
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protein–phenolic interactions and inhibition of glycation – combining a systematic review and experimental models for enhanced physiological relevance
A.
Vlassopoulos
,
M. E. J.
Lean
and
E.
Combet
*
Human Nutrition, School of Medicine, College of Medical, Veterinary & Life Sciences, University of Glasgow, Glasgow, G3 8SJ, UK. E-mail: Emilie.CombetAspray@glasgow.ac.uk; Fax: +44 (0)141 201 0711; Tel: +44 (0)141 201 0768
First published on 4th August 2014
Abstract
Background: while antiglycative capacity has been attributed to (poly)phenols, the exact mechanism of action remains unclear. Studies so far are often relying on supra-physiological concentrations and use of non-bioavailable compounds. Methods: to inform the design of a physiologically relevant in vitro study, we carried out a systematic literature review of dietary interventions reporting plasma concentrations of polyphenol metabolites. Bovine Serum Albumin (BSA) was pre-treated prior to in vitro glycation: either no treatment (native), pre-oxidised (incubated with 10 nM H2O2, for 8 hours) or incubated with a mixture of phenolic acids at physiologically relevant concentrations, for 8 hours). In vitro glycation was carried out in the presence of (i) glucose only (0, 5 or 10 mM), (ii) glucose (0, 5 or 10 mM) plus H2O2 (10 nM), or (iii) glucose (0, 5 or 10 mM) plus phenolic acids (10–160 nM). Fructosamine was measured using the nitro blue tetrazolium method. Results: following (high) dietary polyphenol intake, 3-hydroxyphenylacetic acid is the most abundant phenolic acid in peripheral blood (up to 338 μM) with concentrations of other phenolic acids ranging from 13 nM to 200 μM. The presence of six phenolic acids with BSA during in vitro glycation did not lower fructosamine formation. However, when BSA was pre-incubated with phenolic acids, significantly lower concentration of fructosamine was detected under glycoxidative conditions (glucose 5 or 10 mM plus H2O2 10 nM) (p < 0.001 vs. native BSA). Conclusion: protein pre-treatment, either with oxidants or phenolic acids, is an important regulator of subsequent glycation in a physiologically relevant system. High quality in vitro studies under conditions closer to physiology are feasible and should be employed more frequently.
Introduction
Protein glycation has been implicated in the development of several chronic diseases, particularly diabetic micro and macrovascular complications.1,2 The process of glycation, in diabetes, is mainly driven by the elevated blood glucose concentration through non-enzymatic condensation of a sugar molecule on a protein, lipid or DNA molecule.3,4 Measurement of glycated haemoglobin is the standard method for monitoring diabetes control5 and elevated levels are clearly related to tissue damage. Glycated albumin levels in plasma can vary widely, between 1% to up to 16% in normoglycemic individuals6,7 and the reasons for this wide range are not fully understood. In individuals free of diabetes a 30–50% higher risk of cardiovascular disease is suggested for every one unit higher HBA1c (%).8–10 Although in vitro studies have been employed to study glycation mechanisms, few have used physiologically relevant glucose concentrations. In vitro protein glycation does not easily occur with physiological concentrations of glucose, implying that another factor was necessary. We recently demonstrated that albumin glycation at physiological glucose concentrations (5 and 10 mM) was driven by oxidative stress, and that oxidised albumin is more susceptible to glycation than the native form of the protein.11 We have suggested that the reaction might be considered protein glycoxidation, rather than simply glycation. The study of early glycation, using fructosamine, is of importance for the translational value of the model (HbA1c is commonly used in clinical practice). As the first stable glycation product, its production rate is influential on total AGEs production.12Antioxidants, and (poly)phenols and their metabolites in particular, have been studied for their in vitro antiglycative properties.13–17 (Poly)phenols may offer protection by scavenging ROS produced during the glycation reaction, thereby slowing glycation and inhibiting the formation of AGEs.13,18 Another possible mechanism involves “physical” protection against glycation. This mechanism suggests that (poly)phenols have the capacity to bind to the protein molecule, most likely with a non-covalent bond, and in this way make glycation targets on the protein molecule (usually amino acids like lysine) inaccessible to take part in the glycation reaction.19,20
While these studies may hold value in food science (for example, reduction of AGE formation during cooking in the presence of polyphenols21), their physiological relevance to human health is sometimes questionable. (Poly)phenols are subject to extensive metabolism in the lumen (hydrolysis by the enterocytes' glucosidase system) and after absorption (glucuronidation/sulfation in the liver).22,23 Most high molecular weight dietary polyphenols have low bioavailability and even though aglycones may reach systemic circulation in small amounts, glycosides do not.23–26 Those which do not get absorbed instead accumulate in the colon lumen, where they are subject to bacterial degradation, leading to the formation of phenolic acids. Phenolic acids have a relatively higher bioavailability.22,27 In plasma, an increase in phenolic acid levels is seen 8–10 hours after ingestion, which represents the ‘colonic tide’ of (poly)phenol metabolites.22,28,29 Studies using foodstuff extracts and mixtures containing aglycones and glycosides thus do not replicate physiology25,30 when (poly)phenols in the circulation are mostly phase II metabolites and rarely exceed the 1 μM concentration (and if so, transiently).27,28 Finally, while single compound studies are informative and allow for mechanisms of action to be dissected, polyphenols and their metabolites are not found or consumed in isolation;23,31 they are, also, all consumed within complex food matrices with other nutrients that may modify absorption and metabolism.32,33
