 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The properties and performance of a pH-responsive functionalised nanoparticle
Sandra
Ast
,
Peter J.
Rutledge
and
Matthew H.
Todd
*
School of Chemistry, The University of Sydney, NSW 2006, Australia. E-mail: matthew.todd@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 2180
First published on 24th July 2014
Abstract
We report fluorescence measurements of three quantum dots (QDs) of different sizes functionalised with the same pH responsive naphthalimide dye. QD size strongly influences energy transfer between dye and dot. Using QDs with an emission maximum of 570 nm gives rise to an interesting transfer of energy from dye to dot, while QDs with an emission maximum at 670 nm give unexpected enhancement of the dye emission. Titrations of QDs with the dye provide a means to establish the loading and hence an approximation of the surface dye density, which varies in proportion to QD size. Quenching effects are observed beyond the loading limit, and may indicate non-specific interactions between the excess dye and the nanoparticle. Attachment of the dye to the QD core is achieved by a thiol/disulfide exchange process that has been interrogated with Raman spectroscopy. The stability of these QD–dye conjugates over time and across a physiological pH range has been investigated to provide an assessment of their performance and robustness.
Introduction
Fluorescent nanoparticles, and specifically quantum dots (QDs), have found widespread application in biomedical imaging in recent years.1–4 Much of this work involves tracking the localisation of nanoparticle reporters functionalised with a ligand that binds some cognate biological molecule, and much of the interest is focused on intracellular measurements. We have become interested in two less well-explored areas of sensing that we feel could be addressed with appropriately built fluorescent nanoparticles: (1) monitoring analyte concentrations and dynamics in the extracellular (intercellular) space; and (2) developing nanoparticles that are responsive to analytes, in that their fluorescent output changes with the (varying) local concentration of an analyte – responsive QDs (RQDs).Our initial approach to sensing using nanoparticles targets pH. Intercellular pH regulates a wide range of poorly understood processes including cell adhesion,5 motility,6 myelin repair,7 and cancer progression.8 We felt that proof of concept for a reversible sensing scaffold would be more easily attained with a pH-sensitive system than a prototype based on the metal-responsive probes we have developed recently.9–11 While there is some precedence for pH-responsive nanoparticles12–16 there is considerable scope for the development of new responsive probes that can be simply prepared.
To this end we recently reported an easily-constructed functionalised nanoparticle that responds to pH but which also exhibits an enhanced QD photoluminescence (PL) (Fig. 1).17 This system was designed so that the dye emission overlaps with the absorption band of the nanoparticle, leading to energy transfer and an enhanced PL signal of the QD (Fig. 1B). The conjugate thus exhibits energy transfer from dye-to-dot, in contrast to the majority of the quantum dot literature in which energy transfer is designed and observed to occur to the dye from the excited dot. Dye-to-dot transfer was sought, but not conclusively found, by Mattoussi in 2005,18 who proposed that in their system energy transfer in this direction was precluded by competition between the dye's fast radiative decay pathways and the slower non-radiative Förster Resonance Energy Transfer (FRET) pathways, as well as strong, unavoidable direct excitation of the nanoparticle itself. However, three more recent reports have described dye-to-dot transfer in QDs directly functionalised with simple organic dyes19,20 or a photosynthetic light harvesting complex.21 None of these systems respond to analytes. Our conjugate consists of a naphthalimide dye attached to a QD (via a passive disulfide exchange reaction22) to give a system in which the resulting emission is sensitive to the pH. The conjugate exhibits dye-to-dot energy transfer that is pH-dependent, constituting the first responsive dye-to-dot system. The fluorophore is (2-(dimethyl)ethyl)amino-naphthalimide, a pH-sensitive probe that is switched off at basic pH due to a photoinduced electron transfer (PET) from the non-protonated dimethylamino-group.23 When this kind of dye is attached to the nanoparticle, pH-induced changes to the properties of the dye are communicated to the QD, modulating the intensity of the QD emission.
 | ||
| Fig. 1 Schematic representation of the previously reported pH-responsive QD–dye conjugate using disulfide 1 and water-soluble, core/shell type CdSeS/ZnS QDs.17 | ||
The signal changes observed as a function of pH are significant but not as large as had been anticipated. Further investigation is required to understand the mechanism behind this dye-to-dot fluorescence enhancement. Several aspects of this system in particular require further elucidation. First, we wished to confirm that the spectroscopic performance of the system can indeed be attributed to the spectroscopic overlap between dye and dot as envisaged in the original conjugate design. Second, it was important to ascertain whether the dye interacts directly with the surface of the core of the particle (and not with the outer polymer coating added to the QD to aid aqueous solubility) and if so to what extent, i.e., how many molecules of dye could be installed on the particles. Third it was important to assess the performance and stability of the conjugate with respect to time and changes in pH.
Results and discussion
1 The dot–dye energy transfer changes dramatically with particle size
In our previous studies we found that there was an enhancement of the dye emission as well as the QD PL signal, that might arise from energy transfer of different origin going in both directions, i.e., dye-to-dot as well as dot-to-dye energy transfer. To understand these effects further, QDs of different sizes (QD-460 and QD-670) were employed to complement the originally-used QDs (QD-570) (vide infra for further discussion of the particle sizes).The three types of dot were exposed to increasing concentrations of disulfide 1 under the same conditions (HEPES buffer, pH 7.4, room temperature). The resulting PL spectra for QD-570 (Fig. 2a) show the enhancement of the QD fluorescence at 570 nm, and the growth of the ‘shoulder’ next to the QD arising from the fluorophore emission, which, by virtue of the conjugation to the QD, is blue-shifted to 516 nm with respect to the emission of 1 alone (the lower emission maximum centred at 534 nm). Clearly this blue-shift is accompanied by a substantial increase in PL intensity of the fluorophore emission. A maximum is reached for the QD fluorescence output after the addition of a given amount of 1, beyond which the intensity decreases (Fig. 2b). The spectra are notable in that they show enhancement of both signals.
