 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical characterisation of graphene nanoflakes with functionalised edges
Mailis M.
Lounasvuori
,
Martin
Rosillo-Lopez
,
Christoph G.
Salzmann
,
Daren J.
Caruana†
and
Katherine B.
Holt
*
Department of Chemistry, University College London, 20 Gordon St, London WC1H 0AJ, United Kingdom. E-mail: k.b.holt@ucl.ac.uk
First published on 11th April 2014
Abstract
Graphene nanoflakes (GNF) of diameter ca. 30 nm and edge-terminated with carboxylic acid (COOH) or amide functionalities were characterised electrochemically after drop-coating onto a boron-doped diamond (BDD) electrode. In the presence of the outer-sphere redox probe ferrocenemethanol there was no discernible difference in electrochemical response between the clean BDD and GNF-modified electrodes. When ferricyanide or hydroquinone were used as redox probes there was a marked difference in response at the electrode modified with COOH-terminated GNF in comparison to the unmodified BDD and amide-terminated GNF electrode. The response of the COOH-terminated GNF electrode was highly pH dependent, with the most dramatic differences in response noted at pH < 8. This pH range coincides with partial protonation of the carboxylic acid groups as determined by titration. The acid edge groups occupy a range of bonding environments and are observed to undergo deprotonation over a pH range ca. 3.7 to 8.3. The protonation state of the GNF influences the oxidation mechanism of hydroquinone and in particular the number of solution protons involved in the reaction mechanism. The voltammetric response of ferricyanide is very inhibited by the presence of COOH-terminated GNF at pH < 8, especially in low ionic strength solution. While the protonation state of the GNF is clearly a major factor in the observed response, the exact role of the acid group in the redox process has not been firmly established. It may be that the ferricyanide species is unstable in the solution environment surrounding the GNF, where dynamic protonation equilibria are at play, perhaps through disruption to ion pairing.
1 Introduction
This paper addresses the electrochemical behaviour of well-defined and characterised graphene nanoflakes (GNF) using cyclic voltammetry (CV) in the presence of solution redox species. Previous studies have used graphene from a variety of sources and with differing defect density and impurity content to construct or modify electrodes and to perform voltammetry with standard redox probes.1–9 Electrodes with well-defined exposed surfaces fabricated from single-layer graphene sheets, produced by mechanical exfoliation or by chemical vapour deposition (CVD), have been used to carry out CV using ferrocene-methanol (FcMeOH)3 and ferricyanide ([Fe(CN)6]3−).4 Heterogeneous rate constants for FcMeOH oxidation were found to be one or two orders of magnitude greater than at basal plane graphite for mechanically exfoliated and CVD graphene respectively.3 The enhanced kinetics were attributed to corrugations in the graphene sheet, giving rise to strain and curvature that may enhance reactivity, perhaps through the creation of midgap states.10,11 Likewise, kinetics for the reduction of ferricyanide were found to be improved two-fold for defect-free single layer graphene over those obtained under the same conditions for multi-layer graphite.4 Additionally it was found that defects, such as holes, in the graphene surface made little difference to the voltammetric response of the samples.An alternate approach has been to drop-coat graphene platelets, obtained through reduction of graphene oxide (GO)12–17 or through substrate-free gas-phase synthesis18 onto carbon electrodes and to study the CV response of various redox species at the modified electrode. When graphene flakes of diameter ca. 500 nm were immobilised onto electrodes in this way it was found, in contrast to the studies above, that electron transfer kinetics of a number of solution analyte species (e.g. ferrocyanide, [Ru(NH3)6]3+) were slower at the graphene-modified surfaces. This was attributed to the high proportion of graphene basal plane, where electron transfer kinetics are slower than at edge sites. In addition to these studies there exist many reports of the use of reduced GO materials in the form of composites or pastes for the detection of a range of analytically important species (for reviews see ref. 19–21). Although enhanced electrochemical response and analytical advantages have been cited, it is unclear whether the better performance of these electrodes is due to higher surface areas or the presence of redox active or catalytic impurities.22 A lack of clarity as to purity of material, defect density and identity of oxygen content means in many cases it is difficult to truly assess the electrochemical response of pristine graphene.
Traditional consensus has been that electron transfer at graphitic materials is dominated by the edge plane18,23,24 and it has been shown that the intentional generation of oxygen-containing defects increases reactivity.23 However, recently it has been shown that the basal plane of highly oriented pyrolytic graphite (HOPG) can exhibit fast electron transfer for outer-sphere redox couples.23,25–29 High resolution electrochemical surface imaging studies have shown that electron transfer at carbon nanotube walls and the graphene basal plane surfaces is fast and reversible and limited only by available density of states.28,30–32 This contrasts with previous studies reporting exceedingly sluggish kinetics at the basal plane of graphitic materials. One possible explanation for these discrepancies is in preparation of the materials before electrochemical investigation. Adsorption of organic impurities onto freshly prepared graphene has been shown to take place within minutes on exposure to a typical laboratory atmosphere33 and such a surface layer may lead to inhibition of electron transfer at carbon surfaces.
Notwithstanding the conflicting reports of basal plane activity, it is accepted that the edge plane of graphitic materials shows enhanced electrochemical activity due to the presence of high energy defects such as dangling bonds and oxygen functionalities. The interaction of various redox species with oxygen functionalities at carbon electrodes has been investigated extensively by McCreery and co-workers.34–36 Common redox probes can be classified roughly into three categories: those which are insensitive to surface termination (FcMeOH, [Ru(NH3)6]3+/2+); those which interact with specific oxygen functionalities (such as Fe3+/2+ with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and those which are surface sensitive but apparently do not interact with specific oxygen-containing groups ([Fe(CN)6]3−/4−).37 Given that even the most carefully prepared graphene samples may have some oxygen content, it seems important to determine the influence of these functionalities on the electrochemical response. As graphene is increasingly being manufactured via reduction of graphene oxide, where an array of oxygen groups persist in the final product, the interaction of oxygen moieties with solution species will influence how well the material performs in electrochemical applications.
O) and those which are surface sensitive but apparently do not interact with specific oxygen-containing groups ([Fe(CN)6]3−/4−).37 Given that even the most carefully prepared graphene samples may have some oxygen content, it seems important to determine the influence of these functionalities on the electrochemical response. As graphene is increasingly being manufactured via reduction of graphene oxide, where an array of oxygen groups persist in the final product, the interaction of oxygen moieties with solution species will influence how well the material performs in electrochemical applications.
Our approach in this study is to use novel GNF materials with average diameter of just 30 nm.38 The basal plane of the GNF is predominantly defect free and hence contains negligible oxygen content. In contrast, the edges of the flakes are decorated with carboxylic acid (COOH) functionalities. The high density of edge COOH groups makes this an ideal material with which to study the role of oxygen species on electrochemical response, as their influence is greatly amplified due to the small size of the flakes. Modification of the edge groups by transformation into amide groups also allows us to probe the influence of the edge group acidity and ability to hydrogen bond. A cartoon of the structures of the two types of flakes is shown in Fig. 1 (not to scale). In this paper we show how COOH-terminated GNF (c-GNF) and amide-terminated GNF (a-GNF) modified electrodes interact with the common redox probes FcMeOH, ferricyanide and hydroquinone.
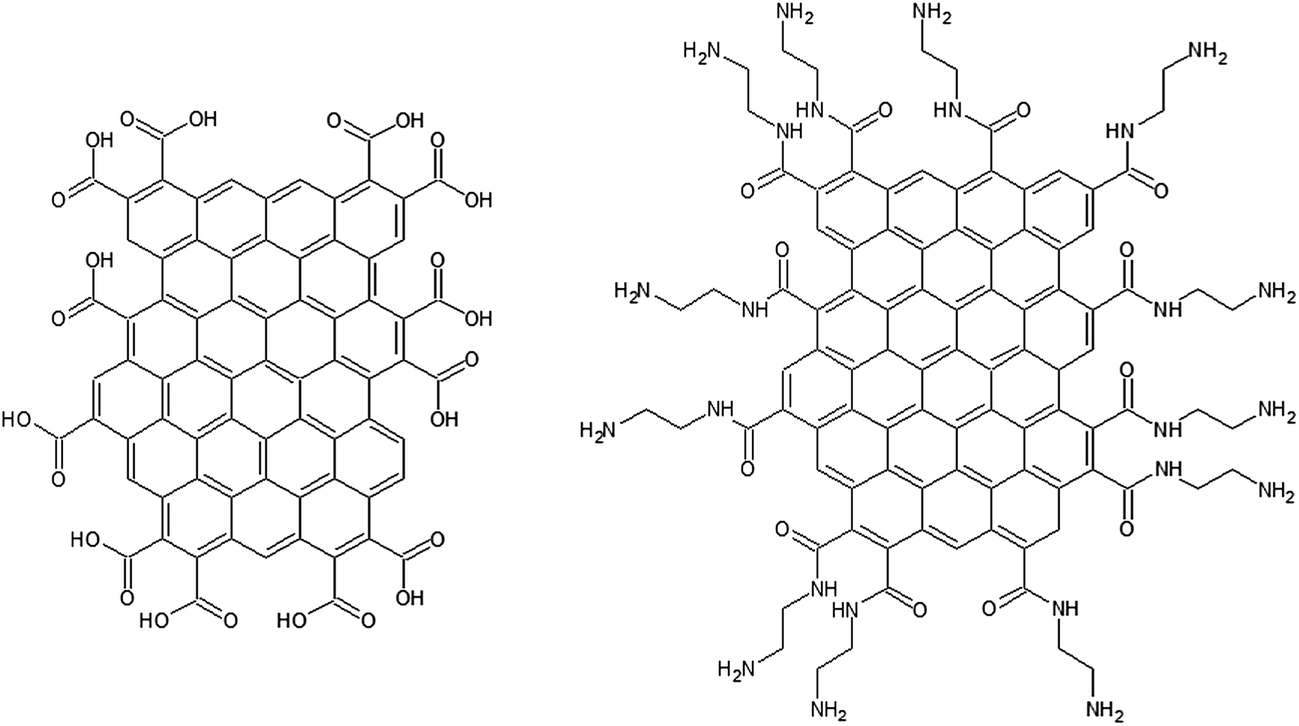 | ||
| Fig. 1 Cartoon of the structure of the COOH-terminated GNF (c-GNF) and amide-terminated GNF (a-GNF) (not to scale). | ||
2 Experimental procedures
2.1 Electrochemical measurements with GNF-modified electrodes
CV was carried out using a μ-Autolab potentiostat (Ecochemie, NL) coupled with GPES software. A boron-doped diamond (BDD) disk, 3 mm in diameter and sealed in PEEK (Windsor Scientific) was used as the working electrode, either unmodified or modified with a layer of adsorbed GNF. A platinum wire, coiled at the end to increase the surface area, served as a counter electrode. The reference electrode was Ag/AgCl in saturated KCl and all potentials are reported relative to it. The BDD electrode was polished using a 1.0 micron alumina suspension, rinsed with ultrapure water, and polished again using a 0.05 micron alumina suspension. Finally, the electrode was rinsed thoroughly with ultrapure water and dried using an ambient air flow. The GNF samples were drop-cast from aqueous suspensions of known concentration onto the freshly polished BDD electrode using a micropipette and allowed to dry under ambient conditions. After drying, the electrode was rinsed thoroughly with ultrapure water to remove any poorly adhered material from the surface and dried using an ambient air flow. The resulting amount of GNF on the electrode was estimated at 1.5 ± 0.5 μg in all experiments, and all CVs were recorded using a freshly modified electrode.Electrochemical experiments were carried out in phosphate buffer solution (PBS) or KCl as background electrolyte (for concentrations refer to text). PBS of different pH in the range 4.5 to 9.2 was prepared by mixing different proportions of the constituent buffer salts KH2PO4 and K2HPO4. Redox probes hydroquinone (H2Q), ferrocenemethanol (FcMeOH) and potassium ferricyanide (K3[Fe(CN)6]) were obtained from Sigma Aldrich and used as received. All solutions were prepared fresh daily with doubly deionised water, taken from a Milli-Q water purification system, with a resistivity of not less than 18.2 MΩ cm at 25 °C. For experiments in deoxygenated solutions and with air-sensitive chemicals such as H2Q, high purity argon was bubbled through electrolyte solutions for 30 minutes to remove dissolved oxygen, and the gas flow was maintained over the surface of the solution during electrochemical experiments.
2.2 Synthesis and characterisation of c-GNF and a-GNF
The c-GNF were prepared from chemical oxidation of carbon nanotubes, as reported previously38 with some modification to the method to allow for scale-up.39 For the amidation reaction, the c-GNF were first treated with oxalylchloride in THF to prepare the acid chlorides, and then reacted with an excess of ethylenediamine in dry THF. The amidated product (a-GNF) was washed with water several times and dried in a vacuum. Characterisation of the flakes was carried out using X-ray photoelectron spectroscopy (XPS) with a Thermo Scientific K-Alpha XPS system with a monochromated Al K-alpha source (E = 1486.6 eV). XPS peak fitting was achieved using XPSPeak software. Fourier Transform Infra-Red (FTIR) spectroscopy was performed using a Bruker Tensor 27 spectrometer and the Opus data collection programme in Attenuated Total Reflectance (ATR) mode with a diamond crystal as the internal reflection element. Average diameter of the flakes was determined using Tapping Mode Atomic Force Microscopy, with the GNF spin-coated onto a smooth HOPG substrate.An equivalence point and approximate pKa for the c-GNF was obtained by titration of an aqueous suspension of dispersed c-GNF with NaOH. The hydrophilic nature of the COOH edge groups means the GNF disperse readily in water and other polar solvents. The NaOH solution was standardised prior to titration using potassium hydrogen phthalate (KHP). NaOH and KHP were placed in a desiccator for 12 hours prior to use. Water was either boiled or deoxygenated with argon before use. All solutions were kept under argon throughout the experiment. A micropipette was used to measure the volume of NaOH additions.
3 Results
3.1 Characterisation of COOH- and amide- terminated GNF by XPS and FTIR
The C1s region XPS spectra of the c-GNF used for this study were similar to the data shown in Fig. 2(b) in ref. 38. The dominant carbon bonding environments are consistent with sp2 carbon (CC, ca. 285 eV) and the presence of carboxylic acid groups (ca. 289 eV). The lack of a peak corresponding to sp3 carbon–carbon bonding confirms the high aromaticity and low defect density of the GNF interior. Thus the XPS data confirm both the defect-free nature of the material and the uniformity and high density of the COOH edge groups. The a-GNF give rise to a C1s XPS spectrum consistent with full conversion of the COOH termination to C(O)NHC2H2C2H2NH2 functionalities. FTIR spectra of c-GNF and a-GNF samples are consistent with the schematic structures shown in Fig. 1 and the C1s XPS data as well as FTIR spectra reported previously.38 Carbonyl stretches at ca. 1720 cm−1 and 1650 cm−1 are observed for the COOH- and amide- terminated GNF respectively, confirming the presence of these functionalities. Given reports that GO materials experience chemical transformation between oxygen functionalities over time in water40 we recorded the IR response of c-GNF suspended in water at regular intervals over six months. There was no change to the IR spectrum over this time period indicating that the COOH functionalities remained intact and stable almost indefinitely in water.
3.2 Determination of acidity of c-GNF using pH titration
Fig. 2 shows the titration curve for the addition of aliquots of (236 ± 6) × 10−4 M NaOH to an aqueous suspension of c-GNF. As the XPS and IR characterisation show no detectable concentration of other acidic functionalities present in the flakes, the observed behaviour can be attributed solely to the COOH edge groups. | ||
| Fig. 2 Titration curve (black) and first derivative (red) for c-GNF. | ||
When the weak acid COOH edge groups of the c-GNF are exposed to water a dynamic equilibrium is established, where the acid groups become deprotonated:
| GNF-COOH + H2O ↔ GNF-COO− + H3O+ | (1) |
Addition of a small amount of strong base to the solution results in reaction of OH− with the solution protons and hence the equilibrium is perturbed. Once the number of mols of OH− added is equal to the number of weak acid groups (the equivalence point, found from the first derivative curve), further addition of base results in a rapid increase in pH. For c-GNF we observe more complex behaviour than would be expected for a single acid species dissolved in water. On addition of ca. 200 to 400 μl NaOH an increase in pH is observed, but this is not the sharp rise expected if all of the acid groups underwent deprotonation with the same pKa. It is probable that different bonding environments or electrostatic/hydrogen-bonding interactions between neighbouring groups result in a range of acid–base behaviours among the edge-group population. After addition of further NaOH an inflection point is observed in the first derivative curve, which we interpret as the point at which all COOH groups are fully deprotonated and which gives us an equivalence point of pH 8.3. This allows us to estimate the number of COOH groups as 7 × 10−3 mol of acid groups per gram of GNF material. The pKa of a weak acid is defined as the pH at which half of the base required to reach the equivalence point is added and for the c-GNF pKa is therefore estimated as 4.5. However given the wide pH range over which deprotonation is observed, the usefulness in reporting a single pKa value for the c-GNF is questionable.
3.3 Electrochemical response of electrode-immobilised GNF in PBS
Fig. 3 shows the response of c-GNF, a-GNF and clean BDD in pH 4.6 and 9.2 solutions over the potential range −0.3 V to 1.0 V. In both solution conditions the response of the GNF layer can be observed over the background response of the BDD at potentials above ca. 0.4 V and below −0.2 V. Currents are larger for the a-GNF than c-GNF under the same conditions. No faradaic peaks are observed for the GNF modified electrodes in the range 0 to 0.4 V. As shown in the inset to Fig. 3, in deoxygenated solution the cathodic currents below −0.2 V are greatly diminished, showing the reduction currents in the main figure can be attributed to oxygen reduction. This indicates that both GNF samples can catalyse oxygen reduction better than the BDD, which is a poor electrocatalyst for this reaction in comparison to sp2 carbon materials. The oxidation response is unaffected by the presence of oxygen in solution and is attributed to direct oxidation of the GNF.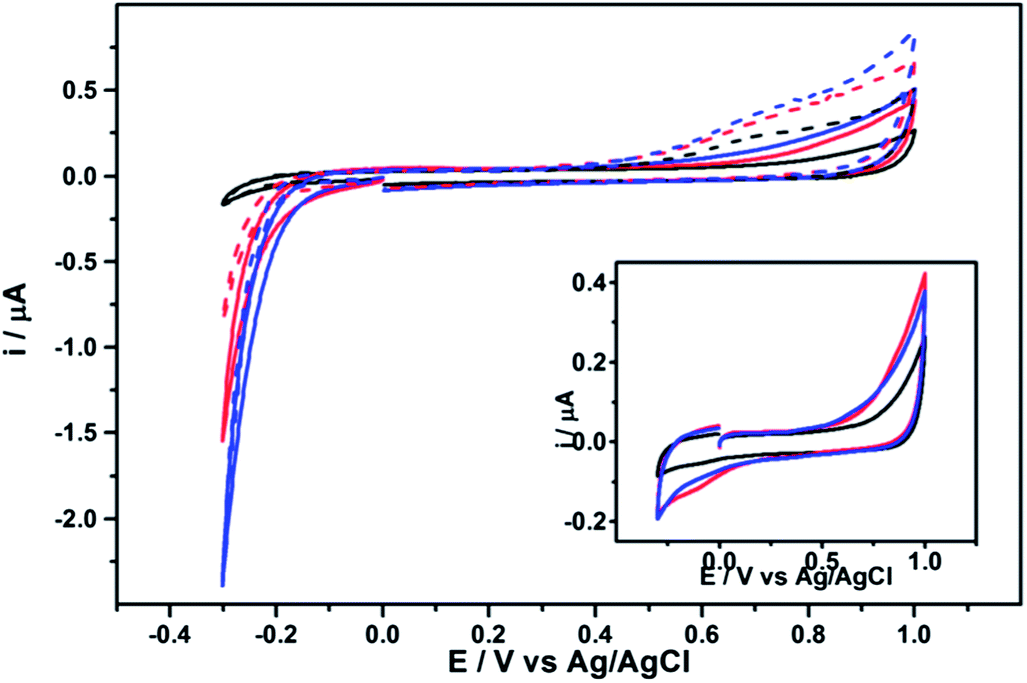 | ||
| Fig. 3 Background response of BDD and GNF in pH 4.6 and 9.2. Black line: clean BDD. Red line: c-GNF. Blue line: a-GNF. Solid line: 0.1 M KH2PO4 (pH 4.6), dashed line: 0.1 M K2HPO4 (pH 9.2). Inset: 0.1 M KH2PO4 solution deoxygenated with argon. Scan rate 50 mV s−1. First scans shown. | ||
These data show that the GNF adhere well to the surface of the BDD electrode for the duration of the experiment. At the present time it is unclear in what orientation the GNF are arranged on the electrode surface, as their small size and transparency makes the immobilised layer difficult to characterise. However as the coverage and flake orientation will clearly be important for fully understanding the electrochemical response, work is ongoing to determine and control the morphology of the electrode layer.
3.4 Electrochemical response of c-GNF and a-GNF towards FcMeOH
The CV response of the c-GNF modified electrode towards 0.5 mM FcMeOH was compared to that obtained at clean BDD under the same conditions, using different pH PBS as the background electrolyte (Fig. 4). No difference in electrochemical response could be discerned between the BDD and the GNF-modified BDD under any conditions. For both electrode types the response was that of a reversible one electron transfer with a ΔEp = |Epox − Epred| = 60 mV at a scan rate of 100 mV s−1, close to the theoretical expected value of 59 mV for a one electron process. At scan rates up to 1 V s−1 the response remained identical at both clean BDD and the GNF modified electrode (ΔEpca. 80 mV at both electrodes). Electrode modification with a-GNF likewise showed no change in the CV response from that of clean BDD. | ||
| Fig. 4 CVs of 0.5 mM FcMeOH (a) at clean BDD in 0.1 M KH2PO4 (pH 4.6) (black line) and 0.1 M K2HPO4 (pH 9.2) (red line); at c-GNF modified BDD in 0.1 M KH2PO4 (pH 4.6) (blue line) and 0.1 M K2HPO4 (pH 9.2) (orange line); (b) at clean BDD in 0.1 M KH2PO4 (pH 4.6) (black line) and 0.1 M K2HPO4 (pH 9.2) (red line); at a-GNF modified BDD in 0.1 M KH2PO4 (pH 4.6) (blue line) and 0.1 M K2HPO4 (pH 9.2) (orange line). Scan rate 100 mV s−1 (solid line), 1 V s−1 (dashed line). | ||
This result is not unexpected, as the FcMeOH/FcMeOH+ redox couple is known to be relatively surface-insensitive and outer-sphere in nature. However, adsorption of this species to graphene3 has been reported, indicating some surface interaction that could influence the electrochemical response. In this case we see no evidence of adsorption and also no indication that the protonation state of the c-GNF plays any role in the redox response of this probe. The GNF materials likewise show no electrode blocking effects that inhibit electrochemical response and no sign of limitation in electron transfer kinetics due to low density of states or lack of surface adsorption sites. On the other hand no enhancement in electron transfer kinetics is noted either, although the response at the underlying BDD is also close to reversible, so it would be difficult to determine any improvement.
3.5 Electrochemical response of c-GNF and a-GNF towards ferricyanide
In this GNF study we have two very clearly defined types of edge functionality with which to probe the interaction of ferricyanide with carbon electrode surfaces. The high density of COOH groups available on the c-GNF allows us to study both the electrostatic interaction between the ferricyanide and acid groups in different protonation states and the effect of acid–base equilibria on the redox response. The a-GNF allows us to probe the influence of the electronegative carbonyl moieties but in the absence of the deprotonation equilibria exhibited by the COOH groups.Fig. 5a shows CVs for 0.5 mM ferricyanide at a clean BDD electrode in background electrolyte of 0.1 M pH 4.6 and 9.2 PBS. At the BDD electrode the peak separation ΔEp remains constant at 65 ± 2 mV over the whole pH range examined (pH 4.6–9.2) indicating close to reversible electron transfer kinetics. The E0′ of the couple, taken as ½(Epox + Epred), shifts towards higher values with increasing pH, being found at ca. 50 mV higher E at pH 9.2 than at pH 4.6. The peak currents for oxidation and reduction also decrease marginally over the same pH range. The ferri/ferrocyanide electron transfer process has been shown to be inhibited at oxygen-terminated BDD surfaces;41 however we observe effectively reversible electron transfer kinetics at the (oxidised) BDD electrodes used in this study. Fig. 5a suggests a small degree of interaction of [Fe(CN)6]3− with the BDD surface, however the effect is very small in comparison with some previous studies.
 | ||
| Fig. 5 The influence of pH on the ferri/ferrocyanide redox reaction at different electrodes. (a) Clean BDD; (b): BDD modified with c-GNF; (c): BDD modified with a-GNF. Supporting electrolyte: 0.1 M KH2PO4 (pH 4.6) (black line); 0.1 M K2HPO4 (pH 9.2) (red line). Potassium ferricyanide concentration 0.5 × 10−3 M. Scan rate: 50 mV s−1. | ||
Fig. 5b shows the response of 0.5 mM ferricyanide at pH 4.6 and 9.2 at a c-GNF modified electrode. The response at this electrode is found to be very dependent on pH, particularly for pH < 8. Peak currents for both oxidation and reduction decrease and ΔEp increases as the pH is lowered: at pH 7 ΔEp = 109 mV; pH 6 ΔEp = 120mV; pH 5 ΔEp = 213 mV and pH 4.6 ΔEp = 250 mV. This indicates that electron transfer becomes slower under these experimental conditions, which could be attributed to a change in the nature of the redox molecule, an unfavourable interaction with the electrode surface (or loss of a favourable interaction) or formation of an adsorbed inhibiting layer on the electrode. The response is not time or potential dependent, as it is observed immediately from the first CV scan and the response does not get worse with cycling (currents rather increase marginally with consecutive scans). This would indicate that the effect cannot be attributed to formation of a surface film that deposits as a function of time or applied potential. However it does not preclude the fast, spontaneous formation of an adsorbed layer, formed independently of applied potential.
Fig. 5c shows the response of the a-GNF modified electrode towards 0.5 mM ferricyanide in 0.1 M pH 4.6 and pH 9.2 PBS. For this electrode there is no pH dependence on the voltammetric response and the electron transfer kinetics appear only slightly less reversible than at clean BDD (ΔEp = 70 ± 1 mV at 50 mV s−1) over pH range 4.6–9.2. In fact the response is less pH dependent than at a clean BDD electrode. Thus it is apparent that carbonyl, amide or amine functionalities have little influence on the electrochemical response of ferri/ferrocyanide at GNF-modified electrodes. The observed inhibition of current at the c-GNF electrode can therefore be attributed specifically to the presence of the acid functionalities.
To investigate the c-GNF electrode further, CVs of ferricyanide were carried out in different ionic strength solution. In these experiments the background electrolyte was KCl so the solutions are not buffered, but are all in the pH range 5.9–6.2. Fig. 6 shows CVs at clean BDD, c-GNF and a- GNF with 0.5 mM ferricyanide in 1.0 M (a), 0.1 M (b) and 0.01 M KCl (c). Increasing the concentration of the supporting electrolyte causes a shift in E0′ to more positive potentials for all electrodes studied here, confirming that the electrolyte plays a role in the redox equilibrium. At high ionic strength (1.0 M KCl) the CV response at all three electrodes is reversible, but at 0.1 M KCl currents at the c-GNF electrode are much reduced and ΔEp is significantly increased. In 0.01 M supporting electrolyte the responses at clean BDD and amide-GNF modified electrodes are still reversible but the CV at the c-GNF electrode shows significant inhibition. The response appears sigmoidal, resembling the CV expected at an array of microelectrodes, or response through pinholes of an electrode partially covered in insulating material. If the experiment is repeated at similarly low ionic strength, but with the addition of KOH to bring the pH to 8.5, the CV returns to a more reversible form (albeit with ΔEp = 231 ± 21 mV) indicating that it is solution acidity as well as ionic strength which is the important factor in this behaviour. Quantitatively similar behaviour is seen when the experiment is repeated in the same concentrations of NaCl and PBS electrolyte.
 | ||
| Fig. 6 The influence of supporting electrolyte concentration on the ferri/ferrocyanide redox reaction at different electrodes. Black line: clean BDD; red line: BDD modified with c-GNF. Blue line: BDD modified with a-GNF. Supporting electrolyte (a) 1.0 M KCl, (b) 0.1 M KCl, (c) 0.01 M KCl. Potassium ferricyanide concentration 0.5 × 10−3 M. Scan rate: 50 mV s−1. | ||
The ferri/ferrocyanide redox couple is often used as a standard probe, even though it has been shown to be inner sphere in nature and very sensitive to the electrode surface.36,42–48 A systematic investigation into the influence of surface oxygen functionalities using glassy carbon electrodes showed that the ferricyanide CV response did not show a dependence on any specific surface oxygen groups, although it was sensitive to the presence of adsorbates.36 However, other studies have shown pH dependence in the electron transfer kinetics of this couple at carbon electrodes, the process becoming slower as pH is increased.42 This effect was attributed to the presence of surface carboxylic acid functionalities that become deprotonated and hence negatively charged in more alkaline solutions; therefore electrostatic repulsion results between the electrode surface and the negatively charged redox species. We estimate from titration of the c-GNF (Section 3.2) that the COOH edge groups are fully deprotonated at pH higher than 8, hence we might expect ferricyanide reduction to be inhibited at more alkaline pH. However we observe relatively reversible electrochemistry at pH 7 and above, and at pH 9.2 (where all of the COOH will be deprotonated and negatively charged) the response is identical to that at a clean BDD electrode. Therefore an electrostatic argument for the observed behaviour is clearly inappropriate in this case. In fact, we see slower kinetics at pH < 8, where according to the titration curve in Fig. 2 a wide range of COOH protonation states may be present. It may be that the dynamic acid–base equilibria in play at the edge of the GNF over this pH range plays a role in the observed electrochemical response.
Study of the ferri/ferrocyanide redox couple is made still more difficult due to its complex solution chemistry, in particular its preference for ion-pairing with solution cations46–48 and propensity to lose ligands and form aggregates that are intermediates to Prussian Blue film deposition.49–52 Additionally acid–base equilibria involving protonation of the nitrogen of the cyanide ligands becomes important over some pH ranges (pKa of H[Fe(CN)6]3− is ca. 4.253). At low ionic strength the electrostatic interaction between the electrode and the redox probe will be enhanced as screening by solution ions in the double layer is less effective. These conditions seem to amplify the inhibiting effect of the COOH groups on the ferricyanide electrochemistry. Additionally the stability of the ferricyanide species may also be affected by the low ionic strength conditions as ion-pairing with K+ will be less effective at the lower cation concentration. Possible factors influencing the observed electrochemistry are discussed further in Section 4 below.
3.6 Electrochemical response of c-GNF and a-GNF towards hydroquinone
The oxidation of hydroquinone (H2Q) to benzoquinone (Q) was studied at clean BDD, c-GNF modified and a-GNF modified electrodes over the pH range 5.0 to 8.5. At BDD the response was very irreversible, as has been reported previously54 with ΔEp being ca. 250 mV at pH 8 and >450 mV at pH 5. Modification of the BDD with a layer of c-GNF resulted in a decrease in peak separation (ΔEpca. 200 mV at pH 8, 250 mV at pH 5). The CV response of the BDD electrode, the c-GNF electrode and the a-GNF electrode towards 0.5 mM H2Q in 0.1 M pH 5.5 PBS is shown in Fig. 7a. A shift in both oxidation and reduction peaks towards reduced overpotential is observed at the c-GNF electrode; however the shift in oxidation peak potential is greater than that for the reduction peak. Indeed it was found that the anodic shift in the reduction peak position of ca. 40 mV compared to the peak at clean BDD was constant and independent of pH over the range tested. In contrast, the cathodic shift in the oxidation peak for c-GNF compared to BDD was pH dependent and was greater at lower pH. A small improvement in electron transfer kinetics is observed for the a-GNF electrode, with respect to the oxidation peak current, although little change to ΔEp is observed on modifying the electrode. | ||
| Fig. 7 (a) CV of 0.5 mM hydroquinone at clean BDD electrode (black line), c-GNF (red line) and a-GNF (blue line). Supporting electrolyte: 0.1 M pH 5.5 PBS. Scan rate: 50 mV s−1. First scans shown. (b): Peak potential of hydroquinone oxidation as a function of pH at clean BDD electrode (black), c-GNF (red) and a-GNF (blue). | ||
The effect of pH on the H2Q oxidation peak potential is plotted in Fig. 7b for BDD, c-GNF and a-GNF. A linear relationship is observed between peak position and pH for oxidation at the BDD electrode. Analysis of the gradient gives a 96 mV per pH unit relationship. The H2Q/Q redox reaction is usually considered a 2e−/2H+ process:
| Q + 2e− + 2H3O+ ↔ H2Q + 2H2O | (2) |
Or in alkaline solution:
| Q + 2e− + 2H2O ↔ H2Q + 2OH− | (3) |
A 59 mV shift in peak position with pH is predicted for a Nernstian 2e−/2H+ process. The deviation from this relationship observed at BDD may be related to the sluggish electrode kinetics for this reaction, which does not allow for such Nernstian analysis. Further elucidation of the reaction mechanism at BDD is beyond the scope of the current study and we confine further discussion to the observed differences at the c-GNF electrode. For the c-GNF electrode two distinct behaviours can be noted. At pH > 7 the relationship between peak position and pH is similar to that seen at BDD (93 mV per pH unit). However at pH < 7 a different gradient of 33 mV per pH unit can be fitted to the data. Clearly a change in reaction mechanism takes place at ca. pH 7, or alternatively the manner in which the H2Q reactant or Q product interacts with the c-GNF changes in this pH range.
A 33 mV shift with pH unit is consistent with a 2e−/1H+ process (predicted as 29.5 mV per pH unit):
| Q + 2e− + H3O+ ↔ HQ− + H2O | (4) |
Such a reaction mechanism is unlikely in acidic, buffered solution as it requires H2Q to be deprotonated, which is not possible given its pKa is 9.9.55 A mechanism with a 2e−/1H+ relationship would however be possible in the presence of an additional, non-solution, source of protons, such as the COOH-terminating groups offered by the c-GNF:
| Q + 2e− + GNF-COOH + H3O+ ↔ H2Q + GNF-COO− + H2O | (5) |
In the case of the H2Q oxidation reaction it has been shown that modification of glassy carbon electrodes with phthalate bases (which contain two COOH groups) shifts the oxidation potential cathodically.56 The proposed mechanism involves surface COO− groups accepting the protons liberated in the oxidation of H2Q and thus stabilising the reaction products. In effect the shift in oxidation potential is a thermodynamic consequence of the change in reaction mechanism rather than an improvement in electron transfer kinetics.
A similar process may be taking place in our system. Some improvement in electron transfer kinetics is observed on modifying the BDD electrode with GNF, as can be seen in the increase in oxidation peak currents for both a-GNF and c-GNF electrodes (Fig. 7a and b). The a-GNF electrode also shows a small cathodic shift in oxidation potential as pH is lowered, indicating improved electron transfer kinetics. H2Q/Q can interact with the GNF via hydrogen bonding or electrostatic interactions with the edge groups or by hydrophobic or π–π interactions with the GNF basal plane. Such surface adsorption has been proposed to explain the improved electron transfer kinetics for this process (both oxidation and reduction) experienced at sp2 carbon materials57 in comparison to BDD, where limited surface adsorption is believed to take place.41 However the marked change in proton concentration dependence noted at pH < 7 is unique to the c-GNF and strongly suggests the COOH groups play a role in the reaction mechanism, as shown in eqn (5). At pH > 7 the solution is sufficiently basic to allow the deprotonation accompanying H2Q oxidation to proceed predominantly via a solution phase mechanism involving OH− as the base (eqn (3)). However at pH < 7 there is a strong thermodynamic driving force for the COOH edge groups of the GNF to protonate, concomitant with conditions where there are fewer basic solution species. At this point a mechanism such as that shown in eqn (5) begins to dominate and is reflected by the change in proton concentration dependence of the oxidation peak position.
4 Discussion
The investigation described above has revealed important differences in the effect of COOH-termination and amide-termination of the GNF on electrochemical response towards ferricyanide and H2Q. Edge termination was shown to have no influence on electron transfer for FcMeOH and indeed GNF-modified and clean BDD electrodes showed no difference in response towards this outer-sphere redox species. However, strong dependence on edge termination was noted for redox processes that are inner sphere or dependent on proton concentration. Some explanation of the role of the deprotonated COOH groups was discussed above with respect to the oxidation of the H2Q species, where a direct function in the reaction mechanism was identified. However the role of the COOH/COO− functionalities in the electrochemical response of the ferri/ferrocyanide is less clear and requires further discussion.Although it is difficult to provide a definitive explanation, it is known that ferri-/ferrocyanide can be unstable in solution, particularly at low ionic strength and low pH. Cyanide ligand loss and subsequent adsorption/decomposition of ferrocyano-species onto metal and carbon electrodes are well documented.43,44,47,50–52,58 The [Fe(CN)6]4−/3− redox reaction is believed to take place via activated ion-paired complexes such as K2[Fe(CN)6]2−/1−. If these ion-pair complexes cannot form, for example at low ionic strength, then the electron transfer rate is much slower.46 The electrochemical response of ferricyanide in the presence of c-GNF at pH < 7 suggests a lack of stability of the ion-paired redox species and hence sluggish electron transfer kinetics. At low ionic strength the response is consistent with a spontaneous deposition of blocking species on the electrode surface, indicating the real lack of stability of the redox molecule in these solution conditions.
In the pH range below 8 the c-GNF are clearly involved in dynamic protonation equilibria. Due to the differing bonding environments of the COOH groups and electrostatic and hydrogen-bonding interactions between neighbouring groups, protonation/deprotonation takes place over a wide pH range, as shown by the titration curve in Fig. 2. The instability of ferricyanide in these conditions may be due to the environment within the diffusion layer surrounding the GNF, where some of the carboxylic acid groups may be acidic enough to protonate the ferricyanide, promoting cyanide ligand loss in the form of HCN and allowing deposition of films similar in nature to Prussian Blue.59 Solutions of ferricyanide at pH 3.6 have been reported to have different UV-Vis spectral features to those at higher pH (indicating protonation or ligand loss) and to develop blue precipitates on standing.60 Although our solution pH values of 4.6–6 would not be considered acidic enough to cause decomposition of ferricyanide, a higher concentration of protons may be present close to the electrode surface due to the high density of carboxylic acid functionalities. The effect is exacerbated in low ionic strength solutions, as ferricyanide is considerably less stable in solution in the absence of ion-pairing to K+.
Interestingly, when experiments with the c-GNF are repeated with the [Ru(CN)6]4− as the redox couple the CV response is found to be independent of pH. The process appears reversible over the pH range 4.5–9.2 with no evidence of the inhibition and proposed surface film formation seen for [Fe(CN)6]3−. This would suggest that a mechanism requiring specific interaction between the COOH groups and the cyanide ligands of the redox species can be ruled out and it is more likely the complex solution chemistry of the ferricyanide molecule that results in the observed response.
5 Conclusion
The influence of edge group termination of GNF materials on electrochemical performance has been studied with respect to interaction with the common redox probes FcMeOH, ferricyanide and H2Q. Neither a-GNF nor c-GNF show a detrimental effect on electron transfer rate with respect to the outer-sphere FcMeOH redox couple or indeed the more surface sensitive ferricyanide at pH > 8 (when c-GNF is fully deprotonated). These results are consistent with the observed fast electron transfer kinetics towards these species obtained using single layer graphene electrodes.3,4 The high density of carboxylic acid or amide functionalities do not appear to perturb the electrochemical response under these reaction conditions.The very high density of carboxylic acid groups of the c-GNF flakes and the absence of other oxygen-containing functionalities allow us to specifically investigate the effect of these highly charged and acidic groups on electrochemical response. Titration experiments reveal that in solution pH of ca. 4 to 8 the edge groups are present in a range of protonation states. In these intermediate states the c-GNF has a dramatic influence on the electrode response towards ferricyanide reduction. The response towards H2Q reduction likewise shows deviation from the response at clean BDD at pH < 7 and in more acidic pH conditions the edge groups clearly play a role in the reaction mechanism. Although more investigations are required to fully understand the role of the partly protonated acid edge groups, it is clear that they have a significant influence on the observed electrochemistry.
Oxidised carbon electrodes will present a range of surface oxygen functionalities. Hence it can be difficult to isolate the interaction of redox probes with a specific surface moiety. The strength of this study are the well-characterised and uniform GNF that enable us to attribute changes in electrochemical response to specific functionalities. However the exact arrangement of the COOH edge groups on the outside of the GNF, the electrostatic interactions with neighbours and degree of hydrogen bonding still remain to be elucidated. Further studies are underway to understand the dynamic environment of the GNF edges in solution conditions.
Acknowledgements
We thank EPSRC/UCL Chemistry Department for studentship for Mailis Lounasvuori and Martin Rosillo-Lopez. Christoph Salzmann thanks the Royal Society for the award of a URF. The work was funded partly by EPSRC grant EP/J010006/1.References
- N. G. Shang, P. Papakonstantinou, M. McMullan, M. Chu, A. Stamboulis, A. Potenza, S. S. Dhesi and H. Marchetto, Adv. Funct. Mater., 2008, 18, 3506–3514 CrossRef CAS PubMed.
- D. A. C. Brownson and C. E. Banks, Phys. Chem. Chem. Phys., 2011, 13, 15825–15828 RSC.
- W. Li, C. Tan, M. A. Lowe, H. D. Abruña and D. C. Ralph, ACS Nano, 2011, 5, 2264–2270 CrossRef CAS PubMed.
- A. T. Valota, I. A. Kinloch, K. S. Novoselov, C. Casiraghi, A. Eckmann, E. W. Hill and R. A. W. Dryfe, ACS Nano, 2011, 5, 8809–8815 CrossRef CAS PubMed.
- A. Bonanni, A. Ambrosi and M. Pumera, Chem. - Eur. J., 2012, 18, 4541–4548 CrossRef CAS PubMed.
- C. Tan, J. Rodríguez-López, J. J. Parks, N. L. Ritzert, D. C. Ralph and H. D. Abruña, ACS Nano, 2012, 6, 3070–3079 CrossRef CAS PubMed.
- E. Kibena, M. Mooste, J. Kozlova, M. Marandi, V. Sammelselg and K. Tammeveski, Electrochem. Commun., 2013, 35, 26–29 CrossRef CAS PubMed.
- N. L. Ritzert, J. Rodríguez-López, C. Tan and H. D. Abruña, Langmuir, 2013, 29, 1683–1694 CrossRef CAS PubMed.
- B. Zhang, L. Fan, H. Zhong, Y. Liu and S. Chen, J. Am. Chem. Soc., 2013, 135, 10073–10080 CrossRef CAS PubMed.
- K. Xu, P. Cao and J. R. Heath, Nano Lett., 2009, 9, 4446–4451 CrossRef CAS PubMed.
- F. Guinea, M. I. Katsnelson and M. A. H. Vozmediano, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 075422 CrossRef.
- A. Ambrosi, A. Bonanni, Z. Sofer, J. S. Cross and M. Pumera, Chemistry (Weinheim an der Bergstrasse, Germany), 2011, 17, 10763–10770 CAS.
- C. Shan, H. Yang, J. Song, D. Han, A. Ivaska and L. Niu, Anal. Chem., 2009, 81, 2378–2382 CrossRef CAS PubMed.
- M. Zhu, C. Zeng and J. Ye, Electroanalysis, 2011, 23, 907–914 CrossRef CAS PubMed.
- Y.-R. Kim, S. Bong, Y.-J. Kang, Y. Yang, R. K. Mahajan, J. S. Kim and H. Kim, Biosens. Bioelectron., 2010, 25, 2366–2369 CrossRef CAS PubMed.
- M. Mallesha, R. Manjunatha, C. Nethravathi, G. S. Suresh, M. Rajamathi, J. S. Melo and T. V. Venkatesha, Bioelectrochemistry, 2011, 81, 104–108 CrossRef CAS PubMed.
- H. Du, J. Ye, J. Zhang, X. Huang and C. Yu, J. Electroanal. Chem., 2011, 650, 209–213 CrossRef CAS PubMed.
- D. A. C. Brownson, L. J. Munro, D. K. Kampouris and C. E. Banks, RSC Adv., 2011, 1, 978–988 RSC.
- K. R. Ratinac, W. Yang, J. J. Gooding, P. Thordarson and F. Braet, Electroanalysis, 2011, 23, 803–826 CrossRef CAS PubMed.
- Q. Y. He, S. X. Wu, Z. Y. Yin and H. Zhang, Chem. Sci., 2012, 3, 1764–1772 RSC.
- S. X. Wu, Q. Y. He, C. L. Tan, Y. D. Wang and H. Zhang, Small, 2013, 9, 1160–1172 CrossRef CAS PubMed.
- M. Pumera, A. Ambrosi and E. L. K. Chng, Chem. Sci., 2012, 3, 3347–3355 RSC.
- C. X. Lim, H. Y. Hoh, P. K. Ang and K. P. Loh, Anal. Chem., 2010, 82, 7387–7393 CrossRef CAS PubMed.
- R. Sharma, J. H. Baik, C. J. Perera and M. S. Strano, Nano Lett., 2010, 10, 398–405 CrossRef CAS PubMed.
- M. A. Edwards, P. Bertoncello and P. R. Unwin, J. Phys. Chem. C, 2009, 113, 9218–9223 CAS.
- S. C. S. Lai, A. N. Patel, K. McKelvey and P. R. Unwin, Angew. Chem., Int. Ed., 2012, 51, 5405–5408 CrossRef CAS PubMed.
- A. N. Patel, M. G. Collignon, M. A. O'Connell, W. O. Y. Hung, K. McKelvey, J. V. Macpherson and P. R. Unwin, J. Am. Chem. Soc., 2012, 134, 20117–20130 CrossRef CAS PubMed.
- N. Ebejer, A. G. Gueell, S. C. S. Lai, K. McKelvey, M. E. Snowden and P. R. Unwin, Annu. Rev. Anal. Chem., 2013, 6, 329–351 CrossRef CAS PubMed.
- A. N. Patel, S.-y. Tan and P. R. Unwin, Chem. Commun., 2013, 49, 8776–8778 RSC.
- A. G. Gueell, N. Ebejer, M. E. Snowden, J. V. Macpherson and P. R. Unwin, J. Am. Chem. Soc., 2012, 134, 7258–7261 CrossRef PubMed.
- T. S. Miller, N. Ebejer, A. G. Gueell, J. V. Macpherson and P. R. Unwin, Chem. Commun., 2012, 48, 7435–7437 RSC.
- A. G. Gueell, K. E. Meadows, P. V. Dudin, N. Ebejer, J. V. Macpherson and P. R. Unwin, Nano Lett., 2014, 14, 220–224 CrossRef PubMed.
- Z. Li, Y. Wang, A. Kozbial, G. Shenoy, F. Zhou, R. McGinley, P. Ireland, B. Morganstein, A. Kunkel, S. P. Surwade, L. Li and H. Liu, Nat. Mater., 2013, 12, 925–931 CrossRef CAS PubMed.
- C. A. McDermott, K. R. Kneten and R. L. McCreery, J. Electrochem. Soc., 1993, 140, 2593–2599 CrossRef CAS PubMed.
- P. Chen, M. A. Fryling and R. L. McCreery, Anal. Chem., 1995, 67, 3115–3122 CrossRef CAS.
- P. Chen and R. L. McCreery, Anal. Chem., 1996, 68, 3958–3965 CrossRef CAS.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS PubMed.
- C. G. Salzmann, V. Nicolosi and M. L. H. Green, J. Mater. Chem., 2010, 20, 314–319 RSC.
- C. G. Salzmann and M. Rosillo-Lopez, in preparation.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576–588 CrossRef CAS PubMed.
- M. C. Granger, M. Witek, J. Xu, J. Wang, M. Hupert, A. Hanks, M. D. Koppang, J. E. Butler, G. Lucazeau, M. Mermoux, J. W. Strojek and G. M. Swain, Anal. Chem., 2000, 72, 3793–3804 CrossRef CAS.
- M. R. Deakin, K. J. Stutts and R. M. Wightman, J. Electroanal. Chem., 1985, 182, 113–122 CrossRef CAS.
- C. Lee and F. C. Anson, J. Electroanal. Chem., 1992, 323, 381–389 CrossRef CAS.
- C. M. Pharr and P. R. Griffiths, Anal. Chem., 1997, 69, 4673–4679 CrossRef CAS.
- M. C. Granger and G. M. Swain, J. Electrochem. Soc., 1999, 146, 4551–4558 CrossRef CAS PubMed.
- L. M. Peter, W. Dürr, P. Bindra and H. Gerischer, J. Electroanal. Chem., 1976, 71, 31–50 CrossRef CAS.
- C. Beriet and D. Pletcher, J. Electroanal. Chem., 1993, 361, 93–101 CrossRef CAS.
- M. Noel and P. N. Anantharaman, Analyst, 1985, 110, 1095–1103 RSC.
- W. Huang and R. McCreery, J. Electroanal. Chem., 1992, 326, 1–12 CrossRef CAS.
- J. Kawiak, P. J. Kulesza and Z. Galus, J. Electroanal. Chem., 1987, 226, 305–314 CrossRef CAS.
- S. Pons, M. Datta, J. F. McAleer and A. S. Hinman, J. Electroanal. Chem., 1984, 160, 369–376 CrossRef CAS.
- A. Więckowski and M. Szklarzyk, J. Electroanal. Chem., 1982, 142, 157–170 CrossRef.
- P. L. Domingo, B. Garcia and J. M. Leal, Can. J. Chem., 1987, 65, 583–589 CrossRef CAS.
- I. Duo, C. Levy-Clement, A. Fujishima and C. Comninellis, J. Appl. Electrochem., 2004, 34, 935–943 CrossRef CAS.
- J. H. Baxendale and H. R. Hardy, Trans. Faraday Soc., 1953, 49, 1140–1144 RSC.
- J. Medina-Ramos, T. M. Alligrant, A. Clingenpeel and J. C. Alvarez, J. Phys. Chem. C, 2012, 116, 20447–20457 CAS.
- S. H. DuVall and R. L. McCreery, Anal. Chem., 1999, 71, 4594–4602 CrossRef CAS.
- A. A. Karyakin, Electroanalysis, 2001, 13, 813–819 CrossRef CAS.
- P. L. Domingo, B. García and J. M. Leal, Can. J. Chem., 1990, 68, 228–235 CrossRef CAS.
- R. Yang, Z. Qian and J. Deng, J. Electrochem. Soc., 1998, 145, 2231–2236 CrossRef CAS PubMed.
Footnote |
| † Presenting author. E-mail: E-mail: d.j.caruana@ucl.ac.uk. |
| This journal is © The Royal Society of Chemistry 2014 |