 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparison of carbon materials as electrodes for enzyme electrocatalysis: hydrogenase as a case study
Jonathan
Quinson
*ab,
Ricardo
Hidalgo
b,
Philip A.
Ash
b,
Frank
Dillon
a,
Nicole
Grobert
a and
Kylie A.
Vincent
b
aDepartment of Materials, University of Oxford, Parks Road, Oxford OX1 3PH, UK. E-mail: nicole.grobert@materials.ox.ac.uk
bDepartment of Chemistry, University of Oxford, Inorganic Chemistry Laboratory, South Parks Road, Oxford, OX1 3QR, UK. E-mail: kylie.vincent@chem.ox.ac.uk
First published on 14th August 2014
Abstract
We present a study of electrocatalysis by an enzyme adsorbed on a range of carbon materials, with different size, surface area, morphology and graphitic structure, which are either commercially available or prepared via simple, established protocols. We choose as our model enzyme the hydrogenase I from E. coli (Hyd-1), which is an active catalyst for H2 oxidation, is relatively robust and has been demonstrated in H2 fuel cells and H2-driven chemical synthesis. The carbon materials were characterised according to their surface area, surface morphology and graphitic character, and we use the electrocatalytic H2 oxidation current for Hyd-1 adsorbed on these materials to evaluate their effectiveness as enzyme electrodes. Here, we show that a variety of carbon materials are suitable for adsorbing hydrogenases in an electroactive configuration. This unified study provides insight into selection and design of carbon materials for study of redox enzymes and different applications of enzyme electrocatalysis.
1. Introduction
Carbon materials with a variety of size, surface features, morphology and arrangement of graphitic structure can offer properties relevant for specific electrochemical applications, ranging from carbon fibre microelectrodes up to high surface area carbon blacks used as supports for metal electrocatalysts in fuel-cells.In this paper, we focus on applications of carbon materials as supports for redox enzymes as electrocatalysts. A number of redox enzymes are amenable to immobilisation on carbon materials (either through covalent attachment or direct adsorption) allowing them to be electrochemically investigated over a wide potential range and in various solution conditions.1 The electrode replaces physiological donors and acceptors, and redox enzymes often show high rates of catalysis when immobilised on an electrode.2 This has led to numerous demonstrations of technological applications of bioelectrocatalysis, including enzyme fuel cells3 and electrochemical biosensors.4
Our model enzyme system in this study is a hydrogenase. This class of enzyme allows micro-organisms to oxidise dihydrogen, or reduce protons to generate H2. Hydrogenases possess an electron relay chain of iron–sulphur redox centres which transport electrons between the surface of the protein framework and the buried catalytic centre, making them well equipped to function as electrocatalysts (Fig. 1). It is well established that immobilisation of hydrogenases onto an electrode makes it possible to probe a submonolayer film of the enzyme in a technique known as protein film electrochemistry. This approach has been used, for example, to understand potential-dependent features of hydrogenase electrocatalysis and their reactions with inhibitors and to compare features of hydrogenases from different organisms or with different genetic modifications.5,6
 | ||
| Fig. 1 (a) Representation of the crystal structure of a hydrogenase molecule showing how the enzyme is equipped for electron exchange with an electrode: the protein framework is shown in ‘ribbon’ format in grey, and metal clusters are shown in elemental colours; (b) schematic representation of direct adsorption of a hydrogenase molecule onto a carbon electrode material (exhibiting exposed graphitic carbon edges) giving rise to efficient electrocatalysis. Representations of the hydrogenase structure (PDB code: 3USC) were prepared using The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC. | ||
Pyrolytic graphite has been used most widely in fundamental studies of hydrogenase electrocatalysis, and it is often possible to adsorb enzymes directly without need for specialised attachment strategies, thus simplifying electrode preparation.7 Pyrolytic graphite has ordered graphitic domains, is very soft, and a fresh surface for enzyme adsorption is readily prepared by mild abrasion.8 Hydrogenases and other redox enzymes have been shown to interact preferentially with the rough ‘edge’ surface and at defects on the basal plane of graphitic materials.7 It has been reported that protein adsorption is aided by fresh abrasion of the pyrolytic graphite prior to protein immobilisation1 and this is likely to be due to changes in the chemical composition of the surface on contact with air or water. Hydrogenases have also been adsorbed onto plate-like particles of pyrolytic graphite, prepared freshly by abrading a piece of graphite with glass paper immediately prior to exposure to enzyme.9–11 The preference for fresh preparation of the particles limits batch processing of larger quantities and the large proportion of basal surface on the plate-like particles probably means that the total surface area of pyrolytic graphite particles prepared in this way is not well utilised for enzyme loading.
The efficiency of hydrogenases as electrocatalysts led rapidly to possible applications for the enzymes in biological fuel-cells,12,13 H2-production devices14 and enzyme-mediated chemical synthesis.9,10 Identifying alternative carbon materials has now become an important challenge for developing and up-scaling applications of electrocatalysis by hydrogenases and other redox enzymes.15–17 In order for enzymes to present an attractive alternative to metal catalysts for fuel cells, for example, high surface-area carbon supports are particularly important to facilitate high enzyme loading due to the relatively large footprint for enzymes (typically ca 5 nanometres in diameter).
A number of different carbon materials in addition to pyrolytic graphite have been explored for adsorption of hydrogenases, including carbon filaments,18 carbon powders,17,19 and various types of carbon nanotubes.14,20–23 However these studies involve hydrogenases from many different organisms and there has not been a systematic study comparing electrocatalysis by the same enzyme on a range of carbon materials with different properties.24 This work is an attempt to explore and compare various carbon materials for hydrogenase electrocatalysis. The particular enzyme we select is hydrogenase-1 (Hyd-1) from the common bacterium Escherichia (E.) coli, a robust O2-tolerant enzyme already demonstrated for application in fuel cell devices.17
In this study we select and characterise eleven readily obtainable carbon-based materials displaying different shape, size, graphitic features, porosity and surface area, and we assess these as supports for H2-oxidation electrocatalysis by Hyd-1. Due to the success demonstrated for pyrolytic graphite ‘edge’ surfaces in hydrogenase electrocatalysis, we selected several particle types with ordered graphitic character. Pyrolytic graphite was abraded with glass paper to produce plate-like particles (APG), already demonstrated in hydrogenase applications,9 and micron-sized graphite particles (MG) were prepared by sonication of commercial graphite flakes to generate a material with similar graphitic character to the APG but with higher surface area and increased proportion of ‘edge’ sites. Multi-wall carbon nanotubes (MWCNT) were selected because they have been previously suggested as conductive networks for high surface area electrodes for hydrogenase immobilisation.25,26 Nitrogen-doped multi-wall carbon nanotubes (N-MWCNT) were also tested because alteration of the carbon lattice by nitrogen doping confers defects and heteroatom sites that may improve hydrophilicity and provide anchor sites for protein attachment. Graphite platelet nanofibers (GNF) from Sigma-Aldrich, consisting of stacked graphitic layers perpendicular to the fibre axis (rather than parallel to the tube as observed in MWCNT) were selected due to the prevalence of graphitic ‘edge’ surface. In order to assess the suitability of amorphous carbon materials, we selected four commercially-available carbon blacks already used in fuel cell catalysis:27 Mogul L (ML), Vulcan XC72R (VX) and Black Pearls 2000 (BP) from Cabot Corporation and carbon nanopowder (CNP) from Sigma-Aldrich. In addition to the commercially available amorphous carbon materials, carbon particles prepared by hydrothermal synthesis from a glucose precursor28 (HNP) were tested because this versatile and well established synthetic approach can be adapted readily for various carbon-based materials and can therefore be tuned for specific applications. To test the applicability of composite materials that could lead to specialised applications, particles with a thin carbon shell surrounding a silicon core were prepared by polymerisation followed by carbonisation (Si@C).29
Here we focus on direct adsorption of the enzyme Hyd-1 onto these raw carbon supports. It may be possible to achieve further improvements to enzyme loading by additional treatments to the materials16,22,26 but in the present study we limit our investigation to as-received or as-synthesised carbon materials in order to identify readily available or easily produced materials for hydrogenase applications and study.
