 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparison of photoelectrochemical water oxidation activity of a synthetic photocatalyst system with photosystem II
Yi-Hsuan
Lai
,
Masaru
Kato
,
Dirk
Mersch
and
Erwin
Reisner
*
Christian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
First published on 12th June 2014
Abstract
This discussion describes a direct comparison of photoelectrochemical (PEC) water oxidation activity between a photosystem II (PSII)-functionalised photoanode and a synthetic nanocomposite photoanode. The semi-biological photoanode is composed of PSII from the thermophilic cyanobacterium Thermosynechococcus elongatus on a mesoporous indium tin oxide electrode (mesoITO|PSII). PSII embeds all of the required functionalities for light absorption, charge separation and water oxidation and ITO serves solely as the electron collector. The synthetic photoanode consists of a TiO2 and NiOx coated nanosheet-structured WO3 electrode (nanoWO3|TiO2|NiOx). The composite structure of the synthetic electrode allows mimicry of the functional key features in PSII: visible light is absorbed by WO3, TiO2 serves as a protection and charge separation layer and NiOx serves as the water oxidation electrocatalyst. MesoITO|PSII uses low energy red light, whereas nanoWO3|TiO2|NiOx requires high energy photons of blue-end visible and UV regions to oxidise water. The electrodes have a comparable onset potential at approximately 0.6 V vs. reversible hydrogen electrode (RHE). MesoITO|PSII reaches its saturation photocurrent at 0.84 V vs. RHE, whereas nanoWO3|TiO2|NiOx requires more than 1.34 V vs. RHE. This suggests that mesoITO|PSII suffers from fewer limitations from charge recombination and slow water oxidation catalysis than the synthetic electrode. MesoITO|PSII displays a higher ‘per active’ site activity, but is less photostable and displays a much lower photocurrent per geometrical surface area and incident photon to current conversion efficiency (IPCE) than nanoWO3|TiO2|NiOx.
1. Introduction
A possible route for the conversion of energy in sunlight into a storable hydrogen-based fuel is solar water splitting.1 Photosynthesis uses sunlight, water and CO2 to generate readily useable carbohydrates. At the heart of this process stands the protein complex PSII, which catalyses efficiently the most thermodynamically demanding process in biology: solar light-driven water oxidation. As such, PSII not only sets the benchmark for photo-water oxidation catalysis, but also provides an invaluable inspiration for the design of artificial photosynthetic systems.2 Essential to the function of PSII is its fine-tuned ability to combine light absorption, multi-charge separation and efficient water oxidation catalysis. Electron transfer in PSII occurs from the excited primary electron donor (P680*), which can be generated upon red light absorption at 680 nm. The electrons are further transferred to the electron acceptor plastoquinones, QA and QB, via a pheophytin (Pheo). The generated holes are captured by a tyrosine (Tyrz) and subsequently transferred to the CaMn4 oxygen evolving catalyst (OEC) of PSII to oxidise water (Scheme 1a).2,3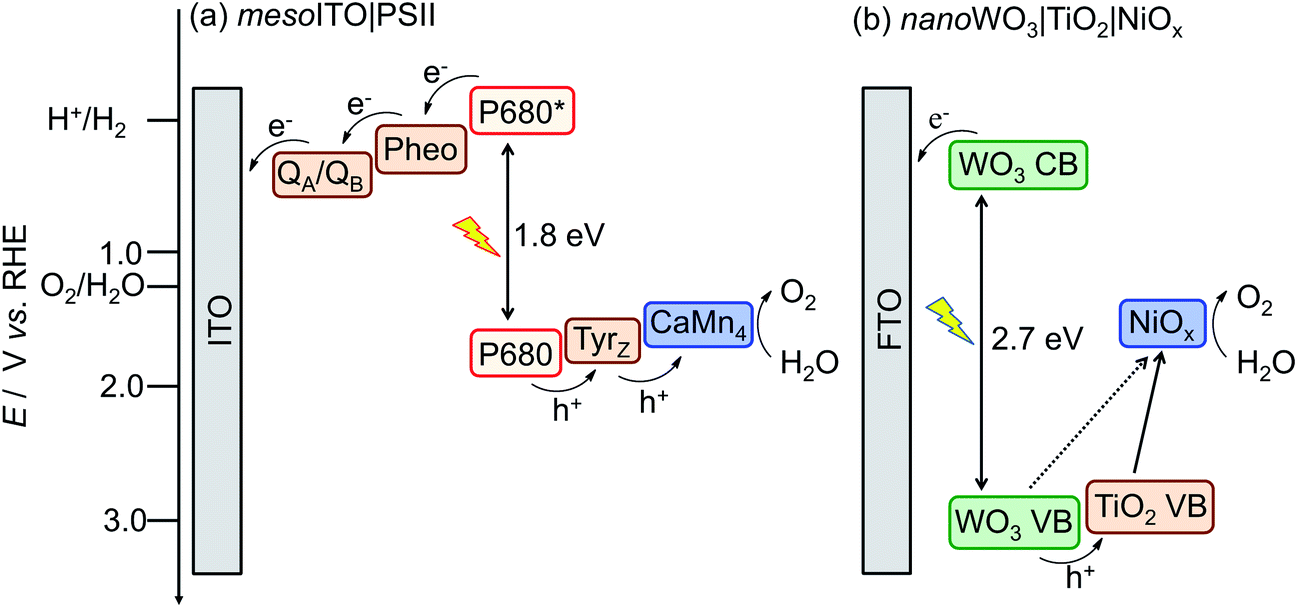 | ||
| Scheme 1 Energy diagrams for solar light driven water oxidation with (a) mesoITO|PSII and (b) the bio-inspired nanoWO3|TiO2|NiOx.4 | ||
Future solar fuel technologies are likely to rely on PEC cells and many electrodes for photo-water oxidation have recently been reported.1b,5 Despite the immense interest in mimicking PSII,6 a direct comparison of a PSII-immobilised electrode with a purely synthetic, PSII-inspired water oxidation photoanode has not yet been presented.
In this discussion, we describe a side-by-side comparison between a PSII-electrode and a purely synthetic, PSII-inspired water oxidation photoelectrode recently developed in our laboratory. The semi-biological photoanode is composed of PSII isolated from the thermophilic cyanobacterium Thermosynechococcus elongatus on a mesoporous indium tin oxide electrode (mesoITO|PSII, Fig. 1a).7 PSII embeds the required functionalities for photo-water oxidation (Scheme 1a) and direct electron transfer is observable from the plastoquinones in PSII to mesoITO, allowing us to use electrochemical methods to study the photocharacteristics of PSII.7
 | ||
| Fig. 1 SEM images of (a) mesoITO and (b) nanoWO3|TiO2|NiOx. The insets in (a) and (b) show photograph images of the mesoITO|PSII and nanoWO3|TiO2|NiOx electrodes, respectively (arrows indicate the exposed working areas). | ||
The PSII-inspired synthetic photoanode consists of a TiO2 and NiOx composite on nanosheet-structured WO3 electrode (nanoWO3|TiO2|NiOx, Fig. 1b). The composite structure of the synthetic electrode allows for partial mimicry of functional key features in PSII: WO3 serves as the visible light harvester, TiO2 acts as a protection and charge separation layer and NiOx behaves as a water oxidation electrocatalyst in borate solution (Scheme 1b).4 NiOx was reported to have a similar cubane-like structure to the CaMn4 OEC of PSII.8 In addition, this synthetic electrode is composed of inexpensive materials and is functional under benign conditions (room temperature and pH 8 to 9 aqueous solution).4 Functional features of the enzymatic and synthetic systems allow us to compare their performance in PEC O2-evolution activity, efficiency and stability.
2. Experimental
Preparation of mesoITO|PSII electrode
MesoITO|PSII was prepared according to literature procedures (Scheme 2a).7a,9MesoITO was loaded onto an ITO-coated glass slide (VisionTek Systems Ltd) by spreading ITO nanoparticles (Aldrich; <50 nm particle size; 27 m2 g−1 surface area; 90% In2O3 and 10% SnO2) with a geometric surface area of 0.25 cm2 (using Scotch® tape (3M) as spacers to control the active surface area and the film thickness), followed by annealing at 450 °C for 30 min.9 PSII dimers were isolated from a thermophilic cyanobacterium, Thermosynechococcus elongatus, and purified following a published protocol.10 To assemble mesoITO|PSII, 2 μL of a 0.54 (mg Chlorophyll a) mL−1 PSII solution was drop-cast onto the mesoITO surface and kept at room temperature in the dark. After 30 min, the PSII-modified mesoITO electrode was used as a working electrode for PEC measurements.7a,9 | ||
| Scheme 2 Schematic representation of the preparation of (a) mesoITO|PSII and (b) nanoWO3|TiO2|NiOx electrodes. The atoms in TiNi are colour-coded as follows (H atoms omitted for clarity): Ni (green), Ti (blue), Cl (orange), O (red), C (grey). | ||
Preparation of nanoWO3|TiO2|NiOx electrode
NanoWO3|TiO2|NiOx was prepared as described previously (Scheme 2b).4 To prepare a WO3 seed layer on a fluoride-doped tin oxide (FTO)-coated glass electrode, a precursor solution containing H2WO4 (0.625 g, 99%; Sigma-Aldrich) and polyvinyl alcohol (0.5 g, 98–99%, medium molecular weight; Alfa Aesar) in H2O2 (10 mL, 30%; Sigma-Aldrich) was spin-coated on the FTO-coated glass (Pilkington; TEC Glass™ 7; sheet resistance 7 ohm sq−1), followed by annealing in air at 500 °C for 2 h. To grow the WO3 nanosheets on the WO3 seed layer-modified FTO-coated glass, a second precursor solution was prepared: a solution of H2WO4 (3 mL of 0.25 M) in H2O2 was added to an aqueous HCl solution (3 mL, 1 M) containing oxalic acid (0.2 g, 99%; Sigma-Aldrich), and then acetonitrile (10 mL, HPLC grade; Fisher Chemicals) was added to this acidic solution. The second precursor solution was put in a 23 mL Teflon-lined stainless steel autoclave (model 4749, Parr). The WO3 seed layer-modified FTO-substrate was immersed vertically into the second precursor solution in the autoclave, whereupon the autoclave was sealed and heated at 180 °C for 2.5 h. After growth of the WO3 nanosheets, the resultant electrodes were rinsed with ethanol and then annealed in air for 1 h at 500 °C.[Ti2(OEt)9(NiCl)]2 (TiNi) was synthesised by heating titanium ethoxide (7.00 mL, 33.4 mmol; 99.99%; Sigma-Aldrich) and nickel(II) chloride (271 mg, 2.09 mmol; >97%; Sigma-Aldrich) in anhydrous ethanol (7.00 mL, 120 mmol) in a Teflon-lined autoclave at 150 °C for 24 h.11 After removal of ethanol, dark yellow crystals of the product were grown from dry toluene at −14 °C. NanoWO3|TiO2|NiOx was assembled by spin coating a fresh solution of TiNi (5 mM in toluene) on nanoWO3. NanoWO3|TiO2|NiOx was dried for at least 30 min in air at room temperature and then washed with water prior to use as a working electrode.
Physical characterisation
The surface morphology of mesoITO|PSII and nanoWO3|TiO2|NiOx was characterised with a Philips XL30-SFGE scanning electron microscope (SEM). UV-vis absorption spectra of the electrodes were recorded on a Varian Cary 50 UV-vis spectrophotometer equipped with an energy diffuse reflectance accessory (Barrelino™). The amount of PSII adsorbed on mesoITO was estimated by scraping off the PSII-modified ITO nanoparticles from the ITO substrate, suspending them in MeOH, centrifuging the suspension and analysing the supernatant by UV-vis spectrophotometry.7a The concentration of Chlorophyll a (Chl a) in the supernatant was determined using an extinction coefficient equal to 79.95 (Chl a mg)−1 mL cm−1 at 665 nm. The amount of PSII on mesoITO was calculated to be approximately 21.2 pmol cm−2 assuming that the PSII dimer contains 35 Chl a molecules.3a This PSII loading allowed us to calculate a turnover frequency (TOF) of 0.03 (mol O2) (mol PSII)−1 s−1 for mesoITO|PSII based on photocurrents obtained at 1.23 V vs. RHE with four electrons per O2 molecule.PEC measurements
PEC measurements were recorded with an Ivium CompactStat potentiostat using a conventional three-electrode system. MesoITO|PSII (exposed projected geometrical area of 0.25 cm2) or nanoWO3|TiO2|NiOx (exposed projected geometrical area of 0.5 cm2) were used as the working electrodes connected to a Pt foil counter electrode and a Ag/AgCl/KCl(sat) reference electrode. All the potentials were converted to the reversible hydrogen electrode (RHE) by using the following equation: E(V vs. RHE) = E(V vs. Ag/AgCl) + 0.197 + 0.059 × pH.12 An aqueous buffered solution (pH 6.5) containing 50 mM KCl, 15 mM CaCl2, 15 mM MgCl2, and 40 mM 2-(N-morpholino)ethanesulfonic acid (MES) was used for the mesoITO|PSII system, whereas the photocurrent responses of nanoWO3|TiO2|NiOx were measured in a potassium borate buffer solution (Bi, pH 9.2). A solar light simulator (Newport Oriel, Xenon 150 W) was used as a light source unless otherwise noted. The light intensity was calibrated to 100 mW cm−2 (1 sun). An air mass 1.5 global (AM 1.5G) filter and an IR water filter were used. A 590 nm cut-off filter (UQG Optics) was used for all PEC experiments with mesoITO|PSII to avoid photoexcitation of ITO. Representative transient photocurrent responses at different potentials of mesoITO|PSII and nanoWO3|TiO2|NiOx with two cycles of a dark period followed by irradiation for 20 s each are shown in Fig. 2a and b, respectively. | ||
| Fig. 2 Representative transient photocurrent response of (a) mesoITO|PSII and (b) nanoWO3|TiO2|NiOx under standardised solar light irradiation (AM 1.5G; ON) and in the dark (OFF) at different applied potentials: (i) 0.54, (ii) 0.64, (iii) 0.74, (iv) 0.84, (v) 0.94, (vi) 1.04 V, (vii), 1.14, (viii), 1.24, (ix) 1.34 and (x) 1.44 V vs. RHE. Measurements with mesoITO|PSII were recorded in an aqueous pH 6.5 electrolyte solution, whereas nanoWO3|TiO2|NiOx was employed in an aqueous Bi buffer (0.1 M, pH 9.2). | ||
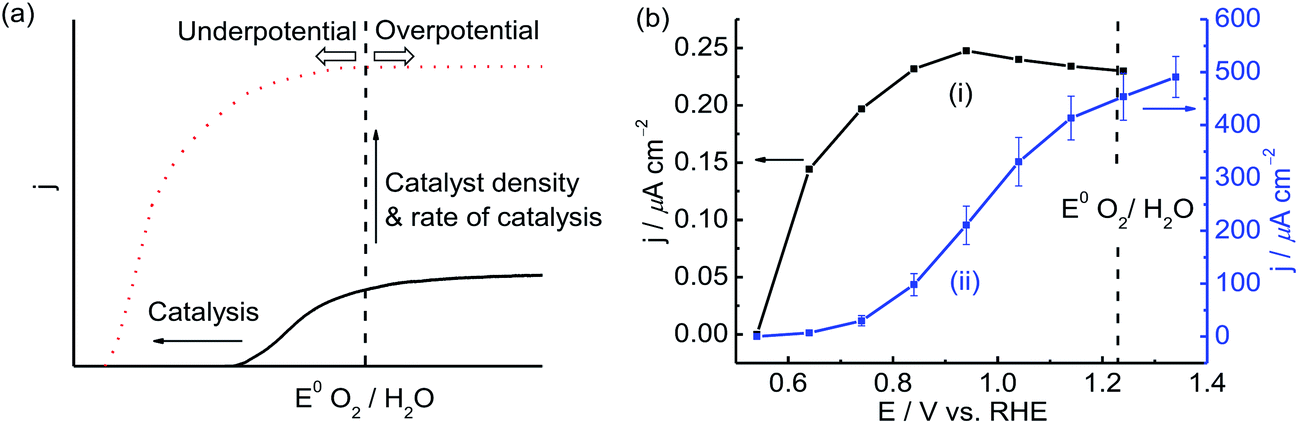 | ||
| Fig. 3 (a) Performance of an idealised photoanode compared with a photoanode deviated from its ideal case. The solid arrows indicate the parameters that need to be addressed to improve the photoanode. (b) Photocurrent responses at various potentials of a (i) mesoITO|PSII electrode and (ii) nanoWO3|TiO2|NiOx under standardised solar light irradiation (AM 1.5G, 100 mW cm−2).2,4 A 590 nm cut-off filter was used for mesoITO|PSII. Measurements with electrode (i) were recorded in an aqueous pH 6.5 MES buffer solution and electrode (ii) were recorded in an aqueous Bi buffer (0.1 M, pH 9.2). The standard deviation for measurements with the mesoITO|PSII electrode (i) was approximately 20%. E(V vs. NHE) = E(V vs. RHE) − 0.059 × pH.12 | ||
Photon to current conversion efficiency measurements
A 300 W Xenon lamp coupled to an MSH300 monochromator (LOT Quantum design) was used for IPCE measurements. The light intensity for the IPCE measurements was measured as a function of wavelength with a photodetector (SEL033/F/QNDS1/W) and power meter (ILT1400). The recorded photocurrents and light intensities at various wavelengths were used to calculate IPCE (η) according to the following equation: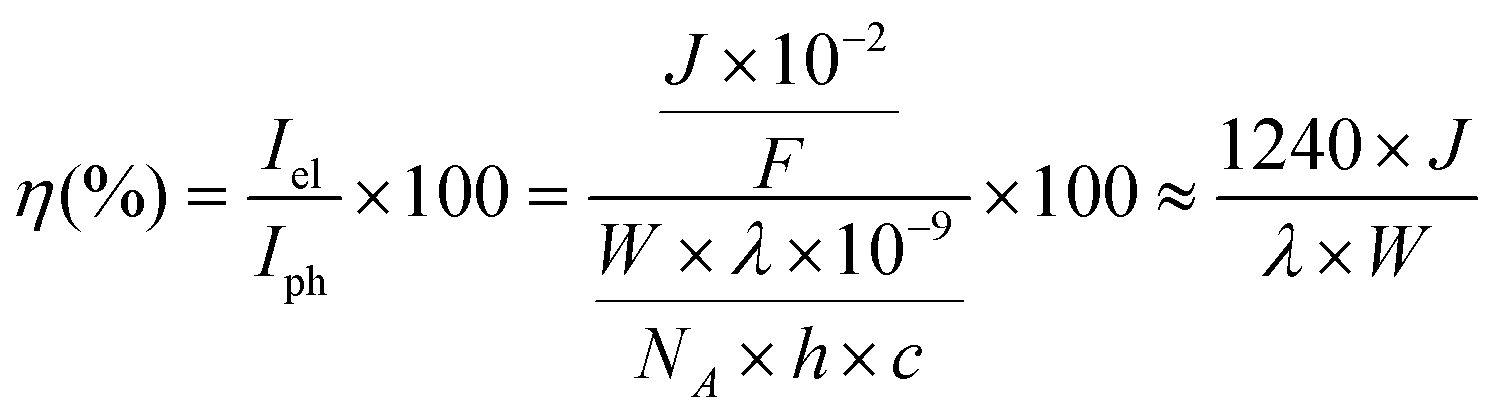 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 484 A s mol−1), λ is the wavelength of light (nm), W is the incident power of the monochromated light (W m−2), NA is Avogadro's number (6.022 × 1023 mol−1), h is Planck's constant (6.626 × 10−34 J s) and c is the speed of light (2.998 × 108 m s−1).
484 A s mol−1), λ is the wavelength of light (nm), W is the incident power of the monochromated light (W m−2), NA is Avogadro's number (6.022 × 1023 mol−1), h is Planck's constant (6.626 × 10−34 J s) and c is the speed of light (2.998 × 108 m s−1).
Absorbed photon to current conversion efficiency (APCE) was obtained by dividing the IPCE by the light harvesting efficiency (LHE) at each wavelength:13
| APCE (%) = IPCE (%)/LHE | (2) |
| LHE = 1 − 10−A(λ) | (3) |
3 Results and discussion
Surface morphologies of mesoITO|PSII and nanoWO3|TiO2|NiOx
MesoITO has a pore diameter of up to 100 nm (Fig. 1a) and therefore provides a high surface area to integrate PSII (approximately 20.5 × 10.5 × 11.0 nm3 for the PSII dimer).3aMesoITO|PSII was assembled by drop-casting the PSII solution on the mesoITO electrode surface (geometric active surface area: 0.25 cm2, inset of Fig. 1a).NanoWO3|TiO2|NiOx was studied as a PSII-inspired synthetic photoanode. This synthetic photoanode consists of green WO3 nanosheets coated with a uniform composite film containing TiO2 and NiOx nanoparticles (Fig. 1b). WO3 is an n-type semiconductor with a suitable band gap (approximately 2.7 eV) and is capable of photo-oxidising water with blue-end visible light (valence band potential at approximately 3 V vs. RHE, Scheme 1b). The nanosheet structure of WO3 gives a high surface area for light absorption, electrocatalyst loading and a decreased hole diffusion length in the photoanode.
A uniform decoration with Ti and Ni containing nanoparticles on WO3 can be obtained by spin-coating the molecular TiNi precursor solution onto nanoWO3 and subsequently hydrolysing the metal-oxide precursor.4 TiNi thereby serves as a single source precursor for the formation of TiO2 and NiOx on nanoWO3. The TiO2 layer protects WO3 from the alkaline electrolyte solution and supports charge separation by receiving holes from the photoexcited nanoWO3. The NiOx is assembled in situ by photo-generated holes in borate buffer (Bi) and promotes water oxidation catalysis. We note that at least some NiOx is likely to be in close contact with WO3 and direct hole transfer from WO3 to NiOx is therefore also possible (Scheme 1b).
PEC characteristics
Fig. 3a shows a schematic J–V curve of an efficient (red dotted line) and inefficient photoanode (black solid line). First, we will discuss (i) the onset potential, (ii) the potential required for reaching plateau current (‘saturation photocurrent’) and (iii) the plateau current as parameters to characterise the photoelectrodes and compare it to an ideal photoanode. An optimal semiconductor photoanode should have an onset potential close to the conduction band position of WO3 during illumination, whereas we would expect an ideal PSII-based electrode to display photocurrents at slightly more positive potentials than the QB redox potential. At the same time the saturation photocurrent should readily be reached well within the “underpotential” region (at more negative potential than the thermodynamic water oxidation).In practice however, the onset and saturation potential are shifted to considerably more anodic potentials due to slow catalysis and charge recombination events at the photoanode. The PEC responses of mesoITO|PSII and nanoWO3|TiO2|NiOx were recorded at different potentials under standardised solar light irradiation. A 590 nm long-pass filter was used for mesoITO|PSII to prevent photoexcitation of ITO. The mean values of the net photocurrent density (J) at specific potentials were obtained by subtracting peak values of the photocurrent density from the background.
The redox potential of plastoquinone QA and QB is approximately 0.3 V vs. RHE (∼−0.1 V vs. NHE in pH neutral environment).14MesoITO|PSII was studied in a pH 6.5 MES buffer solution (in the absence of soluble redox mediator) and has an onset potential of 0.60 V vs. RHE during irradiation, which is approximately 0.3 V more anodic than its theoretical onset potential. We note that direct electron transfer is required from the plastoquinones to ITO for this semi-biological electrode. Coupling of the plastoquinones to the ITO surface is likely to be non-ideal, which would explain the anodic shift of the onset potential. The saturation photocurrent of approximately 0.25 μA cm−2 was reached close to the onset potential at 0.84 V vs. RHE (trace i, Fig. 3b). This small difference between the onset potential and the saturation photocurrent potential may be explained by the efficient charge separation and facile kinetics for water oxidation in PSII (Scheme 1a).
The conduction band of WO3 is situated at approximately 0.3 V vs. RHE,15 which is similar to the redox potential of QA and QB in PSII. Without modification of the surface with TiO2 and NiOx, the onset potential of nanoWO3 is 0.74 V vs. RHE, which is notably larger than the onset potential of mesoITO|PSII due to poor catalysis.4 Interfacing TiO2 and NiOx on nanoWO3 reduces the onset potential to 0.64 V vs. RHE, a potential which is comparable with mesoITO|PSII. A significant difference in the bias potential required to reach the plateau photocurrent is observed between the two electrode systems. A saturation photocurrent of 500 μA cm−2 is reached at 1.34 V vs. RHE with nanoWO3|TiO2|NiOx compared to 0.84 V vs. RHE for mesoITO|PSII (Fig. 3b). TiO2 and NiOx offer only a partial offset for the poor charge recombination kinetics and water oxidation catalytic activity of nanoWO3. The plateau photocurrent density is dominated by the amount of the photogenerated holes reaching the surface and oxidising water. Enhancing the catalyst density on the electrode (the amount of photocatalyst based on the geometrical surface area), using a more efficient catalyst or reducing charge recombination within the photoanode increases the photocurrent.1b,5,13,16MesoITO|PSII therefore displays more efficient catalysis and suffers from less charge recombination below 1.23 V vs. RHE (the underpotential region relative to E°(H2/H2O)). PSII has an evolutionarily well-developed machinery for the photo-oxidation of water and immobilised PSII retains these features and behaves more like an ideal photoanode compared to the synthetic electrode. The amount of immobilised PSII on mesoITO is only 21.2 pmol cm−2 due to the large geometrical footprint of the enzyme photocatalyst, whereas 0.78 μmol cm−2 of NiOx is deposited on nanoWO3 (Table 1). Taking into account the photocurrent density, a much higher single-site catalytic activity was observed for the OEC in PSII with a turnover frequency (TOF) of approximately 0.03 s−1 at 1.23 V compared to 8 × 10−4 s−1 at 1.23 V vs. RHE for NiOx in the synthetic system (Table 1).4 On the other hand, the low PSII loading dramatically limits the photocurrent density with mesoITO|PSII and a three-order of magnitude higher photocurrent density per geometric surface area is observed with nanoWO3|TiO2|NiOx.
| Systema | Onset potential, EO (V vs. RHE) | ΔEO–Pb (V) | J max (μA cm−2) | TOFd(s−1) | Stability, t1/2e(min) | IPCEmaxf (%) | APCEmaxf (%) |
|---|---|---|---|---|---|---|---|
| a MesoITO|PSII was employed as working electrode in a pH 6.5 MES solution. NanoWO3|TiO2|NiOx was studied in an aqueous pH 9.2 Bi buffer (0.1 M). Both electrodes were studied at room temperature with a Pt counter and a Ag/AgCl/KCl(sat) reference electrode. b Potential difference between onset potential (EO) and the potential required to reach the plateau photocurrent (EP). c Saturation photocurrent. d TOF [in mol O2 (mol catalyst)−1 s−1] calculated based on photocurrents obtained at 1.23 V vs. RHE. For PSII, 100% Faradaic efficiency was assumed. For NiOx, the TOF was calculated based on evolved O2 gas.4 e Calculated at an applied potential of 0.94 V vs. RHE under solar light irradiation. f Recorded at an applied potential of 0.94 V vs. RHE (680 nm for mesoITO|PSII and 375 nm for nanoWO3|TiO2|NiOx). | |||||||
| mesoITO|PSII | 0.60 | 0.2 | 0.3 | 0.03 | ∼4 | 0.125 | 0.70 |
| nanoWO3|TiO2|NiOx | 0.64 | 0.7 | 500 | 8 × 10−4 | ∼240 | 50 | 80 |
The electronic absorption spectra in Fig. 4 reveal another key difference between the electrodes. MesoITO|PSII generates O2 during red light irradiation,10b,17 whereas WO3 is only capable of absorbing photons in the blue and UV regions of the solar spectrum. The synthetic electrode therefore requires substantially more energetic photons to oxidise water. The IPCE spectra show a good agreement with the respective absorption spectra for both electrode systems. IPCE is the measure of the ratio of the photocurrent versus the rate of incident photons as a function of wavelength (eqn (1)). IPCE thereby confirms that the photocurrent of mesoITO|PSII is the result of red light excitation (Fig. 4a), whereas the photocurrent of nanoWO3|TiO2|NiOx is generated by blue and UV light (Fig. 4b).
 | ||
| Fig. 4 Diffuse reflectance UV-vis absorption and IPCE spectra of (a) mesoITO|PSII and (b) nanoWO3|TiO2|NiOx. IPCE spectra were recorded at an applied potential of 0.94 V vs. RHE. Measurements were recorded in an aqueous pH 6.5 MES buffer with mesoITO|PSII and in an aqueous pH 9.2 Bi buffer with nanoWO3|TiO2|NiOx. Insets show the corresponding LHE. | ||
An optimum IPCE of PSII on mesoITO of 0.125% was obtained at 680 nm, corresponding to the estimated excited-state energy of P680 (1.825 eV, E680). For any photon absorbed by PSII, the energy driving the photo-water oxidation is equal to E680. The APCE can be readily obtained from the IPCE and the LHE by using eqn (2). LHE quantifies the absorbance of monochromatic light by the electrodes as a function of absorption coefficient (eqn (3)). The LHE of PSII on mesoITO at 680 nm is approximately 17%, and the corresponding APCE is 0.70% (Fig. 4 and Table 1). The low quantum efficiency of mesoITO|PSII presumably stems from the low loading of PSII and the random orientation of PSII on the mesoITO electrode, hindering the injection of electrons from PSII to the mesoITO.7 However, the APCE of mesoITO|PSII is comparable with the APCE of PSII on a Au substrate.18 In contrast, nanoWO3|TiO2|NiOx can only use high-energy photons provided by solar light to promote electron–hole pair separation. NanoWO3|TiO2|NiOx has a maximum IPCE and APCE of 50% and 80% at 375 nm at an applied potential of 0.94 V vs. RHE, respectively. The IPCE drops to zero at wavelengths longer than 465 nm, which is consistent with the band gap of WO3 (2.7 eV, EWO3). Thus, more light energy input (EWO3 − E680 = 0.9 eV) is required to photo-oxidise water and to promote electrons at approximately 0.64 V vs. RHE with the synthetic hybrid electrode. The nanoWO3|TiO2|NiOx electrode requires an additional energy input to compensate for less efficient water oxidation by NiOx compared to CaMn4 OEC in PSII.
Photostability
From a practical standpoint, the long-term photostability of the electrodes in aqueous solution is an important criterion. MesoITO|PSII has a half-life time of approximately 4 min and complete loss of activity is observed within 30 min at an applied potential of 0.94 V vs. RHE (Fig. 5a and Table 1). The short lifetime of mesoITO|PSII can be attributed to the intrinsic photo-instability of PSII in the presence of intense light irradiation (with no repair machinery being available in isolated PSII) as well as the fragility of the PSII-ITO interface (film loss of PSII). Under strong light irradiation, over-accumulation of excited states leads to excessive charge generation in PSII and results in the generation of Chl a triplet states. These react with triplet oxygen to produce singlet oxygen, which is understood to be one of the reasons for the enhanced damage of PSII.19 | ||
| Fig. 5 Chronoamperometric measurements at 0.94 V vs. RHE. Photocurrent profiles of (a) mesoITO|PSII and (b) nanoWO3|TiO2|NiOx under standardised solar light irradiation (AM 1.5G, 100 mW cm−2) are shown. Inset of (b) shows the extended stability trace of nanoWO3|TiO2|NiOx. The activity of the mesoITO|PSII electrode was recorded in a pH 6.5 MES electrolyte solution and the nanoWO3|TiO2|NiOx electrode was recorded in a pH 9.2 Bi buffer solution (0.1 M). | ||
The synthetic nanoWO3|TiO2|NiOx displays much greater photostability. A half-life time of approximately four hours was observed during solar light irradiation, and a complete loss of activity was only observed after 24 h (Fig. 5b and Table 1). Bare WO3 is known to be soluble in alkaline solution and easily damaged by side reactions during photo-O2 evolution.4,20 However, the TiO2 and NiOx coatings stabilise WO3 by acting as a protective layer and an electrocatalyst, respectively.
Comparison of the semi-biological and synthetic electrode
MesoITO|PSII shows more efficient ‘single-site’ catalysis and superior resistance to charge recombination than nanoWO3|TiO2|NiOx. This is evident from the higher TOF of PSII and a 0.5 V more cathodic potential to reach the photocurrent plateau for mesoITO|PSII (Table 1). In this respect, the PEC response of mesoITO|PSII shown in Fig. 3 is closer to an “ideal” photoanode. Moreover, PSII is capable of photo-oxidising water with low energy “red photons”, whereas WO3 requires higher energy photons to compensate for significantly higher charge recombination and less efficient NiOx OEC. Interfacing nanoWO3|TiO2|NiOx with another more efficient co-catalyst might accelerate the water oxidation kinetics and alleviate the surface recombination in the low bias region.Conversely, nanoWO3|TiO2|NiOx has a clear advantage in terms of photocurrent density per geometrical surface area due to its higher catalyst density compared to mesoITO|PSII, which is important for applications. Although nanoWO3|TiO2|NiOx displays a higher photocurrent density than mesoITO|PSII, the photocurrent is still much lower than its theoretical photocurrent (approximately 5 mA cm−2).21 To boost further the photocurrent plateau, charge recombination within the nanoWO3|TiO2|NiOx must be minimised. Combination with another semiconductor to form a p–n junction,22 increasing intrinsic conductivity by elemental doping,23 and suppression of electron back injection with an oxide underlayer24 are possible strategies to enhance further the photocurrent of this synthetic electrode.
Inorganic semiconductors offer a more robust approach to provide the oxidising potentials necessary to photo-oxidise water than semi-biological systems. In this study, WO3 was interfaced with the TiO2 and NiOx composite by a simple and scalable deposition of a molecular precursor to improve its photo-activity and stability. NanoWO3|TiO2|NiOx is considerably more stable than mesoITO|PSII at photo-oxidative conditions (Fig. 5 and Table 1).
4 Conclusions
An enzymatic photoanode consisting of PSII immobilised on mesoITO and a synthetic photoanode consisting of a TiO2 and NiOx composite on a WO3 semiconductor operational under near pH-neutral conditions have been discussed. NanoWO3|TiO2|NiOx mimics the key functionalities of PSII, namely, light absorption, charge separation and water oxidation catalysis. As expected, the highly evolved PSII displays a higher turnover frequency than NiOx on nanoWO3|TiO2|NiOx. Bulk and surface charge recombination is currently a major hurdle in achieving high photon-to-current conversion efficiencies. The resistance to charge recombination of PSII is evident from the lower applied potential required to reach its saturation photocurrent in the mesoITO|PSII system compared with nanoWO3|TiO2|NiOx. Thus the enzymatic system exhibits less surface charge recombination events at a low overpotential region compared with the synthetic system. In this respect, mesoITO|PSII behaves more like an “ideal” photocatalyst. The IPCE spectra confirm that PSII has the ability to promote electron transfer with low energy photons (680 nm), whereas WO3 is only capable of utilising high-energy photons (<465 nm) to achieve water oxidation at a similar onset potential.Nevertheless, the nanoWO3|TiO2|NiOx electrode displays higher IPCEs and a more than three orders of magnitude higher photocurrent density than mesoITO|PSII due to the high NiOx catalyst density on the synthetic electrode compared to the low PSII loading. Another distinct advantage of the synthetic over the enzymatic photoanode is the greater photo-stability under solar light irradiation due to the lack of repair machinery in the isolated PSII. In summary, our synthetic system will show promise for applications in water splitting in the long-term if important lessons from PSII can be learnt: longer wavelength absorption will boost the overall solar energy conversion efficiency and saturation currents at a more negative potential decrease heat losses in an operating device.
Acknowledgements
Financial support from EPSRC (EP/H00338X/2 to E. R. and nanoDTC to D. M.), the Christian Doppler Research Association (Austrian Federal Ministry of Science, Research and Economy and National Foundation for Research, Technology and Development), the OMV Group (to E. R.) and Cambridge Trust (to Y.-H. L.) are gratefully acknowledged. We thank Mr Timothy C. King and Prof. Dominic S. Wright for a kind gift of [Ti2(OEt)9(NiCl)]2. Dr Tanai Cardona and Prof. A. William Rutherford provided us with a sample of photosystem II. We also thank Dr Hyun S. Park, Miss Claire Wombwell and Prof. Rutherford for valuable comments on the manuscript.References
- (a) Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511 CrossRef CAS; (b) S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645 CrossRef CAS PubMed; (c) C.-Y. Lin, Y.-H. Lai, D. Mersch and E. Reisner, Chem. Sci., 2012, 3, 3482 RSC; (d) L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng, Q. Yu, X. Cai, J. Zhao, Z. Ren, H. Fang, F. Robles-Hernandez, S. Baldelli and J. Bao, Nat. Nanotechnol., 2014, 9, 69 CrossRef CAS PubMed; (e) J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407 CrossRef CAS; (f) W. D. Chemelewski, H.-C. Lee, J.-F. Lin, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 2843 CrossRef CAS PubMed; (g) P. M. Rao, L. Cai, C. Liu, I. S. Cho, C. H. Lee, J. M. Weisse, P. Yang and X. Zheng, Nano Lett., 2014, 14, 1099 CrossRef CAS PubMed; (h) B. M. Klepser and B. M. Bartlett, J. Am. Chem. Soc., 2014, 136, 1694 CrossRef CAS PubMed; (i) C.-Y. Lin, D. Mersch, D. A. Jefferson and E. Reisner, Chem. Sci., 2014, 5, 4906 RSC.
- M. Kato, J. Z. Zhang, N. Paul and E. Reisner, Chem. Soc. Rev., 2014, 43, 6485 RSC.
- (a) K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831 CrossRef CAS PubMed; (b) H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861 CrossRef CAS PubMed; (c) Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55 CrossRef CAS PubMed; (d) T. Cardona, A. Sedoud, N. Cox and A. W. Rutherford, Biochim. Biophys. Acta, 2012, 1817, 26 CrossRef CAS PubMed.
- (a) Y.-H. Lai, T. C. King, D. S. Wright and E. Reisner, Chem.–Eur. J., 2013, 19, 12943 CrossRef CAS PubMed; (b) Y.-H. Lai, C.-Y. Lin, Y. Lv, T. C. King, A Steiner, N. M. Muresan, L. Gan, D. S. Wright and E. Reisner, Chem. Commun., 2013, 49, 4331 RSC.
- (a) S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6405 CrossRef CAS PubMed; (b) W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926 CrossRef CAS PubMed.
- (a) Y. Jiang, F. Li, B. Zhang, X. Li, X. Wang, F. Huang and L. Sun, Angew. Chem., Int. Ed., 2013, 52, 3398 CrossRef CAS PubMed; (b) J. D. Megiatto Jr, A. Antoniuk-Pablant, B. D. Sherman, G. Kodis, M. Gervaldo, T. A. Moore, A. L. Moore and D. Gust, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15578 CrossRef PubMed; (c) M. M. Najafpour, T. Ehrenberg, M. Wiechen and P. Kurz, Angew. Chem., Int. Ed., 2010, 49, 2233 CrossRef CAS PubMed; (d) M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Chem. Sci., 2012, 3, 2330 RSC.
- (a) M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2012, 134, 8332 CrossRef CAS PubMed; (b) M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2013, 135, 10610 CrossRef CAS PubMed.
- M. Risch, K. Klingan, J. Heidkamp, D. Ehrenberg, P. Chernev, I. Zaharieva and H. Dau, Chem. Commun., 2011, 47, 11912 RSC.
- P. G. Hoertz, Z. Chen, C. A. Kent and T. J. Meyer, Inorg. Chem., 2010, 49, 8179 CrossRef CAS PubMed.
- (a) A. Boussac, F. Rappaport, P. Carrier, J. M. Verbavatz, R. Gobin, D. Kirilovsky, A. W. Rutherford and M. Sugiura, J. Biol. Chem., 2004, 279, 22809 CrossRef CAS PubMed; (b) M. Sugiura and Y. Inoue, Plant Cell Physiol., 1999, 40, 1219 CrossRef CAS PubMed.
- S. Eslava, M. McPartlin, R. I. Thomson, J. M. Rawson and D. S. Wright, Inorg. Chem., 2010, 49, 11532 CrossRef CAS PubMed.
- (a) A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., NewYork, 2nd edn, 2001 Search PubMed; (b) A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 15714 CrossRef CAS PubMed.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990 CrossRef CAS PubMed.
- T. Shibamoto, Y. Kato, M. Sugiura and T. Watanabe, Biochemistry, 2009, 48, 10682 CrossRef CAS PubMed.
- R. M. Navarro, M. Consuelo Alvarez-Galvan, J. A. V. de la Mano, S. M. Al-Zahrani and J. L. G. Fierro, Energy Environ. Sci., 2010, 3, 1865 CAS.
- L. Zhang, E. Reisner and J. J. Baumberg, Energy Environ. Sci., 2014, 7, 1402 CAS.
- G. Raszewski, W. Saenger and T. Renger, Biophys. J., 2005, 88, 986 CrossRef CAS PubMed.
- O. Yehezkeli, R. Tel-Vered, J. Wasserman, A. Trifonov, D. Michaeli, R. Nechushtai and I. Willner, Nat. Commun., 2012, 3, 742 CrossRef PubMed.
- A. W. Rutherford, A. Osyczka and F. Rappaport, FEBS Lett., 2012, 586, 603 CrossRef CAS PubMed.
- J. A. Seabold and K.-S. Choi, Chem. Mater., 2011, 23, 1105 CrossRef CAS.
- Z. Chen, T. F. Jaramillo, T. G. Deutsch, A. Kleiman-Shwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske, M. Sunkara, E. W. McFarland, K. Domen, E. L. Miller, J. A. Turner and H. N. Dinh, J. Mater. Res., 2010, 25, 3 CrossRef CAS.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456 CrossRef CAS PubMed.
- A. J. E. Rettie, H. C. Lee, L. G. Marshall, J.-F. Lin, C. Capan, J. Lindemuth, J. S. McCloy, J. Zhou, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2013, 135, 11389 CrossRef CAS PubMed.
- T. Hisatomi, H. Dotan, M. Stefik, K. Sivula, A. Rothschild, M. Grätzel and N. Mathews, Adv. Mater., 2012, 24, 2699 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |