 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of chicken extract on ERK/CREB signaling is ApoE isoform-dependent
Shan-May
Yong
,
Qi-Rui
Ong
,
Bei-En
Siew
and
Boon-Seng
Wong
*
Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, 2 Medical Drive MD9, Singapore 117597. E-mail: bswong@nus.edu.sg; Fax: +65-67788161; Tel: +65-65167617
First published on 30th June 2014
Abstract
It is unclear how the nutritional supplement chicken extract (CE) enhances cognition. Human apolipoprotein E (ApoE) can regulate cognition and this isoform-dependent effect is associated with the N-methyl-D-aspartate receptor (NMDAR). To understand if CE utilizes this pathway, we compared the NMDAR signaling in neuronal cells expressing ApoE3 and ApoE4. We observed that CE increased S896 phosphorylation on NR1 in ApoE3 cells and this was linked to higher protein kinase C (PKC) activation. However, ApoE4 cells treated with CE have lowered S897 phosphorylation on NR1 and this was associated with reduced protein kinase A (PKA) phosphorylation. In ApoE3 cells, CE increased calmodulin kinase II (CaMKII) activation and AMPA GluR1 phosphorylation on S831. In contrast, CE reduced CaMKII phosphorylation and led to higher de-phosphorylation of S831 and S845 on GluR1 in ApoE4 cells. While CE enhanced ERK/CREB phosphorylation in ApoE3 cells, this pathway was down-regulated in both ApoE4 and mock cells after CE treatment. These results show that CE triggers ApoE isoform-specific changes on ERK/CREB signaling.
1. Introduction
Chicken extract (CE) is widely consumed as a nutritional supplement in many places1,2 and it is composed of water soluble substances extracted from gently cooked chicken muscle.3 This supplement has been reported to enhance cognition,4–6 by improving attention and working memory during mental task performance.7 However, little is known about the cellular mechanisms underlying this function.The human apolipoprotein E (ApoE) gene is genetically linked to cognitive function in ageing and diseases.8–14 This gene is located on chromosome 19 encoding a 35 kDa protein15 that exists in 3 isoforms, E2, E3 and E4.16,17 These isoforms differ by amino acid substitutions at two positions (residues 112 and 158).18
ApoE is synthesized in various organs15 and high expression is detected in the liver19 and in the brain.20 Non-demented aged ApoE4 carriers experience faster cognitive decline.21–23 Similar impairment is also observed in mice expressing human ApoE4.24,25 This ApoE isoform-dependent effect on cognition is linked to the N-methyl-D-aspartate receptor (NMDAR).26–30
NMDARs are glutamate-gated ion channels comprising an assembly of three major subunits31–33 that are pivotal for learning and memory, and the induction of long-term synaptic plasticity.34–37 NMDAR1 (NR1) is the obligatory subunit of the heterotetramer receptor.31–33 Changes in NMDAR subunits' composition and localization have been detected during ageing.38–41 The NMDAR function is mediated by calcium (Ca2+) ions leading to the activation of the transcription factor cAMP/calcium-dependent response element binding partner (CREB) inside the cells.31,37,42,43 The function of NMDAR is closely associated with AMPAR activation.44,45 Neurons expressing ApoE4 were reported to have lower NMDAR and AMPAR functions,46 leading to lower LTP.30
To understand if CE effect on cognition involves the ApoE–NMDAR pathway, we have conducted this study to compare NMDAR signaling in cells expressing ApoE3 and ApoE4.
2. Materials and methods
2.1. Antibodies and chemicals
All chemicals used in this study were purchased from Sigma-Aldrich. The chicken extract (CE) powder used in this study was provided by Dr. Paramjeet Singh (Cerebos Pacific Ltd) and this health supplement is available under the trade name Brands' Essence of Chicken (BEC). The chemical composition of CE has been characterized,47 and commercial CE preparation was ∼100 μg ml−1.48 CE solution was prepared6 in PBS and stored in aliquots at −80 °C.The primary antibodies used in this study were anti-huApoE (Calbiochem, Cat#178479), anti-NR1 (Cell Signal. Tech., Cat#5704), anti-pNR1(S896) (Cell Signal. Tech., Cat#3384), anti-pNR1(S897) (Cell Signal. Tech., Cat#3385), anti-GluR1 (Cell Signal. Tech., Cat#8850), anti-pGluR1(S831) (Santa Cruz Biotech., Cat#16313), anti-pGluR1(S845) (Cell Signal. Tech., Cat#8084), anti-CaMKII (Cell Signal. Tech., Cat#3357), anti-pCaMKII(T286) (Cell Signal. Tech., Cat#3361), anti-ERK1/2 (Cell Signal. Tech., Cat#9258), anti-pERK1/2 (Invitrogen, Cat#44689G), anti-CREB (Cell Signal. Tech., Cat#9197), anti-pCREB(S133) (Cell Signal. Tech., Cat#9191), anti-PKA-Cα (Cell Signal. Tech., Cat#5842), anti-pPKA-Cα(T197) (Cell Signal. Tech., Cat#5661), anti-PKCα (Abcam, Cat#137807), and anti-pPKCα(T497) (Abcam, Cat#76016).
2.2. Plasmids, cell culture and transfection
The cDNA for human ApoE3 was purchased from Invitrogen, and the ApoE4 cDNA was kindly provided by Drs Katherine Youmans and Mary Jo LaDu (University of Illinois, Chicago, USA). The human ApoE3 and ApoE4 sequences were cloned into the expression vector pcDNA6.2-DEST (Life Technologies).The experimental protocol (#009/10) involving the maintenance and euthanasia of the ApoE-knockout (ApoE KO) mice was approved by the Institutional Animal Care and Use Committees (IACUC) at the National University of Singapore. The ApoE-KO cell line was created using the immortalization method described before.49 Briefly, primary cortical neurons from ApoE KO mice50 were immortalized using the SV40 gene.49,51,52 The cells were grown in DMEM supplemented with 10% fetal bovine serum, 5% penicillin–streptomycin–amphotericin B and 5% sodium pyruvate, and maintained at 37 °C in a humidified incubator supplied with 5% CO2. Expression vectors containing no insert (mock), ApoE3 and ApoE4 were electroporated into the ApoE-KO cells using the Amaxa® Nucleofector® kit V (Lonza) according to manufacturer's instructions. Selection for cells containing the required construct was performed in DMEM with 5 μg ml−1 blasticidin (Life Technologies). Selected clones were maintained in DMEM containing 2 μg ml−1 blasticidin (Life Technologies).
2.3. SDS-PAGE and western blot analysis
In this study, PBS (as a control) or CE (100 μg ml−1) was added to the growing cells and incubated for 24 h in a humidified CO2 (5%) incubator at 37 °C. After CE treatment, cells were lysed in ice-cold 1× RIPA lysis buffer (Cell Signaling Technology) containing detergents such as 1% Nonidet P40 and 1% sodium deoxycholate together with the protease inhibitor cocktail tablet (Roche). This lysis buffer also contains sodium orthovanadate, pyrophosphate and glycerophosphate, which can act as phosphatase inhibitors. The cellular samples were subjected to brief sonication and centrifugation at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes at 4 °C.
000 rpm for 10 minutes at 4 °C.
Cellular samples were resolved on 7.5–10% Tris–glycine sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (BioRad). The Precision Plus protein™ standard (Bio-Rad Laboratories, Hercules, California, USA) was used as a molecular weight standard and ran together with the samples on the same piece of gel. The separated proteins were transferred onto a nitrocellulose membrane, probed with the respective antibodies and exposed to horseradish peroxidase (HRP)-conjugated secondary antibodies. The reactive protein bands were visualized by chemiluminescence on the Image Station 4000R (Carestream Health Inc.) using the SuperSignal® West Dura Substrate (Pierce) system.
Immunoblotting of β-actin (Sigma) was included in all western blot analyses to ensure comparable protein loading. Each immunoblotting was repeated up to four times using different preparations of the same cell line.
2.4. Densitometry analysis
Densitometry analysis was performed53 by measuring the optical densities of the targeted protein bands relative to the endogenous β-actin level from the same cell lysate sample. For protein phosphorylation, the optical densities of the phosphorylated protein bands were measured relative to the targeted total protein level from the same cell lysate sample. The analysis was performed using the NIH ImageJ software.2.5. Statistical analysis
Significant differences were analyzed using the Student's t-test. Confidence levels for statistical significance were set at P < 0.05.3. Results
3.1. Expression ApoE isoforms in the ApoE knockout cell line
ApoE is expressed in many neuronal and non-neuronal tissues15 and cell lines.20 Many human ApoE transgenic mouse models54 were therefore created on the ApoE knock-out (KO) background.55–61To better understand the cellular function of ApoE without the presence of the endogenous mouse ApoE, we have stably transfected human ApoE3 and ApoE4 into the ApoE KO cell line, generated using the immortalization method described before.49 A mock cell line was generated by stably transfecting ApoE KO cells with the same expression vector without any ApoE insert. Immunoblotting detected ApoE protein band only in the ApoE3 and ApoE4 transfected cells but not in the mock transfected cells (Fig. 1A). We also observed higher ApoE levels in ApoE3 cells as compared to ApoE4 cells. This difference in the ApoE level was also detected in ApoE knock-in mice62–64 and in non-demented ApoE4 carriers.64
 | ||
| Fig. 1 CE treatment reduced ApoE4 expression. (A) Western blot analysis of ApoE in the mock-transfected cell line and transfected cells expressing ApoE3 and ApoE4. (B) ApoE expression without (−) and with (+) chicken extract (CE) treatment in mock, ApoE3 and ApoE4-transfected cell lines. The blot in (A) and (B) is a representative of four independent experiments. β-Actin was used as a loading control in each sample. Blot images were cropped for comparison. (C) Densitometry analysis of ApoE relative to the β-actin level was performed using the NIH ImageJ software. Each value represents the mean ± SEM for individual experiments (n = 4). Lower ApoE4 expression was detected after CE treatment (*p = 0.01 using the Student's t-test). | ||
We next examined if the chicken extract (CE) solution will affect ApoE expression in the transfected cell lines. Cells expressing ApoE3 and ApoE4 were incubated with either CE solution (+) or PBS (−). Western blot analysis showed that CE treatment lowered ApoE expression only in the ApoE4 cells (Fig. 1B). Densitometric analysis indicated a reduction of 56% ApoE expression in the ApoE4 cell line (Fig. 1C). No significant change in ApoE expression was detected in the ApoE3 cells after CE treatment.
3.2. Differential NR1 phosphorylation in ApoE expressing cells after CE treatment
ApoE4 was reported to impair synaptic plasticity by reducing the N-methyl-D-aspartate receptor (NMDAR) function.26–30 NMDAR1 (NR1) is the obligatory subunit of the heterotetramer receptor.31–33 We therefore examined if CE treatment can alter the activation of the NR1 subunit.In ApoE3 expressing cells, CE treatment increased the phosphorylation of serine residue 896 (S896) on NR1 by 33%. But in the ApoE4 cells, CE treatment de-phosphorylated serine 897 (S897) on NR1 by 70% (Fig. 2). CE treatment has no effect on NR1 phosphorylation (S896 and S897) in the mock cells.
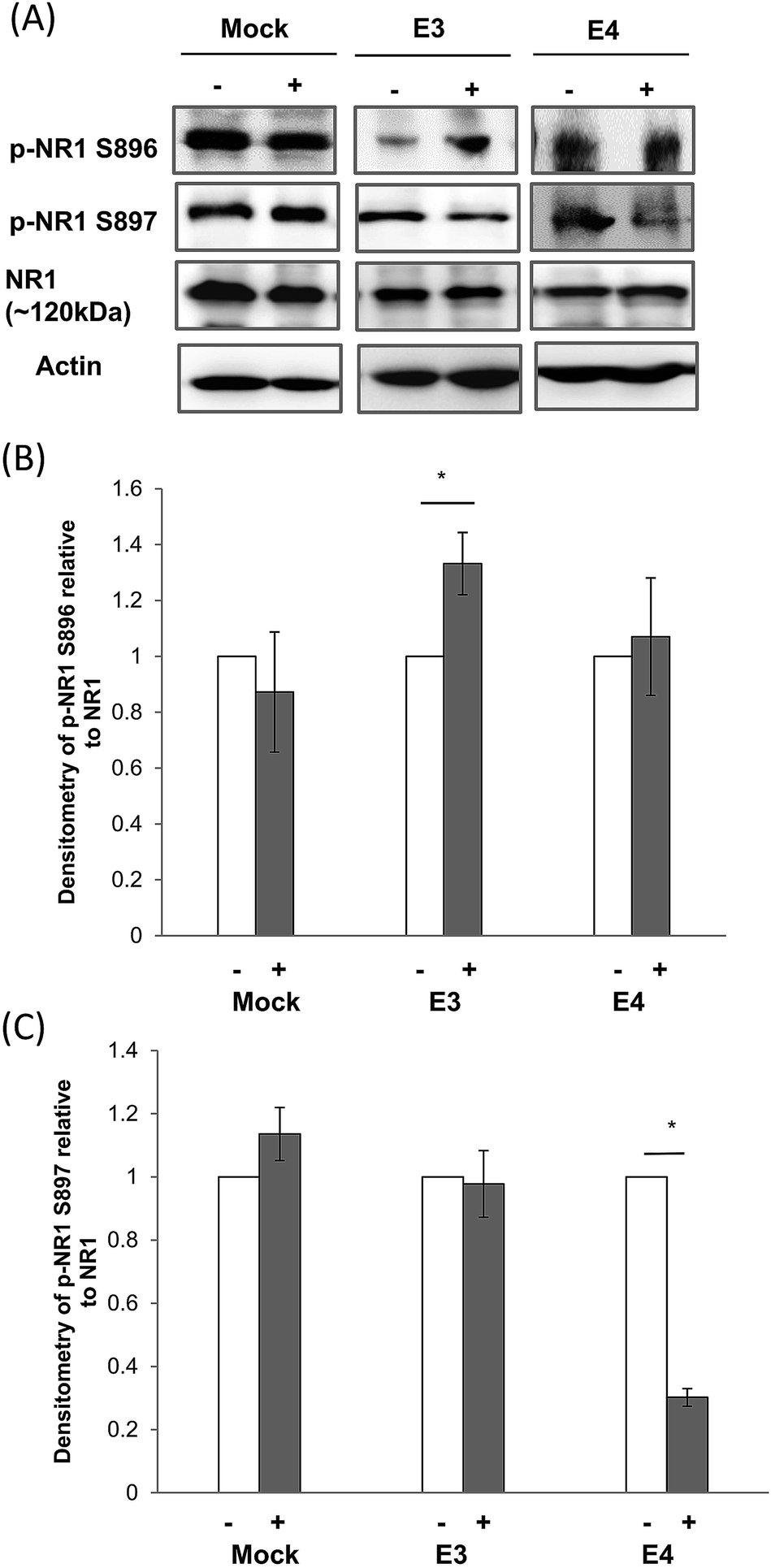 | ||
| Fig. 2 Effect of CE treatment on NR1 expression and phosphorylation in ApoE cell lines. (A) Immunoblotting of total NMDA Receptor subunit 1 (NR1), phosphorylated NR1 (S896) and (S897) in mock, and ApoE3 and ApoE4-transfected cell lines without (−) and with (+) chicken extract (CE) treatment. The blot is a representative of four independent experiments. Blot images were cropped for comparison. Densitometry analysis of (B) phosphorylated NR1 (S896) and (C) NR1 (S897) relative to total NR1 in mock, ApoE3 and ApoE4-transfected cell lines without (−, white bar) and with (+, grey bar) CE treatment was performed using the NIH ImageJ software. Each value represents the mean ± SEM for individual experiments (n = 4). NR1 phosphorylation at S896 in ApoE3 cell lines was increased whereas NR1 phosphorylation at S897 in ApoE4 cells was reduced after CE treatment. (B) *p = 0.04; (C) *p < 0.001 using the Student's t-test. | ||
3.3. Changes on NRI phosphorylation is linked to differential PKA/PKC activation
NR1 phosphorylation on S896 was reported to be regulated by protein kinase C (PKC), whereas NR1 phosphorylation on S897 was regulated by protein kinase A (PKA).65PKA is a heterotetramer composed of a regulatory subunit dimer and a catalytic subunit dimer.66 The catalytic subunit can be spliced into three isoforms (Cα, Cβ, and Cγ). In ApoE4 cells, we observed that the phosphorylation of T197 on the Cα subunit of PKA (PKA-Cα) was reduced by 17% (Fig. 3A and B). No significant change was observed in ApoE3 cells.
 | ||
| Fig. 3 CE treatment reduced PKA phosphorylation in ApoE4 cells but increased PKC phosphorylation in ApoE3 cells. Immunoblotting of the (A) protein kinase A Cα subunit (PKA Cα) and (C) protein kinase C α subunit (PKC α) expression and phosphorylation in mock, ApoE3 and ApoE4-transfected cell lines without (−) and with (+) CE treatment. The blot is a representation of four independent experiments. Blot images were cropped for comparison. Densitometry analysis of phosphorylated (B) PKA (T197) and (C) PKC (T497) relative to total PKA and PKC respectively, in mock, ApoE3 and ApoE4-transfected cell lines without (−, white bar) and with (+, grey bar) CE treatment. The analysis was performed using the NIH ImageJ software. Each value represents the mean ± SEM for individual experiments (n = 4). PKA phosphorylation in ApoE4 cells was reduced while PKC phosphorylation in ApoE3 cells was increased after CE treatment. (B) *p = 0.005; (C) *p = 0.01 using the Student's t-test. | ||
PKC in contrast has more than 12 different isoforms. The PKC isoforms are serine/threonine kinases involved in a wide range of physiological processes including differentiation and brain function.67 PKC α isoform (PKC α) is ubiquitously expressed and is activated in response to many different kinds of stimuli. Here, we detected that T497 phosphorylation on PKC α was increased by 11% in ApoE3 but not in ApoE4 cells (Fig. 3C and D).
3.4. AMPA GluR1 phosphorylation in ApoE expressing cells is altered after CE treatment
Another major glutamate receptor that exists alongside NMDAR is the AMPA receptor (AMPAR).31 These two receptors are found to co-localize in many synapses. Changes in NMDAR phosphorylation therefore could regulate AMPAR activation.44,45We found that CE treatment increased GluR1 S831 phosphorylation by 37% in ApoE3 cells. However, S831 phosphorylation was reduced in mock and ApoE4 cells by 25% and 40% respectively (Fig. 4A and B).
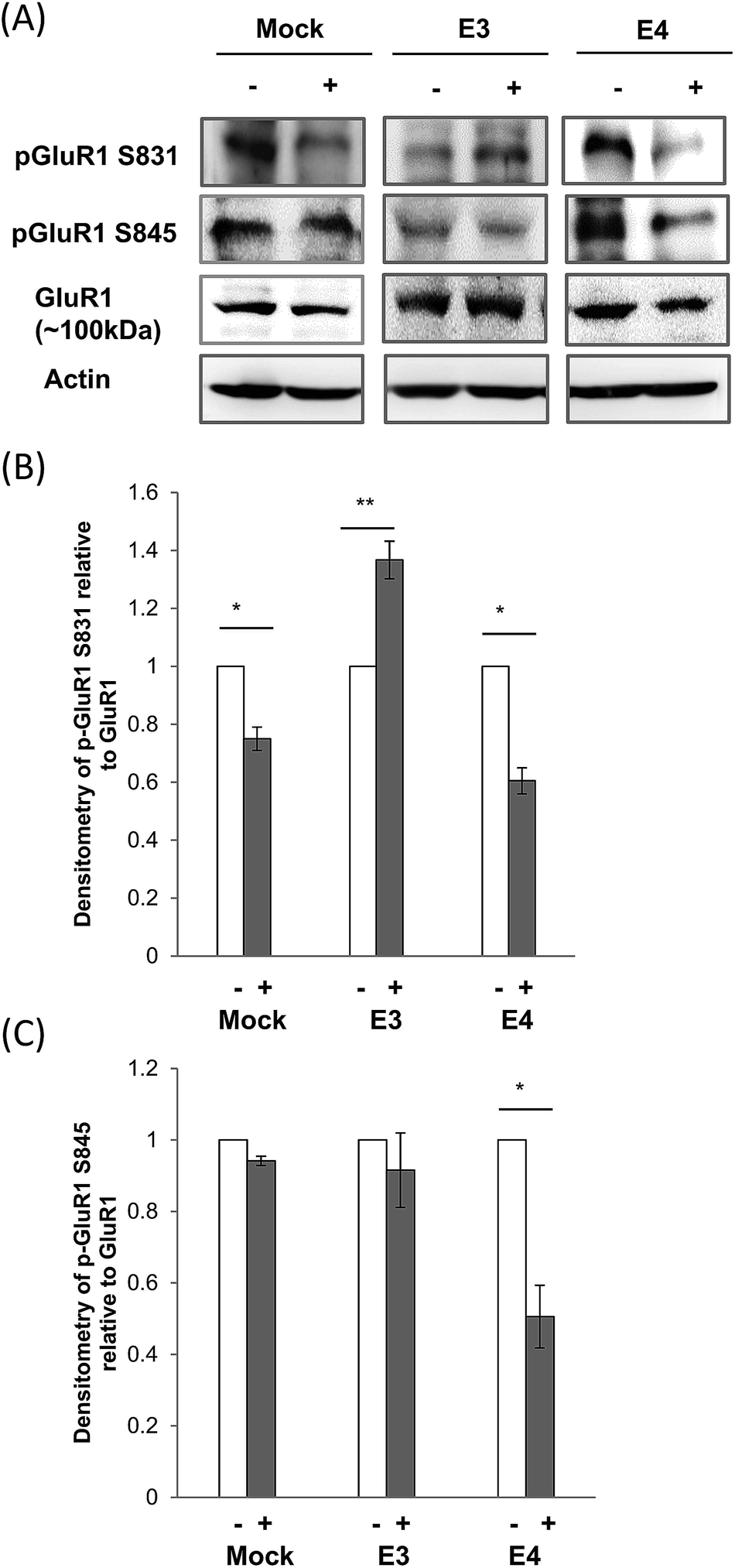 | ||
| Fig. 4 Effect of CE treatment on GluR1 expression and phosphorylation in ApoE cell lines. (A) Immunoblotting of AMPA Receptor subunit GluR1, phosphorylated GluR1 (S831) and (S845) in mock, and ApoE3 and ApoE4-transfected cell lines without (−) and with (+) chicken extract (CE) treatment. The blot is a representative of four independent experiments. Blot images were cropped for comparison. Densitometry analysis of (B) phosphorylated GluR1 (S831) and (C) GluR1 (S845) relative to total GluR1 in mock, ApoE3 and ApoE4-transfected cell lines without (−, white bar) and with (+, grey bar) CE treatment was performed using the NIH ImageJ software. Each value represents the mean ± SEM for individual experiments (n = 4). (B) GluR1 phosphorylation at S831 was increased in ApoE3 cells but reduced in mock and ApoE4 cell lines (*p = 0.02; **p < 0.001, using the Student's t-test). (C) In contrast, GluR1 phosphorylation at S845 was only affected in ApoE4 cells after CE treatment. (*p = 0.01, using the Student's t-test.) | ||
On the other hand, GluR1 phosphorylation at S845 was reduced by 49% after CE treatment in ApoE4 cells. CE treatment however, did not cause any significant change in S845 phosphorylation in both mock and ApoE3 cells (Fig. 4A and C).
3.5. Effect of CE on CaMKII activation in ApoE expressing cells
The ApoE effect on the NMDAR function requires calcium (Ca2+) signaling.68 The secondary messenger effects of Ca2+ are mostly mediated via Ca2+-sensing protein kinases such as calmodulin kinase II (CaMKII)69 that is able to dock with the NMDAR. CaMKII has catalytic and regulatory domains. The binding of Ca2+ to its regulatory domain activates the kinase70 and this involves the autophosphorylation at threonine 286 (T286). CaMKII activation is maintained by PKA by preventing the dephosphorylation of T286.71CE treatment in ApoE3 cells caused a 36% increase in CaMKII phosphorylation at T286 (Fig. 5A and B). But, CaMKII T286 phosphorylation was reduced in mock and ApoE4 cells by 49% and 26% respectively after CE treatment (Fig. 5A and B).
 | ||
| Fig. 5 CE treatment altered CaMKII expression and phosphorylation in ApoE cell lines. (A) Immunoblotting of total CaMKII and phosphorylated CaMKII (T286) in mock, ApoE3 and ApoE4-transfected cell lines without (−) and with (+) CE treatment. The blot is a representative of four independent experiments. Blot images were cropped for comparison. Densitometry analysis of (B) phosphorylated CaMKII (T286) relative to total CaMKII in mock, ApoE3 and ApoE4-transfected cell lines without (−, white bar) and with (+, grey bar) CE treatment was performed using the NIH ImageJ software. Each value represents the mean ± SEM of duplicate assays for individual experiments (n = 4). CaMKII phosphorylation at T286 was increased in ApoE3 cells but reduced in mock and ApoE4 cell lines after CE treatment. (*p = 0.03; **p < 0.001, using the Student's t-test.) | ||
3.6. ERK/CREB signaling in ApoE cells after CE treatment
A major signaling cascade regulated by Ca2+ influx through the NMDAR is the downstream extracellular signal-regulated kinase (ERK) pathway,72,73 which culminates in CREB-mediated gene transcription to influence neuronal survival and plasticity.37,72–74ERK proteins are regulated by the dual phosphorylation of threonine 202 (T202) and tyrosine 204 (Y204) on ERK1 and threonine 185 (T185) and tyrosine 187 (Y187) on ERK2.75 In ApoE3 cells, ERK phosphorylation was increased by 30% after CE treatment. However, CE reduced ERK phosphorylation by 35% in both mock and ApoE4 cells (Fig. 6A and B).
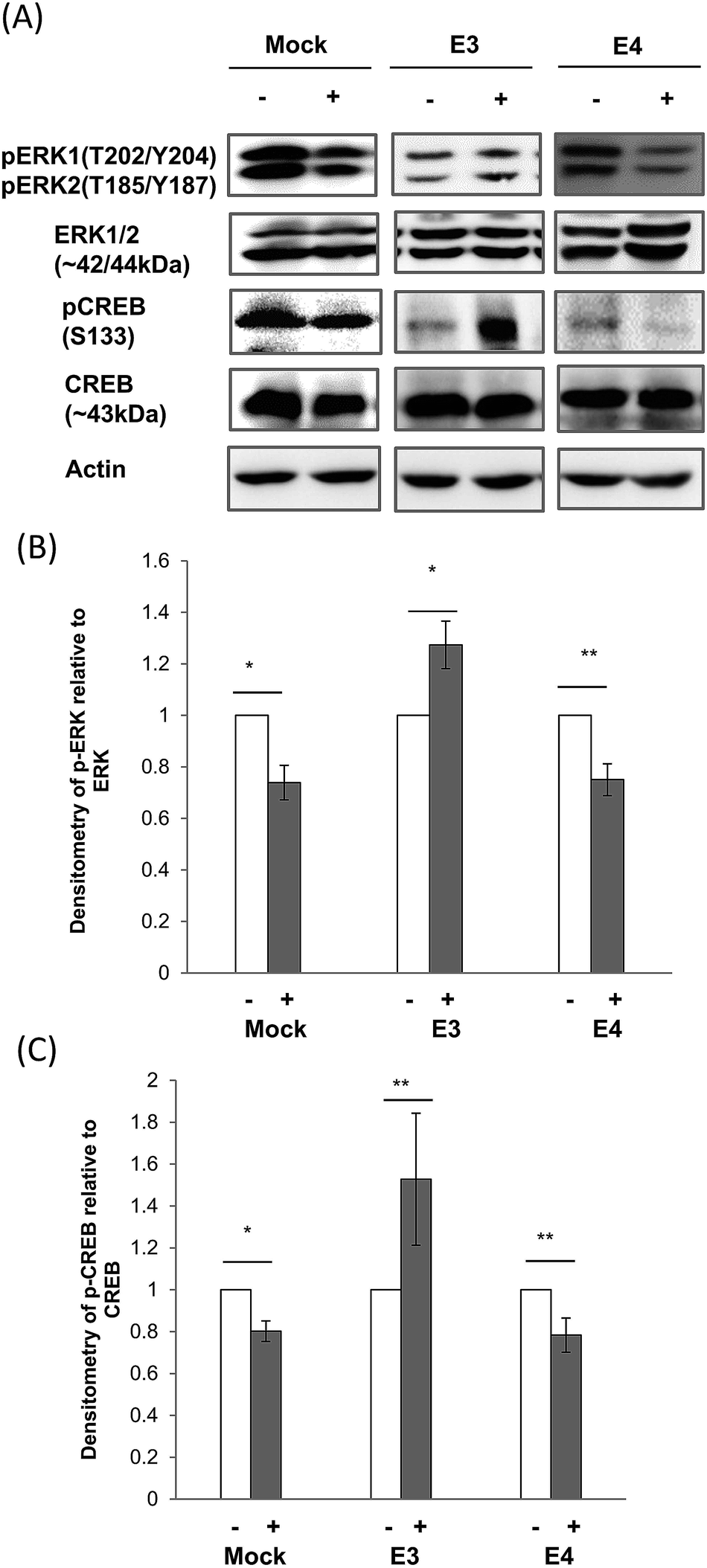 | ||
| Fig. 6 Effects of CE treatment on ERK1/2 and CREB expression and phosphorylation in ApoE cell lines. (A) Immunoblotting of ERK1/2 and CREB expression and phosphorylation in mock, ApoE3 and ApoE4-transfected cell lines without (−) and with (+) chicken extract (CE) treatment. The blot is a representative of four independent experiments. Densitometry analysis of (B) ERK1 (T202/Y204)/ERK2 (T185/Y187) and (C) CREB (S133) phosphorylation relative to total ERK1/2 and CREB respectively, in mock, ApoE3 and ApoE4-transfected cell lines without (−, white bar) and with (+, grey bar) CE treatment was performed using the NIH ImageJ software. Each value represents the mean ± SEM for individual experiments (n = 4). ERK1/2 and CREB phosphorylation was increased in ApoE3 cells but reduced in mock and ApoE4 cell lines after CE treatment. (B) *p = 0.004; **p = 0.03, using the Student's t-test. (C) *p = 0.02; **p = 0.04, using the Student's t-test. | ||
CREB is activated by phosphorylation at serine 133 (S133) by several signaling pathways including ERK.74 In ApoE3 cells, we found that CE treatment increased CREB phosphorylation at S133 by 54%. But, CE treatment increased the de-phosphorylation of CREB S133 by 20% in both mock and ApoE4 cells (Fig. 6A and C).
4. Discussion
Chicken extract (CE) has been reported to enhance memory function.4–6 In this study using cells expressing ApoE3 and ApoE4, we observed that CE can trigger specific activation on the NMDAR and AMPAR, and this is linked to specific changes on the ERK/CREB signaling pathway.While ApoE is mainly expressed by astrocytes, the protein can also be detected in neurons.76,77 Neurons expressing ApoE4 were reported to have lower NMDAR and AMPAR functions,46 leading to lower LTP.30 This association between ApoE and NMDAR/AMPAR could account for the ApoE isoform-dependent effect on cognition.26–30
To understand if this ApoE function underlies the CE effect on cognition, we have stably transfected human ApoE3 and ApoE4 into the immortalized ApoE KO neuronal cell line. We found that CE treatment increased S896 phosphorylation on NR1 in ApoE3 cells. NR1 phosphorylation on S896 was reported to be regulated by protein kinase C (PKC),65 and increased PKC phosphorylation was detected in ApoE3 cells.
In ApoE4 cells however, NR1-S897 phosphorylation was reduced. This phosphorylation was observed to be regulated by protein kinase A (PKA).65 Therefore, the detected lowering of NR1 S897 phosphorylation could be associated with lowered PKA phosphorylation in ApoE4 cells.
NR1 phosphorylation could affect AMPAR activation as these two receptors are found to co-localize in many synapses.44,45 While GluR1 S831 phosphorylation was increased in ApoE3 cells, this residue was significantly de-phosphorylated in mock and ApoE4 cells.
In contrast, GluR1 S845 phosphorylation was only reduced in ApoE4 cells, and this reduction could be linked to lower PKA phosphorylation since a study has reported that PKA activation regulates GluR1 S845 phosphorylation.78 Furthermore, the concomitant decrease in NR1 S897 and GluR1 S845 phosphorylation could be mediated by the phosphatase calcineurin (CaN) inhibiting PKA34,79 and this shifts the direction of synaptic plasticity towards long-term depression (LTD) formation.
In ApoE3 cells, increased CaMKII phosphorylation could up-regulate the phosphorylation of GluR1 S831 but not S845 after CE treatment. Phosphorylation of this residue was shown to induce LTP via PKC.80–82
CE reduced CaMKII phosphorylation in ApoE4 cells. This could contribute to higher de-phosphorylation of both S831 and S845 on GluR1. Notably, the decreased phosphorylation of GluR1 S831 mimics that of CaMKII but not of PKC in CE-treated mock and ApoE4-transfected cells. Interaction between CE and ApoE could mediate the binding of Ca2+ to CaMKII to activate the kinase and/or for sustaining the auto-phosphorylated form of CaMKII. However, the ApoE3 expression level remains unchanged even though CaMKII activation increases in CE-treated ApoE3 cells. This implies that interaction between CE and ApoE may not be the limiting factor in modifying CaMKII activity as it may already have been at a saturated level with the amount of ApoE expressed in the cell lines. Hence, there may be other limiting factors in ApoE3-transfected cells that mediate the increased CaMKII activity.
On the other hand, the percentage of reduction in the phosphorylation of GluR1 S831 in CE-treated ApoE4 cells is almost twice that of CE-treated mock neurons. It is tempting to speculate that the extensive decrease in GluR1 S831 phosphorylation in ApoE4 cells is due to the additive effect of reduced activation of CREB and CaMKII since both are upstream regulators of GluR1 S831. However, mock cells also exhibit a similar decrease in CREB activity and the magnitude of reduction in CaMKII T286 auto-phosphorylation is 2-fold higher compared to that of ApoE4 cells after CE treatment. Hence, it is unlikely that CREB and CaMKII are the main contributors in down-regulating GluR1 S831 in ApoE4 cells. One possibility is that ApoE4 which is still produced at a low level in ApoE4 cells but completely absent in mock neurons after treatment may pose a detrimental effect in activating phosphatases (PP1/PP2) and CaN that can dephosphorylate GluR1 at S831 in conjunction with the decreased phosphorylation by its activators. It is unclear if the gain-of-adverse-function of ApoE4 increases the dephosphorylation of GluR1 at S831 after CE treatment.
ERK1/2 activation can lead to NMDAR-mediated neuroprotection in neurons37,68,72–74 and ApoE is involved in regulating this NMDAR-dependent ERK/CREB signaling.28,83 This regulation involves interaction between ApoE, NR1 and the ApoE receptor ApoEr2.84
CREB-coupled synaptic activity is associated with long-term changes in neuronal plasticity and this is thought to underlie learning and memory.37,74 ApoE3 expression can lead to higher ERK1/2 phosphorylation and CREB activation as compared to ApoE4.28,30,83 Here, we observed that CE treatment enhanced ERK/CREB signaling in ApoE3 cells. But, this treatment reduced ERK/CREB phosphorylation in cells expressing ApoE4 and mock transfected cells. This similarity in ApoE4 and mock cells could be due to the lowered ApoE4 expression after CE treatment.
The lower ApoE4 expression could decrease the binding of ApoE to ApoEr2 and/or reduce the ApoEr2 level.28 This will disrupt the multi-protein complexes comprising ApoE, ApoEr2 and NMDAR,84 reducing CaMKII and NMDA activation,85 and downstream signaling pathways86–89 in CE-treated ApoE4 cells.
CE is abundant in proteins, amino acids and peptides, including bioactive peptides such as carnosine (β-alanyl-L-histidine) and its derivative, anserine (β-alanyl-1-methyl-L-histidine).5 These endogenous imidazole dipeptides are present in high concentrations in the human brain and are neuroprotective.90–93
Carnosine is able to cross the blood brain barrier (BBB),94 enhancing LTP and cognitive performance in rats.95 This function resembles Cerebrolysin, a neuropeptide that mimics the action of endogenous neurotrophic factors to protect synaptic integrity and improves cognition.96
It is interesting to note that another hydrolyzed CE preparation termed chicken meat ingredient-168 (CMI-168) isolated from chicken meat using a proprietary technology was reported to enhance cognition probably via promoting attention and prefrontal cortex functions.5
5. Conclusions
In summary, this study shows that CE triggers ApoE isoform-specific ERK/CREB signaling changes. Although CE has been reported to enhance memory function,4,6 our study suggests that this beneficial effect could be ApoE-isoform dependent. Further studies to examine the impact of ApoE isoform on the neuroprotective effect of CE could benefit and probably slow the age-related cognitive decline process.Conflict of interest
The authors declare no competing financial interests.Author contributions
YSM and QRO performed the experiments. YSM, BES and BSW conceived and designed the experiments. YSM and BSW analyzed the data and wrote the paper.Acknowledgements
We thank Dr. Paramjeet Singh (Cerebos Pacific Ltd.) for providing the chicken extract powder. We also thank Drs Katherine Youmans and Mary Jo LaDu (University of Illinois, Chicago, USA) for providing the human ApoE4 cDNA. This work was supported by grants to BSW from the Biomedical Research Council (BMRC/05/1/21/19/401) and Cerebos Pacific Ltd. YSM and QRO were supported by graduate scholarships from the Singapore Ministry of Education. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.References
- M. Fields, J. Am. Coll. Nutr., 1998, 17, 317–321 CrossRef CAS.
- R. J. Stratton, Proc. Nutr. Soc., 2000, 59, 469–476 CrossRef CAS PubMed.
- M. K. Sim, Y. C. Wong, X. G. Xu, S. Z. Sim and D. Tsi, Biosci., Biotechnol., Biochem., 2009, 73, 2583–2588 CrossRef CAS PubMed.
- M. Z. Azhar, J. O. Zubaidah and K. O. N. Norjan, Malaysian Journal of Medicine and Health Sciences, 2008, 4, 57–68 Search PubMed.
- Z. M. Azhar, J. O. Zubaidah, K. O. Norjan, C. Y. Zhuang and F. Tsang, Nutr. J., 2013, 12, 121 CrossRef PubMed.
- C. L. Xu and M. K. Sim, Int. J. Food Sci. Nutr., 1997, 48, 113–117 CrossRef CAS.
- A. M. Zain and S. Syedsahiljamalulail, Malays. J. Nutr., 2003, 9, 19–29 Search PubMed.
- S. Cosentino, N. Scarmeas, E. Helzner, M. M. Glymour, J. Brandt, M. Albert, D. Blacker and Y. Stern, Neurology, 2008, 70, 1842–1849 CrossRef CAS PubMed.
- S. Bookheimer and A. Burggren, Annu. Rev. Clin. Psychol., 2009, 5, 343–362 CrossRef PubMed.
- E. H. Corder, A. M. Saunders, W. J. Strittmatter, D. E. Schmechel, P. C. Gaskell, G. W. Small, A. D. Roses, J. L. Haines and M. A. Pericak-Vance, Science, 1993, 261, 921–923 CAS.
- R. W. Mahley, K. H. Weisgraber and Y. Huang, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5644–5651 CrossRef CAS PubMed.
- F. Liu, L. M. Pardo, M. Schuur, P. Sanchez-Juan, A. Isaacs, K. Sleegers, I. de Koning, I. V. Zorkoltseva, T. I. Axenovich, J. C. Witteman, A. C. Janssens, J. C. van Swieten, Y. S. Aulchenko, B. A. Oostra and C. M. van Duijn, Neurobiol. Aging, 2010, 31, 1831–1833 CrossRef CAS PubMed.
- C. K. Blair, A. R. Folsom, D. S. Knopman, M. S. Bray, T. H. Mosley and E. Boerwinkle, Neurology, 2005, 64, 268–276 CrossRef CAS PubMed.
- Y. Huang, K. H. Weisgraber, L. Mucke and R. W. Mahley, J. Mol. Neurosci., 2004, 23, 189–204 CrossRef CAS.
- S. C. Rall, Jr, K. H. Weisgraber and R. W. Mahley, J. Biol. Chem., 1982, 257, 4171–4178 Search PubMed.
- R. W. Mahley and S. C. Rall, Jr, Annu. Rev. Genomics Hum. Genet., 2000, 1, 507–537 CrossRef CAS PubMed.
- V. I. Zannis, J. McPherson, G. Goldberger, S. K. Karathanasis and J. L. Breslow, J. Biol. Chem., 1984, 259, 5495–5499 CAS.
- V. I. Zannis, D. M. Kurnit and J. L. Breslow, J. Biol. Chem., 1982, 257, 536–544 CAS.
- R. W. Mahley, Science, 1988, 240, 622–630 CAS.
- J. Herz and U. Beffert, Nat. Rev. Neurosci., 2000, 1, 51–58 CrossRef CAS PubMed.
- M. J. de Leon, A. Convit, O. T. Wolf, C. Y. Tarshish, S. DeSanti, H. Rusinek, W. Tsui, E. Kandil, A. J. Scherer, A. Roche, A. Imossi, E. Thorn, M. Bobinski, C. Caraos, P. Lesbre, D. Schlyer, J. Poirier, B. Reisberg and J. Fowler, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10966–10971 CrossRef CAS PubMed.
- N. Filippini, K. P. Ebmeier, B. J. MacIntosh, A. J. Trachtenberg, G. B. Frisoni, G. K. Wilcock, C. F. Beckmann, S. M. Smith, P. M. Matthews and C. E. Mackay, NeuroImage, 2011, 54, 602–610 CrossRef CAS PubMed.
- P. M. Greenwood, T. Sunderland, J. L. Friz and R. Parasuraman, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11661–11666 CrossRef CAS PubMed.
- R. E. Hartman, D. F. Wozniak, A. Nardi, J. W. Olney, L. Sartorius and D. M. Holtzman, Exp. Neurol., 2001, 170, 326–344 CrossRef CAS PubMed.
- J. Raber, D. Wong, G. Q. Yu, M. Buttini, R. W. Mahley, R. E. Pitas and L. Mucke, Nature, 2000, 404, 352–354 CrossRef CAS PubMed.
- M. Aono, Y. Lee, E. R. Grant, R. A. Zivin, R. D. Pearlstein, D. S. Warner, E. R. Bennett and D. T. Laskowitz, Neurobiol. Dis., 2002, 11, 214–220 CrossRef CAS.
- Z. Qiu, K. A. Crutcher, B. T. Hyman and G. W. Rebeck, Neuroscience, 2003, 122, 291–303 CrossRef CAS PubMed.
- H. S. Hoe, D. C. Harris and G. W. Rebeck, J. Neurochem., 2005, 93, 145–155 CrossRef CAS PubMed.
- J. Herz and Y. Chen, Nat. Rev. Neurosci., 2006, 7, 850–859 CrossRef CAS PubMed.
- K. M. Korwek, J. H. Trotter, M. J. Ladu, P. M. Sullivan and E. J. Weeber, Mol. Neurodegener., 2009, 4, 21 CrossRef PubMed.
- E. E. Benarroch, Neurology, 2011, 76, 1750–1757 CrossRef PubMed.
- R. Dingledine, K. Borges, D. Bowie and S. F. Traynelis, Pharmacol. Rev., 1999, 51, 7–61 CAS.
- G. E. Hardingham and H. Bading, Trends Neurosci., 2003, 26, 81–89 CrossRef CAS.
- Y. S. Lee and A. J. Silva, Nat. Rev. Neurosci., 2009, 10, 126–140 CrossRef CAS PubMed.
- H. K. Lee, S. S. Min, M. Gallagher and A. Kirkwood, Nat. Neurosci., 2005, 8, 1657–1659 CrossRef CAS PubMed.
- K. Nakazawa, T. J. McHugh, M. A. Wilson and S. Tonegawa, Nat. Rev. Neurosci., 2004, 5, 361–372 CrossRef CAS PubMed.
- T. Tully, R. Bourtchouladze, R. Scott and J. Tallman, Nat. Rev. Drug Discovery, 2003, 2, 267–277 CrossRef CAS PubMed.
- D. A. Clayton, D. R. Grosshans and M. D. Browning, J. Biol. Chem., 2002, 277, 14367–14369 CrossRef CAS PubMed.
- A. Hashimoto, T. Nishikawa, T. Oka and K. Takahashi, J. Neurochem., 1993, 60, 783–786 CrossRef CAS PubMed.
- K. R. Magnusson, J. Neurosci., 2000, 20, 1666–1674 CAS.
- K. R. Magnusson, S. E. Nelson and A. B. Young, Mol. Brain Res., 2002, 99, 40–45 CrossRef CAS.
- H. Fukushima, R. Maeda, R. Suzuki, A. Suzuki, M. Nomoto, H. Toyoda, L. J. Wu, H. Xu, M. G. Zhao, K. Ueda, A. Kitamoto, N. Mamiya, T. Yoshida, S. Homma, S. Masushige, M. Zhuo and S. Kida, J. Neurosci., 2008, 28, 9910–9919 CrossRef CAS PubMed.
- S. Cohen and M. E. Greenberg, Annu. Rev. Cell Dev. Biol., 2008, 24, 183–209 CrossRef CAS PubMed.
- B. Li, N. Devidze, D. Barengolts, N. Prostak, E. Sphicas, A. J. Apicella, R. Malinow and E. S. Emamian, J. Neurosci., 2009, 29, 11965–11972 CrossRef CAS PubMed.
- T. Unoki, S. Matsuda, W. Kakegawa, N. T. Van, K. Kohda, A. Suzuki, Y. Funakoshi, H. Hasegawa, M. Yuzaki and Y. Kanaho, Neuron, 2012, 73, 135–148 CrossRef CAS PubMed.
- Y. Chen, M. S. Durakoglugil, X. Xian and J. Herz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12011–12016 CrossRef CAS PubMed.
- Y. J. Zhai, R. R. He, B. Tsoi, Y. F. Li, X. D. Li, N. Tsuruoka, K. Abe and H. Kurihara, Food Funct., 2012, 3, 662–667 CAS.
- H. Kurihara, X. S. Yao, H. Nagai, N. Tsuruoka, H. Shibata, Y. Kiso and H. Fukami, J. Health Sci., 2006, 52, 17–23 CrossRef CAS.
- J. X. Tan, G. Z. Mao, M. Z. Cui, S. C. Kang, B. Lamb, B. S. Wong, M. S. Sy and X. M. Xu, J. Neurochem., 2008, 107, 722–733 CrossRef CAS PubMed.
- A. S. Plump, J. D. Smith, T. Hayek, K. Aalto-Setala, A. Walsh, J. G. Verstuyft, E. M. Rubin and J. L. Breslow, Cell, 1992, 71, 343–353 CrossRef CAS.
- K. Frederiksen, P. S. Jat, N. Valtz, D. Levy and R. McKay, Neuron, 1988, 1, 439–448 CrossRef CAS.
- A. Holme, M. Daniels, J. Sassoon and D. R. Brown, Eur. J. Neurosci., 2003, 18, 571–579 CrossRef PubMed.
- Q. R. Ong, M. L. Lim, C. C. Chua, N. S. Cheung and B. S. Wong, Biochem. Biophys. Res. Commun., 2012, 424, 482–487 CrossRef CAS PubMed.
- Y. Huang, Trends Mol. Med., 2010, 16, 287–294 CrossRef CAS PubMed.
- D. M. Holtzman, K. R. Bales, T. Tenkova, A. M. Fagan, M. Parsadanian, L. J. Sartorius, B. Mackey, J. Olney, D. McKeel, D. Wozniak and S. M. Paul, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2892–2897 CrossRef CAS PubMed.
- Y. Sun, S. Wu, G. Bu, M. K. Onifade, S. N. Patel, M. J. LaDu, A. M. Fagan and D. M. Holtzman, J. Neurosci., 1998, 18, 3261–3272 CAS.
- M. Buttini, M. Orth, S. Bellosta, H. Akeefe, R. E. Pitas, T. Wyss-Coray, L. Mucke and R. W. Mahley, J. Neurosci., 1999, 19, 4867–4880 CAS.
- J. Raber, D. Wong, M. Buttini, M. Orth, S. Bellosta, R. E. Pitas, R. W. Mahley and L. Mucke, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10914–10919 CrossRef CAS.
- C. Knouff, M. E. Hinsdale, H. Mezdour, M. K. Altenburg, M. Watanabe, S. H. Quarfordt, P. M. Sullivan and N. Maeda, J. Clin. Invest., 1999, 103, 1579–1586 CrossRef CAS PubMed.
- P. M. Sullivan, H. Mezdour, S. H. Quarfordt and N. Maeda, J. Clin. Invest., 1998, 102, 130–135 CrossRef CAS PubMed.
- J. A. Piedrahita, S. H. Zhang, J. R. Hagaman, P. M. Oliver and N. Maeda, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 4471–4475 CrossRef CAS.
- Q. R. Ong, E. S. Chan, M. L. Lim, G. M. Cole and B. S. Wong, Sci. Rep., 2014, 4, 8 Search PubMed.
- D. R. Riddell, H. Zhou, K. Atchison, H. K. Warwick, P. J. Atkinson, J. Jefferson, L. Xu, S. Aschmies, Y. Kirksey, Y. Hu, E. Wagner, A. Parratt, J. Xu, Z. Li, M. M. Zaleska, J. S. Jacobsen, M. N. Pangalos and P. H. Reinhart, J. Neurosci., 2008, 28, 11445–11453 CrossRef CAS PubMed.
- P. M. Sullivan, B. Han, F. Liu, B. E. Mace, J. F. Ervin, S. Wu, D. Koger, S. Paul and K. R. Bales, Neurobiol. Aging, 2011, 32, 791–801 CrossRef CAS PubMed.
- W. G. Tingley, M. D. Ehlers, K. Kameyama, C. Doherty, J. B. Ptak, C. T. Riley and R. L. Huganir, J. Biol. Chem., 1997, 272, 5157–5166 CrossRef CAS PubMed.
- M. Montminy, Annu. Rev. Biochem., 1997, 66, 807–822 CrossRef CAS PubMed.
- S. Nakashima, J. Biochem., 2002, 132, 669–675 CrossRef CAS.
- Y. Wang, V. Briz, A. Chishti, X. Bi and M. Baudry, J. Neurosci., 2013, 33, 18880–18892 CrossRef CAS PubMed.
- K. P. Hoeflich and M. Ikura, Cell, 2002, 108, 739–742 CrossRef CAS.
- A. R. Means, Mol. Endocrinol., 2000, 14, 4–13 CrossRef CAS PubMed.
- M. Makhinson, J. K. Chotiner, J. B. Watson and T. J. O'Dell, J. Neurosci., 1999, 19, 2500–2510 CAS.
- X. Z. Wan, B. Li, Y. C. Li, X. L. Yang, W. Zhang, L. Zhong and S. J. Tang, J. Neurosci., 2012, 32, 3910–3916 CrossRef CAS PubMed.
- L. Xiao, C. Hu, C. Feng and Y. Chen, J. Biol. Chem., 2011, 286, 20175–20193 CrossRef CAS PubMed.
- K. Sakamoto, K. Karelina and K. Obrietan, J. Neurochem., 2011, 116, 1–9 CrossRef CAS PubMed.
- W. Xia, R. J. Mullin, B. R. Keith, L. H. Liu, H. Ma, D. W. Rusnak, G. Owens, K. J. Alligood and N. L. Spector, Oncogene, 2002, 21, 6255–6263 CrossRef CAS PubMed.
- Y. Huang, X. Q. Liu, T. Wyss-Coray, W. J. Brecht, D. A. Sanan and R. W. Mahley, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8838–8843 CrossRef CAS PubMed.
- Y. Huang and L. Mucke, Cell, 2012, 148, 1204–1222 CrossRef CAS PubMed.
- K. W. Roche, R. J. O'Brien, A. L. Mammen, J. Bernhardt and R. L. Huganir, Neuron, 1996, 16, 1179–1188 CrossRef CAS.
- K. Baumgartel and I. M. Mansuy, Learn. Mem., 2012, 19, 375–384 CrossRef CAS PubMed.
- J. Y. Lan, V. A. Skeberdis, T. Jover, S. Y. Grooms, Y. Lin, R. C. Araneda, X. Zheng, M. V. Bennett and R. S. Zukin, Nat. Neurosci., 2001, 4, 382–390 CrossRef CAS PubMed.
- A. Nayak, D. J. Zastrow, R. Lickteig, N. R. Zahniser and M. D. Browning, Nature, 1998, 394, 680–683 CrossRef CAS PubMed.
- A. Sen, D. L. Alkon and T. J. Nelson, J. Biol. Chem., 2012, 287, 15947–15958 CrossRef CAS PubMed.
- H. S. Hoe, A. Pocivavsek, H. Dai, G. Chakraborty, D. C. Harris and G. W. Rebeck, Brain Res., 2006, 1112, 70–79 CrossRef CAS PubMed.
- H. S. Hoe, A. Pocivavsek, G. Chakraborty, Z. Fu, S. Vicini, M. D. Ehlers and G. W. Rebeck, J. Biol. Chem., 2006, 281, 3425–3431 CrossRef CAS PubMed.
- Z. Qiu, D. K. Strickland, B. T. Hyman and G. W. Rebeck, J. Biol. Chem., 2002, 277, 14458–14466 CrossRef CAS PubMed.
- S. Qiu, L. F. Zhao, K. M. Korwek and E. J. Weeber, J. Neurosci., 2006, 26, 12943–12955 CrossRef CAS PubMed.
- G. W. Rebeck, M. J. LaDu, S. Estus, G. Bu and E. J. Weeber, Mol. Neurodegener., 2006, 1, 15 CrossRef PubMed.
- H. Y. Chiu, H. H. Lin and C. C. Lai, Regul. Pept., 2009, 158, 77–85 CrossRef CAS PubMed.
- M. Sinagra, D. Verrier, D. Frankova, K. M. Korwek, J. Blahos, E. J. Weeber, O. J. Manzoni and P. Chavis, J. Neurosci., 2005, 25, 6127–6136 CrossRef CAS PubMed.
- B. C. Artun, Z. Kusku-Kiraz, M. Gulluoglu, U. Cevikbas, N. Kocak-Toker and M. Uysal, Hum. Exp. Toxicol., 2010, 29, 659–665 CAS.
- A. Boldyrev, E. Bulygina, T. Leinsoo, I. Petrushanko, S. Tsubone and H. Abe, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2004, 137, 81–88 CrossRef PubMed.
- N. MacFarlane, J. McMurray, J. J. O'Dowd, H. J. Dargie and D. J. Miller, J. Mol. Cell. Cardiol., 1991, 23, 1205–1207 CrossRef CAS.
- R. Kohen, Y. Yamamoto, K. C. Cundy and B. N. Ames, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 3175–3179 CrossRef CAS.
- Y. Shen, P. He, Y. Y. Fan, J. X. Zhang, H. J. Yan, W. W. Hu, H. Ohtsu and Z. Chen, Free Radical Biol. Med., 2010, 48, 727–735 CrossRef CAS PubMed.
- S. Acosta, J. Jernberg, C. D. Sanberg, P. R. Sanberg, B. J. Small, C. Gemma and P. C. Bickford, Rejuvenation Res., 2010, 13, 581–588 CrossRef CAS PubMed.
- E. Masliah and E. Diez-Tejedor, Drugs Today, 2012, 48(Suppl. A), 3–24 CAS.
| This journal is © The Royal Society of Chemistry 2014 |