2. Experimental
2.1 Commercially available carbon materials: ML, VX, BP, CNP and GNF
Carbon black powders: Mogul L (ML), Vulcan XC72R (VX) and Black Pearls 2000 (BP) were obtained from Cabot Corporation and used as received. Carbon nanopowder (less than 50 nm particle size, referred to as CNP) and graphite platelet nanofibers (GNF) were purchased from Sigma-Aldrich and used as received.2.2 Abraded pyrolytic graphite: APG
Pyrolytic graphite blocks were obtained from Momentive Performance Materials. APG flakes were obtained by abrasion of a block of pyrolytic graphite with glass paper (Norton Abrasives, P400 grade). Contrary to previous methods reported, here APG was not freshly prepared prior to enzyme adsorption.302.3 Micron-sized graphite flakes: MG
Micron-sized (<1 μm) graphite flakes were prepared by modifying a method used to prepare graphene through exfoliation.31 In a typical experiment 10 g of millimeter-sized graphite flakes (Aldrich, +100 mesh) were placed in 500 mL isopropanol (≥99.5%, Sigma-Aldrich) and exfoliated and broken down by sonication in an ultrasonic bath (FB11301, Fisherbrand, 600 W, 45kHz) for 200 hours. The mixture was then left for 12 hours to sediment, and a grey suspension containing smaller graphite flakes (<1 μm) was pipetted off from the remaining bulk graphite flakes (>1 μm) at the bottom of the flask and sedimented by centrifugation at 3100 × g (Hettich, Rotina 420R). After centrifugation the supernatant was decanted and the precipitated graphite flakes that were typically less than 1 μm in size were then collected and the solvent was left to evaporate overnight at 80 °C leading to a powder comprising micron-sized graphite flakes (MG) in yield of 0.5–1 g.2.4 Multi-wall carbon nanotube synthesis: MWCNT and N-MWCNT
Multi-wall carbon nanotubes (MWCNT) and nitrogen doped multi-wall carbon nanotubes (N-MWCNT) were synthesised by aerosol-assisted chemical vapour deposition (AACVD) as reported in detail previously.32 Briefly a mixture of 5 wt % ferrocene (98%, Aldrich, purified through sublimation at 90 °C prior to use) and toluene (99.9%, Fluka) for MWCNT, or benzylamine (≥99%, Fluka) for N-MWCNT were injected (for 10 and 20 minutes respectively) into a quartz tube located inside a tube furnace operated at 800 °C using argon (99.999%, BOC) at a flow of 2500 sccm (standard cubic centimetres per minute). The furnace was then left to cool down to <300 °C before the samples of ca 500 mg per experiment were collected from the centre of the quartz tube. Both MWCNT and N-MWCNT contained traces of iron impurities (typically 5 wt %)33 but were used as-synthesised since these impurities were found not to show any electroactivity over the potential range relevant for studies of hydrogenase.2.5 Hydrothermal carbon particle synthesis: HNP
Carbon particles with a size range 50–200 nm were obtained by hydrothermal synthesis, based upon the procedure of Sun et al.28 In a typical experiment 1.27 g glucose (ACS reagent grade, Sigma-Aldrich) was added to distilled water (35 mL) in a PTFE-lined acid digestion vessel (45 mL, Parr Instrument Company) and the closed vessel was then placed into an oven at 180 °C for 8 hours. Parameters such as time, glucose concentration and temperature were selected to optimise the carbon particle size (100–120 nm) and overall yield. The oven was then left to cool down overnight before the sample consisting of the particles was collected from the vessel. The particles were washed and centrifuged at least 5 times using distilled water and 5 times with ethanol (99.8%, Fisher Scientific) for 10 to 15 minutes at 3100 × g per cycle. Subsequently, the particles were left to dry overnight at 80 °C. Typical carbon particle yields were ca 100 mg. In line with previous reports,34,35 we observed that the electrochemical response of materials prepared by this route can be improved by annealing the particles at 1000 °C in argon (100 sccm) for 2 hours.2.6 Silicon core–carbon shell nanoparticles: Si@C
Silicon core–carbon shell nanoparticles (Si@C) were generated through a polymerisation and carbonisation procedure reported by Gao et al.29 Typically 44 mg 4,4′-sulfonyldiphenol (98%, Aldrich), 20 mg phosphonitrilic chloride trimer (99.95%, Aldrich) and 20 mg silicon nanopowder (≥98%, Aldrich) with a particle diameter of ca 100 nm were dispersed using an ultrasonic bath (U1250D from Ultrawave, 200 W, 40 kHz) for 20 hours in 30 mL ethanol (99.8%, Fisher Scientific) and tetrahydrofuran (anhydrous, ≥99.9%, Sigma-Aldrich) in a 1:1 mixture by volume to which 2 mL triethylamine (≥99%, Sigma-Aldrich) had been added. The resulting silicon nanoparticles with a poly(cyclotriphosphazene-4,4′-sulfonildiphenol) shell were washed 3 times with ethanol:tetrahydrofuran solution and left to dry at 80 °C overnight. The particles were then annealed under an argon atmosphere (100 sccm) at 900 °C for 2 hours to form the carbon shell.2.7 Characterisation of materials
For the eleven carbon materials reported here, we investigate the following criteria: the size and shape of the materials, their graphitic structure, their surface area and their mesoporosity (porosity on a scale relevant for enzyme adsorption).The overall morphology and structure of all materials presented here was assessed using scanning and transmission electron microscopy (SEM and TEM) in conjunction with Raman spectroscopy. Specific surface area and porosity were investigated by nitrogen adsorption measurements.
SEM images were recorded using a Jeol 840F microscope with a secondary electron detector operated at 5 kV and a Zeiss NVision FIB microscope equipped with an in-lens detector and operated at 5 kV. TEM images were taken with a Jeol 2000FX and a Jeol 2010 both operated at 200 kV.
The degree of graphitisation of the samples was investigated using a JY Horiba Labram Aramis imaging confocal Raman microscope (532 nm excitation) whereby the ratio of peak intensities of the D band (around 1340 cm−1) and G band (around 1575 cm−1) (ID/IG), and the presence or absence of 2D peak (also called G′, around 2675 cm−1) were identified. In simple terms, for carbon materials the D peak intensity correlates with defects in the carbon lattice and is linked to the extent of sp3 hybridisation while the G peak arises from the graphitic network and extent of sp2 hybridisation. If ID/IG is smaller than 1 the structures are considered to show a higher concentration of sp2 hybridisation (i.e. extended graphitic character). If the ratio is close to 1 then the material is considered to have a more amorphous structure with a mixture of sp2 and sp3 hybridisation. Even in cases where ID/IG is close to 1, the presence of a 2D peak, more related to interactions between graphene-like planes, suggests a strong graphitic component.