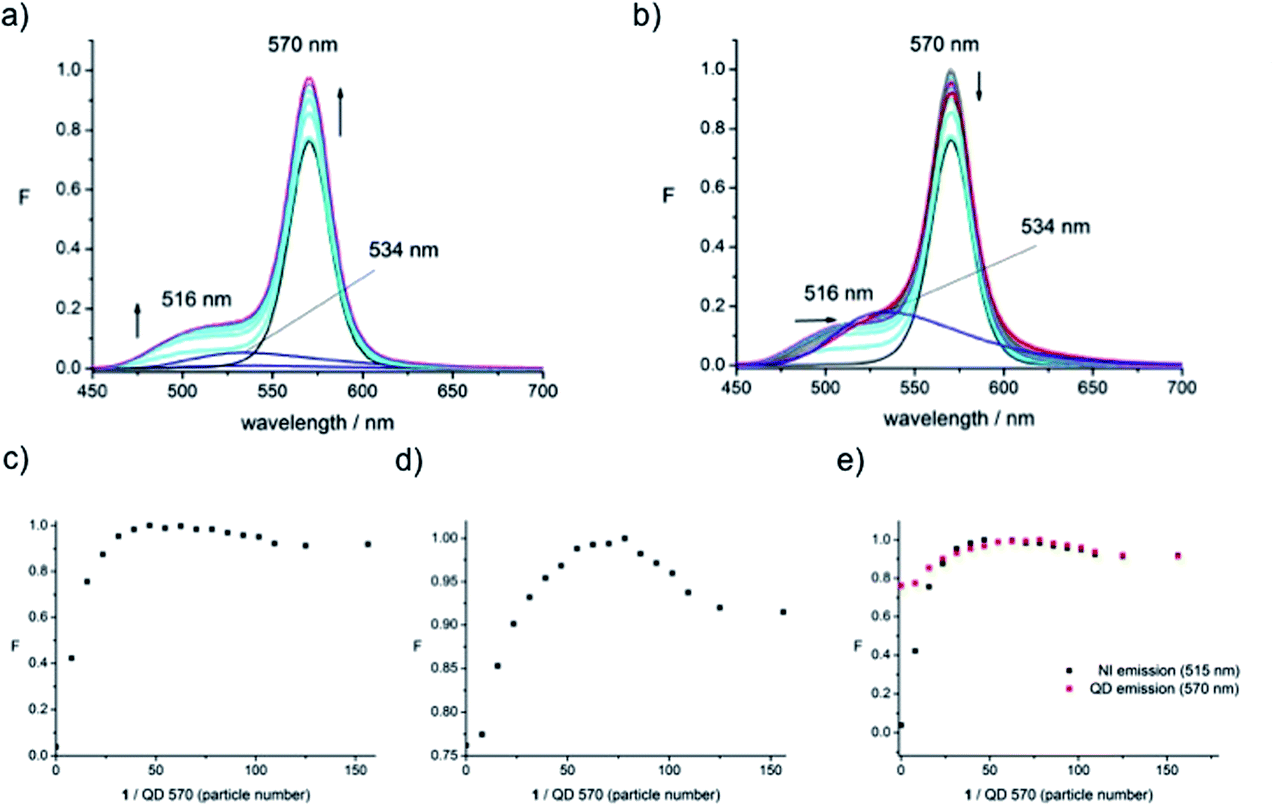 | ||
Fig. 2 Normalised PL emission spectra and plots of emission signal changes as a function of particle number (i.e., ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :QD-570) in HEPES buffer, excitation at 435 nm. (a) Addition of 1 up to saturation of fluorescence emission increase (up to 1 μM), (b) addition of 1 beyond emission signal saturation (up to 4 μM) and 1 alone at the same concentration (4 μM), (c) plot of fluorophore emission intensity at 516 nm, (d) plot of QD emission intensity at 570 nm, and (e) overlay of plots of emission intensities at 516 and 570 nm. :QD-570) in HEPES buffer, excitation at 435 nm. (a) Addition of 1 up to saturation of fluorescence emission increase (up to 1 μM), (b) addition of 1 beyond emission signal saturation (up to 4 μM) and 1 alone at the same concentration (4 μM), (c) plot of fluorophore emission intensity at 516 nm, (d) plot of QD emission intensity at 570 nm, and (e) overlay of plots of emission intensities at 516 and 570 nm. | ||
Plotting the intensity at the emission maximum as a function of the ratio of numbers of dye molecules over number of QDs (“Particle Number”, Fig. 2c – calculated as described below) shows that the PL signal approaches an asymptote around 50, implying further conjugation (or even interaction) of dye and dot is not taking place above a certain concentration of the added dye. This is a strong indicator that the surface of the dot is covered with the maximum amount of dye; each particle thus accommodates ca. 100 fluorophore molecules (since the assumption is made that each molecule of the disulfide 1 delivers two molecules of the fluorophore). The absence of further QD–dye interactions is indicated by the increasing red-shift as the titration of 1 is continued (Fig. 2b) (up to 4 μM). However, the signal intensity arising from the fluorophore remained essentially unchanged in this subsequent titration, i.e., a small increase in dye emission was expected (because more dye is being added) but this was not observed, suggesting some non-specific interaction between the dye and some other component (likely the polymer) of the nanoparticle. This unexpected phenomenon becomes more apparent when comparing the final spectrum (in Fig. 2b) with the emission of 1 alone taken at the concentration that would be expected in the absence of the QD (4 μM, blue line) where the intensities are seen as being approximately equivalent indicating there is no energy transfer from the dot to the dye beyond the surface saturation point.
The plot of the QD's PL signal intensity as a function of particle number shows the same behaviour as that of the dye emission (Fig. 2e) with the only difference being the weaker changes in signal intensity, providing supporting evidence that we are looking at a saturation of the dye on the surface of the dot. We conclude that the communication, presumably FRET, operates from dye to dot. There is no donor quenching in this system, precluding a simple calculation of FRET efficiency. The origin of the fluorescence enhancement of the fluorophore emission in the presence of the dot was unclear. To examine this further, QD-460 and QD-670 were employed in equivalent experiments. The spectra obtained when 1 was titrated into solutions of QD-460 (Fig. 3) and QD-670 (Fig. 4) show very different behaviour.
 | ||
| Fig. 3 Normalised PL emission spectra and plots of emission signal changes as a function of particle number (i.e., ratio of 1:QD-460), in HEPES buffer (pH 7.4), excitation at 400 nm. (a) Addition of 1 up to saturation of fluorophore emission increase of (up to 0.5 μM) and 1 alone at this same concentration (0.5 μM, blue line), (b) addition of 1 beyond emission signal saturation (up to 4 μM) and 1 alone at same final concentration (4 μM), (c) plot of fluorophore emission intensity at 515 nm, (d) plot of QD emission intensity decrease at 459 nm and (e) Stern–Volmer plot (F/F0) of fluorescence emission intensity at 459 nm. | ||
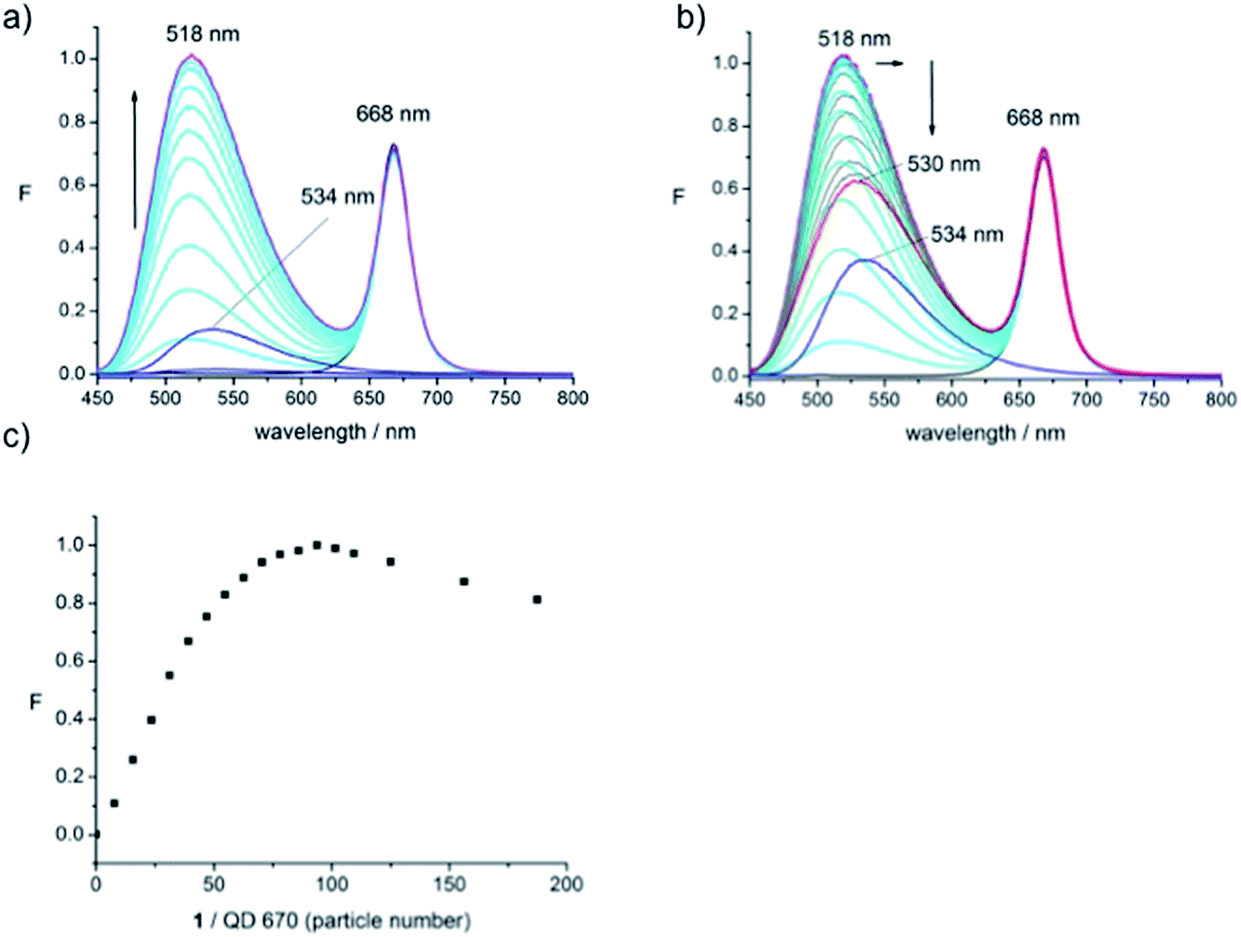 | ||
| Fig. 4 Normalised PL emission spectra and plots of emission signal changes as a function of particle number of 1 per single QD-670 in HEPES-buffer (pH 7.4), excitation at 435 nm. (a) Addition of 1 up to saturation of fluorophore emission increase and 1 alone at the same concentration (1.8–2.2 μM), (b) addition of 1 beyond emission signal saturation (up to 6.6 μM) and 1 alone at the same final concentration (6.6 μM) and (c) plot of fluorophore emission intensity at 518 nm. | ||
In the case of QD-460, with an emission maximum at 459 nm, the combination with 1 (absorption maximum at 435 nm) implies that the nanoparticle is the energy donor, and the dye the acceptor, when the sample of the conjugate is excited at 400 nm. Thus adding 1 to QD-460 results in an immediate decrease in QD PL intensity (unlike the case for QD-570) accompanied by a large enhancement of fluorophore emission (Fig. 3a). The blue-shifted dye emission is centred at 515 nm (pink line), as seen for QD-570.
Interestingly, further addition of dye 1 to the QD–1 conjugate led to a decrease in fluorophore emission intensity and a red-shifted emission signal (Fig. 3b, grey lines). This emission signal experienced a second saturation at a number of particles per dot roughly twice that seen for the first saturation, with the emission centred at 534 nm (the emission maximum of the dye 1 itself), followed by a subsequent increase in emission intensity. The signal increase was accompanied by a further red-shift to 550 nm, suggesting that the free dye is experiencing some interaction with the QD or its surroundings, again potentially with the polymer outer layer. The total increase in dye emission intensity (up to the maximum seen at 515 nm during the initial addition of 1, Fig. 3a) is of about 6 orders of magnitude, as judged by the final intensity compared with the intensity of 1 alone at the same concentration. The quenching of QD emission during addition of 1 is high, with the signal disappearing almost completely after further dye addition (Fig. 3b); the further decrease in intensity at this wavelength for this conjugate was observed to be a partly time-dependent phenomenon (vide infra, Fig. 9a).
From the decrease in QD donor PL intensity, the FRET efficiency (E) can be determined using eqn (1).
 | (1) |
The FRET efficiency is thus defined as the ratio of the fluorescence intensity of the donor in the presence of the quencher (FqD) over the fluorescence intensity of the donor in the absence of the quencher (FD). Here the FRET efficiency is 80% at maximal QD coverage and approaches 96% in the course of further dye addition. Plotting the fluorescence intensity at 515 nm as a function of the number of dye molecules covering the QD, shows that PL signal increase of the dye emission signal is maximal around a dye-to-dot ratio of 25 (Fig. 3c). The same number is obtained by plotting the decrease in QD PL intensity at 459 nm (Fig. 3d) at 80% FRET efficiency.
From the Stern–Volmer plot24 (Fig. 3e), where the rate of the fluorescence intensity quenching (F/F0) is plotted as a function of the quencher concentration (as particle number) it is obvious that a linear relationship only holds at low dye density, i.e., in the concentration range up to the fully covered QD where static quenching is possible. Addition of further dye continues the linear trend up to a particle number of around 50 (corresponding to the second saturation limit) with subsequent addition leading to an upward curving plot indicative of combined static and dynamic quenching processes; one would expect dynamic quenching with dye in solution. The linear range between particle number 25 and 50 is interesting in that this may be supporting evidence for the association of the dye with the polymer coating, as opposed to direct interaction with the dot or free dye in solution.
Also shown (Fig. 3b) is the emission intensity of the dye alone (i.e., the disulfide 1 at a concentration of 4 μM) at the concentration equal to the final concentration reached following addition of 1 into the QD solution, which is observed to be significantly higher (blue line) than in the presence of the QD. A quenching effect appears to operate on the dye even after the surface of the QD is fully covered with dye molecules. However, the increase in dye emission after the point of saturation indicates that these processes are starting to be suppressed and radiative processes of the fluorophore start to dominate. We were not able to observe this for the QD-570 system, but would expect similar behaviour to be operating.
The PL spectra of QD-670 in the presence of disulfide dye 1 (Fig. 4) were acquired. The absorption of the first exciton band is centred around 660 nm and the emission maximum of the QD is centred at 668 nm. Addition of 1 to solutions of QD-670 resulted in a large increase of the fluorophore emission band and a blue shifted emission signal to 518 nm as before, but the QD emission remained unchanged. These results are clearly surprising, since the emission of the QD is far outside the fluorophores' absorption, which stretches out to only 570 nm and thus should not be able to function as an energy donor. It was expected that a QD of this size would have limited capacity for FRET due to FRET's strong distance dependence25 and these QDs were not chosen for our previous studies for precisely this reason. However, the blue-shifted emission suggests very similar interactions to the ones found for the smaller QDs, i.e., energy transfer from the QD donor. However, such a conclusion would need to be verified with, for example, fluorescence lifetime measurements.
Plotting the emission intensity as a function of dye-to-dot density shows that maximal fluorescence enhancement of the dye is achieved at a ratio in the range 80–100. Further addition of 1 led to a decrease in intensity of the dye signal (Fig. 4b) which reached an asymptote (red line) around twice the number of molecules of 1 (200) (Fig. 4c) in a similar way to the QD-460 system. Comparison with the signal intensity of 1 alone (blue line) at 518 nm at this final concentration shows that the final intensity of the dye emission in the conjugate is higher than the dye signal alone; thus the signal of the fluorophore is enhanced in the presence of the QD and remains enhanced in the presence of an excess of the dye. As before it is possible that the first 100 molecules of added 1 interact with the inorganic core of the QD, with the remaining dye interacting with the outer polymer matrix in a non-specific manner.
The Förster radius (R0) represents the distance for 50% of energy transfer and normally falls within the core/shell radius for larger dots due to the 6th power relationship in eqn (2).25,26 However the large number of dye molecules attached to these larger dots appears to change this situation significantly, since the FRET efficiency (E) is dependent on the number of dye molecules (n) as given by eqn (2).27
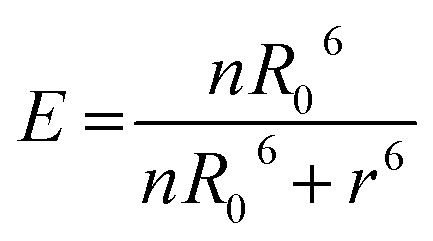 | (2) |
Thus it would appear that the more populous “antenna” of fluorophores surrounding the dot is able to increase the FRET efficiency to allow for energy transfer despite the sub-optimal donor–acceptor distance and spectroscopic overlap.
A question remained: why did the PL of the QD remain unchanged through the addition of the dye? That the dot-to-dye transfer is not accompanied by a quenching in QD PL implies that fluorescence enhancement is competing (perfectly) with fluorescence quenching, leaving the QD signal unchanged – a net zero change of energy transfer at the particle though this would need to be confirmed by independent lifetime measurements of the QD.
From these studies of all three dot–dye conjugates we conclude that attachment of dye 1 to QD-570 (the medium sized particle previously reported) leads to a system involving energy transfer from dye-to-dot. Conjugates based on smaller QDs give instead the expected dot-to-dye FRET because the reverse process is energetically disfavoured. Large QD conjugates only give clear FRET from dot-to-dye but the dye-to-dot transfer is assumed from the lack of any observed QD quenching when the dot transfers energy to the dye, and this may be a feature of FRET efficiency being enhanced by the larger number of dye molecules attached to the core; there remains the possibility that electronic or even molecular mechanisms, rather than FRET, could explain these observations.
2 Raman spectroscopy can probe the nature of dye–dot linkage
The QDs employed in these studies are core shell QDs covered with a capping layer, surrounded by a layer of amphiphilic polymer, functionalised with carboxylic acids. The assumption was made in our previous work that the conjugate is assembled via sulfide exchange between 1 and the “inner” inorganic surface of the dot, requiring the disulfide to diffuse into the particle (through the polymer, without reacting with it) for such exchange to occur. We had previously performed an experiment in which a model compound 2, lacking disulfide functionality, was added to solutions containing QD-570 (Fig. 5a).17 No apparent modification of the QD PL, and certainly no enhancement of the QD fluorescence output, was observed, strongly suggesting that covalent attachment of the dye was necessary for the optical effects we had observed in the conjugate. Additionally, attempts to isolate a potential QD–2 conjugate failed, while 1 formed such an isolable conjugate (Fig. 5b). | ||
| Fig. 5 Emission spectra of QD-570 showing attempts to isolate a QD-conjugate with the model compound 2. (a) Emission spectra of QD-570 (black line), QD + 2 (red line) and deconvoluted PL spectrum of QD after substraction of fluorophore component (blue line) in HEPES-buffer, (b) pictures of attempted isolation of the conjugates with 2 and 1 and (c) structure of 2. This figure is adapted from our previous work and is reproduced here for clarity.17 | ||
It remained possible that the disulfide was engaging in an exchange reaction with the outer polymer layer of the particle, even though such a reaction would not prima facie lead to any PL effects based on FRET due to the inevitably larger distances between donor and acceptor. Nevertheless we had no direct evidence of the formation of new bonds on the particle. We therefore measured Raman spectra of the QD, dye and conjugate to attempt to observe changes in the relevant regions of the spectra. The disulfide S–S stretching vibration occurs in the range of 500–530 cm−1, while the C–S stretching is typically found between 715 and 579 cm−1.28
The individual Raman spectra (overlay, Fig. 6) were acquired from solid samples, using a MultiRAM FT-Raman-spectrometer irradiating at 1064 nm. The QD–dye conjugate was prepared as described previously,17 by precipitation and washing of the pellet and subsequent drying under a stream of nitrogen. The spectrum of the dye 1 (black line) shows distinct peaks indicative of the disulfide bond as well as the aromatic core of the amino-naphthalimide. The spectrum of the QD itself (red) shows less resolved peaks and vibrations originating from the nanoparticle and the encapsulating polymer. The QD–1 conjugate represents the linear combination of the two spectra, except for the disulfide region, which lacks the disulfide vibration peaks at 500 and 660 cm−1. Though not conclusive proof of the attachment of the dye via sulfide exchange on the inner surface of the particle, these results do suggest the expected covalent attachment to the QD in the isolated conjugate.
 | ||
| Fig. 6 Overlay of the Raman spectra of the dye 1 alone (black), QD-570 (red) and the isolated conjugate QD-570–1 (blue). (a) The whole range from 2300 to 300 cm−1 (representative of the disulfide and aromatic vibrations) and (b) the region from 1900 to 1000 cm−1 (representative of the aromatic region). | ||
3 Titration experiments permit calculation of the “loading” of the dye on the particle
The concentration and dimensions of the QDs used in the present experiments are of interest, for the purpose of understanding the loading calculations described above, i.e., to answer the question: how many dye molecules are installed on each particle? The concentration of each QD solution, as provided by the supplier, Ocean NanoTech, was equal to 8 μM. The default volume of QD solution used in each experiment (8 μL, mixed with 3 mL HEPES buffer) implies approximately 3.9 × 1013 particles per experiment, though without weighing the sample this does not permit an estimation of the molecular weight of each particle (and we are unaware of any reports of mass spectrometry applied to quantum dot solutions that might give empirical measures of molecular weights, though this is not an unreasonable experiment). These aqueous-compatible QDs are covered with a layer of polymer that significantly enhances their hydrodynamic radii and which presents a presumably porous barrier between the bulk medium and the covalently functionalised surface of the core particle. The supplier-provided hydrodynamic radii (Table 1) are clearly far larger than the radii normally associated with QDs without the polymer layer encapsulating the particle (1–5 nm), though the hydrodynamic radius may be the value for a polymeric particle encapsulating more than one QD core.29| QD-460 | QD-570 | QD-670 | |
|---|---|---|---|
| a 1 mM stock solution in DMSO. b 1 (0.5 μL) was added to 8 μL QD stepwise into 3 mL aqueous buffer. c Obtained with Avogadro number. d Particle number of dye over particle number of QD. e Size of the dye over surface area. | |||
| Hydro. rad./nm | 4.8 | 6.85 | 8.75 |
| Surface area/nm2 | 290 | 590 | 962 |
| Moles in exp.a,b/mol | 1.25–1.5 × 10−9 | 2.5–3.0 × 10−9 | 5.5–6.5 × 10−9 |
| Number of part. in exp.c | 7.5–9.0 × 1014 | 1.5–1.8 × 1015 | 3.3–3.9 × 1015 |
| Ratio of dye 1:QDd |
19.5–23.4 | 40–46.9 | 85.9–101.5 |
| No. dye per part.e | 40–47 | 80–94 | 172–203 |
The experiments described above (Fig. 2–4) combined with these data provide a means to establish an approximate surface density or loading of the dye on the surface of the conjugate. For each of the three dots a maximum signal is reached at a certain concentration, where this maximum is taken as evidence that the surface of the dot has accepted as many copies of the dye as it is able – though it is unclear whether this limit arises from (i) addition to “vacant” sites on the particle surface, (ii) exchange with a limited number of labile ligands on the surface or (iii) a genuine limit that arises from, e.g., coulombic repulsion between the polar/charged dye molecules installed. From the concentration at the maximal signal change in the sample volume the molarity was calculated, which then allowed for the determination of the number of dye particles.
Assuming perfect correspondence between the empirical limit of fluorescence increase and covalent attachment to a surface (i.e., rather than an equilibrium process of disulfide exchange), the number of dye molecules per dot was calculated: approximately 40–50 dye molecules for QD-460, 80–100 for QD-570 and 170–200 for QD-670. These numbers match well with the comparative surface area of the dots derived from the hydrodynamic radii (Table 1), i.e., with QD-570 having double the surface area, it is able to accommodate twice as many dye molecules. The hydrodynamic radius is, however, likely to represent the overall particle size that includes the polymeric outer layer, rather than the radius of the core QD, which should be much lower. Literature values for estimates of the number of added ligands assembling on the surface of QDs are around the same order of magnitude as those shown above;27,30 these values, derived from QDs without an outer polymeric layer, suggest that the exchange processes occurring in the present work are indeed those taking place on this inner QD surface, rather than the far larger outer polymeric structure, not least because covalent addition to the outer polymeric structure would be expected to give a value for number of dye molecules per particle that is much higher than that calculated here.
If we take an estimated radius for the inner core (CdSe/ZnS) of the QD-570 particle as 2.5 nm,31 we would expect the 100 fluorophore molecules to be spread over an area of 80 nm2. If we (crudely) estimate the cross-sectional area of the dye molecule to be 0.9 nm long by 0.5 nm wide (the approximate dimensions of the naphthalimide dye), each fluorophore would occupy a static cross-sectional area of about 0.5 nm2 meaning this value of loading seems reasonable (coverage of 50 nm2 of the available 80 nm2). If one assumes that each molecule is moving and is solvated, this may imply that the surface of the core has become completely covered with the added dye, which may explain the fairly sharp saturation limit observed above upon addition of the disulfide.
4 The conjugate is moderately robust with respect to time and pH change
In our previous work we were able to show that the QD emission signal varies with pH when the dye is attached. These signal changes were measured as the relative fluorescence signal increase with respect to the QD alone and gave a sigmoidal relationship when plotted as a function of pH. The present studies aimed to address several features of the conjugate performance.:dye ratio was employed that had been found (above) to provide a fully covered nanoparticle.
The measurements for the acidic pH region (Fig. 7) show that the intensity of the QD signal itself is significantly affected by the presence of protons reducing in intensity towards lower pH values (Fig. 7a, black to grey). The emission signals of the conjugate also decrease as the pH decreases. At the lowest pH value assessed, 4.5 (Fig. 7a, blue line), the conjugate's signal intensity has dropped below the intensity of the initial QD emission (Fig. 7a, black line). These effects could be attributed to protonation of the polymer surrounding the dot. Plotting the fluorescence intensity at the signal maximum vs. pH (Fig. 7b) strongly indicates that the conjugate experiences the same effects as the QD. This becomes even more apparent when looking at the fluorophore emission at 517 nm in the conjugate (Fig. 7b, blue), which follows the same trend as the QD (Fig. 7b, black).
 | ||
| Fig. 7 pH-sensitivity of QD-570 and its conjugate with dye 1 in the pH range 4.6 to 7.4. (a) Normalised PL spectra of QD 570 (black and grey lines) and of QD-570–1 conjugate (red, then cyan lines) and (b) plot of normalised intensity at 570 nm vs. pH, sweeping from 7.5 to 4.6. | ||
This situation changes completely when sweeping from neutral to alkaline pH (Fig. 8). In this range the signal from the QD remains perfectly stable while the emission intensity signals (at 516 and 570 nm) of the conjugate decrease towards higher pH values (Fig. 8b). The decrease is expected: it reflects the response of model fluorophore 2 during pH-measurements23 and has been shown by us before for the QD-570–1 conjugate (Fig. 1C).17 Thus, the QD signal changes measured in the alkaline region can be attributed to changes at the dye. Attempts to reverse the pH-sweep were less successful and also appeared to be dominated by a change in QD signal intensity (Fig. 8c and d). Thus, in the reversed sweep, the QD signal of the conjugate (Fig. 8c, pink) shows a trend similar to the signal of the QD alone (Fig. 8c, blue). The signal of the fluorophore at 516 nm is affected in the reverse sweep as well (Fig. 8d, red) and does not retain its original signal intensity (Fig. 8d, black).
 | ||
| Fig. 8 pH-sensitivity of QD-570 and its conjugate with dye 1 in the pH range 7.5–9.6. (a) Normalised PL spectra of QD-570 (black and grey lines) and of QD-570–1 conjugate (red, then cyan lines) and, (b) plot of normalised intensity at 570 nm vs. pH, sweeping from 7.5–9.6, (c) plot of normalised intensity at 570 nm vs. pH, back-sweep from 9.6 to 7.4 (blue and pink) and (d) plot of normalised intensity at 515 nm vs. pH, back-sweep (red) from 9.6 to 7.4. | ||
The fact that the fluorescence signal of the conjugate is reduced at acidic pH would appear to preclude its application as a switch-on probe in that region. However, the pKa of dye 2, reported to be 7.8,23 narrows the window of practical application down to a range between pH 7 and 10, the region where the QD-570 was found to be responsive and stable in the present experiments.
 | ||
| Fig. 9 Normalised PL spectra of QD-460 and QD-570 and their conjugates with 1 over time (14–16 min). (a) QD-460 and QD-460–1 conjugate, (b) QD-460 alone, (c) QD-570 and QD-570–1 conjugate and (d) QD-570 alone. | ||
There is an obvious explanation as to why chemical exchange processes may take a few minutes to complete in these systems: diffusion of the dye through the polymeric outer layer. Indeed it should be remembered that the aqueous solubility of the particle arises from this charged outer layer, while the inner particle itself remains fairly hydrophobic. Though this was not addressed as part of the current study, measurements of pH are made on the assumption that the solvent environment between the QD core and the outer polymeric layer reflects that of the bulk medium, but this has yet to be established in the present case.
To confirm this behaviour, and examine the mechanism of the energy transfer, absorption measurements were undertaken of the QD-570–1 conjugate at varying pH, but these results became unclear at the higher concentrations required for such measurements, possibly arising from issues of solubility. Thus for the absorption measurements the model dye 2 was employed. A solution of 2 (3.33 × 10−5 M) was prepared in buffer and the pH was changed by addition of small amounts of HCl (0.5 M) or NaOH (0.1 M). The resulting UV/vis absorption spectra (Fig. 10a) show that the lowest energy transition around 440 nm changes significantly with pH, leading to both intensity changes and a wavelength shift. Notably, no isosbestic point is found. When absorption at a particular wavelength is plotted as a function of pH (Fig. 9b and c) it is clear that the absorption maxima change significantly depending on the excitation wavelength and that especially large differences are found when moving from neutral to alkaline pH. In the fluorescence studies described above excitation at 435 nm was chosen since the absorption at this wavelength varies least with pH.
 | ||
| Fig. 10 UV/vis absorption measurements of model dye 2 in solutions of different pH and plots of the absorbance vs. pH at different absorption maxima. (a) Absorption spectra in the pH-range 4.15–10.5, (b) plot of the absorbance at wavelengths between 430 and 440 nm and (c) plot of the absorbance at wavelengths between 410 and 460 nm. | ||
The absorption of the model dye therefore clearly changes with pH, and if extrapolated to the conjugate, this could influence the eventual communication between dye and dot and therefore be a possible explanation for the pH responsiveness of the conjugate (as opposed to a feature of the dye's fluorescence changes). However, the proportional absorption change of ca. 10–15% (in Fig. 9b) would seem unable to account for the factor of ca. 30% change observed in the original QD vs. QD–1 conjugate emission. Thus there is clearly communication between dye and dot that is responsive to pH, and a component of this could arise from absorption changes in the dye employed as the pH is altered.
Conclusions
The photophysical measurements described here for three different QDs functionalised with the same naphthalimide dye demonstrate how the nature of energy transfer within such conjugates depends on nanoparticle size. The QD-570 conjugate exhibits clear energy transfer between the dye and dot, an elusive and hitherto rarely reported phenomenon. Titration experiments between QDs and the dye demonstrate a saturation limit with a maximum number of dye molecules accommodated per particle, and dye densities that suggest complete surface coverage. The greater the number of dye molecules attached to the larger dots may be promoting FRET that would not otherwise be expected.The water-soluble QDs used in this study have hydrodynamic radii that are far larger than the core particle size owing to a covering of a solubilising polymer. This coating is assumed to be porous to the dye, and Raman spectra offer some evidence for the loading of the naphthalimide dye directly onto the core surface through disulfide exchange. Temporal changes following addition of the dye suggest that this reaction, though fast, can take several minutes to complete in some cases, consistent with the dye journeying through the polymer coating to reach the dot surface. When greater amounts of the dye are added, at levels above the saturation limit of the core, there appear to be non-specific interactions that lead to quenching of the dye signal.
The QD–dye conjugates (like the QDs themselves) are unstable at low pH, rendering these probes unsuitable for measurements at acidic pH. The conjugates perform well at physiological pH, and in the alkaline region around the pKa of the naphthalimide dye, although the fluorescence output is not perfectly reversible as pH is swept. Towards the goal of biomedical application, it will be of interest to assess the performance of these conjugates in more realistic (i.e., serum-based) media as a precursor to cell-based work, though clearly for these functionalised nanoparticles to be applied in such environments quantitatively, ratiometric probes would be needed. However, of greater interest perhaps is to further characterise the nature of the fluorescence enhancement processes, using intermediate-sized dots to optimise the energy transfer, or acquiring fluorescence decay time measurements to explore the possibility of “antenna-like” FRET behaviour in more detail.
Acknowledgements
This work was supported by a Discovery Grant from the Australian Research Council (DP120104035). We would like to thank Dr Elizabeth Carter of the University of Sydney Vibrational Spectroscopy Facility for assistance with the Raman spectroscopy.References
- P. Zrazhevskiy, M. Sena and X. Gao, Chem. Soc. Rev., 2010, 39, 4326–4354 RSC.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- C. M. Tyrakowski and P. T. Snee, Phys. Chem. Chem. Phys., 2014, 16, 837–855 RSC.
- R. K. Paradise, D. A. Lauffenburger and K. J. Van Vliet, PLoS One, 2011, 6, e15746 CAS.
- C. Stock, F. T. Ludwig, P. J. Hanley and A. Schwab, Compr. Physiol., 2013, 3, 59–119 Search PubMed.
- A. Jagielska, K. D. Wilhite and K. J. Van Vliet, PLoS One, 2013, 8, e76048 CAS.
- B. A. Webb, M. Chimenti, M. P. Jacobson and D. L. Barber, Nat. Rev. Cancer, 2011, 11, 671–677 CrossRef CAS PubMed.
- S. Ast, P. J. Rutledge and M. H. Todd, Eur. J. Inorg. Chem., 2012, 5611–5615 CrossRef CAS.
- Y. H. Lau, J. R. Price, M. H. Todd and P. J. Rutledge, Chem.–Eur. J., 2011, 17, 2850–2858 CrossRef CAS PubMed.
- E. Tamanini, A. Katewa, L. M. Sedger, M. H. Todd and M. Watkinson, Inorg. Chem., 2008, 48, 319–324 CrossRef PubMed.
- C. M. Lemon, P. N. Curtin, R. C. Somers, A. B. Greytak, R. M. Lanning, R. K. Jain, M. G. Bawendi and D. G. Nocera, Inorg. Chem., 2013, 53, 1900–1915 CrossRef PubMed.
- P. T. Snee, R. C. Somers, G. Nair, J. P. Zimmer, M. G. Bawendi and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 13320–13321 CrossRef CAS PubMed.
- A. M. Dennis, W. J. Rhee, D. Sotto, S. N. Dublin and G. Bao, ACS Nano, 2012, 6, 2917–2924 CrossRef CAS PubMed.
- T. Jin, A. Sasaki, M. Kinjo and J. Miyazaki, Chem. Commun., 2010, 46, 2408–2410 RSC.
- R. C. Somers, R. M. Lanning, P. T. Snee, A. B. Greytak, R. K. Jain, M. G. Bawendi and D. G. Nocera, Chem. Sci., 2012, 3, 2980–2985 RSC.
- S. Ast, P. J. Rutledge and M. H. Todd, Phys. Chem. Chem. Phys., 2014, 16, 25255–25257 RSC.
- A. R. Clapp, I. L. Medintz, B. R. Fisher, G. P. Anderson and H. Mattoussi, J. Am. Chem. Soc., 2005, 127, 1242–1250 CrossRef CAS PubMed.
- H. Xu, X. Huang, W. Zhang, G. Chen, W. Zhu and X. Zhong, ChemPhysChem, 2010, 11, 3167–3171 CrossRef CAS PubMed.
- Y. Chang, S. Yueming, S. Bo, T. Wenwen, Q. Qi, Z. Yingping, D. Yunqian and J. Wei, Nanotechnology, 2013, 24, 435704 CrossRef PubMed.
- M. Werwie, X. Xu, M. Haase, T. Basché and H. Paulsen, Langmuir, 2012, 28, 5810–5818 CrossRef CAS PubMed.
- W. Ma, L. X. Qin, F. T. Liu, Z. Gu, J. Wang, Z. G. Pan, T. D. James and Y. T. Long, Sci. Rep., 2013, 3, 1537 Search PubMed.
- A. P. de Silva, H. Q. N. Gunaratne, J.-L. Habib-Jiwan, C. P. McCoy, T. E. Rice and J.-P. Soumillion, Angew. Chem., Int. Ed., 1995, 34, 1728–1731 CrossRef.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum, New York, 1999 Search PubMed.
- I. L. Medintz, J. H. Konnert, A. R. Clapp, I. Stanish, M. E. Twigg, H. Mattoussi, J. M. Mauro and J. R. Deschamps, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9612–9617 CrossRef CAS PubMed.
- A. R. Clapp, I. L. Medintz, J. M. Mauro, B. R. Fisher, M. G. Bawendi and H. Mattoussi, J. Am. Chem. Soc., 2003, 126, 301–310 CrossRef PubMed.
- M. Hardzei, M. Artemyev, M. Molinari, M. Troyon, A. Sukhanova and I. Nabiev, ChemPhysChem, 2012, 13, 330–335 CrossRef CAS PubMed.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, Wiley, 2001 Search PubMed.
- A. M. Smith, H. Duan, M. N. Rhyner, G. Ruan and S. Nie, Phys. Chem. Chem. Phys., 2006, 8, 3895–3903 RSC.
- M. Gonzalez-Bejar, M. Frenette, L. Jorge and J. C. Scaiano, Chem. Commun., 2009, 3202–3204 RSC.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |