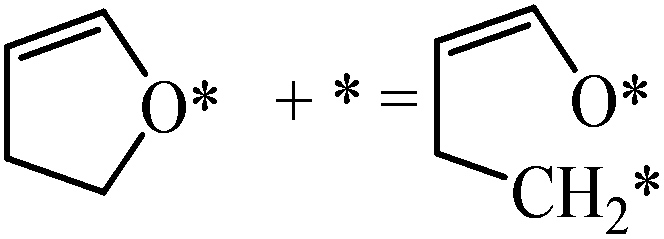
|
0.14 | 2.11/1.97 |
| TriHF (CH2–O scission) |
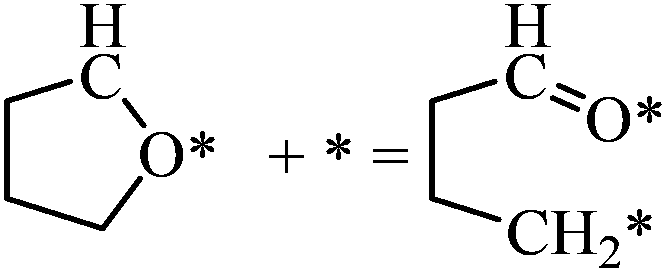
|
0.21 | 1.77/1.56 |
| THF (CH2–O scission) |
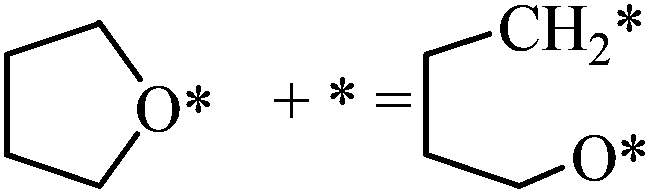
|
0.78 | 2.32/1.54 |
| After HF ring opening | |||
| Linearization |
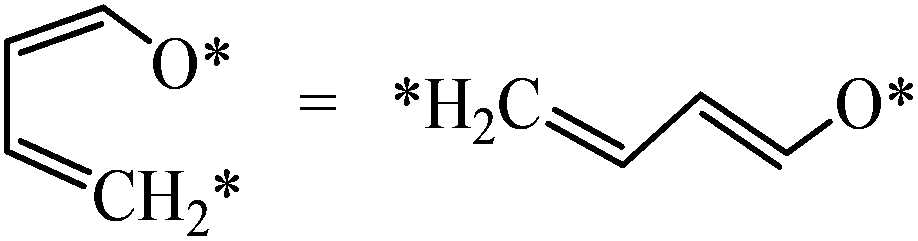
|
0.04 | 2.23/2.19 |
| Hydrogenation |
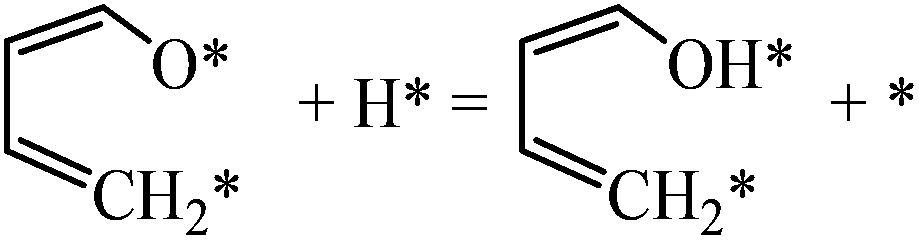
|
0.14 | 1.14/1.00 |
| Dissociation |
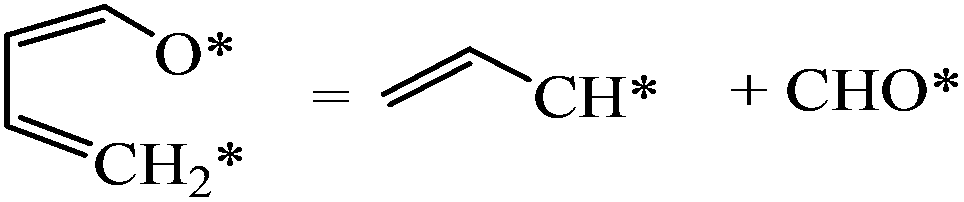
|
0.62 | 2.01/1.38 |
From the potential energy surface, we can clearly see that the hydrogenations of HF and TriHF are energetically neutral (slightly endothermic, ∼0.1 eV), while the hydrogenations of the furan and DHF are both endothermic (∼0.5 eV). The overall reaction is exothermic because of the consumption of H2, i.e., the dissociative adsorption of H2.
The gas-phase reaction energies in Table 1 indicate that 1-butanol is thermodynamically most favorable, followed by THF and DHF. The decarbonylation reaction leading to CO and propene has a very small reaction energy, i.e., this reaction is feasible but not thermodynamically competitive. By comparing the reaction energies, we can conclude that at high temperatures and/or with very active catalysts, 1-butanol will be the major product of furan hydrogenation. This agrees with the experimental results of furan hydrogenation on Pt where it has been reported that with increasing temperature, the major product becomes 1-butanol, instead of THF.
Conclusions
The pathways of hydrogenation and ring opening of furan and its hydrogenated derivatives to DHF and THF, 1-butanol and small hydrocarbons have been researched using density functional theory. While furan's most stable structure is flat with four hydrogen bonds among three Pd atoms, upon hydrogenation, the number of bonds with the Pd surface decreases and the intermediate lifts up into a bended configuration and eventually a configuration that interacts weakly with the surface.Hydrogenations on the α-C and β-C have similar barriers. However, ring distortion upon hydrogenation of the β-C renders subsequent hydrogenation on the α-C more difficult. As a result, THF is formed by sequential hydrogenation of carbon atoms on the furan ring in the order of α-C1, β-C2, β-C3 and α-C4. Due to the double bonds of the ring, ring opening via C–C bond scission of furan is unfavorable. In contrast, C–O bond scission is generally more likely but is energetically favored upon a single hydrogenation of the furan ring.
HF is a reactive intermediate toward both hydrogenation and ring opening. Based on reaction barriers, one would conclude that the HF ring opens under all conditions, in contradiction to experimental data. A simple kinetic analysis reveals that coverage effects are important and specifically the partial pressure of hydrogen is a critical factor in controlling the selectivity toward ring opening vs. further hydrogenation to DHF and THF. The open ring can also close back easily because of the low backward reaction energy barrier. The HF ring opening reaction may be in equilibrium, and the reaction following ring opening appears to be the determining step competing with HF hydrogenation to DHF. Therefore, HF is an important intermediate whose reactions determine the selectivity of the overall reaction. Our calculations support a shortcut strategy whereby understanding the selectivity of the furan family necessitates studies of the singly hydrogenated ring only.
The DHF is a stable intermediate and is observed as a side product in experiments. Once DHF is formed, ring opening is no longer likely because of the high energy barriers associated with ring opening of DHF, TriHF and THF.
Since the forward and backward reactions are generally fast and thermodynamics is dominant at high temperatures, 1-butanol becomes a major product with increasing temperature. At low temperatures, ring opening is slow, making THF the major product. The decarbonylation reaction leading to carbon monoxide and a small hydrocarbon, such as propene, is not competitive due to the high energy barrier but may occur at low H2 partial pressure. Our predicted selectivity trends agree well with experimental results.3
Acknowledgements
SW and DGV were partially supported by an NSF-CDI I grant (award number CBET-940768). VV acknowledges support from an NSF graduate fellowship and from the Catalysis Center for Energy Innovation (CCEI), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under award number DE-SC0001004. DFT calculations were performed in part using the TeraGrid resources provided by the Texas Advanced Computing Center (TACC) of the University of Texas at Austin.References
- H. A. Smith and J. F. Fuzek, J. Am. Chem. Soc., 1949, 71, 415–419 CrossRef CAS.
- S. D. Jackson, A. S. Canning, E. M. Vass and S. R. Watson, Ind. Eng. Chem. Res., 2003, 42, 5489–5494 CrossRef CAS.
- C. J. Kliewer, C. Aliaga, M. Bieri, W. Huang, C. Tsung, J. B. Wood, K. Komvopoulos and G. A. Somorjai, J. Am. Chem. Soc., 2010, 132, 13088–13095 CrossRef CAS PubMed.
- S. Sitthisa and D. E. Resasco, Catal. Lett., 2011, 141, 784–791 CrossRef CAS.
- Y. Xu, Top. Catal., 2012, 55, 290–299 CrossRef CAS.
- V. Vorotnikov, G. Mpourmpakis and D. G. Vlachos, ACS Catal., 2012, 2, 2496–2504 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 15 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter, 1999, 59, 1758 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- P. E. Blochl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter, 1994, 49, 16223–16233 CrossRef CAS.
- F. D. Murnaghan, Proc. Natl. Acad. Sci. U. S. A., 1944, 30, 244–247 CrossRef CAS.
- F. Birch, Phys. Rev., 1947, 71, 809–824 CrossRef CAS.
- R. Lamber, S. Wetjen and N. I. Jaeger, Phys. Rev. B: Condens. Matter, 1995, 51, 10968–10971 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- M. J. Knight, F. Allegretti, E. A. Kroger, M. Polcik, C. L. A. Lamont and D. P. Woodruff, Surf. Sci., 2008, 602, 2524–2531 CrossRef CAS PubMed.
- A. Loui and S. Chiang, Appl. Surf. Sci., 2004, 237, 555–560 CrossRef PubMed.
- R. I. Masel, Principles of adsorption and reaction on solid surfaces, Wiley, New York, 1996 Search PubMed.
- http://webbook.nist.gov/ .
| This journal is © The Royal Society of Chemistry 2014 |