Specific surface areas of samples were evaluated using a Micromeritics Gemini V BET surface area analyser (nitrogen adsorption at 77 K). For data collection the samples were left at 200 °C in dinitrogen overnight prior to the adsorption measurements and the data were analysed using the BET model for relative pressure between 0.05 and 0.3 for all samples. For BP, HNP and Si@C the Langmuir model was more suitable as it gave a better fit to the data, therefore for these materials we calculate both the BET and Langmuir surface areas. The molecular cross sectional area of dinitrogen was taken as 0.1620 nm2. The Barrett–Joyner–Halenda (BJH) model was used to estimate pore characteristics.36
2.8 Adsorption of hydrogenase
The enzyme Hyd-1 was purified from E. coli following a published procedure37 and used at a concentration of 20 μM unless otherwise specified. Each of the 11 carbon materials was dispersed at 20 mg mL−1 by sonication in ultra-high purity water (Millipore MilliQ, 18 MΩ cm). Adsorption of Hyd-1 onto the carbon materials was carried out in a N2-filled glove box (<1 ppm O2, Glove Box Technology Ltd.). A 5 μL aliquot of Hyd-1 solution was added to 5 μL of each material dispersion and the mixture was left overnight at 4 °C to allow Hyd-1 to adsorb. Carbon materials were then washed 3 times with 20 μL potassium phosphate buffer (100 mM, pH 6.0) prepared from potassium phosphate dibasic (≥98%, Sigma-Aldrich) and monobasic (≥99%, Sigma). The enzyme-modified carbon materials were then separated by centrifugation (7000 × g, MiniSpin, Eppendorf). To each sample, 5 μL buffer was added.2.9 Electrocatalytic activity of hydrogenase adsorbed on the different particles
The electrocatalytic activity of adsorbed hydrogenase was assessed in an anaerobic glove box using a water jacketed (25 °C) electrochemical cell sealed onto an electrode rotator (EG&G model 636) with a rotating disc electrode (RDE). A gas inlet and outlet in the cell make it possible to flow 1 bar H2 (99.99%, BOC) at 1000 sccm through the headspace above the cell solution. Measurements were performed with the electrode rotating at 2000 rotations per minute (rpm) unless otherwise stated, with steps to higher rotation rates used to test for mass transport limitation. The working electrode was prepared by drop-casting 0.5 or 1 μL (as specified) of the various material dispersions with adsorbed Hyd-1 onto the surface of a pyrolytic graphite ‘edge’ electrode of geometrical area 0.02 or 0.04 cm2 which had been first polished with 1 μm α-alumina powder and cleaned by sonication in MilliQ water for at least one minute. For each material, an electrode prepared without Hyd-1 showed no Faradaic current response in the presence of H2 over the potential window used for experiments in this study.The counter electrode was a platinum wire and the reference electrode a saturated calomel electrode (SCE, BAS). Potentials were converted to volts (V) against the standard hydrogen electrode (SHE) using the relation E(SHE) = E(SCE) + 0.2412 V at 25 °C. Potentials were applied with an EcoChemie Autolab PGSTAT 128N potentiostat controlled by GPES software. The hydrogenase was activated for at least 5 minutes at −0.559 V vs SHE prior to measurements and further activated by sequences of 5 minutes at this same potential if the current recorded was noticed to increase during cyclic voltammetry measurements indicative of incomplete hydrogenase activation.30 Scans were acquired at 10 mV s−1 between −0.559 and +0.241 V vs SHE. The absence of mediators26 in all experiments means that the catalytic current arises solely from enzyme molecules adsorbed in an electroactive configuration and in direct electronic contact with the carbon.1
3 Results
Structural characterisation using TEM and SEM was carried out to reveal the general morphology and size of the commercial materials (ML, VX, BP, CNP and GNF) and the carbon materials produced in-house following established recipes (APG, MG, MWCNT and N-MWCNT, HNP and Si@C). SEM (Fig. 2 and Fig. 3) and TEM images (Fig. 3 and Fig. 5), Raman spectra (Fig. 4) and surface area and porosity measurements using nitrogen adsorption (Fig. 6) for all 11 materials were found to be in agreement with data provided by suppliers or reported in the literature9,28,29,32,38–42 and results are summarised in Table 1. All materials were tested using electrochemistry for Hyd-1 adsorption and electrocatalysis (Fig. 7 to Fig. 10). | ||
| Fig. 2 Scanning electron micrographs of plate-like and fibre-like carbon materials. Images taken at the same magnification ((a), (c), (e), (g), (i); scale bars = 3 μm) reveal the overall morphology. In order to highlight specific morphological features of each material, images were also taken at different magnifications ((b), (d), (f), (h), (j); scale as indicated). | ||
 | ||
| Fig. 3 Scanning and transmission electron micrographs of particle-like materials. Images taken at the same magnification ((a), (c), (e), (g), (i), (k); scale bars = 3 μm) reveal the overall morphology. In order to highlight specific morphological features of each material, images were also taken at higher magnifications ((b), (d), (f), (h), (j), (l); scale as indicated). | ||
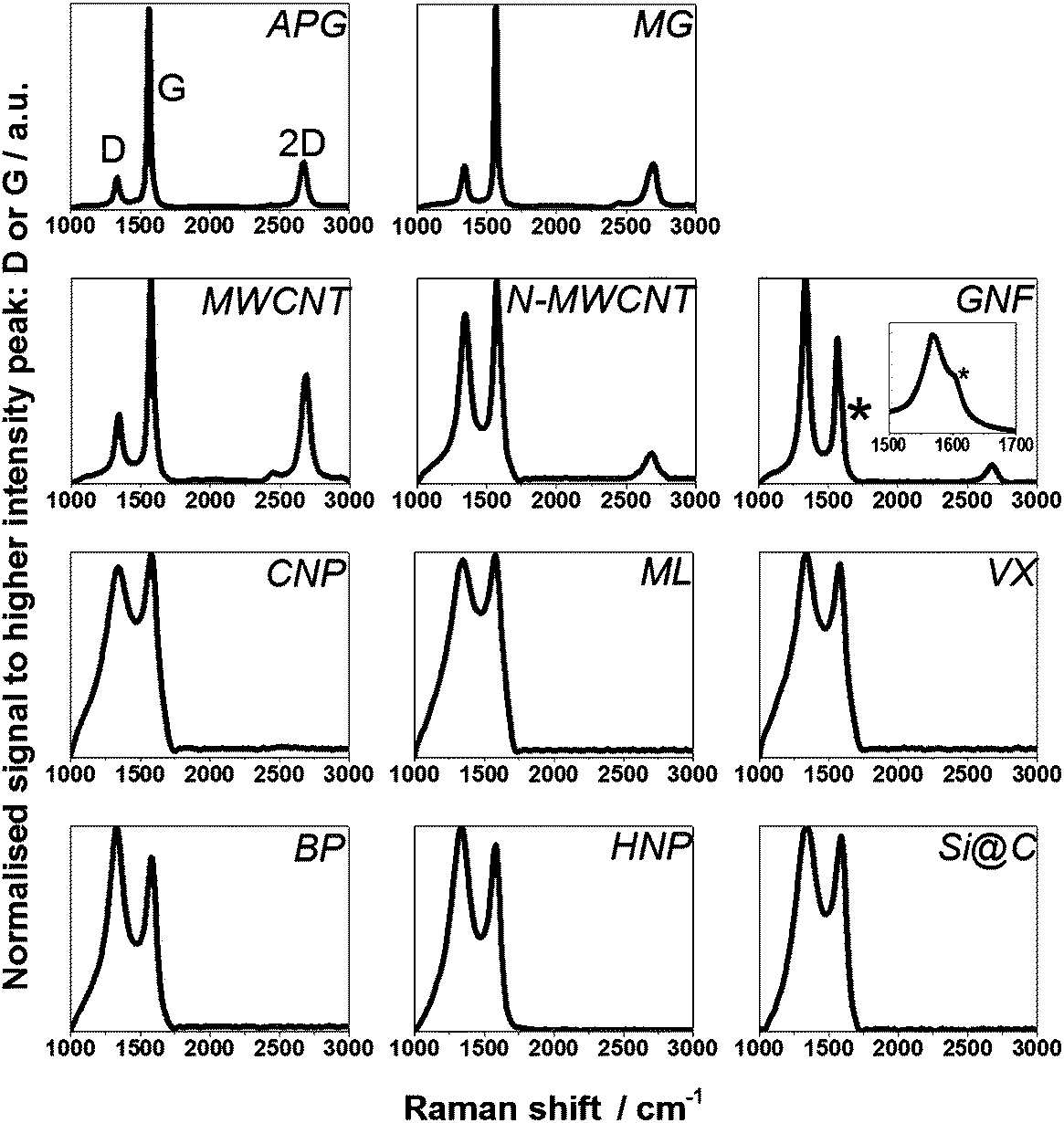 | ||
| Fig. 4 Raman spectra of all 11 carbon samples investigated confirming the graphitic and amorphous nature of individual materials respectively. The * marks the D′ shoulder on GNF (see also the inset). | ||
 | ||
| Fig. 5 High-resolution TEM images revealing the surface morphologies of (a) MWCNT, (b) N-MWCNT, (c) GNF and (d) BP. The image of BP is reprinted from ref. 40, with permission from Elsevier. The image of N-MWCNT is reprinted from ref. 32, with permission from Elsevier. | ||