The present paper systematically reviews the literature reporting plasma levels of phenolic acids as key polyphenol metabolites, following ingestion of polyphenol rich food products (not under “acute” trial settings). The outcomes of this review are then used to test whether phenolic acids can inhibit the early stages of glycation under physiologically relevant experimental conditions, using the bench-top design we previously described.11
Material and methods
Systematic literature review
This review was conducted following the Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) guidelines. A literature study was carried out in PubMed® and ISI Web of Knowledge® for trials reporting plasma phenolic acid levels after a high polyphenol food/diet intake. The search was inclusive of all years up to February 2014. The following search terms were used to identify relevant studies: (phenol*, polyphenol*, phenolic acid*, fruit, vegetable, spice, cocoa, herb, juice, oil, wine, extract, tea or coffee), paired (bolean AND) with (feeding, trial, intervention, consumption OR supplementation). The wild-card term “*” was used to improve the sensitivity of the search by increasing the number of matches. The review was limited to studies utilising chromatographic techniques to identify (poly)phenolic compounds in serum or plasma. Studies on animals were excluded, as well as studies reporting cross-sectional data. Only controlled long-term feeding trials were reviewed. Studies were included in the review if absolute concentrations of phenolic acids were reported.Experimental procedures
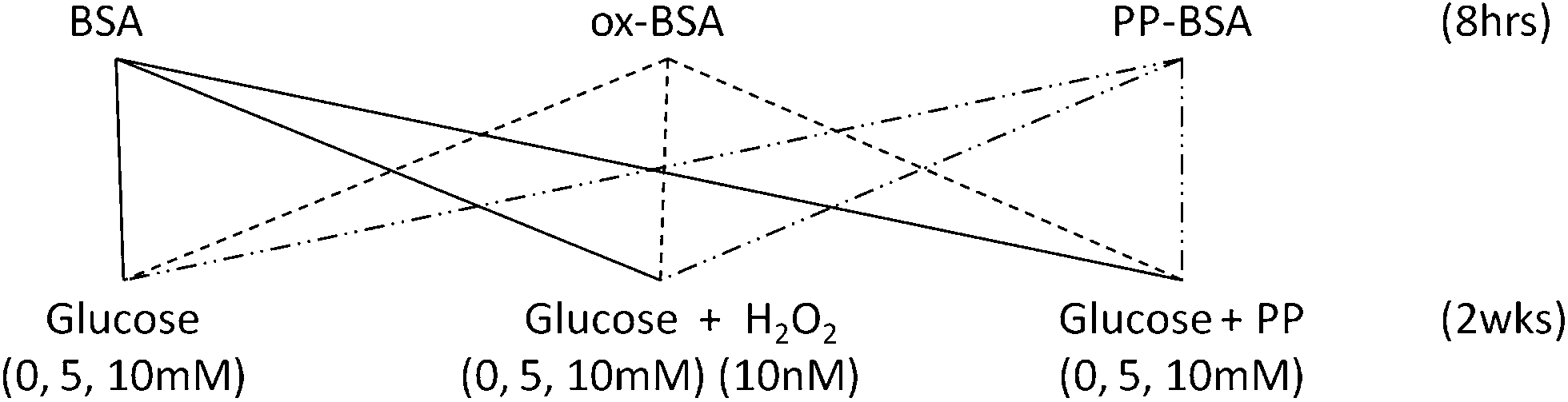 | ||
| Fig. 1 Experimental design for the study of the antiglycative potential of physiologically relevant phenolic acids. BSA: bovine serum albumin, ox-BSA: bovine serum albumin incubated with 10 nM H2O2, PP-BSA: bovine serum albumin incubated with phenolic acid mixtures; PP: caffeic acid 10 nM, p-coumaric acid 8 nM, vanillic acid 21 nM, protocatechuic acid 40 nM, 3-hydroxyphenyl acetic acid 160 nM and 3,4-dihydroxyphenyl acetic acid 40 nM. | ||
Fructosamine measurement
Fructosamine levels were measured at week 2 with the NBT assay, performed in microplates as described previously.34 Briefly, samples (30 μL) were added to sodium carbonate buffer (100 μL, 100 mM, and pH 10.8) with nitro blue Tetrazolium (0.25 mM). Microplates were incubated for 15 min at 37 °C and measured spectrophometrically against controls at 550 nm after 10 and 15 min of incubation. The difference between the two readings was used to calculate concentrations. The fructosamine analog 1-deoxy-1-morpholinofructose (DMF) was used as a standard. All fructosamine measurements were performed in duplicate. The potential interference of H2O2 and phenolic acids with NBT colorization was tested, with no evidence of interference detected. Standards and NBT reagents were made fresh every week and stored at −20 °C and 4 °C, respectively. All samples were stored at −20 °C prior to assay.Measurement of AGE fluorescence
AGE fluorescence was measured in diluted samples (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) at an excitation wavelength of 370 nm and an emission wavelength of 440 nm. AGE fluorescence was measured as arbitrary units and in duplicate using a SpectraMax M2 plate reader.
:5) at an excitation wavelength of 370 nm and an emission wavelength of 440 nm. AGE fluorescence was measured as arbitrary units and in duplicate using a SpectraMax M2 plate reader.
Protein structure analysis
Protein tryptophan fluorescence intensity was recorded with a Shimadzu RF-5301 PC Spectrofluorophotometer, using 5 nm excitation and 10 nm emission slit widths. Proteins at a concentration of 0.2 g L−1 in PBS were used after dialysis and prior to glycation. The UV spectra of the protein, recorded using a Perkin Elmer Lambda 25 UV/VIS Spectrometer, indicated a maximum absorbance at 277 nm for all proteins, which was thereafter used as the excitation wavelength for each protein. Changes in maximum emission were compared between proteins in a semi-quantitative manner and changes in the emission spectra due to pre-treatment were also compared qualitatively. Near UV Circular Dichroism spectra of 1.5 g L−1 protein and far UV Circular Dichroism spectra of 0.2 g L−1 protein were recorded in a 0.5 cm and 0.02 cm path length quartz cuvette, respectively, using a Jasco J-810 spectropolarimeter.35 The UniProt database was used to identify structures, sequences and drug binding location on the BSA molecule (http://www.uniprot.org/uniprot/P02769).Statistical analysis
All combinations of oxidative damage and glycation drivers were tested as six true replicates, according to the experiment. Assays were conducted in duplicate. Differences in fructosamine production between hydrogen peroxide levels were tested using a one-way ANOVA and Tukey's post-hoc tests, at each glucose level separately. The interaction between glucose levels and hydrogen peroxide levels, as well as the overall dose response effect, was studied using two-way ANOVA. Statistical analysis was performed using SPSS statistical software package version 19.0.0 (IBM, SPSS Software, Armonk, NY, USA).Results
Using the primary search terms, 436 papers were identified, with 40 excluded as duplicate entries. Titles from the remaining 396 articles were screened and 43 were excluded as not relevant to the topic of the review (mostly studies focusing on phenylalanine and other phenol ring-containing substances). During abstract screening, 246 reports were excluded from the analysis with the majority being in vitro or animal studies' reports, alongside with reports from cross-sectional studies. Review papers, conference proceedings, and reports written in languages other than English were also excluded. A total of 107 full papers were screened, leading to the exclusion of 28 additional reports. These reports either performed a qualitative analysis of metabolites in plasma or did not provide data on the concentrations of the metabolites measured in plasma. The reference list of each publication was screened in order to identify other publications from the same study, in case the data on the metabolite concentrations were published elsewhere. Finally, two reports36,37 were excluded as phenolic acids were reported as relative concentrations rather than absolute (Fig. 2).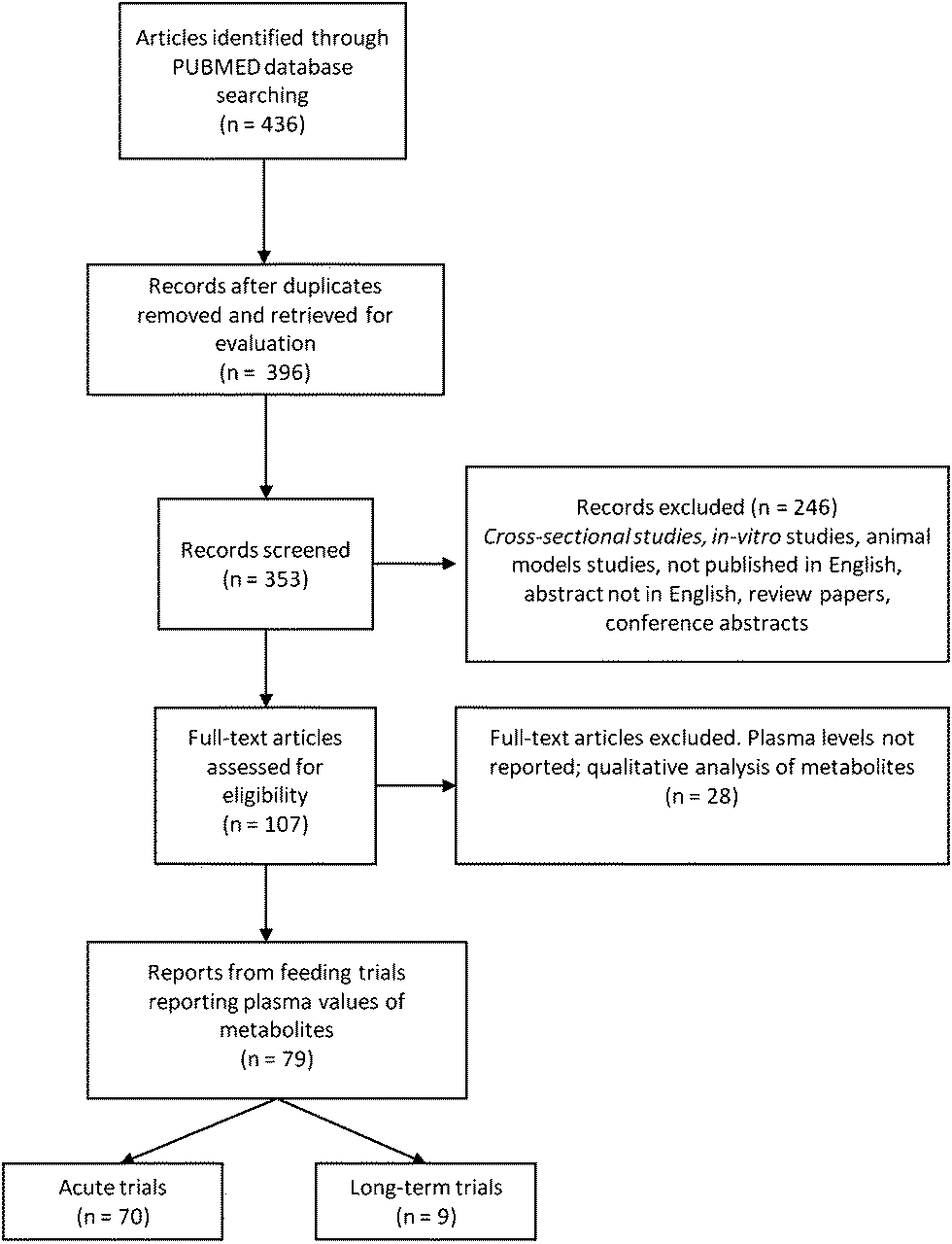 | ||
| Fig. 2 Flow diagram of the study selection process. | ||
A total of 79 published studies satisfied the inclusion criteria and reported plasma levels of polyphenol and polyphenol metabolites following a dietary intervention. From those, 9 reports focused on longer-term feeding studies while the rest covered acute changes in polyphenol levels following a single consumption of a test food (Fig. 1). Acute trials mainly focused on polyphenol bioavailability in the 5–24 hour window following ingestion, while longer-term feeding studies investigated changes in polyphenol metabolite levels after 5 days to 8 weeks supplementations38,39 (Table 1).
| Authors | Food | Study design | Duration | Metabolites | Hydrolysis | Comments |
|---|---|---|---|---|---|---|
| Heinrich et al. (2013) | Blue honeysuckle berry (165 grams per day) | Uncontrolled (n = 10) | 1 week | Below limit | No | Low polyphenol diet throughout |
| Henning et al. (2013) | Black tea (6 cups per day) | Non-blinded RCT, parallel (n = 46) | 3–6 weeks | 3-Hydroxyphenylacetic acid: 261 μM | Yes | No dietary info |
| 4-Hydroxyphenylacetic acid: 668 μM | ||||||
| 3,4-Dihydroxyphenylacetic acid: 117 μM | ||||||
| Hippuric acid: 2305 μM | ||||||
| Homovanillic acid: 176 μM | ||||||
| Henning et al. (2013) | Green tea (6 cups per day) | Non-blinded RCT, parallel (n = 47) | 3–6 weeks | 3-Hydroxyphenylacetic acid: 338 μM | Yes | No dietary info |
| 4-hydroxyphenylacetic acid: 798 μM | ||||||
| 3,4-Di hydroxyphenylacetic acid: 135 μM | ||||||
| Hippuric acid: 1950 μM | ||||||
| Homovanillic acid: 199 μM | ||||||
| Oliveras-Lopez et al. (2012) | Extra virgin olive oil as a fat replacement plus 50 mL raw | Non-blinded cross over (n = 20) | 4 weeks | Hydroxytyrosol: 487 nM | No | Habitual diet as control |
| Karlsen et al. (2010) | Bilberry juice (1 L diluted in water) | Non-blinded parallel RCT (n = 63) | 4 weeks | Quercetin: 43.6 nM | Yes | 3 weeks low antioxidant/berry diet |
| m-Coumaric: 12.8 nM | ||||||
| p-Coumaric: 27.4 nM | ||||||
| Protocatechuic: 99.4 nM | ||||||
| Kempf et al. (2010) | Coffee 4 or 8 cups per day (150 mL) | Single blind cross over (n = 47) | 30 days | 4 cups | Yes | No wash out, all subjects consumed 0 cups (1st month), 4 cups (2nd month), 8 cups (3rd month) |
| Caffeic acid: 38.3 μM | ||||||
| Dihydrocaffeic acid: 47.9 nM | ||||||
| m-Coumaric acid: 26.4 nM | ||||||
| Dihydro-3-coumaric: 716 nM | ||||||
| Ferulic: 55.1 nM | ||||||
| Isoferulic: 23.5 nM | ||||||
| Dihydroferulic: 93.9 nM | ||||||
| Dihydroisoferulic: 56.5 nM | ||||||
| Dimethoxycinnamic: 77 nM | ||||||
| 3-(3,4-Dimethoxyphenyl)-propionic: 203 nM | ||||||
| 8 cups | ||||||
| Caffeic acid: 62.2 μM | ||||||
| Dihydrocaffeic acid: 75.2 nM | ||||||
| m-Coumaric acid: 58.8 nM | ||||||
| Dihydro-3-coumaric: 1583 nM | ||||||
| Ferulic: 67.1 nM | ||||||
| Isoferulic: 49.8 nM | ||||||
| Dihydroferulic: 194.7 nM | ||||||
| Dihydroisoferulic: 90.6 nM | ||||||
| Dimethoxycinnamic: 177.7 nM | ||||||
| 3-(3,4-Dimethoxyphenyl)-propionic: 398 nM | ||||||
| Koli et al. (2010) | 100 g bilberries and nectar containing 50 g lingonberries/100 g black-currant-strawberry puree (80% black currant) | Non blinded RCT, parallel (n = 72) | 8 weeks | Quercetin: 40 nM | Yes | The two supplements were consumed on an alternate day basis |
| Caffeic acid: 100 nM | ||||||
| Protocatechuic acid: 120 nM | ||||||
| p-Coumaric: 15 nM | ||||||
| Vanillic: 70 nM | ||||||
| 3-(3-Hydroxyphenyl)-propionic: 800 nM | ||||||
| 3-Hydroxyphenylacetic: 275 nM | ||||||
| Homovanillic: 90 nM | ||||||
| 3,4-Dihydroxyphenylacetic acid: 140 nM | ||||||
| Urpi-Sarda et al. (2009) | Cocoa powder 40 grams per day with 500 mL skimmed milk | Non-blinded RCT cross-over (n = 42) | 4 weeks | 3,4-Dihydroxyphenylpropionic acid: 0.2 μM | Yes | No wash-out |
| 3-Hydroxyphenylpropionic acid: 0.23 μM | ||||||
| 3,4-Dihydroxyphenylacetic acid: 0.11 μM | ||||||
| 3-Hydroxyphenylacetic acid: 0.12 μM | ||||||
| Phenylacetic acid: 20.32 μM | ||||||
| p-Coumaric acid: 0.03 μM | ||||||
| Caffeic acid: 0.08 μM | ||||||
| Ferulic acid: 0.21 μM | ||||||
| Protocatechuic acid: 10.52 μM | ||||||
| Vanillic acid: 2.71 μM | ||||||
| 4-Hydroxybenzoic acid: 9.73 μM | ||||||
| 4-Hydroxyhippuric acid: 0.11 μM | ||||||
| 3-Hydroxyhippuric acid: 0.48 μM | ||||||
| Grimm et al. (2006) | Pine bark extract (200 milligrams per day) | Non-controlled (n = 5) | 5 days | Catechin: 170 nM | Yes | Measured 4 h after the last dose |
| Caffeic acid: 13.4 nM | ||||||
| Ferulic acid: 103 nM | ||||||
| Boyle et al. (2000) | Rutin supplement (500 milligrams per day) | Double blind RCT parallel (n = 16) | 6 weeks | Quercetin: 166 nM | Yes | Record flavonoid rich food |
| Kaempferol: 5.24 nM | ||||||
| Isorhamnetin: 9.49 nM |
Most studies (Table 1) focused on a single food product, from a diverse range including tea (black and green),40 coffee,41 olive oil,42 cocoa43 and berries, either as a juice44 or a mixed berry diet.39 Two studies used extracts instead of a food product.38,45 Regarding sample treatment prior to phenolic acids' measurements, only two of the studies measured free phenolic acids (i.e. without employing a prior hydrolysis step to derive aglycones from glucuronide and sulfate esters).42,46 One of these two studies did not detect sufficient phenolic acids in plasma for quantification, despite using a HPLC/MS (LCQ Fleet quadripole ion-trap MS) detection system,46 and the other focused on hydroxytyrosol as a marker of compliance (olive oil intake).42
The major phenolic acids identified in the studies were: 3-hydroxyphenylacetic acid (120 nM to 338 μM), 3,4-dihydroxyphenylacetic acid (110 nM to 135 μM), homovanillic acid (90 nM to 199 μM), m-coumaric acid (12.8–58.8 nM), p-coumaric acid (15–30 nM), caffeic acid (13.4 nM to 62.2 μM), protocatechuic acid (99.4 nM to 10.52 μM), ferulic acid (55.1–210 nM) and vanillic acid (70 nM to 2.71 μM). The compounds measured and identified all represent concentrations of aglycones after hydrolysis and not concentrations of free compounds.
A combination of 3-hydroxyphenylacetic acid (160 nM), 3,4-dihydroxyphenylacetic acid (40 nM), vanillic acid (20 nM), p-coumaric acid (10 nM), caffeic acid (10 nM) and protocatechuic acid (40 nM) was selected for the in vitro glycation of BSA. These phenolic acids represent only a selection of all the acids identified in plasma after feeding interventions, but they are the ones for which data on absorption and metabolism are more extensive. The glucuronidation of polyphenols and phenolic acids may impact on the antiglycative capacity of the molecules.13 These concentrations take into consideration the fact that phenolic acid levels in all studies in the literature review were measured after hydrolysis, therefore a factor was applied to estimate what percentage of the reported values would be true for non-conjugated phenolic acids.
This percentage was calculated based on findings from previous studies and more specifically, data from acute feeding interventions identified during the process of the present literature review. From acute feeding interventions measuring both the conjugated and non-conjugated form of phenolic acids, only 20–26% of the total caffeic found in plasma after coffee consumption is in free form.47,48 Similarly only 25% of the total vanillic acid, 40–50% of the total p-coumaric acid and 15–25% of the total ferulic acid are found as free phenolic acids.48,49 In contrast 4-hydroxyphenylacetic acid is less extensively metabolised (80% present as free form).49
The published reports were of variable methodological quality. Although the detection systems used for phenolic acids' measurement were based on mass spectrometry (MS, MS/MS, quadripole MS), some studies used less sensitive detection systems like ultraviolet, fluorimetric and electrochemical detectors.38,41,45 One of the major methodological limitations was that 7 out of 9 studies employed some form of hydrolysis prior to phenolic acids' determination. The measurement of aglycones following hydrolysis leads to a significant increase in the concentrations reported. The only study measuring phenolic acid concentrations without hydrolysis did not detect concentrations high enough for quantification even with a sensitive quadripole MS system.46
The variable duration and dose of the compounds tested also make comparisons between the studies available difficult. The compounds tested were delivered in various matrices, some as supplements,38,45 others as drinks,41,43,50,51 or whole foods46 and even as dietary patterns (combination of various food items).39 Such matrices will have a significant effect on bioavailability and potential long-term changes in absorption through changes in gut microbiota cannot be excluded/controlled for. The lack of successful control conditions is another point to be mentioned, as some studies either had no control groups or used a cross-over design without allowing for sufficient wash-out periods.38,41,43 When bioavailability of (poly)phenolic compounds is the main focus of the study the lack of a control group may be of lesser importance but not allowing for sufficient wash-out periods can introduce a carry-over effect between the interventions making it difficult to compare between doses/groups tested.
Finally, as (poly)phenolic compounds are nearly ubiquitous, controlling the effect of the background diet is important when studying bioavailability. In the studies reviewed, the background diet control was variable ranging from the subjects being requested to abstain from polyphenol sources throughout the experimental period46,51 to being asked to consume their habitual diet.42 One study requested from the participants to avoid food products similar to those provided during the study period39 and another gave no advice but requested from the participants to keep records of the flavonoid rich foods consumed during the study.45 In order to control the effect of background diet, Grimm et al. requested their volunteers to follow a flavonoid free diet 24 hours prior to blood sampling.38 Unfortunately three of the studies did not provide any information on whether and how they attempted to control the effect of the background diet.41,43,50 In this instance lack of adequate control of the background diet was not considered as a bias given the purpose of the review.
In vitro assessment of antiglycative capacity of phenolic acids
The antiglycative capacity of phenolic acids was investigated in a two dimensional design. Phenolic acids were tested for their capacity to reduce fructosamine production when (i) added in the reaction solution alongside glucose and H2O2 and (ii) when pre-incubated with albumin (BSA) prior to the glycation incubation.Effect of addition of hydrogen peroxide and phenolic acids in the reaction solution
Addition of 10 nM H2O2 in the reaction solution throughout the incubation has a significant but opposing impact on fructosamine production of both native and phenolic pre-incubated albumin. Incubation of native albumin in the presence of H2O2 led to significantly higher levels of fructosamine at 10 mM glucose compared to the glucose-only control (11% increase), whereas a reduction in fructosamine production was seen in the phenolic pre-incubated albumin (Table 2). A two-way ANOVA analysis showed that H2O2 affects fructosamine production of native and phenolic pre-incubated albumin independently of glucose (p < 0.001 for both albumin forms; H2O2 plus glucose vs. glucose only control).| Glucose level (mM) | Pre-treatment | ||
|---|---|---|---|
| Native mean (SD) | Pre-oxidised mean (SD) | Phenolic-rich mean (SD) | |
| a p < 0.05 vs native. | |||
| 0 | 0.22 (0.01) | 0.23 (0.02) | 0.23 (0.02) |
| 5 | 0.29 (0.01) | 0.32 (0.01) | 0.32 (0.02) |
| 10 | 0.36 (0.02) | 0.41 (0.03)a | 0.40 (0.03)a |
| 0 + H2O2 (10 nM) | 0.19 (0.04) | 0.20 (0.02) | 0.23 (0.01) |
| 5 + H2O2 (10 nM) | 0.32 (0.03) | 0.31 (0.03) | 0.28 (0.02)a |
| 10 + H2O2 (10 nM) | 0.40 (0.03) | 0.40 (0.04) | 0.31 (0.03)a |
| 0 + phenolic acids | 0.22 (0.01) | 0.22 (0.02) | 0.22 (0.01) |
| 5 + phenolic acids | 0.31 (0.02) | 0.30 (0.02) | 0.29 (0.02) |
| 10 + phenolic acids | 0.38 (0.04) | 0.38 (0.03) | 0.36 (0.03) |
In contrast addition of phenolic acids in the reaction solution throughout the incubation period had no significant effect on fructosamine production compared to glucose alone in any of the three albumin forms used (Table 2).
Effect of protein pre-treatment
Pre-oxidised and phenolic-pre-incubated albumin were more prone to glycation than the native molecule in the presence of 10 mM glucose (p = 0.001 and p = 0.02, respectively) (Table 2). This effect was independent of the glucose concentration, for both the pre-oxidised and the phenolic-pre-incubated albumin (p = 0.001, two-way ANOVA). In the presence of H2O2, native and pre-oxidised albumin showed similar glycation levels (p = 0.52; two-way ANOVA).Pre-incubation with phenolic acids, on the other hand, significantly reduced glycation in the presence of H2O2 (p = 0.001 vs. native; two-way ANOVA). This effect was seen at both 5 and 10 mM glucose (p = 0.01 and p < 0.001, respectively, vs. native albumin) (Fig. 3) and the glycation reduction was greater with increasing glucose levels (p < 0.001 for the interaction pre-treatment* glucose levels).
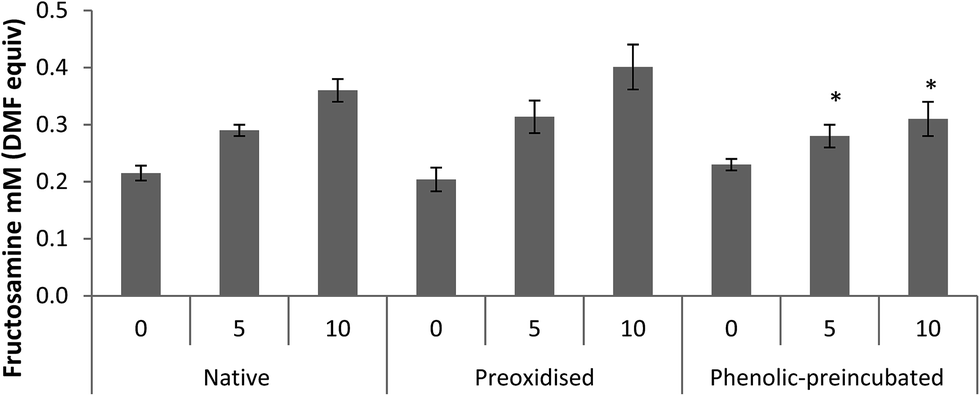 | ||
| Fig. 3 Fructosamine concentration (mM DMF equivalent) after two weeks incubation in the presence of glucose and H2O2 (10 nM) for native, pre-oxidised and phenolic-pre-incubated BSA (PP-BSA). Two-way ANOVA analysis showed a significant effect of phenolic-pre-incubation inhibiting glycation.*p < 0.05. | ||
Effect on AGE production
After two weeks incubation, AGE levels were not higher under any experimental conditions compared to the 0 mM glucose control, but for oxidised BSA exposed to 10 mM glucose with 10 nM H2O2 (p = 0.048, 205 ± 15.8 vs. 284 ± 76.1 AU; ox-BSA 0 mM glucose plus 10 nM H2O2vs. ox-BSA 10 mM glucose plus 10 nM H2O2).Effect of protein pre-treatment on protein structure and characteristics
The circular dichroism (CD) analysis showed no effect of the pre-treatment on the secondary protein structure (Fig. 5 & 6). However, exposure to 10 nM H2O2 for 8 h leads to a 25% decrease in tryptophan fluorescence. Pre-incubation with phenolic acids for 8 h resulted in a 38% reduction, indicative of protein–phenolic acid binding (Fig. 4).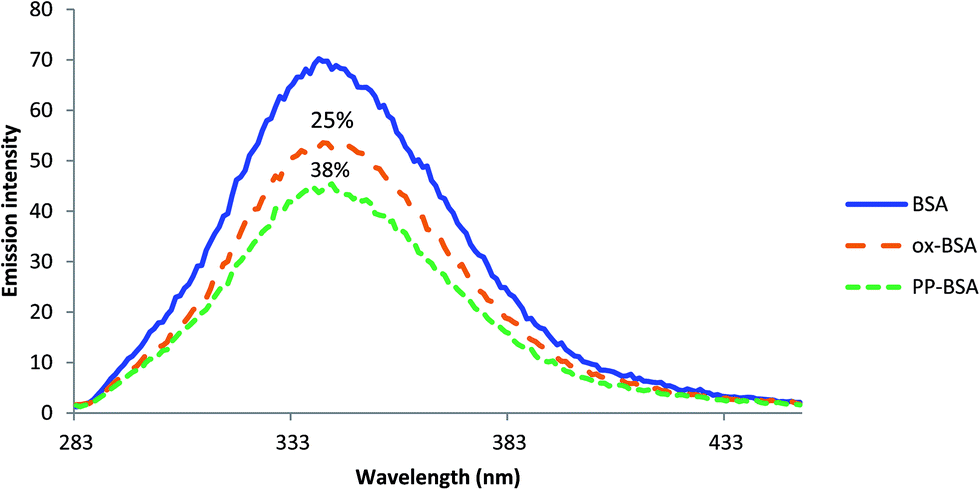 | ||
| Fig. 4 Emission spectra of 0.2 g L−1 BSA, ox-BSA and PP-BSA at λ = 277 nm showing the quenching effect of protein pre-treatment. Spectra were recorded at pH 7.4. | ||
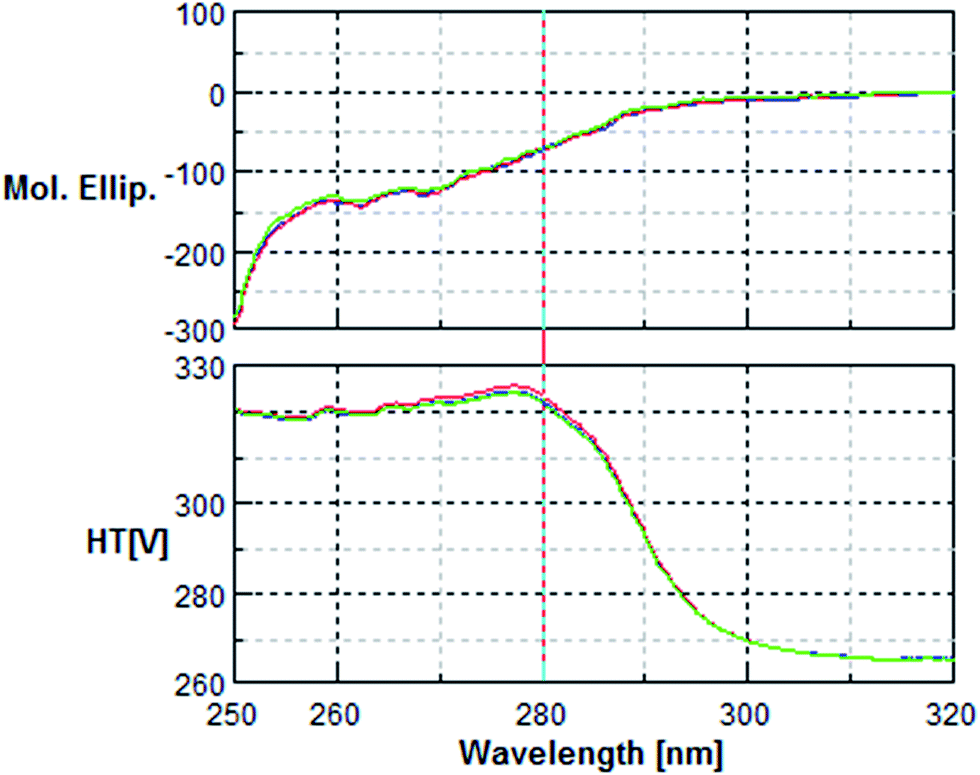 | ||
| Fig. 5 Near UV Circular Dichroism spectra of 1.5 mg mL−1 of native BSA (blue), oxidised BSA (green) and phenolic-treated BSA (red). Spectra were recorded in a 0.5 cm path length quartz cuvette using a Jasco J-810 spectropolarimeter. | ||
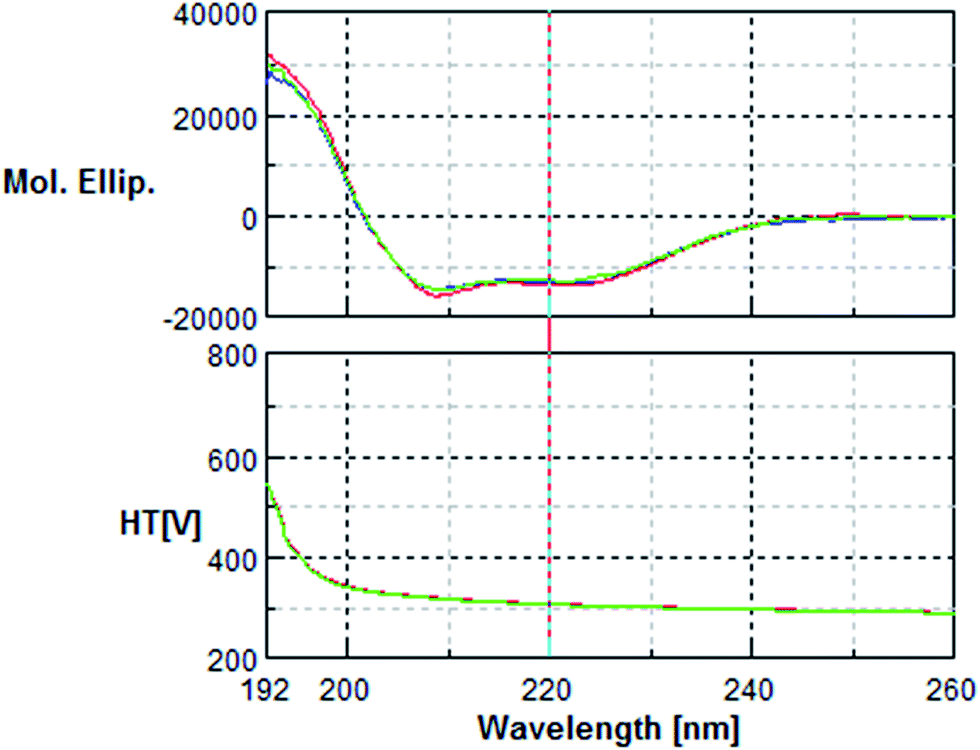 | ||
| Fig. 6 Far UV Circular Dichroism spectra of 0.2 mg mL−1 of native BSA (blue), oxidised BSA (green) and phenolic-treated BSA (red). Spectra were recorded in a 0.02 cm path length quartz cuvette using a Jasco J-810 spectropolarimeter. | ||
Discussion
Our mechanistic study results indicate that pre-treatment of albumin with phenolic acids inhibits fructosamine production, especially in the presence of oxidative stress or oxidative damage. This antiglycative activity was apparent when comparing the glycation achieved using phenolic-enriched albumin to those with the native and pre-oxidised BSA. A two-way ANOVA analysis showed that, in the presence of H2O2, albumin pre-incubated with phenolic acids had a significantly lower fructosamine content compared to the native BSA and the pre-oxidised BSA molecule. Phenolic acid pre-incubation only offered protection against fructosamine production in the presence of H2O2 10 nM: it provided no protection against glycation by glucose alone. In the presence of H2O2, the antiglycative activity of phenolic acid pre-incubation was greater with higher glucose levels (10% vs. 22.5% decrease at 5 and 10 mM glucose, respectively). No effect was seen for AGE production, with the exception of pre-oxidised BSA exposed to a combination of 10 nM H2O2 and 10 mM glucose. This may be due to the duration of the experiment, too short to lead to AGE formation in the given glucose concentrations. In contrast with most of the literature to date, which suggests that polyphenols and phenolic acids added to the incubation solution provide potent antiglycative activity,14,18,24,52,53 our results show that physical protection from glycation through protein–phenolic acid interaction is the most likely antiglycative mechanism especially in oxidative environments. In the previous investigation, concentrations used were non-physiological, with the lowest glucose concentration being 30 mM (>5–6 fold higher than normoglycaemia), generating higher glycation than our use of physiologically relevant 5 and 10 mM concentrations, representative of normoglycaemic and diabetic conditions, respectively.11 Our results suggest that pre-incubation of albumin with phenolic acids is the most likely mechanism to offer protection against glycation in a physiologically relevant system. Pre-incubation of BSA with either 10 nM H2O2 or phenolic acids did not affect the secondary structure of the molecule but both led to the reduction of tryptophan fluorescence. Tryptophan is an established oxidation target in the BSA molecule,54 and oxidation would modify its fluorescence. The BSA molecule has only two tryptophans: one inside a α-helix in domain I and another one inside the hydrophobic binding pocket of domain II.55,56 A proposed mechanism of action from our result involves steric hindrance, with phenolic acids binding in a BSA locum which includes a tryptophan and consequently preventing the amino acids in this locum to participate in oxidation and subsequent glycation reactions (since BSA oxidation increases its susceptibility to glycation).11The literature review highlighted that 3-hydroxyphenylacetic acid, 3,4-dihydroxyphenylacetic acid, homovanillic acid, m-coumaric acid, p-coumaric acid, caffeic acid, protocatechuic acid, ferulic acid and vanillic acid are most commonly detected in plasma after long-term feeding interventions. Concentrations of phenolic acids ranged between 13.4 nM and 338.0 μM. The majority of the studies measured phenolic acid levels as aglycones after hydrolysis of conjugated phenolic acids. This could have led towards a systematic overestimation of phenolic acid levels. Phenolic acids are subject to extensive rapid metabolism22,27 and since the chemical properties of their conjugates are often different, even opposite, from those of the aglycones13 it is important for in vitro studies to take their relative abundance into consideration. More in vivo studies, reporting phenolic acid levels as aglycones and conjugates separately, are needed.
Nonetheless, the results of the literature review showcase that the use of food extracts or food-derived polyphenol molecules in mechanistic studies focusing on human metabolism is likely to be of limited physiological relevance. The parent compounds commonly used for in vitro studies are rarely found in circulation, while metabolites like phenolic acids are found in potentially important concentrations in the nM to μM range. These metabolites are also not seen in isolation, but in fairly consistent combinations of phenylacetic acids, caffeic acid, vanillic acid and protocatechuic acid.
Despite increasing evidence that a very limited fraction of dietary polyphenols are absorbed, with low levels of metabolites circulating for a limited amount of time, in vitro models to-date utilise designs that make them irrelevant to physiology outside the gut lumen.30,57–59 Most of these reports used supra-physiological glucose and albumin concentrations30,57,59 as well as methanolic extracts of food products.60–62 While of importance for the food industry,63 they hold limited translational value for human health and may confuse our understanding of the role of glycation in health and disease. Here, a combination of caffeic acid, p-coumaric acid, vanillic acid, protocatechuic acid, 3-hydroxyphenyl acetic acid, and 3,4-dihydroxyphenyl acetic acid was chosen to replicate physiological conditions, according to the systematic review.
This study has several limitations. The in vitro design is in itself limiting and translation to physiology must still be cautious. Concentrations were chosen to mimic physiology for glucose, H2O2, and phenolic acids but this made it impossible to draw conclusions on the effect of individual phenolic acid on glycation. Not all our results fit a simple mechanistic explanation. It is intriguing that our results suggest that phenolic-pre-incubated albumin was more prone to glycation than native albumin, in the presence of 10 mM glucose. We can offer no plausible mechanism to explain this, which may have been a random or chance effect. Nonetheless, this study provides evidence to extend the published literature, and shows that research into the antiglycative capacity of phenolics using physiological concentrations (for both phenolics and glucose) is achievable and desirable.
Conclusions
Phenolic acids have the capacity to modulate early stages of protein glycation under normoglycaemic, physiologically relevant conditions. Incubation with phenolic acids prior to glycation significantly inhibits the process in the presence of oxidative stress. Designing in vitro studies with a high degree of physiological relevance is very important in order to reach biologically sound conclusions.Acknowledgements
AV is in receipt of a Yorkhill Children Charity PhD scholarship. The authors express their appreciation for the CD spectroscopy analysis to Dr Sharon Kelly. They would also like to thank Dr Stuart Caldwell, Dr Richard C. Hartley and Prof. Graeme Cooke for the help in the tryptophan fluorescence analysis.References
- C. Sabanayagam, G. Liew, E. S. Tai, A. Shankar, S. C. Lim, T. Subramaniam and T. Y. Wong, Diabetologia, 2009, 52, 1279–1289 CrossRef CAS PubMed.
- S. F. Yan, R. Ramasamy, Y. Naka and A. M. Schmidt, Circ. Res., 2003, 93, 1159–1169 CrossRef CAS PubMed.
- J. W. Baynes, Ann. N. Y. Acad. Sci., 2002, 959, 360–367 CrossRef CAS PubMed.
- P. J. Thornalley, Biochem. Soc. Trans., 2003, 31, 1341–1342 CrossRef CAS.
- A. American Diabetes, Diabetes Care, 2011, 34(suppl. 1), S62–S69 CrossRef PubMed.
- N. Shaklai, R. L. Garlick and H. F. Bunn, J. Biol. Chem., 1984, 259, 3812–3817 CAS.
- E. Selvin, M. W. Steffes, C. M. Ballantyne, R. C. Hoogeveen, J. Coresh and F. L. Brancati, Ann. Intern. Med., 2011, 154, 303–309 CrossRef PubMed.
- E. Selvin, M. W. Steffes, H. Zhu, K. Matsushita, L. Wagenknecht, J. Pankow, J. Coresh and F. L. Brancati, N. Engl. J. Med., 2010, 362, 800–811 CrossRef CAS PubMed.
- E. L. Barr, E. J. Boyko, P. Z. Zimmet, R. Wolfe, A. M. Tonkin and J. E. Shaw, Diabetologia, 2009, 52, 415–424 CrossRef CAS PubMed.
- E. Selvin, J. Coresh, E. Shahar, L. Zhang, M. Steffes and A. R. Sharrett, Lancet Neurol., 2005, 4, 821–826 CrossRef CAS.
- A. Vlassopoulos, M. E. Lean and E. Combet, Free Radical Biol. Med., 2013, 60, 318–324 CrossRef CAS PubMed.
- J. Venkatraman, K. Aggarwal and P. Balaram, Chem. Biol., 2001, 8, 611–625 CrossRef CAS.
- Y. Xie and X. Chen, Curr. Drug Metab., 2013, 14, 414–431 CrossRef CAS.
- H. Y. Kim and K. Kim, J. Agric. Food Chem., 2003, 51, 1586–1591 CrossRef CAS PubMed.
- T. H. Hou, J. P. Chung, S. S. Chen and T. L. Chang, Food Sci. Biotechnol., 2013, 22, 839–844 CrossRef PubMed.
- J. W. Wu, C. L. Hsieh, H. Y. Wang and H. Y. Chen, Food Chem., 2009, 113, 78–84 CrossRef CAS PubMed.
- W. Sompong, A. Meeprom, H. Cheng and S. Adisakwattana, Molecules, 2013, 18, 13886–13903 CrossRef CAS PubMed.
- C. H. Wu and G. C. Yen, J. Agric. Food Chem., 2005, 53, 3167–3173 CrossRef CAS PubMed.
- J. B. Xiao and G. Y. Kai, Crit. Rev. Food Sci. Nutr., 2012, 52, 85–101 CrossRef CAS PubMed.
- E. Verzelloni, D. Tagliazucchi, D. Del Rio, L. Calani and A. Conte, Food Chem., 2011, 124, 1430–1435 CrossRef CAS PubMed.
- X. Zhang, F. Chen and M. Wang, J. Agric. Food Chem., 2014, 62, 1643–1648 CrossRef CAS PubMed.
- A. Scalbert, C. Morand, C. Manach and C. Rémésy, Biomed. Pharmacother., 2002, 56, 276–282 CrossRef CAS.
- C. Manach, G. Williamson, C. Morand, A. Scalbert and C. Remesy, Am. J. Clin. Nutr., 2005, 81, 230S–242S CAS.
- T. Nagasawa, N. Tabata, Y. Ito, N. Nishizawa, Y. Aiba and D. D. Kitts, Mol. Cell. Biochem., 2003, 249, 3–10 CrossRef CAS.
- G. Williamson and C. Manach, Am. J. Clin. Nutr., 2005, 81, 243s–255s CAS.
- A. Crozier, I. B. Jaganath and M. N. Clifford, Nat. Prod. Rep., 2009, 26, 1001–1043 RSC.
- A. Scalbert and G. Williamson, J. Nutr., 2000, 130, 2073s–2085s CAS.
- C. Manach, A. Scalbert, C. Morand, C. Remesy and L. Jimenez, Am. J. Clin. Nutr., 2004, 79, 727–747 CAS.
- P. Vitaglione, G. Donnarumma, A. Napolitano, F. Galvano, A. Gallo, L. Scalfi and V. Fogliano, J. Nutr., 2007, 137, 2043–2048 CAS.
- C. Harris, A. Cuerrier, E. Lamont, P. Haddad, J. Arnason, S. L. Bennett and T. Johns, Plant Foods Hum. Nutr., 2014, 69, 71–77 CrossRef CAS PubMed.
- A. Crozier, I. B. Jaganath and M. N. Clifford, Nat. Prod. Rep., 2009, 26, 1001–1043 RSC.
- M. Serafini, M. F. Testa, D. Villaño, M. Pecorari, K. van Wieren, E. Azzini, A. Brambilla and G. Maiani, Free Radical Biol. Med., 2009, 46, 769–774 CrossRef CAS PubMed.
- W. Mullen, M.-A. Archeveque, C. A. Edwards, H. Matsumoto and A. Crozier, J. Agric. Food Chem., 2008, 56, 11157–11164 CrossRef CAS PubMed.
- J. R. Baker, D. V. Zyzak, S. R. Thorpe and J. W. Baynes, Clin. Chem., 1993, 39, 2460–2465 CAS.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, 2005, 1751, 119–139 CrossRef CAS PubMed.
- D. C. Nieman, N. D. Gillitt, A. M. Knab, R. A. Shanely, K. L. Pappan, F. Jin and M. A. Lila, PLoS One, 2013, 8, e72215 CAS.
- B. A. Stracke, C. E. Ruefer, A. Bub, S. Seifert, F. P. Weibel, C. Kunz and B. Watzl, Eur. J. Nutr., 2010, 49, 301–310 CrossRef CAS PubMed.
- T. Grimm, R. Skrabala, Z. Chovanova, J. Muchova, K. Sumegova, A. Liptakova, Z. Durackova and P. Hogger, BMC Clin. Pharmacol., 2006, 6, 4 CrossRef PubMed.
- R. Koli, I. Erlund, A. Jula, J. Marniemi, P. Mattila and G. Alfthan, J. Agric. Food Chem., 2010, 58, 3927–3932 CrossRef CAS PubMed.
- S. M. Henning, P. Wang, N. Abgaryan, R. Vicinanza, D. M. de Oliveira, Y. Zhang, R.-P. Lee, C. L. Carpenter, W. J. Aronson and D. Heber, Mol. Nutr. Food Res., 2013, 57, 483–492 CAS.
- K. Kempf, C. Herder, I. Erlund, H. Kolb, S. Martin, M. Carstensen, W. Koenig, J. Sundvall, S. Bidel, S. Kuha and J. Tuomilehto, Am. J. Clin. Nutr., 2010, 91, 950–957 CrossRef CAS PubMed.
- M.-J. Oliveras-Lopez, M. Innocenti, F. Martin Bermudo, H. Lopez-Garcia de la Serrana and N. Mulinacci, Eur. J. Lipid Sci. Technol., 2012, 114, 999–1006 CrossRef CAS PubMed.
- M. Urpi-Sarda, M. Monagas, N. Khan, R. Llorach, R. Ma Lamuela-Raventos, O. Jauregui, R. Estruch, M. Izquierdo-Pulido and C. Andres-Lacueva, J. Chromatogr. A, 2009, 1216, 7258–7267 CrossRef CAS PubMed.
- A. Karlsen, I. Paur, S. Bøhn, A. Sakhi, G. Borge, M. Serafini, I. Erlund, P. Laake, S. Tonstad and R. Blomhoff, Eur. J. Nutr., 2010, 49, 345–355 CrossRef CAS PubMed.
- S. P. Boyle, V. L. Dobson, S. J. Duthie, D. C. Hinselwood, J. A. M. Kyle and A. R. Collins, Eur. J. Clin. Nutr., 2000, 54, 774–782 CrossRef CAS.
- J. Heinrich, K. Valentova, J. Vacek, I. Palikova, M. Zatloukalova, P. Kosina, J. Ulrichova, J. Vrbkova and V. Simanek, J. Agric. Food Chem., 2013, 61, 4526–4532 CrossRef CAS PubMed.
- M. Nardini, E. Cirillo, F. Natella and C. Scaccini, J. Agric. Food Chem., 2002, 50, 5735–5741 CrossRef CAS PubMed.
- M. Nardini, M. Forte, U. Vrhovsek, F. Mattivi, R. Viola and C. Scaccini, J. Agric. Food Chem., 2009, 57, 2711–2718 CrossRef CAS PubMed.
- M. Nardini, F. Natella, C. Scaccini and A. Ghiselli, J. Nutr. Biochem., 2006, 17, 14–22 CrossRef CAS PubMed.
- S. M. Henning, P. W. Wang, N. Abgaryan, R. Vicinanza, D. M. de Oliveira, Y. J. Zhang, R. P. Lee, C. L. Carpenter, W. J. Aronson and D. Heber, Mol. Nutr. Food Res., 2013, 57, 483–492 CAS.
- A. Karlsen, I. Paur, S. K. Bohn, A. K. Sakhi, G. I. Borge, M. Serafini, I. Erlund, P. Laake, S. Tonstad and R. Blomhoff, Eur. J. Nutr., 2010, 49, 345–355 CrossRef CAS PubMed.
- N. Lunceford and A. Gugliucci, Fitoterapia, 2005, 76, 419–427 CrossRef PubMed.
- I. Bousova, J. Martin, L. Jahodar, J. Dusek, V. Palicka and J. Drsata, J. Pharm. Biomed. Anal., 2005, 37, 957–962 CrossRef CAS PubMed.
- S. Guedes, R. Vitorino, R. Domingues, F. Amado and P. Domingues, Rapid Commun. Mass Spectrom., 2009, 23, 2307–2315 CrossRef CAS PubMed.
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A. J. Stewart, M. Chruszcz and W. Minor, Mol. Immunol., 2012, 52, 174–182 CrossRef CAS PubMed.
- N. Tayeh, T. Rungassamy and J. R. Albani, J. Pharm. Biomed. Anal., 2009, 50, 107–116 CrossRef CAS PubMed.
- W. Daiponmak, C. Senakun and S. Siriamornpun, Int. J. Food Sci. Technol., 2014, 49, 1805–1810 CrossRef CAS PubMed.
- H. Ito, P. Li, M. Koreishi, A. Nagatomo, N. Nishida and T. Yoshida, Food Chem., 2014, 152, 323–330 CrossRef CAS PubMed.
- P. G. Dorsey and P. Greenspan, J. Med. Food, 2014, 17, 447–454 CrossRef CAS PubMed.
- S.-C. Ho, P.-W. Chang, H.-T. Tong and P.-Y. Yu, Int. J. Food Prop., 2013, 17, 617–628 CrossRef.
- G. Cervato, M. Carabelli, S. Gervasio, A. Cittera, R. Cazzola and B. Cestaro, J. Food Biochem., 2000, 24, 453–465 CrossRef CAS PubMed.
- P. Hegde, G. Chandrakasan and T. Chandra, J. Nutr. Biochem., 2002, 13, 517 CrossRef CAS.
- X. Zhang, F. Chen and M. Wang, J. Agric. Food Chem., 2014, 62, 1643–1648 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |