 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Potential bioavailability enhancement of bioactive compounds using food-grade engineered nanomaterials: a review of the existing evidence
Kathleen
Oehlke
*,
Marta
Adamiuk
,
Diana
Behsnilian
,
Volker
Gräf
,
Esther
Mayer-Miebach
,
Elke
Walz
and
Ralf
Greiner†
Max Rubner-Institut, Department of Food Technology and Bioprocess Engineering, Haid-und-Neu-Str. 9, 76131 Karlsruhe, Germany. E-mail: kathleen.oehlke@mri.bund.de; Fax: +49-721-6625303; Tel: +49-721-6625308
First published on 26th February 2014
Abstract
The development of engineered nanometre sized materials (ENM) produced with food-grade ingredients and designed as delivery systems for organic and inorganic materials has gained increasing interest. The major reason for this trend is the aim to overcome problems associated with the low bioavailability of many bioactive compounds (BC) which are usually claimed to benefit human health. In this review, outcomes of studies investigating the potential bioavailability enhancement of BC using ENM as delivery systems are summarised and discussed. It focuses on in vitro and in vivo studies carried out with ENM produced with food-grade materials and designed for the delivery of vitamins, other secondary plant metabolites and minerals. Furthermore, the physical and physicochemical aspects governing the preparation of the systems, the loading of the BC, the stability of the delivery systems in food applications and finally the release of the BC in the gastrointestinal tract are also considered. The mechanisms leading to an enhanced bioavailability are based on (i) improved solubility of the BC under gastrointestinal conditions, (ii) the protection of the BC from the chemical conditions in the gastrointestinal tract (GIT), (iii) the controlled release within the GIT or (iv) an improved transfer through the intestinal wall. The main outcome of the review is that particle size, surface properties and physical state of the ENM are key parameters to be controlled aiming at an enhanced nutritional value of food materials. Furthermore, the bioavailability classification scheme (BCS) can help to understand the efficacy of different ENM for the delivery of specific BC.
Introduction
During the last few years, numerous engineered nanometre sized materials (ENM) have been proposed aiming at the improvement of technological properties and the enhancement of the nutritional and health value of food products.1–6 In the food area the interest in ENM intended for human consumption relies mainly on the possibility (i) to overcome solubility incompatibilities between ingredients, e.g. bioactive compounds (BC), and the food matrices, (ii) to protect sensitive ingredients, e.g. aroma, antimicrobial, antioxidant or nutritionally relevant compounds from degradation e.g. by oxidation, (iii) to increase the bioavailability of BC including the controlled release of encapsulated compounds or (iv) to modify physical properties of food materials, e.g. rheological properties.7–11As nanotechnology has been a strongly emerging key technology in various industrial fields, during the last few years, several commissions were concerned with a global definition of “nano”.12–15 Mostly, it is stated that ENM are manufactured intentionally and comprise a broad range of structures characterised by length scales of approximately 1–100 nm in one or more dimensions. The innovative character of these structures is determined by at least one new property and/or functionality arising from their nanometre-scale size and active surface as compared to the chemically identical bulk material or basic molecular structures. However, due to the diversity of potential applications and possible manufacturing processes for ENM, a lack of consensus for a definition still exists. Nevertheless, the European Commission recently included the labelling of ENM in food products into the regulation concerning consumer information, defining materials with 50% of the number size distribution below 100 nm as “nanomaterials”.16,17
ENM can be produced from single molecules via chemical reactions or by the self-assembly of the individual components. This procedure, the bottom-up approach, typically results in the creation of capsules, fibres or tubes that can be used as delivery systems for smaller molecules, e.g. bioactive or aroma compounds. Various lipid, polysaccharide or protein based ENM are described in the literature including e.g. micelles, liposomes or biopolymer particles. ENM preparation processes based on the fragmentation of larger into smaller structures usually by the input of mechanical and/or thermal energy, e.g. milling or high pressure homogenisation, are known as the top-down approach. The starting material can consist of e.g. plant materials rich in secondary plant metabolites, minerals or trace elements. An overview of ENM which have been proposed to enhance the bioavailability of BC is presented in Table 1. The structures of interest as well as their possible applications and formulation techniques have been recently reviewed elsewhere.18–25
| ENM | Preparation/composition | Structure | Ref. |
|---|---|---|---|
| Lipid and surfactant based ENM | |||
| Micelles | Self assembly after dissolving of surface active compounds ternary mixture of emulsifiers, oil, water |
|
26 |
| Microemulsions/swollen micelles | |||
| Nanoemulsions | High pressure homogenisation, ultrasound-assisted homogenisation |

|
27 |
| Solid lipid nanoparticles (SLN) | Hot emulsification of high melting lipids |
|
28 |
| Lipid nanocarriers (LNC), nanostructured lipid carriers (NLC) | Hot emulsification of high melting lipids with certain proportion of low melting lipids |

|
29 |
| Liposomes/vesicles | Mixture of phospholipids, evaporation of solvent under reduced pressure |
|
30 |
| Polysaccharide based ENM | |||
| Molecular complexes: cyclodextrin inclusion complexes, amylose complexes | Solubilisation under appropriate conditions |
|
10 and 31–33 |
| Biopolymeric nanogels: chitosan particles, alginate gels | Ionic or covalent cross-linking of polymers |
|
34 and 35 |
| Protein based ENM | 33 | ||
| Protein inclusion complexes (e.g. with β-lactoglobulin) | Solubilisation under appropriate conditions |

|
36 and 37 |
| Protein nanotubes | Self-assembly |

|
38 |
| Re-assembled casein micelles | Self-assembly |

|
33 |
| Plant and inorganic materials, nanocrystals | |||
| Plant materials, minerals | Comminution of larger materials | 39 and 40 | |
| Nanocrystals | 41 | ||
| Minerals (iron) | Flame spray pyrolysis | 42 | |
This review focuses on in vitro and in vivo studies investigating the effect of the formulation and the structure of ENM on the bioavailability of substances which may present benefits for human health beyond basic nutrition. The ENM will be discussed based on their principle composition, i.e. lipid, polysaccharide and protein based ENM, plant materials and minerals.
Each chapter dealing with delivery systems is preceded by a summary of the physical and physicochemical aspects governing the preparation of the systems, the loading of the BC, the stability of the delivery systems in food applications and the release of the BC in the gastrointestinal tract.
With few exceptions mainly studies carried out with ENM delivery systems prepared from food-grade ingredients, milled plant materials and minerals with particle size distributions within the range up to 300 nm are included. In many papers only the intensity-weighted arithmetic average particle diameter obtained from dynamic light scattering (DLS) measurements is given. This value is usually larger than the 50th percentile of the number based distribution, so that many apparent “sub-micron materials” reviewed would probably fall under the given definition.
After a short discussion of methodological approaches, aspects concerning the possible mechanisms for bioavailability enhancement of BC are presented and discussed.
Methodological considerations
Characterisation of ENM
The ENM intended as delivery systems should be comprehensively characterised in order to understand the relationships between structure and function and in this way elucidate the mechanisms leading to an enhanced bioavailability of the BC to be delivered. Several properties of ENM may influence their behaviour in biological systems. Therefore, it is generally accepted, irrespective of the kind of ENM, that several characterisation methods should be combined to provide representative information on their physicochemical properties, e.g. agglomeration/aggregation state, chemical composition, crystallinity/crystal structure, particle size/size distribution, purity, shape, surface area, surface charge and surface chemistry.43 The relevant methods can basically be divided into two major groups: sample preparation and separation methods (e.g. centrifugation, ultra-/nanofiltration, dialysis, different chromatography techniques, field-flow fractionation) and methods to characterise ENM with regard to their physical, chemical or physicochemical properties (e.g. dynamic light scattering, electrophoretic mobility). Nevertheless, most of the published data lack such a detailed description of the ENM which results in limited comparability of outcomes derived from different studies. Furthermore, the characterisation should be carried out for both, the initial ENM and the ENM incorporated into the food matrix. However, many of the available methods suitable for the analysis of ENM in simple matrices are not easily transferable to complex food matrices because they require extensive sample preparation that may cause artefacts and hence misleading results. Thus, despite the progress in the field, the analysis of ENM in complex food matrices or simulated gastric and intestinal fluids is still a challenging topic.44–48 Since the food matrix is of particular importance for the efficiency of delivery systems the development of suitable methods to understand interactions between ENM and the food matrix is indispensable.In vitro tests: simulated gastric and intestinal conditions and cell studies
The knowledge of the fate of ENM in the gastrointestinal tract (GIT) is essential for the evaluation of the effect of the formulation parameters and the structure on the bioavailability of encapsulated BC. Therefore, the stability of the ENM along the GIT and the release of encapsulated substances under GIT conditions ought to be studied together with the evaluation of the amount of substance of interest that reaches the plasma.Typical approaches to assess and understand the bioavailability in nutritional studies include in vitro dissolution and disintegration assays under simulated gastrointestinal conditions.49–51 Suitable digestion media for in vitro models, i.e. simulated gastric and intestinal fluids (SGF and SIF), are usually prepared according to the European or the United States Pharmacopoeia.52–54 Unlike many pharmaceutical formulations the ENM discussed here would typically be ingested as part of a meal. Therefore, the simultaneous uptake of other nutrients inducing the secretion of digestive enzymes and bile salts should be taken into consideration when preparing the digestive media for the investigation of food related applications.55 However, there is no consensus about the addition of digestive enzymes or bile salts to the media, which explains the different results obtained in some studies presented here. Furthermore, the applied models should be designed with increasing degrees of complexity to take into account factors such as the interactions with other food components, the interactions with biological surfaces and different materials present in the GIT.
During in vitro digestion experiments precipitation of the BC may occur in the GIT media although it was fully solubilized in the initial formulation. Although the dietary mixed micelles contribute to the solubilisation of the poorly water-soluble compounds they cannot always compensate for the disappearance of the initial solubilisation site (e.g. oil droplets) upon digestion as demonstrated for polymethoxyflavone loaded nanoemulsions.56 The release of BC can also be studied by using dialysis membranes and respective diffusion models. It has to be considered that difficulties in providing sink conditions can occur due to the low solubility of many BC. Therefore, aqueous/ethanolic solutions or micellar solutions are commonly used as receptor media.29,57,58
With regard to the measurement of the particle stability the increasing complexity of the fasted and fed state GIT media causes difficulties. For example the size of 51 nm large lipid nanocarriers (LNC) did not change under simulated gastric and fasted-state intestinal conditions. However, the LNC were not detectable by dynamic light scattering under fed-state intestinal conditions in the presence of pancreatic enzymes and bile salts, which reflects the limitations of the current methodological approaches.59 The application of a separation technique like field-flow fractionation prior to particle size measurements could possibly overcome these problems. In studies with lipid based formulations, the measurement of free fatty acids during and after digestion has become common practice to determine the particle stability instead of or in addition to particle size measurements.60–62 In cases where the particle integrity is not directly measureable, the release of the encapsulated compound is sometimes used to give at least some indirect information about physical changes of the particle. Since the release of encapsulated compounds is not necessarily caused by particle disruption, such conclusions should be avoided.
The transfer of particles or released BC into cells or through the intestinal wall is investigated using cell layer covered membranes like Franz diffusion cells or transwell-systems or Ussing chambers separated by pieces of the jejunum from animals.29,57,63–67 To take into account the dynamic environment of the intestinal lumen, three-dimensional models of the intestine prepared e.g. from pig intestinal tissue and human endothelial cells have recently been developed.68 The complexity of the intestinal environment also includes the presence of mucus that covers the intestinal wall. Interaction of ENM with mucus was shown to have a great effect on the mobility of ENM and due to this interaction, diffusion of ENM through the mucus layer could be very slow or ENM could be removed by increased mucus secretion.69 Thus, the use of mucus producing cell lines in in vitro test systems may yield additional information compared to other cell lines.
Bioavailability enhancement through delivery systems
The Biopharmaceutical Classification System (BCS), commonly used for pharmaceutical applications, has been proposed as a useful classification scheme for nutrients and other BC when trying to elucidate the impact of formulation on their bioavailability.70,71 It was developed for correlating in vitro dissolution and in vivo bioavailability studies of substances assuming that their solubility and dissolution in the gastrointestinal milieu and their effective permeability across the intestinal mucosa are the fundamental parameters controlling the rate and extent of their absorption.72 The BCS is used to classify substances into four classes with respect to their bioavailability as shown in Table 2. Accordingly, a low bioavailability of BC can result from a low solubility and/or a low resorption rate due e.g. to their low intestinal permeability. In addition, a low stability under GIT conditions or a fast clearance rate as a result of the liver first-pass metabolism can further be responsible for low plasma levels. Hence, ENM based delivery systems can increase the bioavailability of BC based on one or more of the following mechanisms: (i) solubility enhancement, e.g. by reduction of the particle size and increase of the surface area for dissolution or by formulating lipid or biopolymer based delivery systems; (ii) enhancement of the intestinal permeation e.g. by selection of the formulation surfactants; (iii) chemical stabilisation of the BC in the GIT, e.g. by encapsulation in gastric milieu resistant materials; (iv) controlled release and increased residence time within the GIT e.g. by encapsulation in mucoadhesive polymers.| Class | Solubility | Permeability | Example |
|---|---|---|---|
| I | High | High | Theophylline |
| II | Low | High | CoQ10, tocopherol |
| III | High | Low | Catechin |
| IV | Low | Low | Curcumin, quercetin, soy flavonoids |
The absorption of encapsulated BC along the GIT, including the mouth, the stomach, the small intestine and the colon, has been the subject of several studies and was recently reviewed.55,73,74 The impact of ENM based formulations on the bioavailability of BC in in vivo and in vitro studies and the way in which their structural characteristics contribute to the observed effects will be discussed in the following section. The outcomes of the in vivo studies with focus on the bioavailability of secondary plant metabolites, minerals and trace elements formulated as ENM reviewed here are summarised in Table 3. In the in vivo studies the bioavailability and biokinetic parameters are generally compared based on the time to reach the maximum concentration in the blood (tmax), the maximum plasma levels (cmax) and/or the area under the curve of plasma levels vs. time (AUC).
| Compound | Delivery system/ENM preparation | Increase in bioavailability (based on AUC unless stated otherwise) | Model | Ref. |
|---|---|---|---|---|
| a In formulation names A is for alginate and C for chitosan. The control was not further specified. b The dispersed powder was applied with or without a test meal. All other formulations were given together with the test meal. c Different matrices: casein micelles were dispersed in 1% fat milk while Tween micelles were in aqueous dispersion (commercial supplement). d As estimated from Fig. 3a from that publication. e Standard feed as the matrix. f As estimated from Fig. 2 from that publication. g Specific surface areas were given instead of mean diameters and were 105–197 m2 g−1 except electrolyte Fe (0.35 m2 g−1). h Infant cereal as the food matrix. i Yoghurt drink as the food matrix. j Apple juice as the food matrix. k As estimated from Fig. 5 from that publication. | ||||
| Curcumin | Suspension/lecithin mixture/liposomes (263 nm) | 5-fold compared to suspension | In vivo with rats (100 mg kg−1 BW) | 75 |
| Curcumin | Nanocrystal dispersion (250 nm)/amorphous solid dispersion (<1 mm)/nanoemulsion (196 nm)/crystalline curcumin | 16-/12-/9-fold higher plasma levels | In vivo with rats (formulations: 20 mg kg−1 BW or control: 100 mg kg−1 BW) | 76 |
| Curcumin | Solid lipid nanoparticles (135 nm)/Tween 20 micellar solution | 39-fold higher plasma levels | In vivo with rats (50 mg kg−1 BW) | 58 |
| Curcumin | Organogel-based nanoemulsion (218 nm)/aqueous dispersion | 9.3-fold | In vivo with mice (240 mg kg−1 BW) | 77 |
| Curcumin | Tween 80 micelles/native powder | 185-fold | In vivo with women and men (410 mg per person) | 78 |
| Quercetin | SLN (155 nm)/suspension | 5.7-fold | In situ perfusion method with rats (50 mg kg−1 BW) | 66 |
| Quercetin | Microemulsion (39 nm)/Tween micelles (5–20 nm) | 1.6-fold | In situ perfusion method with rats (50 mg kg−1 BW) | 79 |
| Capsaicin | Nanoemulsion A1 (50 nm)/nanoemulsion A2 (100 nm)/nanoemulsion A3 (150 nm)/nanoemulsion C (100 nm)/nanoemulsion CA (100 nm)/oleoresin capsicuma | 132-/81-/72-/77-/8-fold | In vivo with rats (10 mg kg−1 BW) | 80 |
| Hoodia gordonii steroid glycoside | Colloidal particles (110 nm)/nanoemulsion (200 nm)/mesophase (100 nm)/SEDDS/dispersed powder + Tween 80b | No differences | In vivo with pigs (7.9 mg kg−1 BW) | 81 |
| EGCG | Lipid complex (50 nm)/EtOH/H2O solution | 2.5-fold | In vivo with rats (100 mg kg−1 BW) | 82 |
| CoQ10 | Nanoemulsion (60 nm)/dry emulsion (770 nm)/dry emulsion (1700 nm)/cyclodextrin/crystalline CoQ10 | 1.7-/1.3-/1.3-/1.2-fold | In vivo with rats (60 mg kg−1 BW) | 83 |
| CoQ10 | Micelles (“Nanosolve”, 30–60 nm)/gelatine capsules | 5-fold | In vivo with humans (100 mg per person) | 84 |
| Vitamin E | Micelles (“Nanosolve”, 30–60 nm)/gelatine capsules | 10-fold | In vivo with humans (120 mg per person) | 84 |
| Tocopherol acetate | Micelles (“Aquanova”, 50 nm)/soft gel capsule | 1.5-fold | In vivo with humans via gummi bear model food matrix (100 IU per person) | 85 |
| Vitamin D | Re-assembled casein micelles/Tween-80 based commercial product | No differencec |
In vivo with humans (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 IU per person) 000 IU per person) |
86 |
| Vitamin D3 | β-Lactoglobulin/vit. D3 complex/free vit. D3 | 1,4-fold plasma level of 25(OH)D3 | In vivo with rats | 87 |
| Lignan glycosides | Nanoparticles (200 nm)/2 μm particles (top-down approach) | Approx. 1.7-foldd | In vivo with rats (80 mg kg−1 BW) | 88 |
| Sitosterol | Crystalline commonscale sitosterol. (100 μm)/crystalline microscales. (1.9 μm)/crystalline nanoscales. (550 nm)/emulsified nanoscales. (100–130 nm) | No differences in plasma levelse | In vivo with guinea pigs (160/200/180/140 mg/animalf) | 89 |
| Iron | Amorphous FePO4 particles: 11 nm/31 nm/64 nm vs. FeSO4 | Hb repletion: 0.96/0.70/0.61 relative to FeSO4 | In vivo with Fe depleted rats (170 μg per day – 484 μg per day for 15 days) | 90 |
| Iron | FePO4/Zn3(PO4)2, Fe2O3/ZnO/MgO, Fe2O3/ZnO/CaO, Fe2O3/ZnO, FePO4/Fe2O3, electrolytic Fe vs. FeSO4g | Hb repletion: 0.96/0.91/0.82/0.77/0.78/0.60 relative to FeSO4 | In vivo with Fe depleted rats (146 μg per day – 455 μg per day for 13 days) | 91 |
| Iron | Emulsifier coated FePO4 particles (300 nm, “SunactiveFe™”)/FePO4 particles (5.2 μm)/Fe(II)citrate/FeSO4 | Iron absorption: 1.2-/0.6-/1.1-fold relative to FeSO4Hb repletion:1.05-/0.78-/1-fold relative to FeSO2 | In vivo with rats (single oral dose of 2 mg Fe per kg BW) and with anaemic rats (10 mg g−1 diet ad libitum for 4 weeks) | 92 |
| Iron | Emulsifier coated FePO4 particles (300 nm, “SunactiveFe™”)/FeSO4 | Iron absorption: 0.83h-/0.94i relative to FeSO4 | In vivo with humans (5 mg Fe per person) | 93 |
| Iron | Emulsifier coated FePO4 particles (160–218 nm, “SunactiveFe™”)/FeSO4 | Iron absorption: 0.66-foldj relative to FeSO4 | In vivo with humans (9 mg Fe per person) | 94 |
| Calcium | Pearl powder: 84 nm vs. 29 μm | 1.4-fold increase of serum calcium by nanometre sized particles | In vivo with humans (780 mg per person) | 39 |
| Calcium | Pearl powder: nanoparticles (40–80 nm) vs. microparticles | 2-fold increase in calcium content in bones | In vivo with rats (10 mg per day – 70 mg per day for 4 weeks) | 95 |
| Calcium | Micrometre sized carbonate (3.7 μm)/nanometre sized carbonate (398 nm)/micrometre sized citrate (1.8 μm)/nanometre sized citrate (151 nm) | 1.28-/1.38-/1.38-/1.53-foldk increased bone mass density compared to normal diet | In vivo with ovariectomised mice (1.3 g kg−1 BW or 2.3 g kg−1 over 4 weeks) | 96 |
| Calcium | Commercial nanometre sized Ca supplemented diet (30–900 nm)/normal diet | No difference in Ca-balance, but reduced hydroxyproline | In vivo with rats (20 mg for 18 weeks) | 97 |
| Chromium | Nanometre sized chromium picolinate (70 nm) vs. chromium picolinate | 2-fold increase in serum levels | In vivo with rats (300 μg kg−1 BW for 18 days) | 98 |
Lipid and surfactant based delivery systems
The use of lipid and surfactant based systems is currently by far the most pursued approach to enhance the bioavailability of BC. Various structures can be obtained such as micellar structures, microemulsions, liposomes, nanoemulsions, solid lipid nanoparticles (SLN) or lipid nanocarriers (LNC)/nanostructured lipid carriers (NLC). Examples of the different structures are given in Table 1 and have recently been well reviewed e.g. by Tamjidi et al., 2013.99 Preparation techniques generally include emulsification methods or simply mixing the components and allow obtaining various stable formulations with exclusively food grade materials that offer high encapsulation rates for lipophilic BC.Emulsifier micelles and oil/water (o/w) microemulsion droplets are formed by the spontaneous self-assembly of emulsifier molecules based on hydrophobic interactions, leading the amphiphilic molecules to form a hydrophobic core and a more hydrophilic shell that is orientated towards the aqueous phase. Reversed micelles form a hydrophilic core and a hydrophobic shell that it orientated towards the lipid phase. While micelles consist solely of the emulsifier, o/w or w/o, microemulsions consist of an additional oily or aqueous core, respectively, and may thus be considered as swollen micelles. The term microemulsion (ME) was introduced in the late 1950s and refers to thermodynamically stable systems with nanometre sized micelle-like structures also known as Winsor-type systems. Emulsifier micelles and ME droplets are typically in the size range of approximately 5–20 nm and are characterised by a very narrow size distribution. Both, diameter and size distribution depend on the emulsifier(s) and eventually co-surfactants used and are determined under sterical and electrostatic conditions.100
While ME and micelles form spontaneously, the formation of liposomes requires the input of some energy, e.g. by stirring or leading the preparation mixture through a microfluidizer (high pressure homogenizer). Liposomes are thermodynamically stable vesicular structures consisting of a phospholipid double layer surrounding an aqueous core and hence are suitable to solubilise both hydrophilic and hydrophobic compounds.
The formation of emulsion based ENM usually requires the input of energy which is often achieved by high pressure homogenisation, micro-fluidization or sonication with ultrasound. Low-energy input methods like phase inversion or solvent evaporation methods are less common and the presence of organic solvents sometimes limits their use in food products. The resulting ENM can be categorised depending on the type of lipid, i.e. liquid lipid (nanoemulsions, NE), a solid lipid phase (solid lipid nanoparticles, SLN) or a mixture of solid and liquid lipid (nanostructured lipid carriers, NLC). The resulting droplets can be as small as 100 nm or below and the small droplet size leads to a high kinetic stability. However, these systems are not thermodynamically stable.25 While NE contain spherical oil droplets, SLN are known to form platelet shaped particles when the lipid crystallises upon cooling.28 Hence, the curvature and the packing of the emulsifier molecules at the respective o/w interface differ between these systems. This should be taken into account when discussing the encapsulation and release of BC and the digestibility of the system.
Lipid and surfactant based ENM consist of a more hydrophobic (core) and more hydrophilic (shell) regions with different water contents‡ and are thus capable to solubilise compounds of different hydrophilicities. Hence, the solubility or hydrophobicity of the BC determine their encapsulation rates and locations in the system. The properties of the individual solubilisation sites are largely determined by properties of the emulsifiers. The loading efficiency largely depends on the emulsifier and its interactions with the BC, i.e. hydrophobic interactions, electrostatic interactions and hydrogen bonds. The concurrence of these mechanisms can then be observed as partitioning behaviour. If diffusion between the emulsifier phase and the continuous phase is not hindered, a partitioning equilibrium will evolve. The partitioning equilibrium of BC allows adding the BC at different steps during the preparation of the micellar formulation, ME or NE. The BC obey preferences with regard to their solubility and will therefore locate at the respective solubilisation site.
The solubilisation of BC with relatively high solubilisation rates with respect to volume is possible with micelles and ME, but to achieve this, high amounts of surfactants are necessary.71
Generally, the solubilisation in emulsion droplets is mostly determined by the emulsifier layer at the o/w interface. Exceptions are very oily and lipophilic compounds that also partition into the core of an oil droplet.101,102 Due to the partitioning of the BC between the oil droplets, the aqueous phase and the co-existent micellar phase NE are more efficient for the encapsulation of strongly hydrophobic compounds.
The solubilisation site, i.e. the presence of a distinct pH-value, water content and accessibility by radicals, also impacts the reactivity and stability of encapsulated BC. E.g. the large head groups of non-ionic emulsifiers or the palisade layer provide a certain physical barrier against adverse conditions.103 Liposomes are considerably larger than micelles or ME droplets. Therefore, the curvature is less pronounced and thus the lipid bilayer is more densely packed than the emulsifier layer of micelles or ME droplets. This could result in a better protection of encapsulated compounds from adverse conditions or e.g. oxidizing agents.
The liquid lipid phase and the emulsifier interface of “soft” ENM is connected with a relatively high diffusivity of the BC and e.g. oxidizing agents which may result in the release of the BC or a low chemical stability. A solid lipid matrix as in SLN was thought to overcome these problems, because the BC is more or less entrapped in the solid lipid matrix. However, crystallisation of the lipid typically leads to low encapsulation rates and eventually to the expulsion of the BC during storage. One further development to address these shortcomings is the incorporation of a liquid lipid, leading to the formation of a solid/liquid matrix called nanostructured lipid carriers (NLC).99 In terms of encapsulation rates, NLC are regarded to be superior over SLN, because imperfections in the crystalline lipid matrix of NLC allow for a better encapsulation/retention of BC.104
Usually micelles, liposomes, NE, SLN and NLC are characterised by a high stability under moderate conditions with respect to the pH value, temperature or salt concentration. However, their addition to a liquid food matrix may lead to a significant dilution which can become critical with respect to the stability.
Micelles are formed above the critical micelle concentration (cmc). In liquid food matrices their size and shape depend on the pH value, salt concentration and solute (i.e. BC) concentration. However, if they are too highly diluted, they disaggregate into single emulsifier molecules in solution and the solubilised BC is released. The achievement of the dilutability of ME is a difficult task, because of the sometimes small lyotropic regions of the mixture of oil, emulsifiers and co-surfactants. However, some examples of dilutable, food grade ME have been reported in the literature. The stability of liposomes decreases at low pH-values, but is relatively high at neutral pH-values. Also NE, SLN and NLC can be destabilized by dilution, which may be an effect of a reduced concentration of the excess emulsifier molecules that are necessary to stabilise the droplets. Despite a very high kinetic stability of NE and SLN, the creaming of NE or the sedimentation of SLN may occur at prolonged storage times. The formation of a polysaccharide or a protein shell around the oil droplets, e.g. by co-acervation, can increase the stability of the NE, can increase encapsulation rates and can retard the release of BC in the GIT (see below). The crystallisation and melting behaviour of SLN and NLC has to be well known, since for their incorporation into a food matrix and during the subsequent storage the temperature must be kept below the melting point of the respective ENM. Otherwise, unpredictable changes to the system have to be expected. In this context, it has to be taken into account that the melting point of SLN is usually lower than that of the high melting bulk lipid. However, the crystallisation process, the degree of crystallinity and polymorphic transitions may cause the expulsion of the BC from the delivery system. This may reduce the protective effect of the encapsulation on the one hand and may later also lead to a burst release in the GIT.29,57,59,77,105,106 Therefore, phase changes during the storage period have to be controlled as precisely as possible.
The partitioning equilibrium of BC that facilitates their incorporation into lipid- and surfactant-based ENM can also represent a disadvantage: “leakage” of the encapsulated compound into the continuous phase is frequently reported. If a micellar or liposomal solution, a ME or a NE is diluted, the partitioning equilibrium will shift towards the continuous phase. Depending on the hydrophilicity of the compound this can result in its release. The addition of liposomes to milk during cheese manufacture resulted in a slow release of encapsulated proteolytic enzymes.107 Leakage of the BC from liposomes has been reported to be connected to a phase transition of the phospholipids21 and is intended to be overcome by e.g. the application of a polysaccharide layer. In addition, in order to stabilise or to add liposomal formulations, NE, SLN or NLC to dry food products they can be spray dried.108
Apart from concentration effects, interactions with other food components occur when incorporating ENM into food matrices. For instance, when added to o/w emulsions interactions of emulsifiers with the oil droplets are likely to occur and depend on the types of emulsifiers used. It is known that the emulsifiers of the o/w interface are in dynamic equilibrium with the coexistent micellar phase. Thus, if two emulsions are mixed, re-organisation and re-structuration of the respective interfaces and the formation of mixed micelles are expected, unless electrostatic or steric repulsion are too strong. Furthermore, the coexistent micellar pseudophase is considered as means of transport for lipophilic compounds in o/w emulsions, resulting in the partitioning of the BC between the continuous aqueous phase, the coexistent micellar pseudophase and the oil droplets. Electrostatic attraction and hydrophobic interactions would facilitate such an exchange while electrostatic and steric repulsion would make it less favourable. Furthermore, Ostwald ripening may lead to destabilisation, especially if a NE is composed of oil with a relatively high water solubility,109 SLN have been demonstrated to accumulate at the surface of emulsion droplets, which is an effect known from Pickering emulsions.110 The surface tension, the surface charge/zeta potential and to a certain extent the cristallinity/polymorph behaviour count among the properties of the SLN that affect such interactions. While the former properties are determined by the emulsifier, the latter is more complex and determined by the lipid, the combination of emulsifiers and also storage conditions. In addition, it was recently reported that SLN can be incorporated into oil droplets of o/w emulsions leading to the release of the encapsulated compounds in the oil phase.111
The stability and interactions of emulsion-based ENM in the GIT are of the same nature as in food matrices, but the surrounding conditions are even more challenging: varying pH values, high ionic strengths, mechanical stress and the presence of lipase and bile salts lead to a destabilisation of ENM. Coalescence, flocculation, Ostwald ripening and the activity of digestive enzymes alter the droplet size distribution of initial NE in the GIT and it is highly questionable whether they reach the intestine as initially ingested. Even without the addition of enzymes the gastric media destabilised SLN formulations due to their high ionic strength or low pH-values. Factors leading to high stability are a zeta potential in the range of 8–9 mV or higher, a sufficiently thick sterically stabilising emulsifier layer and an appropriate combination of the lipid phase and the emulsifier.59,112 It has to be taken into account that protein stabilised emulsions can already be destabilised in the stomach by proteolysis by pepsin.113 In this respect it is interesting to note that e.g. β-lactoglobulin is more digestible when it is adsorbed to an o/w interface than in solution which was ascribed to partial unfolding at the interface.114
The above-mentioned interactions of ENM with other food components also occur in the GIT and may be even more pronounced. This would result in a re-organisation of the ENM under post-prandial conditions if the meal contained fat and/or surface active compounds. The dilution of micellar solutions, ME, NE or liposomes by gastric and intestinal fluids and the sink conditions in the GIT can shift the partitioning equilibrium of the solubilised BC and hence promote the release of compounds (see above). Otherwise, the release is initiated by the break-down of the ENM or interactions with bile salt micelles.
The digestibility of lipid based ENM by lipase can be influenced or controlled in various ways. For instance it was reported to be enhanced with decreasing droplet size.61 Nevertheless, if normalised to the surface area the digestibility decreased which was ascribed to a less favourable packing order of lipase at the droplet surface. The digestibility of an emulsion can furthermore be controlled by the choice of the lipid phase, more specifically, by the chain length of the lipid: medium (MCT) and short chain triglycerides (SCT) are digested by lipase more rapidly than long chain triglycerides (LCT).115
Lipase digestibility was reduced when the lipid was crystallised.116 This effect could be diminished when the lipid crystalline structure was disturbed by surfactants at the particle surface.105 The activity of pancreatic lipase is further inhibited by many emulsifiers (e.g. small molecule emulsifiers like Tween 80). Bile salts can displace Tween 80 from o/w surface and hence aid lipolysis.53 If this displacement is hindered, e.g. because the emulsifier is anchored in a solid lipid matrix, lipolysis may be reduced. Recently, the application of a shell of indigestible fibres was developed to further reduce the lipolysis by representing a physical barrier or by chemical interactions. Each of the factors that lead to a reduced or sustained lipolysis can also lead to a retarded release of the encapsulated BC. The release of the encapsulated BC is positively correlated with its diffusivity within the ENM. Thus, a slower release can be expected from solid matrices like in SLN than from NE, provided that expulsion of the BC during storage is effectively prevented (see above).
Similar to the particle digestibility, the release of encapsulated compounds from different lipid nanoparticles depends on the pH-value of the media. It increases in the presence of digestive enzymes, especially pancreatin, and is characterised by an initial burst. Since the release of a BC is also determined by its partitioning behaviour, hydrophilic BC will be released earlier and more readily than lipophilic BC from lipid based ENM. Furthermore, in the intestine, lipid and surfactant-based delivery systems undergo interactions with dietary micelles resulting in the formation of mixed micelles with bile salts. Colloidal phytosterols (160 nm) replaced cholesterol from dietary mixed micelles more quickly and to a larger extent than larger phytosterol particles (100 μm) due to improved solubilisation.117 It has also been reported that o/w ME droplets interact with dietary micelles to form mixed micelles which present physicochemical properties that differ from that of the initial structures contributing to the delivery of lipophilic BC.118 More specifically, the bioavailability of β-carotene from emulsifier micelles depended on the different phospholipids used to prepare the micelles (phosphatidyl choline or lysophosphatidylcholine) and their respective absorption or digestion routes.119
The bioaccessibility of BC in NE also depends on the chain length of carrier oil. For instance the bioaccessibility of encapsulated BC increases with increasing chain length, as demonstrated for curcumin, β-carotene or tocopherol acetate. This effect was related to the ability of long chain fatty acids to form mixed micelles with bile salts that in turn solubilise released BC more efficiently.62,120,121
Unless particle absorption by epithelium cells, although inefficient, or by M-cells occurs, the encapsulated BC must be transferred into mixed micelles or has to undergo paracellular transfer to reach systemic circulation. Micelles are not absorbed intact, but dissociate and release the lipophilic compound near the enterocyte membrane.122 Apart from solubilisation kinetics lipid based ENM structures may increase the absorption of BC by acting as intestinal absorption promoters. Emulsifiers can increase the membrane fluidity facilitating transcellular absorption of BC. The impact of the presence of surfactants, e.g. Tween 80, on the cellular uptake of BC was pointed out by Benzaria et al., 2013. While Tween 80 increased the permeability of TC-7 cells protein stabilised emulsions did not have an impact on membrane integrity. This resulted in faster uptake and metabolisation of retinyl acetate from Tween 80 micelles than from emulsions (300 nm droplets).123 Emulsifiers can also cause an opening of tight junctions resulting in increased paracellular transport. Furthermore, the inhibition of efflux transporters by emulsifiers is also discussed. In addition, emulsifiers stimulate lipoprotein and chylomicron production, substances which transport lipophilic compounds and hence increase the BC concentrations in enterocytes, and in this way possibly enabling an increased lymphatic transport.122 A direct uptake of BC into the lymphoid system means bypassing the first-pass metabolism in the liver and hence higher plasma concentrations of the unmetabolised BC.75
A higher bioavailability from ENM compared to larger delivery systems may be a result of the larger surface area and hence better bioaccessibility of the BC. However, in studies designed to investigate the bioavailability of BC encapsulated in emulsified vs. non-emulsified lipids in the GIT it has to be taken into account that e.g. nanoemulsions may destabilise and form significantly larger droplets than the initial formulation whereas the presence of bile salts and lipolysis products may lead to an in situ emulsification of bulk oil.124
The encapsulation, release and bioavailability of curcumin have been extensively studied so that several in vitro and in vivo studies can at least to a certain extent be compared regarding different encapsulation approaches. After a single oral dose of 263 nm large curcumin-loaded liposomes the bioavailability of curcumin in rats was 5-fold increased, with a significantly shorter tmax, as compared to a curcumin suspension. Furthermore, it was demonstrated that this effect was not attributable to the mere presence of lecithin in the liposomes.75 Somewhat larger enhancements of the bioavailability compared to a solid crystalline dispersion were reached when curcumin was administered either via (i) a NE (196 nm, 9-fold), (ii) a solid dispersion of the amorphous material (>1 mm, 12-fold) or (iii) a solid crystalline dispersion with nanometre sized crystals (nanodispersion) obtained by a wet-milling process (250 nm, 16-fold). The tmax from the nanoemulsion was greatly reduced as compared to the control, whereas all solid dispersions led to a sustained release of curcumin.76 A 9-fold increase in the bioavailability of curcumin was also obtained with a Tween-20 stabilized nanoemulsion (218 nm) with an organogel-based lipid phase. Tmax was not affected and in vitro cell studies indicated that the uptake of intact nanoemulsion droplets seems unlikely. Thus, the increased bioavailability is a result of an improved bioaccessibility due to the facilitated lipolysis because of the larger surface area.77 A noticeable 39-fold increase in the bioavailability but not significantly changed tmax was reported after rats received curcumin loaded SLN (135 nm) instead of a Tween 20 micellar solution. The micellar solution was chosen as the control to ensure that observed effects were not solely attributable to the emulsifier.58 The considerable effect of the emulsifier was clearly presented by Schiborr et al., 2014, who administered native curcumin powder or curcumin-loaded Tween 80 micelles to men and women. A considerable increased bioavailability was observed among all participants (average 185-fold), however, the bioavailability enhancement was significantly higher in women (276-fold) than in men (113-fold).78
The absorption of quercetin from a ME with a droplet size of 39 nm (in situ intestinal perfusion method with rats) was 1.6-fold higher and presented a shorter tmax than the absorption from quercetin loaded Tween micelles (approx. 10 nm). The small difference in bioavailability reflects the structural and physicochemical similarities between these systems. The inclusion of a quercetin dispersion as a control sample would have been interesting in terms of comparability.79 The biokinetic parameters of quercetin within SLN (155 nm) and in the suspension were investigated by an in situ perfusion method in rats. The inclusion into SLN led to an increased uptake of quercetin, e.g. the maximum plasma concentration was 5.7-fold with SLN as compared to the suspension. Furthermore, the tmax in plasma was delayed by 3 h. It was suggested, but not demonstrated, that the higher plasma levels may also be attributed to decreased degradation and clearance rates. However, the authors emphasized that some care must be taken when interpreting these results due to the possible presence of emulsifier micelles which can also solubilise quercetin and thus impact its absorption.66 While only approx. 6% of the quercetin from SLN was absorbed in the stomach, it was efficiently absorbed in all segments of the intestine by a passive transport mechanism. Absorption rates were generally higher in lower parts of the intestine than in upper parts. This was explained by the fluidity of the cell membranes and the presence of M-cells in the ileum which affect the absorption rates differently depending on the formulation. Solid particles may be transported more efficiently by M-cells in the ileum, whereas the fluidity of the membranes is more important when interactions between lipids and surfactants with membranes are more pronounced.66,79
The relative bioavailability of epigallocatechin gallate (EGCG) was about 2.5-fold enhanced in rats when EGCG was incorporated into 50 nm large ENM by co-solubilisation with phospholipids. A low lipid to EGCG ratio led to slightly higher plasma concentrations. It was suggested that the higher bioavailability resulted from the modified metabolism of EGCG when embedded in a lipid matrix that prevents the EGCG from glucuronidation in the intestinal wall.82
The bioavailability of encapsulated co-enzyme Q10 (CoQ10) was studied in rats from a single oral dose using crystalline CoQ10 as control. The bioavailability decreased in the following order: NE (60 nm) > high pressure homogenised and spray dried emulsion (770 nm) = spray dried emulsion (1700 nm) > crystalline CoQ10.83 This study showed again that the bioavailability of an encapsulated compound can be increased with smaller droplets when they are in the nanometre scale.
Increased absorption of CoQ10 and vitamin E in humans when ingested incorporated within a commercial micellar formulation (NanoSolve®, Lipoid GmbH, Ludwigshafen, Germany) as compared to gelatine capsules containing the BC was reported by Wajda et al.84 This ENM consists of phospholipids and was stated, but not demonstrated, to be within the size range of 30–60 nm. While plasma levels increased 5-fold, the tmax of CoQ10 was unaffected. In contrast, with vitamin E, plasma levels increased 10-fold with a 3 h shorter tmax.84 Similarly, the plasma concentration of α-tocopherol acetate was significantly increased after the ingestion of a water soluble micellar formulation with a particle size of 50 nm (Aquanova AG, Darmstadt, Germany) which was incorporated into gummy bears as a food matrix and compared to gelatine capsules containing α-tocopherol acetate.85 However, the authors did not investigate the effect of the processing, e.g. integrating the micelles in a model food system, on the particle characteristics. The presence of a test meal was also included in a study investigating the bioavailability of the plant steroid glycoside rich extracts of Hoodia godornii in pigs. The BC was administered via different Tween 80 containing formulations, i.e. colloidal particles (110 nm), NE (200 nm), a mesophase (100 nm), a self-emulsifying drug delivery system (no size given) or via a non-formulated extract. The absolute bioavailability of the BC increased 46-fold upon formulation, irrespective of the formulation or the presence of a test meal. In this case, dissolution and/or incorporation into dietary mixed micelles were sufficient from the formulations alone. However, in the presence of a test meal the slower gastric emptying resulted in a delayed tmax.81
Capsaicin loaded NE with different droplet sizes and different stabilizers (alginate, chitosan, alginate + chitosan) in addition to Tween 80 as an emulsifier were given to rats. The bioavailability was increased up to 131-fold compared to the capsaicin oleoresin with unchanged tmax. A tendency towards higher AUC with decrease in the droplet size from 150 nm to 50 nm could be observed. However, the combination of biopolymers (triple-layer nanoemulsion) resulted in comparably lower bioavailability with respect to the double-layer nanoemulsions. The role of the biopolymers within the release and absorption rates was discussed but could not be experimentally elucidated.80
The different types of delivery systems influenced the biokinetic parameters differently, e.g. tmax was longer with SLN66 but was shorter with liposomes,75 micelles,84,85 ME79 and NE.76 This could be attributed to different interactions with cell membranes in the intestinal wall and different release kinetics. A sustained release of the BC may be preferable in order to avoid peak concentrations in the blood plasma. With respect to the BCS it can be concluded that lipid based ENM can be used to overcome solubility problems of lipophilic compounds (class II, IV) and that their action as absorption promoter would be beneficial for class III and IV compounds. Nevertheless, lipidic formulations may not be able to overcome vitamin deficiency caused e.g. by lipid malabsorption.
Controlling the impact of lipid crystallinity on digestibility, expulsion of active compounds and release kinetics is challenging. Formulation parameters need to be very carefully adjusted to ensure a stable conformation of the crystalline lipid matrix and hence constant properties over the entire shelf life of a product.
Polysaccharide-based delivery systems
Polysaccharides are predominantly used in combination with other macromolecules to form delivery systems in the microscale, e.g. in spray dried microcapsules they act as wall materials and in emulsions they are used to form structured/multi-layered o/w interfaces. The increasing interest in using polysaccharides to design delivery systems for BC has arisen from the knowledge of non-covalent interactions between polyphenols and polysaccharides.125 Such interactions were first observed in relation with functional and structural modifications of the polysaccharides and are now investigated targeting the delivery of BC. It has been reported that flavonoids may alter starch digestion by interfering with gut digestive enzymes and that the interactions of polyphenols with non-digestible polysaccharides in turn may lead to the transport of phenolic compounds to the lower gut. However, it has to be stretched that results reported in the literature vary greatly with the compounds studied and general conclusions or assumptions should be avoided.125Hydrophobically modified starch (HMS), cyclodextrines and chitosan are the most frequently used polysaccharides for the design of delivery systems.
HMS is an amphiphilic polymer which forms micelles based on mechanisms similar to those described before for emulsifier micelles. The low bioavailability of curcumin results from its low solubility and low membrane permeability. The incorporation of curcumin in HMS micelles (14 nm) enhanced its solubility considerably (>1600-fold). The authors reported that the enhanced solubility resulted from the hydrophobic interactions and hydrogen bonding between curcumin and HMS.126 HMS is known for its emulsifying capacity. Therefore, it is expected that it participates in the formation of emulsions under gastric conditions. It can also be assumed that the micelles disintegrate upon digestion by amylase and hence release the BC in the GIT. However, HMS is partially resistant to digestion.127 Thus, intact micelles may reach the intestine after ingestion where they might interact with dietary mixed micelles. Interactions with micelles on the molecular level have been reported for dietary soluble fibres128 and amphiphilic polymers.129 Furthermore, it has been reported that dietary soluble fibres reduce the resorption of bile salt micelles.130 It should be further investigated whether such an effect could reduce the practicability of starch micelles as delivery systems for BC. On the other hand the transfer of encapsulated compounds to the lower parts of the GIT can be beneficial. Their release after microbial degradation of the dietary soluble fibres can be expected.
Cyclodextrins (CD) are cyclic ring molecules and may include BC into their hydrophobic cavity by hydrophobic interactions, van der Waals forces and hydrogen bonds. With both components in a dynamic equilibrium, association and dissociation rates of the inclusion complexes are negatively correlated with molecular size and the ionisation degree and positively correlated with the hydrophilicity and electron-donor character of the BC.31 Unsaturated fatty acids, CoQ10 and sensitive aroma compounds are reported to form stable nanometre sized CD inclusion complexes spontaneously.31 The well-documented improved aroma retention after adding the latter complexes to different food matrices may indicate that the stability of these CD inclusion complexes is only marginally affected by food components. The stability of CD is provided in an environment with a pH above 3, but can also be influenced by inorganic salts. Due to their acid sensitivity CD are expected to be hydrolysed under gastric conditions. Under in vitro GIT conditions a β-CD inclusion complex was partially hydrolysed. This was explained by both acidic hydrolysis and enzymatic degradation resulting in a partial release of complexed conjugated linoleic acid.131 Since CD inclusion complexes reach and possibly pass the small intestine, the knowledge/understanding of their interactions with bile salts is also necessary. Inclusion phenomena with surfactants have been reported so that similar interactions between CD and bile salts are expected.132 However, the impact of such interactions on the inclusion or release of BC cannot be predicted. The transfer of CD inclusion complexes to the colon seems possible to a certain extent, microbial amylases will then finally degrade CD thereby causing the release of the BC.133
Nanometre sized chitosan particles or nanogels can be formed by ionic gelation with polyanions, e.g. sodium tri-poly-phosphate (TPP) and peptides, and may be loaded with BC by ionic interactions, if the BC is present as an ion. Non-ionic molecules like flavonoids are loaded into the biopolymeric nanogels predominantly by hydrogen bonds, with lower loading capacities (e.g. rutin).134 Covalent bonds between chitosan nanogels and tea catechins have also been reported.135 At pH values below approx. 5.5, i.e. under gastric conditions, dispersed chitosan nanogels are stable. Higher pH values lead to the hydrogenation of NH-groups of the chitosan macromolecules, reducing its solubility and resulting in precipitation and particle disintegration. Freeze-drying or spray-drying of chitosan nanogels in order to provide storage stability is applicable with an acceptable re-dispersibiltiy degree of the dried material, provided that suitable concentrations of cryoprotectants and wall materials, respectively, are used. It can be deduced from these findings, that chitosan nanogels would be stable if mixed with other food components in the dry state or if added to acidic food products (beverages) with low or moderate ionic strength. After rehydration of dried particles and following glass transition of the polysaccharides a burst release of the encapsulated compound is typical73,136,137 pointing out a very loose association of BC and chitosan nanogels. Freeze drying would enhance such behaviour because the dried material is characterised by a porous structure that facilitates diffusion processes. In such cases the use of chitosan particles/nanogels in liquid food is questionable. Furthermore, the release of the BC from a dry food matrix has to be expected as soon as it is ingested, while chitosan nanogels, stable under gastric but not intestinal conditions, will be transported to and degraded in the intestine. Despite these shortcomings, chitosan nanogels have gained considerable attention for several reasons. Due to the mucoadhesive properties of chitosan, the contact time of the BC with the intestinal mucosa would be prolonged, potentially resulting in an increased intestinal absorption.34 Furthermore, chitosan is able to increase the permeability of cell layers by the reversible opening of tight junctions. Sadeghi et al.138 have shown, that the chitosan dependent permeability of cell layers is less pronounced in a caco-2 cell system when chitosan is present as a nanometre sized material than in solution due to fewer positive charges available in nanometre sized materials. In contrast, in a mucus producing cell line no differences in cell permeability were detected between chitosan solution and nanometre sized chitosan nanogeles139 thus illustrating the importance of the mucoadhesive properties of chitosan. The permeability of cell layers for chitosan–lecithin-encapsulated melatonin depended on the formulation, especially the type of lecithin which was associated with chitosan. No direct evidence for trans- or endocytosis was found.140 This is in accordance with results obtained with freeze dried nanometre sized epigallocatechin gallate (EGCG) loaded chitosan particles/nanogels (432 nm) and murine jejunum samples. The significantly increased in vitro intestinal absorption of EGCG after association with chitosan nanogels was not attributable to an increased active or passive transport by the chitosan nanogels but to the increased intestinal stability of EGCG. In addition, it was shown that the transport of EGCG through the intestinal wall was initially slower after association with chitosan nanogels.63 This indicated that the chitosan nanogels offer the possibility for a retarded release of associated BC which is related to the swelling of the glassy material and the transition from the glassy to the rubbery state.137 The burst release of EGCG could be slowed down with peptides as cross-linking agents instead of TPP. Furthermore, it could be demonstrated by fluorescence microscopy that these nanogels can be taken up by living HepG2 cells resulting in a considerable in vitro antioxidant activity of EGCG.136 The low stability of chitosan nanogels at pH values higher than 5.0 to 5.5 is a limiting factor for their use as delivery systems.35
In summary, these few studies showed the potential of polysaccharide based delivery systems to overcome solubility or permeability problems of BC that would otherwise reduce their bioavailability in the GIT. Thus, such delivery systems could be useful at least for class II or class III compounds. However, available data are too little to draw general conclusions.
Protein based ENM
The development of protein based ENM for food applications is less advanced and most studies have a technological background, without addressing delivery/bioavailability aspects as the main topic. However, some promising examples of potentially useful formulations have recently been presented.141 With respect to the mechanisms of delivery system formation and the association and release of BC, it is helpful to differentiate between BC inclusion into molecular complexes (e.g. with β-lactoglobulin, BLG), BC incorporation into self-assembled structures (casein micelles) and BC embedding into polymeric gel particles. The latter can be formed by cation induced gelling (e.g. soy protein isolate; SPI142) or by enzymatic (trans-glutaminase) or chemical (glutaraldehyde) cross-linking of gels.143 The association of BC with the protein complex results from hydrophobic interactions, van-der-Waals forces, electrostatic interactions, hydrogen bonds and covalent binding.The ability to form hydrogen bonds and a high hydrophobicity are favourable properties for BC in terms of the inclusion into protein complexes. The presence of aromatic amino acids and proline are positively correlated with high binding capacities only if they are well accessible. The formation of helices makes proteins less flexible and therefore less able to form inclusion complexes with BC.144 Proteins with random coil or random helical character are generally considered to have higher binding capacities than globular proteins. However, Bovine Serum Albumin (BSA) and BLG possess specific binding sites for phenolic compounds.
Conditions affecting the formation of molecular BLG/BC inclusion complexes usually also determine their release since non-covalent interactions are reversible. Thus, some points regarding the binding, stability and release are discussed concurrently.
Depending on the pH value of the environment, a reversible transition of BLG into monomers and dimers can be observed. The BLG monomer possesses three binding sites with different properties and affinities for BC. The polar aromatic binding site near the AB-loop at the surface has a high affinity e.g. for resveratrol,145 folic acid,146 EGCG36,147 and other flavonoids.148 Complexation typically occurs via hydrophobic interactions and hydrogen bonding. In addition, the reversible or irreversible covalent binding of allylthiocyanate via amino-, thiol- and disulfide-bonds to BLG has recently been proved.149 This binding site is not affected by lowering the pH value of the environment, thus allowing a stable binding also in acidic beverages and under gastric conditions. Thermal treatment of the protein increased the affinity to resveratrol.145
A hydrophobic pocket or groove between an α-helix and β-sheet is known as the retinol binding site and has also an affinity for folic acid,146 vitamin D150 and possibly for tocopherol.145,151 This binding site is also pH-independent. The BLG/vitamin D3-complex is stable over a wide range of pH values from 1.2 to 8 i.e. also under gastric and intestinal conditions.150
The tertiary structure of BLG forms an internal cavity (calyx) that is known to incorporate fatty acids like docosahexaenoic acid (DHA),152 vitamin D150 and tocopherol.151 Binding in the calyx is most efficient at pH values between 6 and 8. At more acidic pH the EF loop which acts as a lid for the calyx closes which is associated with the release of fatty acids. However, the dissociation is reversible so that the reformation of the complex upon increase in the pH value can occur.153 Furthermore, it has been postulated that small hydrophobic molecules inside the calyx may be protected from the environment by a closed EF-loop.150
Thus, complexation is affected by the size and chemical structure of the BC. Generally, the complexation of the above mentioned BC to BLG increased their stability during storage. The application of high temperature resulted in the release and loss of vitamin A but not of vitamin D, which was ascribed to the binding of the latter on the surface and hence is independent of unfolding.176
The surface of the BLG molecule also bears binding sites which leads to the formation of BLG/pectin complexes presenting a diameter of about 100 nm. BC complexation in the presence of pectin results in additional protection and increased stability of the complexed BC as demonstrated for DHA.152 The formation of such complexes could allow retarded release because the late digestion of pectin in the intestine may lead to a transfer of the BLG-complex into lower regions of the intestine.152
In contrast to other milk proteins, native BLG is resistant against enzymatic digestion due to its globular structure with inaccessible binding sites. Thus, intact BLG can reach the upper intestine.154 For instance, BLG/EGCG inclusion complexes released the EGCG very slowly during in vitro gastric digestion. However, intestinal digestion experiments are still needed to evaluate the potential of these delivery systems.155 Under the alkaline conditions of the intestine BLG loses its tertiary structure resulting in the release of bound BC.154 A small portion of BLG (5–10%) can be resorbed intact by transcellular or paracellular routes.156,157 The similarity of BLG with the retinol binding protein (RBP) leads to the assumption that a specific transporter in the brush border membrane allows transcellular uptake of BLG.177 Nevertheless BLG did not improve the transport of retinol or palmitic acid across caco-2 cell layers.157 Despite the high pH value in the intestinal lumen, a release of substances located in the calyx can occur near cell surfaces where a lower pH is present and therefore dissociation of the complex is favoured.153
Caseins have a large number of proline residues, a high net charge and low intrinsic hydrophobicity leading to a unique unfolded structure under native conditions. In milk, caseins are usually present in the form of micelles which are formed by hydrophobic interactions and colloidal calcium phosphate bridges and are 50–500 nm in diameter.158 Beta-casein is more hydrophobic than alpha or kappa-casein, reversibly forms micelles of about 12 nm with a hydrophobic core159 and was demonstrated to be very efficient to encapsulate EGCG.144
The spontaneous binding of resveratrol to sodium caseinate is based on hydrogen bonds and hydrophobic interactions.160 In the presence of calcium, re-assembled casein micelles (rCM) can be prepared from sodium caseinate. In contrast, the co-assembly of casein with DHA in the absence of calcium was based on hydrophobic interactions and lead to the formation of clusters of about 290 nm in diameter.10 rCM have been demonstrated to incorporate e.g. DHA,10 vitamin D86 or curcumin.161 Different measurement techniques in these studies revealed that the respective BC was located in the hydrophobic core of the micelles which lead to its protection against degradation during storage.
rCM are stable during thermal or high pressure treatment and would therefore be suitable for the incorporation into food matrices as long as the pH value is near neutral.10,86 However, at pH values below 4.6 casein micelles are destabilised.158 Such destabilisation should thus occur in the stomach leading to the release of incorporated BC. Furthermore, the open structure of casein makes it accessible for digestive enzymes. Thus, retarded release of BC from casein based delivery systems cannot be expected unless a protective coating is applied. Interestingly, the renneting properties of casein micelles were altered by the association of EGCG.162
Mixtures of proteins and polysaccharides, rather than pure proteins, are used to prepare nanometre sized delivery systems. Incorporation of astaxanthin for example into 100 nm large colloidal dispersions consisting of polysorbate 20, gum Arabic and sodium caseinate resulted not only in a higher chemical stability of astaxanthin, but also in an increased in vitro cellular uptake into HT-29 cells. The cellular uptake was further increased by the addition into skimmed milk as compared into water or orange juice. Although the model food systems were not subjected to in vitro digestion before application, this study highlighted the impact of the food matrix on the efficacy of delivery systems.163
EGCG from ENM formed on the basis of caseino-phosphopeptides and chitosan particles was more readily taken up by caco-2 cells than an EGCG solution.164 It was also possible to form nanometre sized lactoferrin particles coated with pectin or carrageenan. The polysaccharide shell delayed in vitro gastric proteolysis which would allow some of the protein to reach the duodenum.165 Furthermore, the coating of different ENM with a protein layer has gained increased interest. In the case of ENM that are sensitive to gastric fluids, a coating with poorly digestible proteins, e.g. zein or BLG, enables retaining the incorporated compound in the ENM until it reaches the intestine.166–168
The protection of incorporated or associated compounds from gastric conditions could be one of the major advantages of protein based ENM as delivery systems. However, much more research is needed to evaluate their potential for food applications. A drawback of the use of proteins as delivery systems may be their potential allergenicity, which, however could also be reduced due to conformational changes. Furthermore, proteins provide essential amino acids and are therefore interesting to investigate from a nutritional point of view. However, it has to be kept in mind that amounts of proteins added to a food product as delivery systems for BC are probably too low to significantly contribute to the uptake of essential amino acids. Interactions between the carrier and BC could lead to interesting effects. For instance the interactions between proteins and phenolic compounds cannot only influence the bioavailability of the phenolics but probably also impact the nutritional value of the protein. It has been reported that covalent bonds between the protein and the phenolics in tea, coffee and cocoa extracts lead to increased digestibility by pancreatic enzymes while non-covalent bonds may decrease the digestion by pepsin.169,170 Digestibility and allergenicity can also be affected by cross-linking of proteins, e.g. by trans-glutaminase. Cross-linked casein and BLG showed increased digestibility. The digestion of BLG also showed lower in vitro immunoglobuline E (IgE) antigenicity.171,172 Furthermore, it was recently shown that the digestibility of BLG in emulsions depends on the composition of the oil phase and the presence of other surfactants. This effect was related to conformational changes of the protein at the o/w interface and hence the accessibility for digestive enzymes.114 Different peptide patterns arising from enzymatic digestion, in turn, might play a role with respect to the allergenicity of the protein.173
To our knowledge, the only studies investigating the effect of purely protein-based delivery systems for food applications concerned the delivery of vitamin D. The encapsulation of vitamin D in rCM resulted in a protection of vitamin D from degradation superior to a commercial Tween 80-based supplement while the bioavailability of vitamin D from both preparations was similar. Conclusions about possible differences with respect to the fate of the preparations in the GIT and their impact on the absorption of vitamin D cannot be drawn from that study, because the test systems were comprised of very different constituents and of different complexities (the rCM were added to 1% fat milk while the Tween-80 micelles were given in the form of an aqueous suspension).86 The complexation of vitamin D3 with β-lactoglubulin (BLG/D3) did not only improve the vitamin D3 stability during storage and under GIT conditions but also increased the intestinal absorption of vitamin D3 compared to an aqueous solution/suspension of vitamin D3. The 1.4-fold increase in the bioavailability of vitamin D3, measured as 25(OH)D3 plasma concentrations, was ascribed to the complex crossing the intestinal epithelium membrane.150
Despite the large potential of protein based delivery systems in terms of BC stabilisation, bioavailability enhancement and/or retarded release properties, general assumptions should be avoided, because the mentioned effects depend on the combination of the protein and BC. For instance, the presence of different (milk and soy) proteins reduced the bioavailability of galloylated catechins from green tea extracts, but increased bioavailability of non-galloylated catechins.174
Plant materials, nanocrystals and minerals
The effect of particle size reduction on bioavailability has been studied mainly for plant materials, minerals and trace elements.The impact of the particle size on the permeation of BC from the plant material through cell layers was studied with caco-2 cells. The transport and absorption of lignan glucosides was increased 1.7-fold with decreasing size of the plant material (2 μm vs. 200 nm), but the direct transport of the plant material could not be demonstrated.40 The same material was administered to rats through a stomach tube. The concentrations of sesaminol triglucoside in all major organs were higher when the rats received the ENM meal, whereas the organic particulate material was not detected in organ tissue.88 However, a study towards the effect of the particle size and crystallinity on the bioavailability of sitosterol carried out using crystalline commonscale (∼100 μm), microscale (1.9 μm), nanoscale (550 nm) and emulsified nanoscale (100–130 nm) sitosterol showed no differences in cholesterol or sitosterol concentrations in plasma and liver of guinea pigs. The authors suggested a similar solubilisation, micellisation and uptake in the small intestine enterocytes with regard to particle size.89 This could be a result of alterations of particle characteristics after their inclusion into the diet or after ingestion.
Nanocrystals have been discussed to be advantageous for BCS class II compounds because they can help to overcome their low solubility. The nanocrystals dissolve quickly in the GIT after oral consumption and hence their uptake from the gut is unlikely.41 However, it is worth noting that amorphous curcumin presented a similar bioavailability as in ENM formulations despite the large difference in material size.76 This reflects the role of the crystallinity of the particle matrix on the dissolution and how this can influence the bioavailability. A general issue during the processing of plant materials with high mechanical and/or thermal energy input is the in situ formation of emulsion droplets consisting of plant material components. Therefore, it should be kept in mind that in situ emulsification may be the reason for the uptake effect recorded for ENM obtained by a top-down approach.40
Pearl powder is used as an anti-inflammatory and detoxification agent in traditional Chinese medicine. A single oral dose of nanometre sized pearl powder (NPP, 84 nm, 470 nm) or micrometre sized pearl powder (MPP, 29 μm, 172 μm) of similar composition was given to adults. Calcium from the NPP had a 1.4-fold higher bioavailability as compared to MPP and reduced the Ca2+ dependent serum intact parathyroid hormone to 54% and 39%, respectively. No difference was observed in the urine calcium concentration.39 When similar particles were administered to rats over four weeks, the absorption and retention rates of calcium were significantly higher from nanometre than from micrometre sized material, resulting in a higher body weight and increased bone length and weight.95 Similarly, the bone mass density of ovariectomised rats increased after four weeks in the following order: micrometre sized carbonate < nanometre sized carbonate (398 nm) = micrometre sized citrate < nanometre sized citrate (151 nm). This order results from overlapping effects from the solubility of the different salts and the particle sizes, but clearly indicates that calcium from smaller particles of the same material is better metabolised.96 In a different study, rats were fed for 18 weeks either with a normal diet or a diet enriched with a commercial nanometre sized calcium containing product. The particles were not characterised by the authors, but according to the product specifications, the particle diameter was in the range of 30–900 nm. Although there was no difference in the calcium balance between the groups, hydroxyproline as a marker for bone resorption was reduced by nanometre sized calcium.97
Amorphous ferric pyrophosphate of different size ranges (dBET = 64 nm, 31 nm, 11 nm) prepared by flame spray pyrolysis or a commercial ferrous sulphate preparation was administered to Fe depleted rats in concentrations between 170 μg per day to 484 μg per day for 15 days. The bioavailability of the respective formulation was monitored via the haemoglobin concentration in the blood and was evaluated by dose–response curves. These revealed that the small particles and the FeSO4·H2O formulations were more efficient in Fe repletion than the medium and the large particles. This result was explained by the high solubility of the small ferric phosphate particles but not by the direct uptake of intact particles via paracellular uptake or the lymph.90 Ferric phosphate (FePO4) and ferric oxide (Fe2O3) materials prepared in a similar way and size range were used for experiments investigating the effect of additional ZnO or MgO. The relative bioavailability of iron increased from 77% from Fe2O3/ZnO particles to 91% from Fe2O3/ZnO/MgO particles. The MgO forms a solid solution within the Fe2O3/ZnO/MgO particles, thereby reducing the bond strength and thus increasing the solubility resulting in an enhanced bioavailability. The enhanced bioavailability of iron from nanometre sized ferric pyrophosphate did not result in iron deposition in tissue or translocation in the mucosa or submucosa in the GIT of rats.91
A commercial formulation of ferric pyrophosphate with an average size of 240–300 nm, coated with an emulsifier layer (SunActive Fe™, Taiyo International Inc., Minneapolis, USA), has been compared to other iron containing materials in acute and long-term animal and acute human studies. The bioavailability of the nanometre sized ferric pyrophosphate relative to ferrous sulphate and micrometre sized ferric pyrophosphate was increased and the release was retarded resulting in longer tmax. Contrary to the larger ferric pyrophosphate particles, the nanometre sized ferric pyrophosphate could regenerate haemoglobin values in anaemic rats to normal levels within four weeks.92 When the same materials were given to humans in two different test meals (yoghurt drink and infant cereal) on two consecutive days, no difference was detected in the bioavailability of the nanometre sized ferric pyrophosphate and ferrous sulphate from neither of the meals.93 The same nanometre sized iron was absorbed to somewhat lower extents when it was added to apple juice.94 From the outcomes of these studies it can be concluded that nanometre sized ferric iron materials may reach the same bioavailability as ferrous irons without having the same negative sensory side effects. When comparing the commercial product “SunActiveFe™” to other formulations, it has to be kept in mind that the iron particles are coated with an emulsifier layer and are therefore not chemically and structurally equivalent to the other formulations. Furthermore, the particle diameters given in the different studies using this material vary between 160 nm and 300 nm which may reflect batch to batch variations and the differences dependent on the method to obtain the particle size.
Chromium has been reported to increase the lean body mass and to stabilise the blood glucose level, but it is also known for its very low bioavailability. The digestibility of nanometre sized chromium picolinate (55–100 nm) was 1.7 times as high as the bulk chromium picolinate resulting in 2-fold increased serum concentrations of chromium in rats after administration over 18 days.98 Furthermore, high density lipoprotein (HDL) was increased while low density lipoprotein (LDL) was decreased as compared to the other test materials (CrCl3 and non-nanometre sized chromium picolinate). The abdominal fat pad was significantly larger in the group that received the nanometre sized material as compared to the bulk material. The authors concluded that the chemistry of chromium was independent of the formulation, but that the higher bioavailability resulted in greater effects on blood lipoproteins. Zha et al. (2007) proved experimentally that nanometre sized particulate chromium had dose dependent effects on body composition and some blood lipid parameters in rats. However, since no control group receiving chromium in another formulation was studied, it is not possible to postulate that the observed effects were the result of a higher bioavailability.175
The reviewed studies indicate that the bioavailability of calcium and chromium is enhanced by 1.4- to 2-fold from nanometre sized sources as compared to larger particles. The higher calcium bioavailability from the smaller sized material could be explained by its better solubility in the GIT. In contrast, the higher chromium bioavailability was suggested to result from the transfer of the particles through cells in the intestinal wall.
Summary and concluding remarks
Many types of ENM are known from pharmaceutical applications and nowadays efforts focus on the knowledge transfer to food related applications, mainly aiming at the enhancement of the nutritional and health value of food products. One of the main difficulties hereby is the formulation of ENM intended as delivery systems for BC using exclusively food-grade ingredients. In vitro and especially in vivo studies with regard to ENM based on food-grade materials are scarce and, so far, mostly lipid based ENM have been investigated. Each type of system has its advantages and drawbacks. With lipid based ENM the crystallinity of the lipid phase is the most challenging issue, since it affects the encapsulation and release of BC as well as the stability and digestibility of the ENM. Generally, a slower release can be expected from ENM with a crystalline lipid matrix. Polysaccharide based ENM are typically characterised by a burst release of encapsulated compounds, which can to a certain extent be avoided by formulation parameters and possibly by drying. Dried particles require swelling and glass transition before the active compound is released so that these systems are potentially useful if a retarded release is desired. Protein based ENM can host BC via the formation of covalent or non-covalent bonds. The type of interaction will determine release rates and may also influence the digestibility and potential allergenicity of the protein. Optimal formulations with respect to particle stability, BC stability, encapsulation rates and release rates will most likely consist of more than one type of material. Recently, research has been directed towards mixed formulations, e.g. by the application of coatings onto the initial particles.According to the studies reviewed here, potentially health promoting BC formulated within nanometre sized systems may be used to provide functional food and nutraceuticals with an enhanced bioavailability within the GIT. In most of the reviewed in vivo studies the bioavailability of BC was enhanced and/or the absorption of BC from ENM was characterised by more favourable biokinetic properties. Furthermore, the delivery by ENM had other advantages e.g. with respect to the chemical stability of the BC in the food matrix or sensory properties compared to conventional formulations. It was pointed out that the type of ENM used for the delivery of a certain BC should address the limiting factor for its bioavailability, e.g. by taking into account the different BCS classes. The modes of action include enhanced solubility of the BC, improved uptake by dietary micelles, prolonged contact time with the intestinal wall and improved chemical stability under GIT conditions. The bioavailability enhancement achieved by the use of ENM is not only strongly supported by their nanometre sized structures, but also by their surface properties and the presence of crystalline structures. Many authors claim that the ENM formulations may lead to an increased bioavailability of BC due to the transfer of intact ENM through cell layers. However, there is no experimental proof of the passage of intact food-grade ENM through intestinal walls in the literature. There are numerous studies that demonstrate the permeability of cell membranes for certain types of ENM. Most of these studies concern pharmaceutical formulations in which the ENM contain substances allowed for this use but are generally not food-grade materials. Due to the different surface and binding properties of ENM prepared with different materials the transfer of such results to food-grade formulations would rather be speculation. Therefore, this review was focused on ENM prepared using food grade materials. The optimisation of ENM formulations with respect to the permeability through the intestinal walls would have to be directed to the particle size, digestibility and surface properties. It can be concluded that the enhancement of the bioavailability of BC but not the targeted transport of BC to certain organs is expected to remain in the focus of the development of ENM for food use. Although exclusively food grade materials can be used for the preparation, risk assessment of the use of ENM in food is crucial. Actual or perceived risks include the transfer of intact particles through the intestinal walls into the systemic circulation and accumulation of particles or BC in organs and the occurrence of very high peak concentrations of BC in the blood. Since reliable data regarding toxicity or risks in general are not present in the literature, this is a discussion with many unknowns. It is true that particles below a certain size can pass the intestinal wall, as has been shown for many inorganic particles and also for pharmaceutical formulations, but this has not yet been shown for food grade formulations that have undergone gastric and intestinal conditions. If structures are soluble (e.g. nanocrystals) or digestible, the components will be part of regular absorption and metabolism. Absorption of intact food grade ENM is therefore unlikely. Nevertheless, until analytical methods are more advanced, more research is necessary to understand the fate of ENM in the GIT. With respect to plasma concentrations of BC it can be summarized that up to 184-fold enhanced bioavailability of BC and up to 39-fold maximum plasma levels are reported. The possibility of undesired effects due to intakes exceeding acceptable daily intake values (ADI) of encapsulated BC or very high peak plasma levels is not reliably excluded for all delivery systems reviewed. Therefore, manufacturers of fortified food products should take into account the increased bioavailability of the encapsulated compounds and adjust the added amount accordingly. Moreover, from the results of the acute studies it is not possible to predict the consequences for long-term intake of food products fortified with BC-loaded ENM. Studies including nanometre sized delivery systems in direct comparison to natural food matrices containing the BC of interest are still lacking and would be interesting for further research. As reviewed here, many efforts are made towards the design of delivery systems and the in vitro or in vivo study of nutrient release. However, any potential application of ENM produced with food-grade materials and designed for the delivery of nutrients depends not only on their success as the delivery system per se, but also on their stability during processing to the final food products, their stability in the food matrices along storage-life and their stability and release of the nutrients along the GIT (Fig. 1). It deserves special attention that in several studies where the administration of BC and BC-loaded ENM was done via a food or feed matrix, only a little or no difference in the bioavailability of the BC was observed between BC and BC-loaded ENM or between different delivery systems for the same BC. Unfortunately studies targeting the interactions of the ENM with the food matrices and their fate in the GIT are still very scarce. We strongly recommend investing much more research efforts towards understanding the interactions between ENM and the food matrix as well as the GIT to develop efficient delivery systems for BC for the use in food.
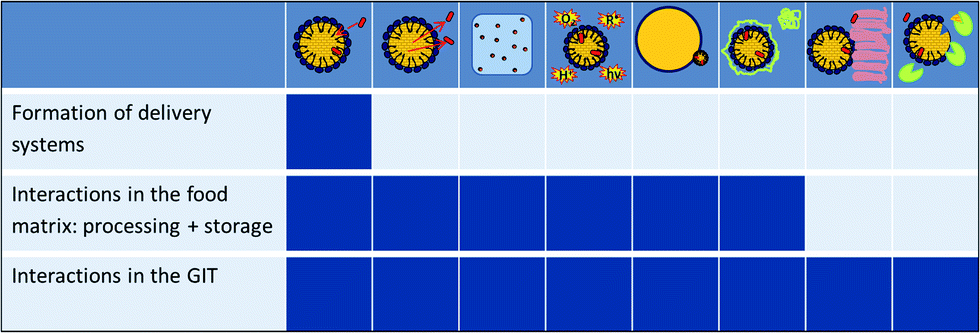 | ||
| Fig. 1 Factors needed to be investigated to fully understand the oral delivery of BC via ENM in food matrices: formation, release, solubility, protection of BC, interactions with the food matrix, molecular interactions with food components or bile salts, interactions with the intestinal wall, absorption, (enzymatic) digestion/hydrolysis and release. | ||
References
- N. Sozer and J. L. Kokini, Trends Biotechnol., 2009, 27, 82 CrossRef CAS PubMed.
- M. A. Augustin and Y. Hemar, Chem. Soc. Rev., 2009, 38, 902 RSC.
- P. R. Srinivas, M. Philbert, T. Q. Vu, Q. Huang, J. L. Kokini, E. Saos, H. Chen, C. M. Peterson, K. E. Friedl, C. Dade-Ngutter, V. Hubbard, P. Starke-Reed, N. Miller, J. M. Betz, J. Dwyer, J. Milner and S. A. Ross, J. Nutr., 2010, 140, 119 CrossRef CAS PubMed.
- E. Acosta, Curr. Opin. Colloid Interface Sci., 2009, 14, 3 CrossRef CAS PubMed.
- Q. Huang, H. Yu and Q. Ru, J. Food Sci., 2010, 75, R50–R57 CrossRef CAS PubMed.
- Q. Chaudhry, M. Scotter, J. Blackburn, B. Ross, A. Boxall, L. Castle, R. Aitken and R. Watkins, Food Addit. Contam., 2008, 25, 241 CrossRef CAS PubMed.
- T. Helgason, T. S. Awad, K. Kristbergsson, E. A. Decker, D. J. McClements and J. Weiss, J. Agric. Food Chem., 2009, 57, 8033 CrossRef CAS PubMed.
- A. Hentschel, S. Gramdorf, R. H. Müller and T. Kurz, J. Food Sci., 2008, 73, N1–N6 CrossRef CAS PubMed.
- A. Dube, K. Ng, J. A. Nicolazzo and I. Larson, Food Chem., 2010, 122, 662 CrossRef CAS PubMed.
- P. Zimet, D. Rosenberg and Y. D. Livney, Food Hydrocolloids, 2011, 25, 1270 CrossRef CAS PubMed.
- J. W. Liao, L. Z. Hong, M. F. Wang, S. C. Tsai, Y. J. Lin and Y. C. Chan, J. Food Sci., 2010, 75, T82–T90 CrossRef CAS PubMed.
- Nanotechnologies – Terminology and definitions for nano-objects – Nanoparticle, nanofiber and nanoplate, ISO/TS 27687, International Organization for Standardization (ISO), 2008.
- Nanotechnologies – Vocabulary – Part 1: Core terms; ISO/TS 80004–1, International Organization for Standardization (ISO), 2010.
- Nanotechnologies – Methodology for the classification and categorisation of nanomaterials; ISO/TR 11360, International Organization for Standardization (ISO), 2010.
- Food and Drug Administration (FDA) Nanotechnology Task Force, Nanotechnology – A Report, 2007.
- Regulation (EU) No. 1169/2011 on the provision of food information to consumers , Official Journal of the European Union, L304/18, 2011.
- Comission recommendation of 18 October 2011 on the definition of nanomaterial (2011/696/EU), 2012.
- J. Weiss, P. Takhistov and J. McClements, J. Food Sci., 2006, 71, R107–R116 CrossRef CAS.
- M. R. Mozafari, J. Flanagan, L. Matia-Merino, A. Awati, A. Omri, Z. E. Suntres and H. Singh, J. Sci. Food Agric., 2006, 86, 2038 CrossRef CAS.
- L. Sagalowicz and M. E. Leser, Curr. Opin. Colloid Interface Sci., 2010, 15, 61 CrossRef CAS PubMed.
- T. M. Taylor, P. M. Davidson, B. D. Bruce and J. Weiss, Crit. Rev. Food Sci. Nutr., 2005, 45, 587 CrossRef CAS PubMed.
- J. Ubbink and J. Krüger, Trends Food Sci. Technol., 2006, 17, 244 CrossRef CAS PubMed.
- J. Weiss, S. Gaysinsky, M. Davidson and J. McClements, in Global Issues in Food Science and Technology, ed. G. Barbosa-Canovas, A. Mortimer, D. Lineback, W. Spiess, K. Buckle and P. Colonna, Academic Press, 2009, ch. 24 Search PubMed.
- D. McClements, J. Food Sci., 2010, 75, R30–R42 CrossRef CAS PubMed.
- M. Fathi, M. R. Mozafari and M. Mohebbi, Trends Food Sci. Technol., 2012, 23, 13 CrossRef CAS PubMed.
- D. J. McClements, E. A. Decker, Y. Park and J. Weiss, Crit. Rev. Food Sci. Nutr., 2009, 49, 577 CrossRef CAS PubMed.
- D. J. McClements and J. Rao, Crit. Rev. Food Sci. Nutr., 2011, 51, 285 CrossRef CAS PubMed.
- J. Weiss, E. A. Decker, D. J. McClements, K. Kristbergsson, T. Helgason and T. Awad, Food Biophysics, 2008, 3, 146 CrossRef.
- C. H. Liu and C. T. Wu, Colloids Surf., A, 2010, 353, 149 CrossRef CAS PubMed.
- M. R. Mozafari, C. Johnson, S. Hatziantoniou and C. Demetzos, J. Liposome Res., 2008, 18, 309 CrossRef PubMed.
- G. Astray, C. Gonzalez-Barreiro, J. C. Mejuto, R. Rial-Otero and J. Simal-Gándara, Food Hydrocolloids, 2009, 23, 1631 CrossRef CAS PubMed.
- U. Lesmes, S. H. Cohen, Y. Shener and E. Shimoni, Food Hydrocolloids, 2009, 23, 667 CrossRef CAS PubMed.
- Y. D. Livney, Curr. Opin. Colloid Interface Sci., 2010, 15, 73 CrossRef CAS PubMed.
- A. R. Dudhani and S. L. Kosaraju, Carbohydr. Polym., 2010, 81, 243 CrossRef CAS PubMed.
- G. A. Morris, M. S. Kök, S. E. Harding and G. G. Adams, Biotechnol. Genet. Eng. Rev., 2010, 27, 257 CrossRef CAS.
- R. Zorilla, L. Liang, G. Remondetto and M. Subirade, Dairy Sci. Technol., 2011, 91, 629 CrossRef CAS.
- J. Keppler and K. Schwarz, Dtsch. Med. Wochenschr., 2012, 3, 260 Search PubMed.
- J. F. Graveland-Bikker and C. G. de Kruif, Trends Food Sci. Technol., 2006, 17, 196 CrossRef CAS PubMed.
- H. S. Chen, J. H. Chang and J. S. B. Wu, J. Food Sci., 2008, 73, H246–H251 CrossRef CAS PubMed.
- C. D. Liao, W. L. Hung, K. C. Jan, A. I. Yeh, C. T. Ho and L. S. Hwang, Food Chem., 2010, 119, 896 CrossRef CAS PubMed.
- R. H. Müller, S. Gohla and C. M. Keck, Eur. J. Pharm. Biopharm., 2011, 78, 1 CrossRef PubMed.
- F. M. Hilty, A. Teleki, F. Krumeich, R. Buchel, R. F. Hurrell, S. E. Pratsinis and M. B. Zimmermann, Nanotechnology, 2009, 20, 457101 CrossRef PubMed.
- J. W. Card and B. A. Magnuson, J. Food Sci., 2009, 74, vi–vii CAS.
- D. M. A. M. Luykx, R. J. B. Peters, S. M. van Ruth and H. Bouwmeester, J. Agric. Food Chem., 2008, 56, 8231 CrossRef CAS PubMed.
- K. Tiede, A. B. A. Boxall, S. P. Tear, J. Lewis, H. David and M. Hassellov, Food Addit. Contam., 2008, 25, 795 CrossRef CAS PubMed.
- M. Hassellov, J. W. Readman, J. F. Ranville and K. Tiede, Ecotoxicology, 2008, 17, 344 CrossRef PubMed.
- C. Blasco and Y. Pico, TrAC, Trends Anal. Chem., 2011, 30, 84 CrossRef CAS PubMed.
- R. Peters, G. ten Dam, H. Bouwmeester, H. Helsper, S. Weigel, G. Allmaier, F. vd Kammer, R. Ramsch, C. Solans, M. Tomaniova and J. Hajslova, TrAC, Trends Anal. Chem., 2010, 30, 100 CrossRef PubMed.
- E. Fernández-García, I. Carvajal-Lérida and A. Pérez-Gálvez, Nutr. Res., 2009, 29, 751 CrossRef PubMed.
- R. P. Heaney, J. Nutr., 2001, 131, 1344S CAS.
- R. J. Wood and T. Tamura, J. Nutr., 2001, 131, 1396S CAS.
- A. G. Oomen, A. Hack, M. Minekus, E. Zeijdner, C. Cornelis, G. Schoeters, W. Verstraete, T. Van de Wiele, J. Wragg, C. J. M. Rompelberg, A. n. J. A. M. Sips and J. H. Van Wijnen, Environ. Sci. Technol., 2002, 36, 3326 CrossRef CAS.
- M. Golding and T. J. Wooster, Curr. Opin. Colloid Interface Sci., 2010, 15, 90 CrossRef CAS PubMed.
- R. C. Benshitrit, C. S. Levi, S. L. Tal, E. Shimoni and U. Lesmes, Food Funct., 2012, 3, 10 CAS.
- E. Roger, F. Lagarce, E. Garcion and J. P. Benoit, Nanomedicine, 2010, 5, 287 CrossRef CAS PubMed.
- Y. Li, H. Xiao and D. J. McClements, Encapsulation and Delivery of Crystalline Hydrophobic Nutraceuticals using Nanoemulsions: Factors Affecting Polymethoxyflavone Solubility, Food Biophys., 2012, 7, 341 CrossRef PubMed.
- C. H. Liu, C. T. Wu and J. Y. Fang, Drug Dev. Ind. Pharm., 2010, 36, 751 CrossRef CAS PubMed.
- V. Kakkar, S. Singh, D. Singla and I. P. Kaur, Mol. Nutr. Food Res., 2011, 55, 495 CAS.
- E. Roger, F. Lagarce and J. P. Benoit, Int. J. Pharm., 2009, 379, 260 CrossRef CAS PubMed.
- C. Qian, E. A. Decker, H. Xiao and D. J. McClements, Food Chem., 2012, 135, 1440 CrossRef CAS PubMed.
- E. Troncoso, J. M. Aguilera and D. J. McClements, Food Hydrocolloids, 2012, 27, 355 CrossRef CAS PubMed.
- K. Ahmed, Y. Li, D. J. McClements and H. Xiao, Food Chem., 2012, 132, 799 CrossRef CAS PubMed.
- A. Dube, J. A. Nicolazzo and I. Larson, Eur. J. Pharm. Sci., 2010, 41, 219 CrossRef CAS PubMed.
- J. Shaikh, D. D. Ankola, V. Beniwal, D. Singh and M. N. V. Kumar, Eur. J. Pharm. Sci., 2009, 37, 223 CrossRef CAS PubMed.
- W. Tiyaboonchai, W. Tungpradit and P. Plianbangchang, Int. J. Pharm., 2007, 337, 299 CrossRef CAS PubMed.
- H. L. Li, X. B. Zhao, Y. K. Ma, G. X. Zhai, L. B. Li and H. X. Lou, J. Controlled Release, 2009, 133, 238 CrossRef CAS PubMed.
- C. Prego, M. García, D. Torres and M. J. Alonso, J. Controlled Release, 2005, 101, 151 CrossRef CAS PubMed.
- J. Pusch, M. Votteler, S. Gähler, J. Engl, M. Hampel, H. Walles and K. Schenke-Layland, Biomaterials, 2011, 32, 7469 CrossRef CAS PubMed.
- L. M. Ensign, R. Cone and J. Hanes, Adv. Drug Delivery Rev., 2012, 64, 557 CrossRef CAS PubMed.
- S. W. Hoag and A. S. Hussain, J. Nutr., 2001, 131, 1389S CAS.
- A. R. Patel and K. P. Velikov, LWT–Food Sci. Technol., 2011, 44, 1958 CrossRef CAS PubMed.
- G. Amidon, H. Lennernäs, V. Shah and J. Crison, Pharm. Res., 1995, 12, 413 CrossRef CAS.
- A. des Rieux, V. Fievez, M. Garinot, Y. J. Schneider and V. Préat, J. Controlled Release, 2006, 116, 1 CrossRef CAS PubMed.
- J. J. Powell, N. Faria, E. Thomas-McKay and L. C. Pele, J. Autoimmun., 2010, 34, J226–J233 CrossRef CAS PubMed.
- M. Takahashi, S. Uechi, K. Takara, Y. Asikin and K. Wada, J. Agric. Food Chem., 2009, 57, 9141 CrossRef CAS PubMed.
- S. Onoue, H. Takahashi, Y. Kawabata, Y. Seto, J. Hatanaka, B. Timmermann and S. Yamada, J. Pharm. Sci., 2010, 99, 1871 CAS.
- H. Yu and Q. Huang, J. Agric. Food Chem., 2012, 60, 5373 CrossRef CAS PubMed.
- C. Schiborr, A. Kocher, D. Behnam, J. Jandasek, S. Toelstede and J. Frank, Mol. Nutr. Food Res., 2014, 58, 516 CAS.
- Y. Gao, Y. Q. Wang, Y. K. Ma, A. H. Yu, F. Q. Cai, W. Shao and G. X. Zhai, Colloids Surf., B, 2009, 71, 306 CrossRef CAS PubMed.
- A. Y. Choi, C. T. Kim, H. Y. Park, H. O. Kim, N. R. Lee, K. E. Lee and H. S. Gwak, J. Agric. Food Chem., 2013, 61, 2096 CrossRef CAS PubMed.
- B. A. Graf, C. J. van Platerink, G. A. M. Ten Have, N. E. P. Deutz, K. P. Velikov, L. M. Flendrig, S. M. Melnikov, U. M. Garczarek, S. L. Abrahamse and G. S. M. J. Duchateau, J. Funct. Foods, 2011, 3, 135 CrossRef CAS PubMed.
- A. Smith, B. Giunta, P. C. Bickford, M. Fountain, J. Tan and R. D. Shytle, Int. J. Pharm., 2010, 389, 207 CrossRef CAS PubMed.
- J. Hatanaka, Y. Kimura, Z. Lai-Fu, S. Onoue and S. Yamada, Int. J. Pharm., 2008, 363, 112 CrossRef CAS PubMed.
- R. Wajda, J. Zirkel and T. Schaffer, J. Med. Food, 2007, 10, 731 CrossRef CAS PubMed.
- E. I. Back, C. Frindt, E. Ocenášková, D. Nohr, M. Stern and H. K. Biesalski, Eur. J. Nutr., 2006, 45, 1 CrossRef CAS PubMed.
- M. Haham, S. Ish-Shalom, M. Nodelman, I. Duek, E. Segal, M. Kustanovich and Y. D. Livney, Food Funct., 2012, 3, 737 CAS.
- F. Diarrassouba, G. Garrait, G. Remondetto, P. Alvarez, E. Beyssac and M. Subirade, Food Chem., 2014, 145, 646 CrossRef CAS PubMed.
- C. D. Liao, W. L. Hung, W. C. Lu, K. C. Jan, D. Y. C. Shih, A. N. I. Yeh, C. T. Ho and L. S. Hwang, J. Agric. Food Chem., 2010, 58, 563 CrossRef CAS PubMed.
- G. Jahreis, S. Wohlgemuth, G. Grünz, L. Martin, M. Knieling, R. Engel, M. Türk and S. Keller, Nanomedicine, 2013, 9, 1027 CrossRef CAS PubMed.
- F. Rohner, F. O. Ernst, M. Arnold, M. Hilbe, R. Biebinger, F. Ehrensperger, S. E. Pratsinis, W. Langhans, R. F. Hurrell and M. B. Zimmermann, J. Nutr., 2007, 137, 614 CAS.
- F. M. Hilty, M. Arnold, M. Hilbe, A. Teleki, J. T. N. Knijnenburg, F. Ehrensperger, R. F. Hurrell, S. E. Pratsinis, W. Langhans and M. B. Zimmermann, Nat. Nanotechnol., 2010, 5, 374 CrossRef CAS PubMed.
- N. Sakaguchi, T. P. Rao, K. Nakata, H. Nanbu and L. R. Juneja, Int. J. Vitam. Nutr. Res., 2004, 74, 3 CrossRef CAS PubMed.
- M. C. Fidler, T. Walczyk, L. Davidsson, C. Zeder, N. Sakaguchi, L. R. Juneja and R. F. Hurrell, Br. J. Nutr., 2004, 91, 107 CrossRef CAS.
- M. Roe, R. Collings, J. Hoogewerff and S. Fairweather-Tait, Eur. J. Nutr., 2009, 48, 115 CrossRef CAS PubMed.
- H. Y. Gao, H. I. Chen, W. X. Chen, F. Tao, Y. H. Zheng, Y. M. Jiang and H. J. Ruan, Food Chem., 2008, 109, 493 CrossRef CAS PubMed.
- S. Huang, J. C. Chen, C. W. Hsu and W. H. Chang, Nanotechnology, 2009, 20, 375102 CrossRef PubMed.
- H. S. Park, J. Ahn and H. S. Kwak, J. Med. Food, 2008, 11, 454 CrossRef CAS PubMed.
- T. Lien, H. Yeh, F. Lu and C. Fu, J. Sci. Food Agric., 2009, 89, 1164 CrossRef CAS.
- F. Tamjidi, M. Shahedi, J. Varshosaz and A. Nasirpour, Innovative Food Sci. Emerging Technol., 2013, 19, 29 CrossRef CAS PubMed.
- E. Acosta, Nanotechnology in the Food, Beverage and Nutraceutical Industries, 2012, p. 257 Search PubMed.
- K. Oehlke, A. Heins, H. Stöckmann and K. Schwarz, Food Chem., 2010, 118, 48 CrossRef CAS PubMed.
- S. Losada-Barreiro, V. Sánchez-Paz and C. Bravo-Díaz, J. Colloid Interface Sci., 2013, 389, 1 CrossRef CAS PubMed.
- A. Heins, D. B. McPhail, T. Sokolowski, H. Stöckmann and K. Schwarz, Lipids, 2007, 42, 573 CrossRef CAS PubMed.
- S. Das and A. Chaudhury, AAPS PharmSciTech, 2011, 12, 62 CrossRef CAS PubMed.
- C. Olbrich, O. Kayser and R. H. Müller, Int. J. Pharm., 2002, 237, 119 CrossRef CAS.
- L. Rong, C. Shoemaker, Y. Xiaoqing, Z. Fang and H. Qingrong, J. Agric. Food Chem., 2013, 61, 1249 CrossRef PubMed.
- R. O. Benech, E. E. Kheadr, C. Lacroix and I. Fliss, Appl. Environ. Microbiol., 2002, 68, 5607 CrossRef CAS.
- P. N. Ezhilarasi, P. Karthik, N. Chhanwal and C. Anandharamakrishnan, Food Bioprocess Technol., 2013, 6, 628 CrossRef CAS.
- A. S. Kabalnov, Langmuir, 1994, 10, 680 CrossRef CAS.
- J. A. Gallego-Urrea, J. Tuoriniemi and M. Hassellöv, TrAC, Trends Anal. Chem., 2011, 30, 473 CrossRef CAS PubMed.
- J. Heisler, K. Oehlke, R. Greiner and A. Steffen-Heins, Characterization of SLN in o/w emulsions, presented at Inside Food Symposium, 2013.
- E. Zimmermann and R. H. Müller, Eur. J. Pharm. Biopharm., 2001, 52, 203 CrossRef CAS.
- A. Mackie and A. Macierzanka, Curr. Opin. Colloid Interface Sci., 2010, 15, 102 CrossRef CAS PubMed.
- J. Maldonado-Valderrama, P. J. Wilde, F. Mulholland and V. J. Morris, Soft Matter, 2012, 8, 4402 RSC.
- D. J. McClements and Y. Li, Adv. Colloid Interface Sci., 2010, 159, 213 CrossRef CAS PubMed.
- L. Bonnaire, S. Sandra, T. Helgason, E. A. Decker, J. Weiss and D. J. McClements, J. Agric. Food Chem., 2008, 56, 3791 CrossRef CAS PubMed.
- L. Rossi, J. W. ten Hoorn, S. M. Melnikov and K. P. Velikov, Soft Matter, 2010, 6, 928 RSC.
- S. Rozner, D. E. Shalev, A. I. Shames, M. F. Ottaviani, A. Aserin and N. Garti, Colloids Surf., B, 2010, 77, 22 CrossRef CAS PubMed.
- R. Marisiddaiah, L. Rangaswamy and B. Vallikannan, Eur. J. Nutr., 2011, 50, 531 CrossRef CAS PubMed.
- C. Qian, E. A. Decker, H. Xiao and D. J. McClements, Food Chem., 2012, 135, 1440 CrossRef CAS PubMed.
- Y. Yang and D. J. McClements, Food Chem., 2013, 141, 473 CrossRef CAS PubMed.
- C. M. O'Driscoll, Eur. J. Pharm. Sci., 2002, 15, 405 CrossRef CAS.
- A. Benzaria, D. Chevalier-Lucia, L. Picart-Palmade, P. Hue, T. Lopez-Pedemonte and E. Dumay, Food Res. Int., 2013, 51, 679 CrossRef CAS PubMed.
- H. Pool, S. Mendoza, H. Xiao and D. J. McClements, Food Funct., 2013, 4, 162 CAS.
- N. Bordenave, B. R. Hamaker and M. G. Ferruzzi, Food Funct., 2014, 5, 18 CAS.
- H. Yu and Q. Huang, Food Chem., 2010, 119, 669 CrossRef CAS PubMed.
- B. W. Wolf, T. M. S. Wolever, C. Bolognesi, B. A. Zinker, K. A. Garleb and J. L. Firkins, J. Agric. Food Chem., 2001, 49, 2674 CrossRef CAS PubMed.
- P. Gunness, B. M. Flanagan and M. J. Gidley, J. Cereal Sci., 2010, 52, 444 CrossRef CAS PubMed.
- T. Dey and A. R. Das, Z. Phys. Chem., 2012, 226, 315 CrossRef CAS.
- P. Gunness, B. M. Flanagan, K. Shelat, R. G. Gilbert and M. J. Gidley, Food Chem., 2012, 134, 2007 CrossRef CAS PubMed.
- Y. Yang, Z. Gu and G. Zhang, J. Agric. Food Chem., 2009, 57, 7125 CrossRef CAS PubMed.
- Y. Yan, L. Jiang and J. Huang, Phys. Chem. Chem. Phys., 2011, 13, 9074 RSC.
- E. M. M. Del Valle, Process Biochem., 2004, 39, 1033 CrossRef CAS.
- K. Konecsni, N. H. Low and M. T. Nickerson, Food Chem., 2012, 134, 1775 CrossRef CAS PubMed.
- B. Hu, C. L. Pan, Y. Sun, Z. Y. Hou, H. Ye, B. Hu and X. X. Zeng, J. Agric. Food Chem., 2008, 56, 7451 CrossRef CAS PubMed.
- B. Hu, Y. Ting, X. Zeng and Q. Huang, J. Agric. Food Chem., 2013, 61, 875 CrossRef CAS PubMed.
- S. A. Agnihotri, N. N. Mallikarjuna and T. M. Aminabhavi, J. Controlled Release, 2004, 100, 5 CrossRef CAS PubMed.
- A. M. M. Sadeghi, F. A. Dorkoosh, M. R. Avadi, M. Weinhold, A. Bayat, F. Delie, R. Gurny, B. Larijani, M. Rafiee-Tehrani and H. E. Junginger, Eur. J. Pharm. Biopharm., 2008, 70, 270 CrossRef CAS PubMed.
- D. Vllasaliu, R. Exposito-Harris, A. Heras, L. Casettari, M. Garnett, L. Illum and S. Stolnik, Int. J. Pharm., 2010, 400, 183 CrossRef CAS PubMed.
- A. Hafner, J. Lovric, D. Voinovich and J. Filipovic-Grcic, Int. J. Pharm., 2009, 381, 205 CrossRef CAS PubMed.
- L. Y. Chen, G. E. Remondetto and M. Subirade, Trends Food Sci. Technol., 2006, 17, 272 CrossRef CAS PubMed.
- J. Zhang, L. Liang, Z. Tian, L. Chen and M. Subirade, Food Chem., 2012, 133, 390 CrossRef CAS PubMed.
- T. G. Shutava, S. S. Balkundi, P. Vangala, J. J. Steffan, R. L. Bigelow, J. A. Cardelli, D. P. O'Neal and Y. M. Lvov, ACS Nano, 2009, 3, 1877 CrossRef CAS PubMed.
- M. C. Bohin, J. P. Vincken, H. T. W. M. Van Der Hijden and H. Gruppen, J. Agric. Food Chem., 2012, 60, 4136 CrossRef CAS PubMed.
- L. Liang and M. Subirade, Food Chem., 2012, 132, 2023 CrossRef CAS PubMed.
- L. Liang and M. Subirade, J. Phys. Chem. B, 2010, 114, 6707 CrossRef CAS PubMed.
- X. Wu, R. Dey, H. Wu, Z. Liu, Q. He and X. Zeng, Int. J. Dairy Technol., 2013, 66, 7 CrossRef CAS.
- L. H. Riihimäki, M. J. Vainio, J. M. S. Heikura, K. H. Valkonen, V. T. Virtanen and P. M. Vuorela, J. Agric. Food Chem., 2008, 56, 7721 CrossRef PubMed.
- J. K. Keppler, T. Koudelka, K. Palani, M. C. Stuhldreier, F. Temps, A. Tholey and K. Schwarz, J. Biomol. Struct. Dyn., 2013, 1 Search PubMed.
- F. Diarrassouba, G. Remondetto, L. Liang, G. Garrait, E. Beyssac and M. Subirade, Food Res. Int., 2013, 52, 515 CrossRef CAS PubMed.
- L. Liang, V. Tremblay-Hébert and M. Subirade, Food Chem., 2011, 126, 821 CrossRef CAS PubMed.
- P. Zimet and Y. D. Livney, Food Hydrocolloids, 2009, 23, 1120 CrossRef CAS PubMed.
- S. Le Maux, A. Brodkorb, T. Croguennec, A. A. Hennessy, S. Bouhallab and L. Giblin, J. Dairy Sci., 2013, 96, 4258 CrossRef CAS PubMed.
- S. Roufik, S. F. Gauthier and S. L. Turgeon, Int. Dairy J., 2007, 17, 471 CrossRef CAS PubMed.
- A. Shpigelman, Y. Cohen and Y. D. Livney, Food Hydrocolloids, 2012, 29, 57 CrossRef CAS PubMed.
- I. Caillard and D. Tomé, Reprod., Nutr., Dev., 1995, 35, 179 CrossRef CAS.
- L. Riihimäki, A. Galkin, M. Finel, J. Heikura, K. Valkonen, V. Virtanen, R. Laaksonen, J. P. Slotte and P. Vuorela, Int. J. Pharm., 2008, 347, 1 CrossRef PubMed.
- P. F. Fox and A. Brodkorb, Int. Dairy J., 2008, 18, 677 CrossRef CAS PubMed.
- D. G. Dalgleish, Soft Matter, 2011, 7, 2265 RSC.
- D. P. Acharya, L. Sanguansri and M. A. Augustin, Food Chem., 2013, 141, 1050 CrossRef CAS PubMed.
- K. Pan, Q. Zhong and S. J. Baek, J. Agric. Food Chem., 2013, 61, 6036 CrossRef CAS PubMed.
- S. Haratifar and M. Corredig, Food Chem., 2014, 143, 27 CrossRef CAS PubMed.
- N. Anarjan and C. Tan, Food Chem., 2013, 139, 527 CrossRef CAS PubMed.
- B. Hu, Y. Ting, X. Zeng and Q. Huang, Carbohydr. Polym., 2012, 89, 362 CrossRef CAS PubMed.
- T. David-Birman, A. Mackie and U. Lesmes, Impact of dietary fibers on the properties and proteolytic digestibility of lactoferrin nano-particles, Food Hydrocolloids, 2012, 31, 33 CrossRef PubMed.
- Y. Luo, B. Zhang, W. H. Cheng and Q. Wang, Carbohydr. Polym., 2010, 82, 942 CrossRef CAS PubMed.
- L. Chen and M. Subirade, Biomaterials, 2005, 26, 6041 CrossRef CAS PubMed.
- E. Troncoso, J. M. Aguilera and D. J. McClements, J. Colloid Interface Sci., 2012, 382, 110 CrossRef CAS PubMed.
- G. Budryn and D. Rachwal-Rosiak, Food Rev. Int., 2013, 29, 217 CrossRef CAS.
- M. Stojadinovic, J. Radosavljevic, J. Ognjenovic, J. Vesic, I. Prodic, D. Stanic-Vucinic and T. Cirkovic Velickovic, Food Chem., 2013, 136, 1263 CrossRef CAS PubMed.
- M. B. Villas-Boas, M. A. Fernandes, R. D. L. Zollner and F. M. Netto, Int. Dairy J., 2012, 25, 123 CrossRef CAS PubMed.
- R. Havenaar, A. De Jong, M. E. Koenen, J. Van Bilsen, A. M. Janssen, E. Labij and H. J. M. Westerbeek, J. Agric. Food Chem., 2013, 61, 7636 CrossRef CAS PubMed.
- J. Maldonado-Valderrama, J. A. H. Terriza, A. Torcello-Gómez and M. A. Cabrerizo-Vílchez, Soft Matter, 2013, 9, 1043 RSC.
- S. Egert, J. Tereszczuk, S. Wein, M. J. Müller, J. Frank, G. Rimbach and S. Wolffram, Eur. J. Nutr., 2013, 52, 281 CrossRef CAS PubMed.
- L. y. Zha, X. Zi-rong, W. Min-qi and L. y. Gu, J. Zhejiang Univ., Sci., B, 2007, 8, 323 CrossRef CAS PubMed.
- Y. Liu, PhD Dissertation, NC State Univ., 2003, http://www.lib.ncsu.edu/resolver/1840.16/4845.
- S. E. Dew, Arch. Biochem. Biophys., 1997, 338, 233 CrossRef CAS PubMed.
Footnotes |
| † Ralf Greiner is chairman of the working group “engineered nanomaterials” of the senate of the federal research institutes of the Federal Ministry of Food, Agriculture and Consumer Protection. |
| ‡ For simplicity, mechanisms are described for “conventional micelles”, o/w microemulsions, and o/w nanoemulsions. They also apply for the reversed types of ENM but in “opposite directions”. |
| This journal is © The Royal Society of Chemistry 2014 |