 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic and atomic force studies of the functionalisation of carbon surfaces: new insights into the role of the surface topography and specific chemical states†
Carlo
Buono
,
Philip R.
Davies
*,
Robert J.
Davies‡
,
Thomas
Jones
,
Jiří
Kulhavý
,
Ryan
Lewis
,
David J.
Morgan
,
Neil
Robinson
and
David J.
Willock
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: daviespr@cardiff.ac.uk; Fax: +44 (0)29 2087 4030; Tel: +44 (0)29 2087 4072
First published on 12th May 2014
Abstract
The utility of carbon materials in applications as diverse as drug delivery and photocatalysis is often undermined by the complexity of their surface chemistry; different sources of carbon give rise to a varied mixture of functional groups and hence different properties. Considerable efforts have been made to identify specific groups at these surfaces and elucidate the complex interactions that take place but even on materials such as graphene and carbon nanotubes there remains uncertainty about the nature of the components present and their role in the nucleation of other functional materials at the surface. The present study uses highly ordered pyrolytic graphite (HOPG) as a model on which the fundamental properties of specific functional groups and their interactions with deposited nanoparticles can be characterised. We have shown that treatment of HOPG surfaces with low concentrations of hydrochloric acid results in significant topographic changes to the surface and a low concentration of oxygen containing species. From selective derivatization and a comparison of their XP spectra, the latter can be unambiguously identified as surface hydroxyls. DFT calculations have shown that these groups are stable in close proximity to each other. Heating to 573 K leads to conversion of the hydroxyls to mixture of two states, one of which is identified as a ketone whilst the other is proposed to be an ether. Gold deposition on the surface from aqueous solutions of chloroauric acid is shown to be strongly influenced by the nature of the oxygen species present.
Introduction
In addition to their unique electronic properties carbon nano-materials have advantageous physical properties that have been utilised in a variety of different applications. Their high surface area, high physical strength and low weight makes them ideal support surfaces for uses ranging from nano-oscillators to heterogeneous catalysts. A common factor in all such applications is the need to control the interface between the carbon allotrope involved and the components that need to bind to it. The nature of that interface will be dependent on the structure and functionality of the carbon surface and many applications of carbon materials apply empirically developed treatments to modify the surfaces concerned.1,3–7 Carbon nanotubes are a good example, modifications to their surface chemistry being used to improve their dispersal in polymers and solvents as well as to tailor their electronic properties.6,8Typically, surface modifications involve the use of strong oxidants to create, or functionalise, defect sites; sonication in 8 M HNO3 for 1 hour7 or refluxing in 2.6 M HNO3 for 48 h9 being representative examples. However, determining the effects of such treatments is not always straightforward and there remains considerable uncertainty about the functional groups present at carbon surfaces; reviews by Boehm10 and Rodríguez-reinoso11 for example highlight the difficulty of unravelling this chemistry. Methods used to identify functional groups include temperature programmed desorption (TPD) and infrared spectroscopy (IR) but the former is a rather indirect probe and IR is only reliable for the identification of functional groups with strong transition dipoles. It is possible for minority species to dominate the infrared spectra whilst the majority state is undetected. A relevant example is the work of Goncalves et al.12 who studied the attachment of gold nanoparticles to oxygen functionalised graphene. Although they were able to demonstrate the importance of the oxygen functionality at the surface the authors were unable to distinguish between the roles of the different possible oxygen groups in the attachment of the gold.
X-ray photoelectron spectroscopy (XPS) is a surface sensitive and quantitative tool that has great potential in the characterisation of functional groups at carbon surfaces13–15 but because of the huge complexity of the materials involved there remains uncertainty about species assignments in XPS. One approach to address this complexity is the selective chemical derivatization of functional groups discussed by Everhart & Reilley in 1981 (ref. 16) and Batich in 1988.17 The method, which has previously been applied to the study of the functionality of polymer and carbon nanotube surfaces,18–20 is semiquantitative and involves the selective reaction of specific functional groups with a distinctive marker species, in this case fluorine, which is easily detectable in XP spectra.
Our approach to this complex area has been to focus on surfaces that are as reproducible as possible and HOPG is an ideal starting point, being readily available in well characterised orientations and quality. HOPG is also an ideal substrate for comparison with high quality DFT modelling, enabling us to probe the way in which functional groups are formed at HOPG surfaces. In our initial investigations2 we explored changes in the topography of the surface after treatment with water and dilute acids under ambient conditions (a parallel investigation into HOPG under UHV conditions is also underway).21 We have shown that dilute HCl and HNO3 cause areas of local delamination at the surface of the HOPG and speculate that these are due to the formation of local defects with associated oxygen containing functional groups that reduce the interlayer bonding. In the present paper, we explore the identity of the functional groups formed by the acids and the effects of heat treatment on the surfaces and on the deposition of gold nanoparticles from aqueous solution. The experimental protocol we have adopted mimics that of the preparation of gold supported catalysts for the hydrochlorination of ethyne and we hope eventually to explain the effects that acid washing has on the performance of these and other catalysts.4,5,22
Experimental
HOPG samples (∼10 mm square, ZYH quality, supplied by NT-MDT ltd, Europe) were cleaned before each experiment by peeling off the top few graphene monolayers using adhesive tape. Acid treatment involved placing a 100 μl droplet of fresh diluted HCl (Fischer Chemicals Ltd, 99%) and leaving to stand for 30 minutes before drying under a pure helium stream. The acid wet the surface very effectively and so larger droplets were not needed to provide complete coverage of the crystal surfaces. Gold deposition was achieved in a similar fashion: a 100 μl droplet of a gold precursor solution (HAuCl4 × H2O, Sigma-Aldrich, 99.999%) was placed on the pre-treated sample and left for 1 minute. The sample was subsequently dried under a stream of helium for 2 minutes.Ultra-pure water (UPW; Millipore) was used for dilution of the acids before surface treatment and also for control experiments that replicated dosing and analysis conditions. Selected samples were heated to 423 K and 573 K for 1 hour after surface treatment and allowed to cool to ambient temperature before analysis.
Derivatization reagents trifluoroacetic anhydride (TFAA, 99%), trifluoroethyl hydrazine (TFH, 70 wt% in water), triflouroethanol (TFE, 99%) and N,N′-di-tert-butylcarbodiimide (DTBC, 99%) were purchased from Sigma-Aldrich and used as received. Derivatization reactions were carried out in a sealed glass chamber pumped with a rotary vane pump to a base pressure of ∼10−2 mbar. (Initial attempts at derivatization using a stainless steel vacuum chamber were abandoned because subsequent analysis by XPS showed transport of copper from the metal vacuum seals to the HOPG surfaces. No contamination issues occurred in the glass chamber.) 2 ml of TFAA and TFH liquids were frozen and placed into a reservoir of liquid nitrogen inside the chamber; this enabled the evacuation of the chamber without pumping away the reactant. TFE on the other hand was dosed from a 2 ml mixture of TFE, pyridine and DTBC in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:8 ratio, respectively, according to the procedure reported by Everhart & Reilley.16 HOPG samples were placed into the chamber avoiding contact with the reservoir and walls. The chamber was evacuated until the liquid nitrogen reservoir had evaporated, at which point it was isolated from the pump and allowed to equilibrate for ∼24 hours. The chamber was opened to the air and the samples typically analysed by XPS within a few hours of removal. However, control experiments in which the derivatised samples were allowed to stand in air for up to 7 days showed no significant change in the surface chemistry.
:4:8 ratio, respectively, according to the procedure reported by Everhart & Reilley.16 HOPG samples were placed into the chamber avoiding contact with the reservoir and walls. The chamber was evacuated until the liquid nitrogen reservoir had evaporated, at which point it was isolated from the pump and allowed to equilibrate for ∼24 hours. The chamber was opened to the air and the samples typically analysed by XPS within a few hours of removal. However, control experiments in which the derivatised samples were allowed to stand in air for up to 7 days showed no significant change in the surface chemistry.
It is worth noting here that the concentration of different oxygen components in the XP spectra of the treated HOPG surfaces were slightly variable from sample to sample (±20%) meaning that quantitative estimates between samples is difficult. However, binding energies of the components within the spectra were reproducible from sample to sample.
XP spectra were recorded with a Kratos Axis Ultra-DLD photoelectron spectrometer using a monochromatic Al Kα X-ray source and the “hybrid spectroscopy” mode, resulting in an analysis area of 700 × 400 μm at a pass-energy of 40 eV for high resolution scans and 160 eV for survey scans. The XPS data was analysed using CasaXPS23 with all binding energies referenced to the C(1s) peak at 284.5 eV. Curve fits were made using Gaussian line profiles and surface concentrations were calculated using the method described in detail by Carley et al.24,25 AFM images were acquired on a Bruker Veeco Multimode probe in tapping mode using silicon nitride tips. AFM images were analysed using WSxM software.26
The periodic density functional theory (DFT) calculations presented here were carried out with the VASP code.27–30 Convergence was found for the bulk structure of graphite using a 500 eV energy cut off for the plane-wave basis and a 5 × 5 × 1 Monkhorst–Pack k-point mesh.31 We use the PBE generalised gradient approximation32 functional with Grimme's parameterised dispersion interaction33 included (PBE-D2) in order to model the van der Waals interactions which are the major force operating between graphitic layers. The core 1s states for C and O are represented using the projector augmented wave (PAW) approach while the ultrasoft pseudopotential is employed for H atoms.34,35 The cell dimensions were optimised by systematic variation of the equivalent a and b lattice vectors followed by variation of the c-parameter. This process gave a lower cell energy than the alternative of simple cell volume scaling and resulted in hexagonal cell parameters of a = b = 2.465 Å and c = 6.297 Å. The c-lattice vector is controlled by the interlayer spacing and we were only able to obtain a stable minimum with respect to lattice expansion in this direction when the D2 corrected functional is employed. The pure graphite structure is hexagonal with an ABA… stacking sequence so that the c-vector represents two inter-layer separations. The inter-layer separation found here (3.149 Å) is shorter than that observed experimentally36,37 (3.354 Å) by 6% although it is in line with earlier simulations using similar functionals38 (3.21 Å).
The graphite (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 5) plane was used to model step edge features and the convergence of results with respect to supercell size and slab thickness was assessed using the calculated edge energies (see eqn (5)) for hydrogenated step edges. Slabs containing two graphite layers and doubled in the direction parallel to the edge were constructed so that the opposite edges of graphite sheets on different sides of the slab were separated by at least four aromatic rings.
5) plane was used to model step edge features and the convergence of results with respect to supercell size and slab thickness was assessed using the calculated edge energies (see eqn (5)) for hydrogenated step edges. Slabs containing two graphite layers and doubled in the direction parallel to the edge were constructed so that the opposite edges of graphite sheets on different sides of the slab were separated by at least four aromatic rings.
Results and discussion
XPS and AFM studies of acid treated HOPG
AFM imaging shows that HOPG surfaces treated with 0.2 M HCl under ambient conditions become decorated with raised approximately circular features 50–100 nm in diameter and ∼15–20 nm in height above the surrounding lattice, Fig. 1. We have previously demonstrated that these features are due to local areas of delamination, possibly due to the functionalization of defects sites and the reduction in inter-layer bonding and shown that the size of the islands is related to acid concentration.2 Control experiments using ultra-pure water, in contrast, showed neither the local delamination effects nor any significant change in the species present at the surface.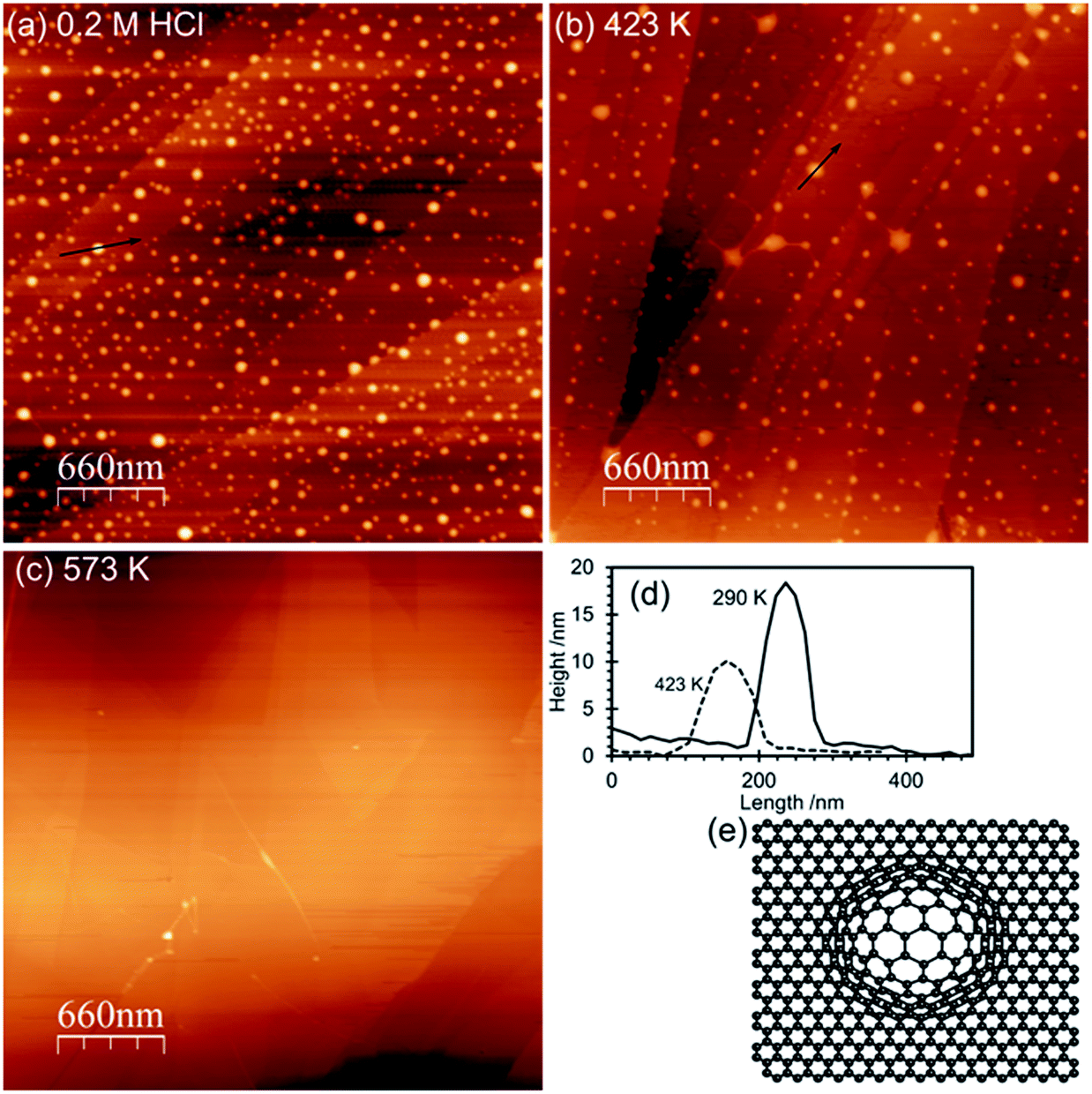 | ||
| Fig. 1 AFM images of the acid treated surfaces: (a) HOPG surface treated with 0.2 M HCl for 30 minutes; (b) after heating to 423 K for 1 hour; (c) after heating to 573 K for 1 hour; (d) line profiles from the AFM images; (e) model illustrating the local delamination proposed to be the cause of the features in the AFM images. | ||
The corresponding XP spectra are shown in Fig. 2, and reveal a single peak in the O(1s) region at 532.6 eV with a calculated surface concentration of 1.1 × 1015 cm−2 corresponding to ∼0.27 monolayers. Changes in the C(1s) spectrum are too small to be detected against the background but there are small increases in the Cl(2p) spectra (∼3.5 × 1013 cm−2) and, perhaps surprisingly, in the N(1s) region with the development of a peak at 400.2 eV with a surface concentration corresponding to 1.6 × 1014 cm−2. The N(1s) binding energy of 400.2 eV is relatively low, more indicative of an amine than a nitrate.
 | ||
| Fig. 2 XP spectra of HOPG after treatment with 0.5 M HCl and subsequent heating to 573 K. (a) Clean surface; (b) after treatment with 0.5 M HCl for 30 minutes; (c) after heating to 573 K for 1 hour and cooling to ambient. | ||
Heating the sample in stages causes a decrease in the number of features in the AFM images with an accompanying decline in the overall volume of the features. By 573 K virtually all of the features in the AFM images have disappeared. In the XP spectra the peaks in the Cl(2p) and N(1s) region vanish by 573 K whereas the single peak in the oxygen region splits into two with binding energies of 531.5 and 533.3 eV. Repeated experiments suggest only a small reduction in surface oxygen concentration occurs on heating from 290 K to 573 K.
Identification of functional groups at acid treated HOPG surfaces by selective derivatisation
:1 stoichiometry and high selectivity, generating an adsorbed fluoroacetate and an acetic acid that desorbs or reacts with other hydroxyl groups to give the same product:17,39| CF3C(O)OC(O)CF3 (g) + OH (a) → CF3C(O)O (a) + CF3CO2H (g) | (1) |
HOPG surfaces, freshly cleaved in air, always show a small concentration (∼1 × 1014 cm−2) of oxygen dominated by a peak at 532.6 eV. Exposure of this surface to TFAA leads to a similarly weak peak in the F(1s). When the HOPG surface treated with 0.5 M HCl was exposed to TFAA the strong O(1s) peak at 532.7 eV is broadened with the development of a shoulder to higher binding energy; curve-fitting of the spectra show that the resulting envelope is consistent with peaks at 532.6 eV and 534.0 eV with area ratios of ∼3:1. In the F(1s) region a strong peak at 688.8 eV indicates the presence of fluorine with a surface concentration that is a little less than twice that of the oxygen peak at 534.0 eV, Fig. 3.
 | ||
| Fig. 3 XP spectra showing O(1s) and F(1s) regions of HOPG after treatment with HCl and TFAA. (a) Clean HOPG exposed to TFAA; (b) (in O(1s) spectra only for comparison), HOPG treated with 0.5 M HCl for 30 minutes; (c) surface in (b) after exposure to TFAA; (d) after heating (b) to 573 K for 1 hour followed by exposure to TFAA. | ||
The data suggest that the HCl treatment of graphite surfaces generates a high concentration of hydroxyl species, with a characteristic binding energy of ∼532.7 eV. The fluoroacetate produced from the reaction of the TFAA with this hydroxyl will have two distinct oxygen species, a carbonyl and a methoxy type functionality (C–O–CF3). We show below that a carbonyl functionality on the graphite surface gives rise to an O(1s) binding energy of 531.6 eV. In the case of the fluoroacetate the functionality is closely associated with the CF3 group and we might have expected a higher binding energy but Holländer's study39 of TFAA derivatised poly(vinyl alcohol) also assigns the fluoroacetate carbonyl to a peak at 531.6 eV whilst the methoxy type functionality appeared at 534.8 eV. The larger intensity of the 531.6 eV peak compared with the 534.0 peak suggests that some intensity from the original peak before TFAA treatment remains. This implies that either unreacted OH groups remain at the surface after TFAA treatment or a second species is present with a binding energy similar to the OH. Exposure of the heated HCl treated surface to TFAA results in negligible reaction; the small amount of fluorine that is present is probably due to hydroxyl groups formed at the HOPG surface whilst the sample was transferred from the oven to the treatment chamber. Clearly heating to 573 K is sufficient to convert all of the surface hydroxyl groups to other functionalities, confirming the observations from Fig. 2.
:1 stoichiometry and high selectivity:16–18| CF3CH2OH (g) + –CO2H (a) → CF3C(OH)O (a) + H2O (g) | (2) |
Clean, HCl treated and heated HOPG surfaces were exposed to the TFE–DTBC mixture for 24 hours and the resulting XP spectra are shown in Fig. 4. No reaction was observed between the TFE and the clean HOPG surface and exposure of the HCl treated surface to the TFE–DTBC mixture for 24 hours also results in only a minimal uptake in fluorine containing molecules. The corresponding O(1s) spectra are also within 0.1 eV of the spectra observed without TFE treatment. We can conclude that mild HCl treatment of the HOPG surface does not lead to carboxylic functionality. Fig. 4(c) shows that there is also no reaction between TFE and either of the states generated by heating the acid treated surface. We can therefore rule out the assignment to carboxylic groups of either of the two species with O(1s) binding energies of 531.5 and 533.3 eV.
 | ||
| Fig. 4 XP spectra of HCl treated HOPG surface exposed to TFE vapour. (a) Clean HOPG surface exposed to TFE; (b) HOPG surface treated with 0.5 M HCl and subsequently exposed to TFE; (c) HOPG surface treated with 0.5 M HCl and heated for 1 h to 573 K before exposure to TFE. | ||
:1 stoichiometry:16–18CF3CH2NH–NH2 (g) + –CO (a) → CF3CH2NH–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) (a) + H2O (g) (a) + H2O (g) | (3) |
The F(1s) spectra in Fig. 5 demonstrate that there is no reaction between TFH and the clean HOPG surface and there is also only minimal reaction when the HOPG washed with HCl is exposed to TFH. However, exposure of the HOPG surface to TFH after HCl washing and subsequent heating to 573 K results in much more extensive reaction. A strong peak develops in the fluorine region at 688.4 eV accompanied by a strong peak at 400.5 eV in the N(1s) region. Interestingly, although the reaction stoichiometry (eqn (3)) indicates complete removal of the O(1s), two oxygen features remain after TFH treatment; the peak at 533.3 eV is unaffected whereas the peak at 531.6 eV is shifted up to 531.9 eV. A possible explanation is that the water is not completely desorbed but rather reacts with the lattice in a concerted reaction with the hydrazine reaction, furthermore a hydroxyl group would be a possible assignment for the peak at 531.9 eV since its close proximity to the hydrazine group could be sufficient to shift the OH binding energy significantly from that of the hydroxyl group observed after HCl treatment.
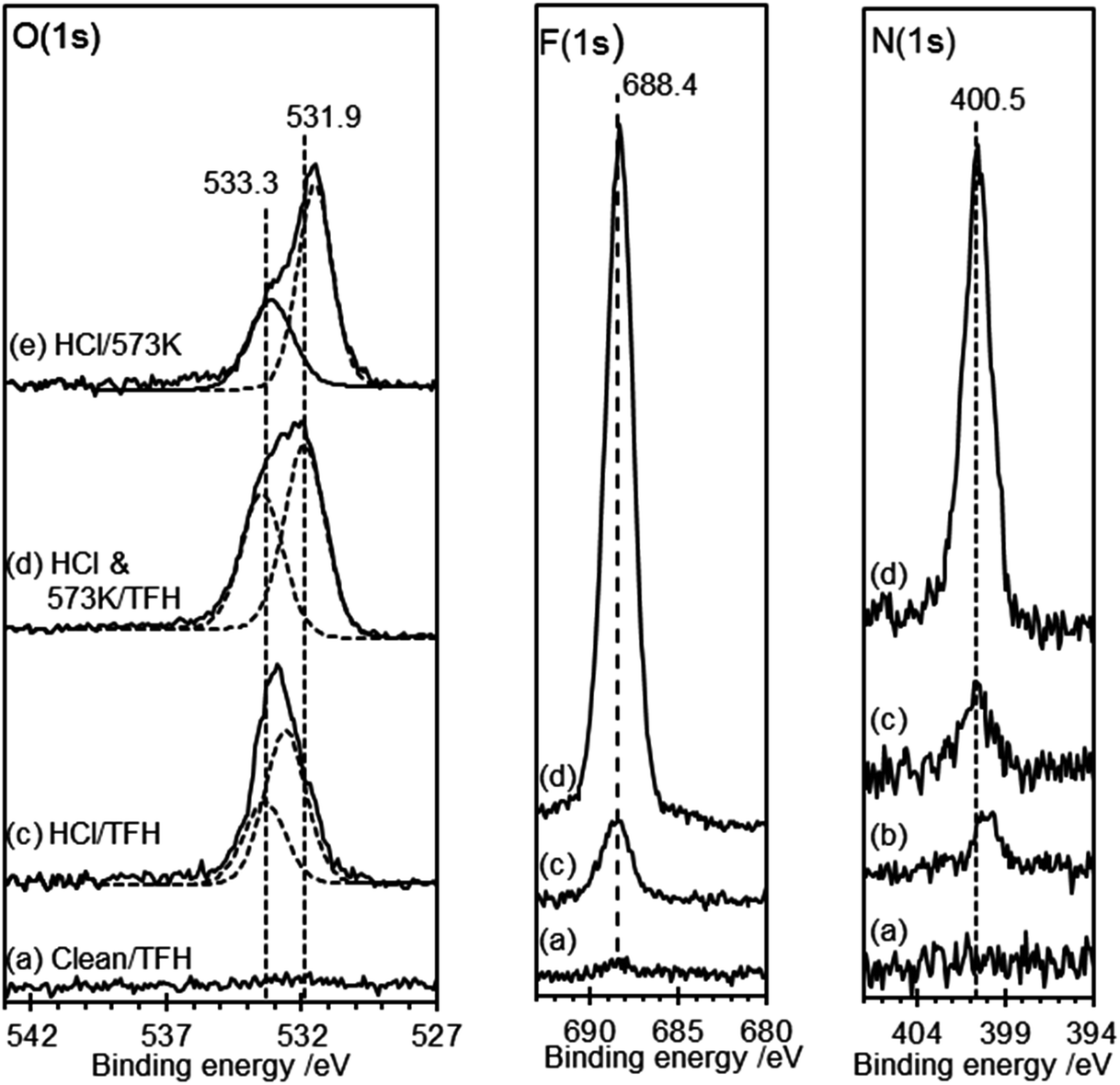 | ||
| Fig. 5 XP spectra of HCl treated HOPG surface exposed to TFH vapour. (a) Clean HOPG surface exposed to TFH; (b) (N(1s) spectra only), HOPG surface treated with 0.5 M HCl; (c) HOPG surface treated with 0.5 M HCl and subsequently exposed to TFH; (d) HOPG surface treated with 0.5 M HCl and heated for 1 h to 573 K before exposure to TFH. (e) (O(1s) spectra only), HCl treated surface heated to 573 K surface without TFH exposure for comparison. | ||
Gold deposition at acid treated and heated HOPG surfaces
The AFM images of the gold deposited on heated and non-heated HCl treated HOPG (Fig. 6) show similar topography to each other with neither surface exhibiting the raised features originally created by the HCl treatment. This is consistent with the flat surface reported in Fig. 1 for the heated sample but implies that the gold deposition process can also remove these features. The AFM images of the gold deposited on a non-heated surface, Fig. 6(a), show evidence for a discontinuous film, where the gaps between sections of the film indicate a thickness of approximately 2 nm. The films deposited on the heated surface are much smoother, broken only by what appear to be the step edges in the graphite substrate. XP spectra of the surfaces are shown in Fig. 7, and from the attenuation of the C(1s) signal we estimate at least a monolayer of gold or gold chloride is present in each case.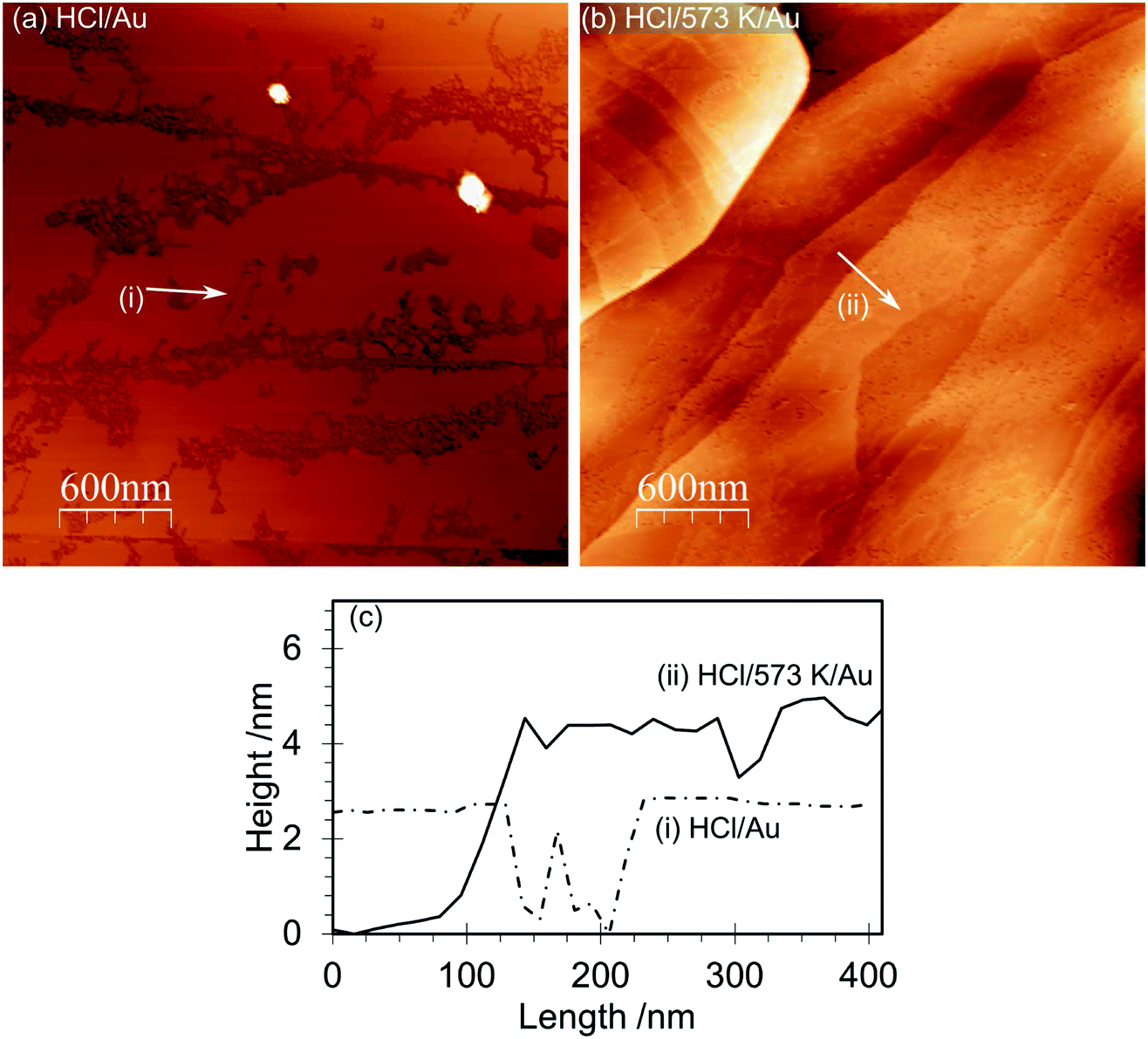 | ||
| Fig. 6 AFM images of the HOPG surface after gold deposition from a solution of chloroauric acid: (a) HOPG surface treated with 0.5 M HCl, dried and then exposed to a 6 × 10−6 M solution of HAuCl4. (b) HOPG surface treated with 0.5 M HCl, dried and heated to 573 K (1 hour) before cooling and being exposed to 2 × 10−6 M solution of HAuCl4. (c) Line profiles from the AFM images. | ||
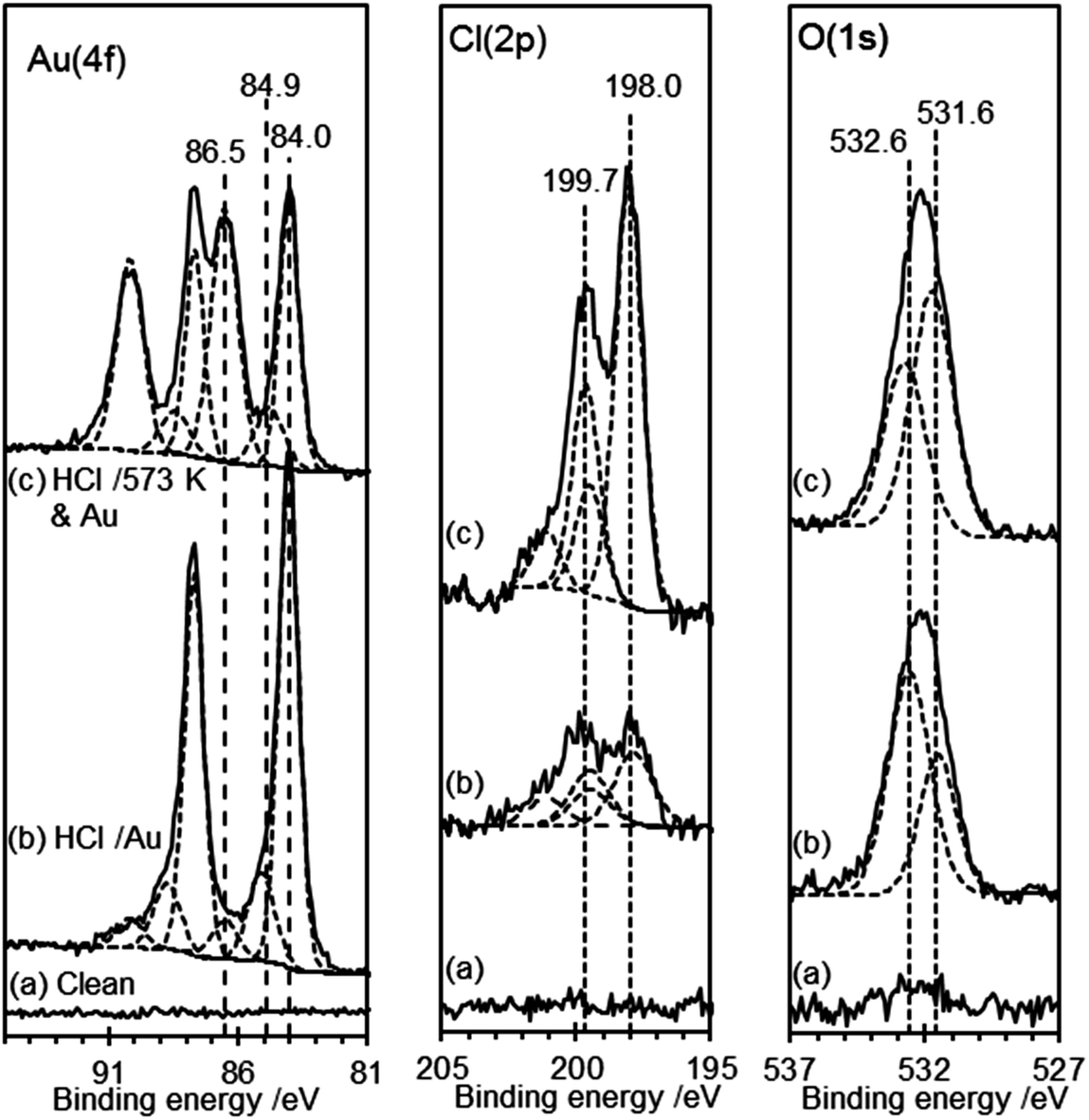 | ||
| Fig. 7 XP spectra of the HOPG surface after gold deposition from a solution of chloroauric acid: (a) clean surface; (b) HOPG surface treated with 0.5 M HCl, dried and then exposed to a 6 × 10−6 M solution of HAuCl4. (c) HOPG surface treated with 0.5 M HCl, dried and heated to 573 K (1 hour) before cooling and being exposed to 2 × 10−6 M solution of HAuCl4. | ||
In both types of experiment the XP spectra show at least two gold states with characteristic Au(4f7/2) peaks at 84.0 and 86.5 eV, which can be assigned to Au0 and Au3+, respectively.40 Curve fitting of the data unambiguously indicates the presence of a third state in both experiments with a Au(4f7/2) peak at 84.9 (±0.2) eV. This state has two likely origins, a Au+ state or a nano-particulate Au0 (since small gold islands are expected to show a shifted binding energy41,42). In view of the relatively smooth films evident in the AFM images, the former explanation is more likely in the present case. Support for these assignments comes from the Cl(2p) spectra, which show two chlorine species with characteristic binding energies of 198.0 and 199.7 eV. Kishi reported similar binding energies for chlorine bound to the Au3+ and Au+ species after exposure of gold metal to chlorine gas.43
DFT studies of the stability of functional groups on HOPG surfaces
The functionalization of graphene as a function of chemical potential of reactants has been studied extensively using DFT approaches by Mauri and co-workers covering hydrogenation, and reaction with O2 and water.44–46 As part of the present study we have calculated the structure of the most favourable terminations of step edges on graphite using PBE-D2.Graphite has a hexagonal crystal structure with the graphitic sheets parallel to the (0001) surfaces. A step edge can be represented by introducing higher Miller indices at the expense of increasing the cell size and so the computational cost of the calculations. We choose to use a Miller index of 5 in the c-direction so that functional groups at a step edge have a terrace region of at least one aromatic ring separating them from the next edge in the simulation cell.
We initially considered two possible step edge structures: the zigzag (z), in which the step edge is formed with single C atoms from individual 6-rings exposed, and the armchair (a), in which two C atoms from each individual 6-ring appear (see Fig. S1†). The calculated energy required to create a non-functionalised graphite step edge with z-termination is much higher (1.5 eV Å−1) than that for the a arrangement (1.1 eV Å−1), an effect which can be understood in terms of Clar's rules:47 on hydrogenation the a-edge leads to CC double bonds that are effectively isolated from the aromatic system of the graphite sheet whereas the aromatic system is disrupted to a greater extent for the z edge.
The experimental procedure begins with materials with no oxygen present and so we take the a-edge as the best representation of the steps on the unreacted surface. For comparison with the experimental data presented here we considered the reaction of the isolated double bonds of the armchair step edge with water:
| C(105) + NH2OH2ONH2OC(105)(−OH)(−H) | (4) |
The energy required for this process can be calculated using the PBE-D2 energies following:
 | (5) |
Eight possible terminations of the slab edges were considered (Fig. S2 and S3†) but the calculated edge energies showed that these could be classified into two sets. Fig. 8(a) and (c) show the lowest energy structure obtained with each of the isolated CC bonds of the armchair edge functionalised with an OH group and H atom such that the OH group is roughly in the plane of the graphite sheet and orientated toward the H on the same CC bond. This structure has a calculated formation energy, E(edge), of 0.282 eV Å−1, a little higher than that reported for a single graphene sheet by Mauri and co-workers45 (0.247 eV Å−1). This difference is likely due to the interaction of the functional groups at the step edge with C atoms in the terrace below.
 | ||
| Fig. 8 PBE-D2 calculated structures for the hydroxylated step edge on graphite using the (105) surface. (a) The most stable arrangement and orientation of hydroxyl groups at the step edge found. (b) An example structure with the hydroxyl groups rotated through roughly 180° resulting in a higher energy structure. (c) Space fill representation of the stepped surface from (a) showing an expansion of the simulation cell by 2 into the plane of the figure and 2.5 across the page. Atoms coloured according to C: grey; O: red; H: white. | ||
Rotation of the OH group by roughly 180° results in a member of the higher energy set (Fig. 8(b), E(edge) = 0.331 eV). It can be seen in the figure that there is a steric clash between the OH and an H atom on the neighbouring CC section that leads to the higher energy of this arrangement.
It is interesting that the edge energy is positive for all terminations studied, which indicates that in pure water functionalization will not take place; this may be why acid conditions are required to stabilise the OH functional groups experimentally. In addition, the close proximity of the OH functional groups along the step edge suggests that the addition of the probe molecules used in our experimental work will lead to significant crowding and so can explain the residual population of OH groups observed in the XPS spectrum.
Conclusions
We have shown previously that washing HOPG surfaces with low concentrations of HCl gives rise to areas of local delamination. In the present work, using selective derivatization, we have established that the effect of the acid washing is to generate hydroxyl groups almost exclusively, and we can now identify a characteristic O(1s) binding energy of 532.7 eV for OH(a) on the HOPG surface. Even allowing for the slight difference in calibration used between studies this is slightly lower than that observed13 with aliphatic polymers and significantly lower than that of hydroxyl groups associated with aromatic polymers, which might have been expected to be a good match to the HOPG surface. It does, however, lie within the ranges (albeit at the lower end) suggested by Yue15 and Zielke14 for hydroxyls on carbon fibres. The XP data suggest that either exposure to TFAA did not remove all of the hydroxyl groups present after acid treatment or that a second unreactive species is present. Our DFT calculations, which show that the hydroxyls tend to be closely aggregated, support the former model with steric hindrance preventing complete reaction.Acid treatment of carbon surfaces has previously been associated with the formation of carboxylic functionality but those studies48,49 have generally utilised much stronger concentrations of acid than used here. In this work no evidence for carboxylic acid surface groups was obtained, suggesting that the hydroxyl groups are the first stage in the oxidation process and perhaps carboxylic acid functionality develops later. This aspect will be explored in future work.
Heating the hydroxyls leads to the formation of two different oxygen species. The first, with a characteristic O(1s) binding energy of 533.3 eV, is unaffected by treatment with any of the reagents used in this study. We provisionally assign this state to an ether type group (–C–O–C–). The second, which has a characteristic O(1s) binding energy of 531.6 eV, reacts strongly with TFH and can be assigned to a surface ketone.
Conte et al.4 have recently pointed out the importance of the metal/functional group interaction in deposition onto carbon surfaces and the particular paucity of information on the deposition of gold on carbon. Whilst it is clear that we have deposited far more gold in the present experiments than would be desirable in most applications, the striking differences seen here between the oxidation states of gold deposited on the hydroxylated and ketone rich surfaces demonstrate that this is a rich area to be explored. The results provide an interesting glimpse into the anchoring of metal nanoparticles onto carbon surfaces and suggest several future pathways for investigation.
Acknowledgements
This research was supported by EPSRC Grant EP/I038748/1. TJ thanks Cardiff University for provision of a summer studentship under the CUROP scheme. Computing resources were obtained locally from the Cardiff ARCCA division and via our membership of the UK's HPC Materials Chemistry Consortium (EPSRC EP/L000202). This work made use of the facilities of HECToR and ARCHER, the UK's national high-performance computing service, which is funded by the Office of Science and Technology through EPSRC's High End Computing Programme.References
- P. Serp and J. L. Figueiredo, Carbon materials for catalysis, John Wiley & Sons, Hoboken, N.J, 2009 Search PubMed.
- E. Bouleghlimat, P. R. Davies, R. J. Davies, R. Howarth, J. Kulhavy and D. J. Morgan, Carbon, 2013, 61, 124–133 CrossRef CAS PubMed.
- X. Zhang, Y. C. Guo, Z. Cheng Zhang, J. S. Gao and C. M. Xu, J. Catal., 2012, 292, 213–226 CrossRef CAS PubMed.
- M. Conte, C. J. Davies, D. J. Morgan, T. E. Davies, D. J. Elias, A. F. Carley, P. Johnston and G. J. Hutchings, J. Catal., 2013, 297, 128–136 CrossRef CAS PubMed.
- M. Conte, A. F. Carley, C. Heirene, D. J. Willock, P. Johnston, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 250, 231–239 CrossRef CAS PubMed.
- K. Balasubramanian and M. Burghard, Small, 2005, 1, 180–192 CrossRef CAS PubMed.
- M. N. Tchoul, W. T. Ford, G. Lolli, D. E. Resasco and S. Arepalli, Chem. Mater., 2007, 19, 5765–5772 CrossRef CAS.
- S. Banerjee, T. Hemraj-Benny and S. S. Wong, Adv. Mater., 2005, 17, 17–29 CrossRef CAS.
- J. Zhang, H. Zou, Q. Qing, Y. Yang, Q. Li, Z. Liu, X. Guo and Z. Du, J. Phys. Chem. B, 2003, 107, 3712–3718 CrossRef CAS.
- H. P. Boehm, Carbon, 2002, 40, 145–149 CrossRef CAS.
- F. Rodríguez-reinoso, Carbon, 1998, 36, 159–175 CrossRef.
- G. Goncalves, P. A. A. P. Marques, C. M. Granadeiro, H. I. S. Nogueira, M. K. Singh and J. Grácio, Chem. Mater., 2009, 21, 4796–4802 CrossRef CAS.
- G. Beamson and D. Briggs, High resolution XPS of organic polymers: the Scienta ESCA300 database, Wiley, Chichester, England, New York, 1992 Search PubMed.
- U. Zielke, K. J. Hüttinger and W. P. Hoffman, Carbon, 1996, 34, 983–998 CrossRef CAS.
- Z. R. Yue, W. Jiang, L. Wang, S. D. Gardner and C. U. Pittman Jr, Carbon, 1999, 37, 1785–1796 CrossRef CAS.
- D. S. Everhart and C. N. Reilley, Anal. Chem., 1981, 53, 665–676 CrossRef CAS.
- C. D. Batich, Appl. Surf. Sci., 1988, 32, 57–73 CrossRef CAS.
- A. Chilkoti, B. D. Ratner and D. Briggs, Chem. Mater., 1991, 3, 51–61 CrossRef CAS.
- L. A. Langley, D. E. Villanueva and D. H. Fairbrother, Chem. Mater., 2006, 18, 169–178 CrossRef CAS.
- K. A. Wepasnick, B. A. Smith, K. E. Schrote, H. K. Wilson, S. R. Diegelmann and D. H. Fairbrother, Carbon, 2011, 49, 24–36 CrossRef CAS PubMed.
- M. Bowker, P. R. Davies, R. J. Davies and D. J. Morgan, Phys. Chem. Chem. Phys., 2014 Search PubMed , in preparation.
- J. K. Edwards, B. Solsona, E. N. N., A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- N. Fairley, CasaXPS.
- A. F. Carley and M. W. Roberts, Proc. R. Soc. London, Ser. A, 1978, 363, 403–424 CrossRef CAS.
- A. F. Carley, P. R. Davies, R. V. Jones, K. R. Harikumar, G. U. Kulkarni and M. W. Roberts, Surf. Sci., 2000, 447, 39–50 CrossRef CAS.
- I. Horcas, R. Fernandez, J. Gomez-Rodriguez, J. Colchero, J. Gomez-Herrero and A. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- R. Franklin, Acta Crystallogr., 1951, 4, 253–261 CrossRef CAS.
- G. Bacon, Acta Crystallogr., 1951, 4, 558–561 CrossRef CAS.
- G. Graziano, J. Klimes, F. Fernandez-Alonso and A. Michaelides, J. Phys.: Condens. Matter, 2012, 24, 424216 CrossRef PubMed.
- F. Pippig, S. Sarghini, A. Holländer, S. Paulussen and H. Terryn, Surf. Interface Anal., 2009, 41, 421–429 CrossRef CAS.
- Y.-Y. Fong, B. R. Visser, J. R. Gascooke, B. C. C. Cowie, L. Thomsen, G. F. Metha, M. A. Buntine and H. H. Harris, Langmuir, 2011, 27, 8099–8104 CrossRef CAS PubMed.
- T. K. Sham, A. Hiraya and M. Watanabe, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 7585–7592 CrossRef CAS.
- H. Altass, A. F. Carley, P. R. Davies, and R. J. Davies, 2013, in preparation.
- K. Kishi and S. Ikeda, J. Phys. Chem., 1974, 78, 107–112 CrossRef CAS.
- A. P. Seitsonen, A. M. Saitta, T. Wassmann, M. Lazzeri and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 115425 CrossRef.
- T. Wassmann, A. P. Seitsonen, A. M. Saitta, M. Lazzeri and F. Mauri, Phys. Status Solidi B, 2009, 246, 2586–2591 CrossRef CAS.
- T. Wassmann, A. P. Seitsonen, A. M. Saitta, M. Lazzeri and F. Mauri, Phys. Rev. Lett., 2008, 101, 096402 CrossRef.
- T. Wassmann, A. P. Seitsonen, A. M. Saitta, M. Lazzeri and F. Mauri, J. Am. Chem. Soc., 2010, 132, 3440–3451 CrossRef CAS PubMed.
- I. W. Chiang, B. E. Brinson, R. E. Smalley, J. L. Margrave and R. H. Hauge, J. Phys. Chem. B, 2001, 105, 1157–1161 CrossRef CAS.
- N. Dementev, X. Feng and E. Borguet, Langmuir, 2009, 25, 7573–7577 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4fd00061g |
| ‡ Present address: Imperial College, London SW7 2AZ, UK. |
| This journal is © The Royal Society of Chemistry 2014 |