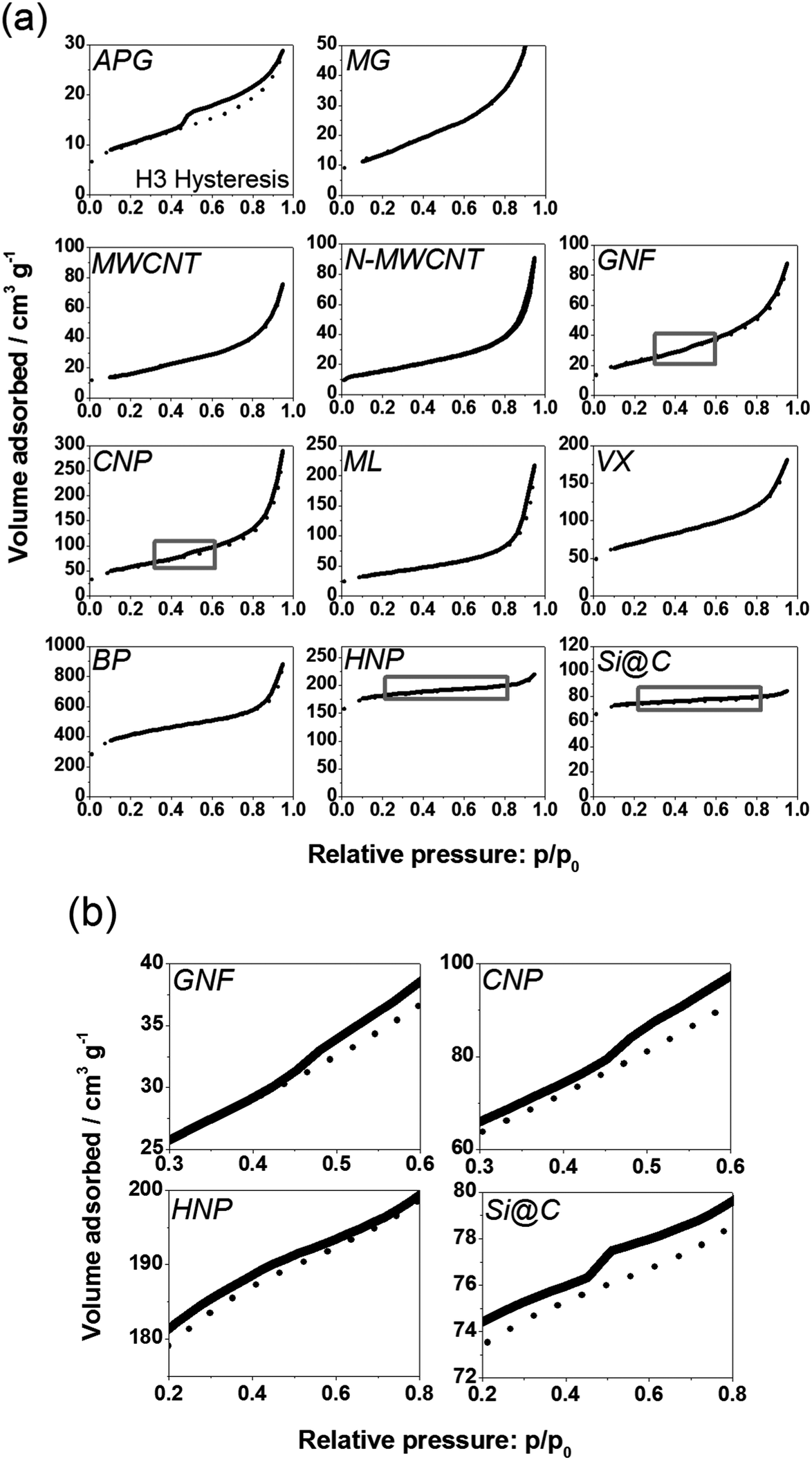 | ||
| Fig. 6 (a) Nitrogen adsorption (dotted) and desorption (solid) isotherms at 77 K for each carbon material investigated. Grey boxes indicate the part of the isotherm which is enlarged in panel (b) for GNF, CNP, HNP and Si@C to highlight the H3 hysteresis for GNF, CNP, and the H4 hysteresis observed in the HNP and Si@C sample. | ||
| Material | Abbreviation | Source | Typical size | Raman ID/IG | Model used for specific surface area | Specific surface area (m2 g−1) | Pore diametera (nm) | Pore volumeb (cm3 g−1) |
|---|---|---|---|---|---|---|---|---|
| a average pore diameter estimated by BJH desorption (4V/A). b single point measurement at relative pressure 0.95 for pores less than 40 nm; t: thickness; L: length; D: diameter, in the cases where the Langmuir model gives a better fit to the data than the BET model, values obtained with the Langmuir model are specified in addition to the results obtained using the BET model. | ||||||||
| Abraded Pyrolytic Graphite | APG | Abrasion | D: 3–5 μm | 0.17 ± 0.04 | BET | 35.2 ± 0.1 | 4.6 | 0.05 |
| t > 500 nm | ||||||||
| Micron-sized Graphite | MG | Exfoliation | D: 500 nm–1μm | 0.19 ± 0.05 | BET | 52.5 ± 0.5 | 5.9 | 0.11 |
| t < 300 nm | ||||||||
| Graphite platelet NanoFibres | GNF | Sigma-Aldrich | D: 20–250 nm | 1.20 ± 0.11 | BET | 81.0 ± 0.4 | 5.7 | 0.14 |
| L < 3 μm | ||||||||
| Carbon Nanotubes | MWCNT | AACVD | D: 50–100 nm | 0.36 ± 0.04 | BET | 60.0 ± 0.6 | 6.1 | 0.12 |
| L: 50–150 μm | ||||||||
| Nitrogen doped Carbon Nanotubes | N-MWCNT | AACVD | D: 50–100 nm | 0.89 ± 0.11 | BET | 57.1 ± 0.2 | 7.2 | 0.14 |
| L: 40–120 μm | ||||||||
| Hydrothermal Nanoparticles | HNP | Hydrothermal synthesis | D: 100–120 nm | 1.08 ± 0.01 | Langmuir | 816 ± 4 | 3.6 | 0.34 |
| BET | 550 ± 14 | |||||||
| Carbon NanoPowder | CNP | Aldrich | D: 30–50 nm | 0.94 ± 0.08 | BET | 198.0 ± 0.5 | 6.9 | 0.45 |
| Mogul L | ML | Cabot Corporation | D: 25–40 nm | 0.96 ± 0.05 | BET | 127.6 ± 0.4 | 8.7 | 0.34 |
| Vulcan XC72R | VX | Cabot Corporation | D: 12–63 nm | 1.06 ± 0.05 | BET | 237 ± 3 | 5.1 | 0.28 |
| Black Pearls 2000 | BP | Cabot Corporation | D: 17–31 nm | 1.18 ± 0.03 | Langmuir | 2053 ± 24 | 4.7 | 1.36 |
| BET | 1332 ± 22 | |||||||
| Silicon Core Carbon Shell Nanoparticles | Si@C | Polymerisation and Carbonisation | Core D: 50–150 nm | 1.05 ± 0.03 | Langmuir | 328.9 ± 0.8 | 3.8 | 0.13 |
| Shell D: 10–20 nm | BET | 221 ± 5 | ||||||
 | ||
| Fig. 7 Cyclic voltammograms of Hyd-1 (a) adsorbed directly on a pyrolytic graphite RDE with the ‘edge’ surface exposed or (b) on APG deposited on the RDE. The electrode was rotated at 2000 rpm in pH 6.0, 100 mM potassium phosphate buffer under 1 bar H2 at 25 °C. The dotted vertical lines mark the thermodynamic potential of H+/H2 couple under the conditions of the experiment. The grey voltammograms are obtained under identical conditions with no hydrogenase deposited on the RDE. | ||
3.1 SEM/TEM characterisation of material size and morphology
Fig. 2 shows SEM images of materials which can be classed as plate-like (APG and EG) or fibre-like (MWCNT, N-MWCNT and GNF). Images presented in the left hand panel were recorded at the same magnification and are presented to show differences in the overall morphology of the samples (Fig. 2(a), (c), (e), (g) and (i)), while images presented in the right hand panel show magnifications selected to highlight more specific aspects of the material shapes and sizes, (Fig. 2 (b), (d), (f), (h) and (j)). Further SEM and TEM images (not shown here) were also used in analysis of particle size and shape distributions.We found that the diameter of the flat surface of the APG plates is in the range 3–5 μm whereas the MG plates are smaller, falling mostly in the range 0.5–1 μm. A rough estimate of the thickness (t) of the plates (Table 1) was also evaluated from examination of the SEM images (see Table 1). The MG plates are thinner (<300 nm) than APG (>500 nm). These observations confirm that exfoliation of graphite flakes is a relevant synthetic route to obtain small-size graphite plate-like materials (MG).
MWCNT and N-MWCNT have typical diameters below 100 nm, and a length up to 150 μm and 120 μm respectively. TEM images confirm that they are both hollow materials and qualify as tubes.32 The hollow nature of the N-MWCNT can also be observed by looking at the catalyst-free tips of the N-MWCNT at higher magnification SEM images (see circular features in Fig. 2(h)). In contrast GNF are not hollow and consist of stacked layers (discussed further in section 3.2) with diameters in the range 20–250 nm, and are typically shorter than 3 μm.
The structural nature of the materials that have particle-like morphologies (CNP, ML, VX, BP, HNP and Si@C) is displayed in Fig. 3 which is split into SEM images in the left hand panel (Fig. 3(a), (c), (e), (g), (i) and (k)) and TEM images in the right hand panel (Fig. 3(b), (d), (f), (h), (j) and (l)). The electron microscopy studies showed that all the commercially available materials (CNP, ML, VX, BP) possess an average particle size less than 50 nm (Table 1) while the particle size for in-house synthesised HNP ranged between 100 nm and 120 nm. TEM images of the Si@C particles are consistent with a silicon core (50–150 nm) coated with a thin carbon shell (10–20 nm). The core-shell structure of the Si@C particles was indicated by the difference in contrast in the TEM images and the presence of the carbon shell was confirmed by Raman analysis (see section 3.2).
3.2 Characterisation of graphitic structure by Raman spectroscopy and high-resolution TEM
Raman spectra for each material are shown in Fig. 4, and ID/IG values are summarised in Table 1. Three main peaks in the Raman spectrum were considered in order to assess the degree of graphitisation in the materials: the D, G and 2D peaks at ca 1340, 1575 and 2675 cm−1, respectively, as discussed in section 2.7. APG and MG display ID/IG less than 1 and show a strong 2D peak, confirming that the graphitic structure (sp2 network) of the parent materials is retained during preparation of these plate-like particles. Similarly, the ID/IG of the MWCNT is less than 1 and the spectrum also shows a strong 2D peak. A high-resolution TEM (HRTEM) image of a MWCNT edge, Fig. 5(a), confirms an ordered graphitic structure and the majority of the exposed MWCNT surface is basal-plane type carbon. The presence of ‘edge’-like structures can be expected at the open end of the tube only and are therefore not likely to be abundant in the material.43 N-MWCNT shows ID/IG close to 1 and the presence of a 2D peak. Production of nitrogen-doped carbon nanotubes is known to introduce defects in the graphitic carbon network,44 consistent with the observed increase in ID/IG and smaller 2D peak compared to MWCNT. A HRTEM image of N-MWCNT (Fig. 5(b)) confirms that this material has a less ordered graphic structured compared to the MWCNT. The presence of nitrogen sites has also been demonstrated.32 The Raman spectrum of GNF shows ID/IG greater than 1 and a 2D peak confirming graphitic character. In contrast to MWCNT, HRTEM study of GNF, Fig. 5(c), shows the predominance of graphitic ‘edge’-like features making up the majority of the GNF surface. The Raman spectrum of GNF also displays a D′ peak (shoulder on the D peak, see inset in Fig. 4) which is also related to an abundance of stacked platelets making up the fibre with their edges oriented perpendicular to the fibre axis and therefore responsible for the ‘edge’-like features (rather than basal plane-like walls) along the fibre surface.42As expected the remaining materials (CNP, ML, VX, BP, HNP, Si@C) have a Raman spectrum indicative of a rather amorphous structure without extended crystalline domains, with ID/IG close to 1 and no 2D peak. A HRTEM image of the BP particles reported by Zhu et al. (see Fig. 5(d)) confirms that these particles do not exhibit obvious ‘edge’-like character.40
3.3 BET characterisation: surface area
The surface area and porosity of the materials was assessed by nitrogen adsorption,36 as shown in Fig 6(a), and respective values are summarised in Table 1. The observed porosity results from two factors: the surface properties of the material itself but also from aggregation of the carbon particles leading to porous networks in the material powder.APG, MG, MWCNT, N-MWCNT, GNF, CNP, ML and VX all display a type II isotherm. This is an isotherm concave to the pressure axis, since at relative pressure 0 the volume of gas adsorbed must be 0, and displaying a B point marking the beginning of a linear relationship between adsorption and relative pressure. The presence of the type II isotherm is indicative of the predominance of rather large pores (macropores with diameter greater than 50 nm). Pores in this size range are likely to be due to aggregation of the materials, which can also be seen in the SEM and TEM images, Fig. 2 and Fig. 3.
Fitting the isotherms according to the BET model gives a specific surface area lower than 100 m2 g−1 for APG, MG, MWCNT, N-MWCNT and GNF. CNP, ML and VX have a specific surface area between 100 and 250 m2 g−1. Thus the plate-like and fibre-like particles fall into a lower surface-area range, consistent with the larger sizes of these materials. APG display a pronounced H3 hysteresis: the desorption isotherm does not follow exactly the adsorption isotherm at high relative pressure, Fig 6(a). GNF and CNP show a less pronounced H3 hysteresis, as evident from the enlargement of part of the isotherm in Fig 6(b). The combination of a type II isotherm and presence of H3 hysteresis is designated as a type II(b) isotherm. A type II(b) isotherm is usually observed in powders and aggregates, and thus is consistent with the nature of these materials.
The nitrogen adsorption isotherm for BP, HNP and Si@C differs slightly from the previously described type II isotherms. A more pronounced plateau at high relative pressure is observed indicative of a type I isotherm, suggesting the presence of micropores (pore diameter less than 2 nm). This is in agreement with the fact that for these three materials the Langmuir model gives a better fit than the BET model since the Langmuir model is known to give a better fit for materials with micropores.36 The specific surface area of these materials is greater than 300 m2 g−1 (as determined from the Langmuir model). Values obtained from the BET model are also included in Table 1 for comparison. HNP and Si@C show evidence of H4 hysteresis as shown in Fig 6(b). This behaviour is usually associated with activated carbons and materials displaying slit-shaped pores especially in the micropore range.36 This is also consistent with the better fit of the Langmuir model for data from HNP and Si@C.
A convenient estimation of the average size of pores in a material can be inferred by dividing the pore volume (V) in the material by the surface area (A) of the material. Assuming the pores can be modelled by perfect cylinders, the average pore diameter is equal to 4V/A.36 For all materials considered in this study, the average mesopore size is within the range 3.5 to 9 nm (Table 1), although pores of smaller and larger diameter can also be found in the materials. Since all materials have a pronounced mesoporous structures (type II isotherm) and mesoporous volume (Table 1) most of the porosity of the whole material should be accessible to hydrogenase molecules (ca 5 nm diameter). The materials with smallest average pore size (<4 nm) are the synthesised materials, HNP and Si@C.
3.4 Electrocatalytic activity of adsorbed Hyd-1
A typical cyclic voltammogram for Hyd-1 adsorbed onto a rotating disc electrode (RDE) made of pyrolytic graphite with the edge surface exposed is reported in Fig. 7(a). The electrode is immersed in buffered aqueous solution (pH 6.0) equilibrated with 1 bar H2 (ca 0.8 mM). Hyd-1 does not show significant activity for proton reduction under a H2 atmosphere,45 so no Faradaic current is observed at potentials more negative than –0.3 V. At higher potentials a clear oxidative catalytic wave is observed. No Faradaic current is observed under H2 for a bare electrode without enzyme adsorbed. Each cycle of catalytic turnover by a molecule of hydrogenase leads to electron flow into the electrode, and thus the catalytic current is directly proportional to the number of electroactive enzyme molecules, provided that the current is not limited by availability of H2 to the enzyme.7 The electrode is rotated rapidly (2000 rpm) to maintain the H2 concentration at the electrode surface close to that in the bulk solution. Hyd-1 exhibits an onset potential for H2 oxidation just positive of –0.3 V, consistent with the slight overpotential requirement (50–60 mV) of this enzyme relative to the E(H+/H2) potential at 1 bar H2.45 The thermodynamic potential for E(H+/H2) under the experimental conditions is indicated by a vertical line in Fig. 7. The residual slope in the cyclic voltammogram at higher potentials is characteristic of immobilised enzyme electrocatalysts engaged in direct electron transfer, and has been explained by a dispersion of orientations of the enzyme on the electrode leading to a range of electron transfer rates.46 A similar waveshape, although with higher electrocatalytic current, is obtained for Hyd-1 adsorbed on plate-like particles of pyrolytic graphite (APG) deposited onto the RDE (Fig. 7(b)). This is consistent with an earlier report in which Hyd-1 on APG was deposited on a RDE in a Nafion binder.30In order to assess the potential benefit of alternative materials to APG we deposited an equal amount of each material, modified with Hyd-1, onto a RDE and recorded the associated catalytic current in a cyclic voltammetric sweep in solution equilibrated with H2 (Fig. 8). For the results reported in this figure, electrodes were prepared by drop-casting 0.5 μL of a 20 mg mL−1 dispersion of the hydrogenase-modified particles onto a RDE of surface area 0.02 cm2. Thus the current per geometric area of electrode is directly proportional to the current per gram of material. It is observed that the commercial materials with higher specific surface area (CNP, VX, ML and BP) give higher current densities. However the shape of the cyclic voltammogram for those materials yields a plateau at high potentials, characteristic of a film in which H2 is not efficiently provided to the enzyme, and thus current is limited by mass transport. Rotation at a higher rate of 5000 rpm increases the observed current by improving the H2 supply to the film (see the dashed line in Fig. 8 for BP). Further increase in rotation rate to 7000 rpm (not shown) provides negligible improvement in current and the plateau shape is still present, suggesting that fast rotation fails to provide efficient mass transport through the particle film to buried hydrogenase molecules.
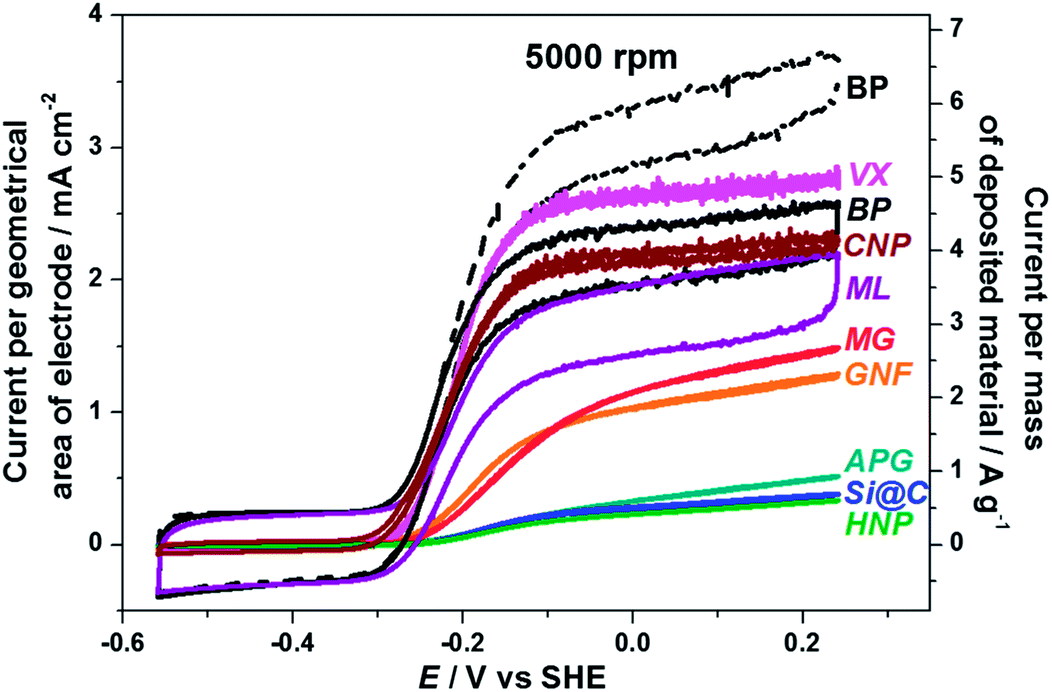 | ||
| Fig. 8 Cyclic voltammograms of Hyd-1 adsorbed on various carbon materials in 100 mM potassium phosphate buffer, pH 6.0, 25 °C, equilibrated with 1 bar H2. Materials were deposited on a 0.02 cm2 pyrolytic graphite RDE with the ‘edge’ surface exposed, and the electrode was rotated at 2000 rpm. The left hand axis shows the current normalised to the geometric area of electrode and the right hand axis indicates the current per gram of deposited carbon material. In all cases 0.5 μL of 20 mg mL−1 dispersion of carbon material with adsorbed enzyme were deposited on the RDE. The current associated with BP material is reported at 2000 rpm (BP solid line) and 5000 rpm (BP dashed line). | ||
For CNP, VX, ML and BP, to obtain conditions that are not limited by mass transport at the RDE we diluted the particle dispersions to 5 mg mL−1, applying 0.5 μL to the RDE. This allowed us to assess the suitability of each carbon material for adsorption of Hyd-1 in an electroactive configuration. Results are shown in Fig. 9. Also shown on this figure are MWCNT and N-MWCNT modified with Hyd-1. Difficulty in dispersing the relatively hydrophobic nanotubes in water means that the mass of nanotubes deposited cannot be controlled reliably, so the absolute current is not comparable with the other materials. Importantly, the similarity in waveshape (general form and onset potential) for Hyd-1 on all the carbon materials suggests that the carbon support has little effect on enzyme electroactivity. Therefore, despite their intrinsic differences all materials investigated accommodate Hyd-1 in a suitable configuration to perform H2 oxidation electrocatalysis.
 | ||
| Fig. 9 Cyclic voltammograms of adsorbed Hyd-1 on the various carbon materials deposited onto a pyrolytic graphite RDE with the edge surface exposed, in 100 mM potassium phosphate buffer, pH 6.0, 25 °C, equilibrated with 1 bar H2. Dilution of the dispersion of certain particles (CNP, ML, VX and BP, see text) means that currents are not significantly mass-transport limited. The dashed lines mark the thermodynamic potential of the H+/H2 couple under the experimental conditions. | ||
The mass transport limitation observed for CNP, ML, VX and BP in Fig. 8 is likely to be a problem for realistic applications of hydrogenase electrocatalysis. We therefore investigated Hyd-1 activity on BP in a wall-jet electrochemical cell configuration where mass transport can be improved. In Fig. 10, scans (a) and (b) were recorded at an RDE electrode, whereas scan (c) was recorded with the same amount of material in the wall-jet cell. At high potentials a higher current is reached in the wall-jet cell and the plateau is absent suggesting that mass transport to enzyme molecules in the particle film is no longer significantly limiting the current. In order to test whether enzyme loading on BP could be improved by longer adsorption times from more concentrated enzyme solution, an electrode was prepared in the wall-jet cell using particles that had been incubated with 0.2 mM Hyd-1 for 48 hours at 4 °C, scan (d). In this case an improved current per mass of material of 12 A g−1 (at +0.24 V vs SHE) was recorded.
 | ||
| Fig. 10 Cyclic voltammograms of Hyd-1 adsorbed on BP assessed on a pyrolytic graphite RDE (grey) at (a) 2000 rpm or (b) 5000 rpm; and cyclic voltammograms for the same quantity of BP studied in a wall-jet cell configuration (with H2-saturated solution flowing at 50 mL min−1) for (c) the same set of particles as (a) and (b); and (d) for particles prepared with longer adsorption time (48 hours) from more concentrated Hyd-1 solution (0.2 mM). All were prepared by deposition of BP (1 μL) from a 20 mg mL−1 suspension. | ||
4 Discussion and conclusions
The materials considered in this study fall into three broad categories with respect to their general shape and morphology. APG and MG are micron-sized platelets with low specific surface area compared to the other materials. They are highly graphitic, and it is expected that the thin edges of the plates expose graphite ‘edge’-like regions that have been associated with effective protein adsorption. The fibre-like materials, MWCNT, N-MWCNT and GNF, have diameters in the nanometer range and up to 250 nm for GNF and have slightly higher surface area than the plate-like materials and are also graphitic. Whereas MWCNT and N-MWCNT show a predominance of basal-like surface, GNF has mainly ‘edge’-like surface structure. CNP, ML, VX, BP, HNP and Si@C are particle-like materials that do not show an ordered graphitic structure but have the advantage of a high surface area. All materials show some extent of mesoporosity, on an appropriate scale for enzyme adsorption.In this study we have investigated all of these materials as supports for electrocatalysis by the enzyme Hyd-1 from E. coli in order to present a unified comparison of different types of carbon with a single enzyme. The results demonstrate that a range of carbon materials can be used for adsorption of Hyd-1 in an electroactive configuration. The catalytic waveshapes in voltammograms recorded under conditions where mass transport is not significantly limiting (Fig. 9) are very similar to the well-characterised waveshape reported for Hyd-1 adsorbed on a pyrolytic graphite electrode (Fig. 7(a)). Importantly, the onset potential for H2 oxidation is independent of material, showing that interfacial electron transfer to and from hydrogenase molecules is effective on each carbon material. The residual slope in the electrocatalytic voltammogram at high potential is similar for each material (Fig. 9), suggesting that there are not significant differences in the distribution of orientations of Hyd-1. The results of this study suggest that any conductive carbon-based material is likely to be a candidate for hydrogenase study or application.
All of the carbon materials give an increase in catalytic current per geometrical area of the electrode relative to Hyd-1 on a bare pyrolytic graphite electrode (Fig. 7(a)). In this set of experiments, the same mass of particles was applied to each electrode and so the magnitude of the current also gives a direct indication of the electrocatalytic activity per gram of carbon. A significant increase in current is observed for the commercially available carbons, BP, VX, ML and CNP but in each case the voltammogram is severely mass transport limited, showing a plateau at high potential. As demonstrated for BP (the highest surface area material with the most pronounced mesoporous structure), mass transport can be improved slightly by increasing the electrode rotation rate but the plateau remains. The mesopores, with an average diameter in the range 3.5–9 nm (Table 1) for all materials used in this study, are of similar dimension to Hyd-1 which has a diameter of roughly 5 nm. This is advantageous since the mesoporous network should be accessible to Hyd-1. Enzyme molecules inside the pores are likely to be almost completely surrounded by carbon, favouring contact of their electron relay centres with the electrode surface so that the majority of molecules are electroactive. Templated ordered mesoporous carbons and other inorganic materials have been developed specifically for the adsorption of enzymes in biotechnology, but usually involve use of a sacrificial template, multi-step synthesis, and may display relatively poor conductivity.47,48 The success of Hyd-1 adsorption on BP and other commercial carbon blacks shows that it is possible to make use of readily available mesoporous carbons for enzyme electrocatalysis.
In general the materials which are most affected by mass transport limitations are those which also have the most developed mesoporous structures (Table 1), suggesting poor transport of H2 within electrodes constructed from these materials. Mass transport limitation at mesoporous carbon electrodes modified with hydrogenase has been noted by other authors.15 In Fig. 10 we compare the same preparation of Hyd-1 modified BP on a RDE (scans (a) and (b)) or in a cell with a wall-jet configuration (scan (c)). In the wall-jet cell, the current per gram of BP is substantially increased and the waveshape no longer shows a plateau, resembling that at the pyrolytic graphite electrode, Fig. 7(a). In the voltammograms of Fig. 8 electrodes formed from larger or less mesoporous particles, such as APG, MG and GNF show less evidence of mass transport limitation, although the relative currents are lower. This illustrates that in addition to selecting carbon materials to improve enzyme loading, another important consideration is optimisation of mass transport at the electrode.
Fig. 11 shows the voltammetric data from Fig. 8 recalculated to show the current density with respect to the surface area (BET) of each material. Fig. 11 gives an indication of how effectively the total surface area of each material is being used for electrocatalysis. In this case the highest current densities occur for materials with larger size or lower specific surface area. In particular APG, MG and GNF compare more favourably with the other materials when evaluated on the basis of current per surface area of material, showing that the surface of these materials is used efficiently for electrocatalysis. The fact that these materials with exposed graphitic ‘edge’ surfaces show high currents per surface area of material is in line with the successful use of pyrolytic graphite ‘edge’ electrodes for protein film electrochemistry. Results associated with MG suggest that even more favourable current densities could be obtained from smaller particles of these graphitic materials. Conversely, materials with larger specific surface areas (BP, Si@C and HNP) show very low current densities in Fig. 11. This is particularly the case for BP which has a specific surface area (BET) of 1332 m2 g−1. Even after improving mass transport to Hyd-1 on BP by use of a wall-jet cell, the high surface area of the material is clearly being under-utilised (dashed line, Fig. 11). Slow diffusion of Hyd-1 into the mesoporous network of BP will lead to very long equilibration times for adsorption of the enzyme. Restricted or hindered diffusion of macromolecules through pores of a similar diameter is a well-documented phenomenon,49 and is the basis of size exclusion chromatography. Enzyme loading on BP was improved by allowing Hyd-1 to adsorb over a longer time and from more concentrated solution (Fig. 10(d) and Fig. 11). In this case an improved current per surface area of material of 0.9 μA cm−2 was obtained. Hyd-1 is a particularly robust hydrogenase, and for some metalloproteins it would be unfeasible to allow such long adsorption times due to protein degradation. The fact that the current for BP in the wall-jet cell with longer adsorption time for Hyd-1 exceeds that obtained for the initial particle preparation (Fig. 10) at all potentials during H2 oxidation implies that a significant amount of enzyme can adsorb within the mesopores of the material where efficient mass transport becomes particularly important.
 | ||
| Fig. 11 Voltammetric data from Fig. 8 reported as current per surface area (BET) of material. For comparison, the scan obtained in the wall-jet configuration cell from Fig. 10(d) is also reported (dotted BP voltammogram). | ||
Carbon nanotubes have been widely used in development of applications of hydrogenases.25 Attractive features of MWCNT or N-MWCNT are the opportunity to develop relatively high surface area electrodes with a mesoporous network (Table 1, Fig. 2). We observed that direct adsorption of Hyd-1 is possible on MWCNT and N-MWCMT, but their dispersion in aqueous solution is challenging due to their intrinsic hydrophobicity. A slight improvement in dispersion is observed for N-MWCNT compared to MWCNT, but we were still unable to make accurate comparisons of current per mass of material deposited for the nanotubes. It might be expected that the defects and heteroatom sites brought about by nitrogen incorporation within the carbon lattice in N-MWCNT should offer anchor-points for specific attachment of enzymes, but this is beyond the scope of the present study.
In other hydrogenase electrocatalysis studies, nanotubes have been handled in specific ways, for example, multi-wall carbon nanotubes grown directly on a gold substrate in a study by De Lacey and co-workers.20 Carbon nanotubes have also been further modified for covalent attachment of hydrogenase, for example commercially available multi-wall carbon nanotubes modified by adsorption of a pyrene derivative which was covalently linked to Hyd-1 via an amide linkage were demonstrated in a high surface-area electrode for a hydrogenase H2/O2 fuel cell by Armstrong and co-workers.16 Lojou and co-workers used chemically modified single and multi-wall carbon nanotubes for covalent attachment of hydrogenase from Aquifex aeolicus; stabilising the nanotubes network in the polymer electrolyte, Nafion.26 These examples highlight the need for specialised handling of carbon nanotubes if they are to be used successfully for hydrogenase electrocatalysis.
The fact that high currents per gram of material can be achieved by Hyd-1 on commercial carbon blacks (BP, VX and ML) is promising for applications of hydrogenase electrocatalysis, for example in enzyme fuel cells or H2-production devices, provided that mass transport issues can be properly addressed in the design of the device.
Successful adsorption of hydrogenase is not limited to commercially available materials. In-house generated particles HNP and Si@C investigated in this study are good examples of materials that can be easily tailored to accommodate hydrogenases for specific purposes. HNP material can be obtained by the versatile and simple hydrothermal synthesis:50–52 a green synthetic route using glucose (a renewable feedstock) as starting material and requiring readily-available laboratory equipment. The high surface area material obtained has also been demonstrated in various other electrochemical devices35,53 and so is a promising platform for tailoring of materials for enzyme electrocatalysis. Si@C may have interesting applications as an electrode material for coupling to in situ infrared spectroscopy as it has an IR-transparent silicon core but a sufficiently conductive outer carbon shell (10 to 20 nm) to support enzyme adsorption and electrochemistry. These examples illustrate opportunities to select and develop simple carbon materials for specific applications of hydrogenases.
Acknowledgements
We are grateful for financial support from Engineering and Physical Sciences Research Council [INSPIRE grant EP/J015202/1 (J.Q., N.G. and K.A.V.), EPSRC DTA award EP/J500495/1 (J.Q.), EPSRC Pathways to Impact Block Awards (N.G.) and EPSRC Small Equipment Award EP/K031503/1 (K.A.V.)], The Royal Society (N.G.), the European Research Council (ERC) [EnergyBioCatalysis-ERC-2010-StG-258600 (K.A.V. and P.A.A.) and DEDIGROWTH-ERC-2009-StG-240500 and DEVICE ERC-2012-PoC-309786 (N.G.)]. J.Q. is also grateful for support from The Keble Association and Keble College Oxford. R.H. is supported by Ministerio de Ciencia y Tecnología and Univesidad de Costa Rica and Lincoln College Oxford. The authors thank Miss Judy Britton and Dr Antal A. Koós for their help in the acquisition of high resolution TEM images, and Prof. Fraser A. Armstrong and Elena Nomerotskaia for providing a sample of E. coli Hyd-1.References
- C. Léger and P. Bertrand, Chem. Rev., 2008, 108, 2379–2438 CrossRef PubMed.
- F. A. Armstrong and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14049–14054 CrossRef CAS PubMed.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- W. Yang, K. R. Ratinac, S. P. Ringer, P. Thordarson, J. J. Gooding and F. Braet, Angew. Chem., Int. Ed., 2010, 49, 2114–2138 CrossRef CAS PubMed.
- S. V. Hexter, F. Grey, T. Happe, V. Climent and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11516–11521 CrossRef CAS PubMed.
- A. A. Hamdan, P.-P. Liebgott, V. Fourmond, O. Gutiérrez-Sanz, A. L. D. Lacey, P. Infossi, M. Rousset, S. Dementin and C. Léger, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19916–19921 CrossRef PubMed.
- K. A. Vincent, A. Parkin and F. A. Armstrong, Chem. Rev., 2007, 107, 4366–4413 CrossRef CAS PubMed.
- C. F. Blanford and F. A. Armstrong, J. Solid State Electrochem., 2006, 10, 826–832 CrossRef CAS PubMed.
- K. A. Vincent, X. Li, C. F. Blanford, N. A. Belsey, J. H. Weiner and F. A. Armstrong, Nat. Chem. Biol., 2007, 3, 761–762 CrossRef CAS PubMed.
- H. A. Reeve, L. Lauterbach, P. A. Ash, O. Lenz and K. A. Vincent, Chem. Commun., 2012, 48, 1589–1591 RSC.
- O. Lazarus, T. W. Woolerton, A. Parkin, M. J. Lukey, E. Reisner, J. Seravalli, E. Pierce, S. W. Ragsdale, F. Sargent and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 14154–14155 CrossRef CAS PubMed.
- A. A. Karyakin, S. V. Morozov, E. E. Karyakina, S. D. Varfolomeyev, N. A. Zorin and S. Cosnier, Electrochem. Commun., 2002, 4, 417–420 CrossRef CAS.
- K. A. Vincent, J. A. Cracknell, J. R. Clark, M. Ludwig, O. Lenz, B. Friedrich and F. A. Armstrong, Chem. Commun., 2006, 5033–5035 RSC.
- D. Svedruzic, J. L. Blackburn, R. C. Tenent, J.-D. R. Rocha, T. B. Vinzant, M. J. Heben and P. W. King, J. Am. Chem. Soc., 2011, 133, 4299–4306 CrossRef CAS PubMed.
- A. de Poulpiquet, H. Marques-Knopf, V. Wernert, M. T. Giudici-Orticoni, R. Gadiou and E. Lojou, Phys. Chem. Chem. Phys., 2014, 16, 1366–1378 RSC.
- S. Krishnan and F. A. Armstrong, Chem. Sci., 2012, 3, 1015–1023 RSC.
- L. Xu and F. A. Armstrong, Energy Environ. Sci., 2013, 6, 2166–2171 CAS.
- S. V. Morozov, E. E. Karyakina, N. A. Zorin, S. D. Varfolomeyev, S. Cosnier and A. A. Karyakin, Bioelectrochemistry, 2002, 55, 169–171 CrossRef CAS.
- A. I. Yaropolov, A. A. Karyakin, S. D. Varfolomeev and I. V. Berezin, Bioelectrochem. Bioenerg., 1984, 12, 267–277 CrossRef CAS.
- M. A. Alonso-Lomillo, O. Rudiger, A. Maroto-Valiente, M. Velez, I. Rodriguez-Ramos, F. J. Munoz, V. M. Fernandez and A. L. De Lacey, Nano Lett., 2007, 7, 1603–1608 CrossRef CAS PubMed.
- J. Baur, A. Le Goff, S. Dementin, M. Holzinger, M. Rousset and S. Cosnier, Int. J. Hydrogen Energy, 2011, 36, 12096–12101 CrossRef CAS PubMed.
- F. J. M. Hoeben, I. Heller, S. P. J. Albracht, C. Dekker, S. G. Lemay and H. A. Heering, Langmuir, 2008, 24, 5925–5931 CrossRef CAS PubMed.
- E. Lojou, X. Luo, M. Brugna, N. Candoni, S. Dementin and M. T. Giudici-Orticoni, JBIC, J. Biol. Inorg. Chem., 2008, 13, 1157–1167 CrossRef CAS PubMed.
- S. V. Morozov, E. E. Karyakina, O. A. Zadvornyi, N. A. Zorin, S. D. Varfolomeev and A. A. Karyakin, Russ. J. Electrochem., 2002, 38, 97–102 CrossRef CAS.
- E. Lojou, Electrochim. Acta, 2011, 56, 10385–10397 CrossRef CAS PubMed.
- A. De Poulpiquet, A. Ciaccafava, K. Szot, B. Pillain, P. Infossi, M. Guiral, M. Opallo, M.-T. Giudici-Orticoni and E. Lojou, Electroanalysis, 2013, 25, 685–695 CrossRef CAS PubMed.
- M.-x. Wang, Q. Liu, H.-f. Sun, N. Ogbeifun, F. Xu, E. A. Stach and J. Xie, Mater. Chem. Phys., 2010, 123, 761–766 CrossRef CAS PubMed.
- X. M. Sun and Y. D. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- P. Gao, J. Fu, J. Yang, R. Lv, J. Wang, Y. Nuli and X. Tang, Phys. Chem. Chem. Phys., 2009, 11, 11101–11105 RSC.
- A. J. Healy, H. A. Reeve, A. Parkin and K. A. Vincent, Electrochim. Acta, 2011, 56, 10786–10790 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- A. A. Koós, M. Dowling, K. Jurkschat, A. Crossley and N. Grobert, Carbon, 2009, 47, 30–37 CrossRef PubMed.
- S. S. Meysami, F. Dillon, A. A. Koós, Z. Aslam and N. Grobert, Carbon, 2013, 58, 151–158 CrossRef CAS PubMed.
- Z. Wen, Q. Wang, Q. Zhang and J. Li, Electrochem. Commun., 2007, 9, 1867–1872 CrossRef CAS PubMed.
- X. XiaoHong, S. Lei, L. HongBo, Y. Li and H. YueDe, J. Phys. Chem. Solids, 2012, 73 Search PubMed.
- F. Rouquerol, J. Rouquerol, K. S. W. Sing, P. Llewellyn and G. Maurin, Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, Elsevier, 2nd edn, 2014 Search PubMed.
- M. J. Lukey, A. Parkin, M. M. Roessler, B. J. Murphy, J. Harmer, T. Palmer, F. Sargent and F. A. Armstrong, J. Biol. Chem., 2010, 285, 20421–20421 CrossRef CAS PubMed.
- D. Pantea, H. Darmstadt, S. Kaliaguine and C. Roy, Appl. Surf. Sci., 2003, 217, 181–193 CrossRef CAS.
- M. Li, W. Li and S. Liu, Carbohydr. Res., 2011, 346, 999–1004 CrossRef CAS PubMed.
- W. Z. Zhu, D. E. Miser, W. G. Chan and M. R. Hajaligol, Carbon, 2004, 42, 1841–1845 CrossRef CAS PubMed.
- M. Maja, C. Orecchia, M. Strano, P. Tosco and M. Vanni, Electrochim. Acta, 2000, 46, 423–432 CrossRef CAS.
- A. Ambrosi, T. Sasaki and M. Pumera, Chem. – Asian J., 2010, 5, 266–271 CrossRef CAS PubMed.
- C. E. Banks and R. G. Compton, Analyst, 2006, 131, 15–21 RSC.
- A. A. Koós, F. Dillon, E. A. Obraztsova, A. Crossley and N. Grobert, Carbon, 2010, 48, 3033–3041 CrossRef PubMed.
- M. J. Lukey, A. Parkin, M. M. Roessler, B. J. Murphy, J. Harmer, T. Palmer, F. Sargent and F. A. Armstrong, J. Biol. Chem., 2010, 285, 3928–3938 CrossRef CAS PubMed.
- C. Léger, A. K. Jones, S. P. J. Albracht and F. A. Armstrong, J. Phys. Chem. B, 2002, 106, 13058–13063 CrossRef.
- P. F. Fulvio, R. T. Mayes, X. Wang, S. M. Mahurin, J. C. Bauer, V. Presser, J. McDonough, Y. Gogotsi and S. Dai, Adv. Funct. Mater., 2011, 21, 2208–2215 CrossRef CAS PubMed.
- D. I. Fried, F. J. Brieler and M. Froeba, ChemCatChem, 2013, 5, 862–884 CrossRef CAS PubMed.
- M. G. Davidson and W. M. Deen, Macromolecules, 1988, 21, 3474–3481 CrossRef CAS.
- X. M. Sun and Y. D. Li, J. Colloid Interface Sci., 2005, 291, 7–12 CrossRef CAS PubMed.
- M. Liu, C. Wang and X. Wang, J. Mater. Chem., 2011, 21, 15197–15200 RSC.
- Z. Liu, C. Zhang, L. Luo, Z. Chang and X. Sun, J. Mater. Chem., 2012, 22, 12149–12154 RSC.
- M. Liu, Y. Yan, L. Zhang, X. Wang and C. Wang, J. Mater. Chem., 2012, 22, 11458–11461 RSC.
| This journal is © The Royal Society of Chemistry 2014 |