 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Studying chemical reactivity in a virtual environment
Moritz P.
Haag
and
Markus
Reiher
*
Laboratorium für Physikalische Chemie, ETH Zürich, Vladimir-Prelog-Weg 2, CH-8093 Zürich, Switzerland. E-mail: markus.reiher@phys.chem.ethz.ch; Fax: +41 44 633 15 94; Tel: +41 44 633 43 08
First published on 28th February 2014
Abstract
Chemical reactivity of a set of reactants is determined by its potential (electronic) energy (hyper)surface. The high dimensionality of this surface renders it difficult to efficiently explore reactivity in a large reactive system. Exhaustive sampling techniques and search algorithms are not straightforward to employ as it is not clear which explored path will eventually produce the minimum energy path of a reaction passing through a transition structure. Here, the chemist's intuition would be of invaluable help, but it cannot be easily exploited because (1) no intuitive and direct tool for the scientist to manipulate molecular structures is currently available and because (2) quantum chemical calculations are inherently expensive in terms of computational effort. In this work, we elaborate on how the chemist can be reintroduced into the exploratory process within a virtual environment that provides immediate feedback and intuitive tools to manipulate a reactive system. We work out in detail how this immersion should take place. We provide an analysis of modern semi-empirical methods which already today are candidates for the interactive study of chemical reactivity. Implications of manual structure manipulations for their physical meaning and chemical relevance are carefully analysed in order to provide sound theoretical foundations for the interpretation of the interactive reactivity exploration.
1 Introduction
A detailed understanding of the atomic rearrangements occurring during a chemical reaction is at the heart of chemistry. Computational investigations based on first principles of quantum mechanics can provide such a microscopic picture. They also complement the findings of experiments by making information on reaction steps accessible for which experimental data are not available.Usually, the elucidation of reaction mechanisms starts with the set up of structures which are likely to be close to either the reactants, the products or the transition state structures. Subsequent optimisation of the electronic structure and of the atomic arrangement is computationally expensive, and it is by no means guaranteed that these optimisations produce the desired mechanism. A trial-and-error protocol has to be repeated until all potentially relevant reaction steps have been identified. In most cases the build-up of the starting structures is carried out in conventional molecule editors using a computer mouse and a three dimensional ball-and-stick representation of the structure.
Despite the remarkable progress in the development of fast algorithms for electronic structure calculations and the continuously increasing computer power, it is still a challenge to predict reaction mechanisms of molecular systems consisting of a few hundred atoms. The reason for this is the inherently high complexity induced in the potential energy surface by the large number of degrees of freedom of such systems. Already for an elementary reaction step, a transition-state search requires starting structures from which local optimisations can be initiated and such optimisations may easily fail. Then, only little additional information is provided on how to improve on such a failed search.
In addition, technical hurdles need to be overcome. Firstly, the build-up of three dimensional chemical structures with two dimensional input devices is a slow and cumbersome procedure. Secondly, manipulating the system and receiving the resulting response from the calculations are sequential and delayed processes that often require different computer programs. This separation is not only due to the electronic structure optimisation being computationally expensive, but also due to the fact that there exists currently no general implementation to have the manipulation of the system and the instantaneous presentation of the quantum chemical reactivity response in the same program.
Both issues can efficiently be resolved by having all the processes united in a single virtual environment. Such a virtual environment allows for a deeper immersion of the scientist into the problem. In addition to the visual presentation the complex output of quantum chemical calculations can be perceived more intuitively by addressing further human senses such as the haptic sense. The main advantage then is the seamless combination of manipulation and output presentation. It allows the chemist to exploit prior knowledge about the system and his/her ‘chemical intuition’ for the exploration of reaction mechanisms.
Studying complex phenomena in a more intuitive way is not entirely new to chemists and biologists. For molecular dynamics,1,2 protein docking3 and structural biology4 interactive approaches and even haptic interactions have been applied. The idea of an interactive build-up of molecular assemblies with continuous energy minimisation in the background for a quick assembly of reasonable structures has recently been implemented for parametrised analytical potentials and classical force fields in the molecule editors SAMSON5 and AVOGADRO.6
In 2007, we initiated a research program to explore chemical reaction mechanisms interactively with quantum chemical approaches. We started with the haptic exploration of interpolated Born–Oppenheimer potential energy surfaces within the framework of what we call Haptic Quantum Chemistry.7 During such a haptic exploration scientists are able to feel the quantum mechanical forces exerted on the atoms, when they are moved with an input device capable of providing force-feedback. We further extended this approach to larger systems and presented a protocol to refine potential energy surfaces guided by the haptic exploration.8 To be able to treat structural relaxation in complex systems we also investigated the possibility to calculate the necessary forces directly.9 Recently, we have investigated real-time reactivity exploration10 in the framework of the SAMSON environment, for which a fast semi-empirical method for structure optimisations of hydrocarbons is already available.11,12
In this work, we explore the physical meaning of externally controlled configurational changes in molecular assemblies. We work out the requirements for treating molecular assemblies in virtual environments and how much of the underlying physical theory can be recovered in an interactive exploration of chemical reactivity. The goal is to provide an intuitive and at the same time chemically meaningful experience of a reactive molecular system.
The interactive study of reaction mechanisms in a virtual environment as described in this work is envisaged as being part of an even larger framework comprising additional automatic or semi-automatic tools supporting the investigation of complete reaction networks. We, therefore, start in section 2 with a description of this bigger picture to clarify the context of the methodology described in this work. The main ingredient for an interactive study of chemical reactivity is a real-time availability of first-principles forces, which will be detailed in section 3. We continue then by dissecting the main components of a reactivity exploration in a virtual environment in section 5. In section 6 the manipulation and the presentation of molecular systems are discussed in detail. This is then followed by an analysis of the physical laws governing a molecular system in a virtual environment in section 7. We then conclude the work and provide an outlook at the end of this paper.
2 Virtual environment for the investigation of reaction mechanisms
The interplay between the various components of the virtual framework introduced in this work for the investigation of reaction networks is summarised in Fig. 1. The figure highlights the essential components of the framework and serves as a blue print for a quantum chemical tool for the exploration of reaction networks.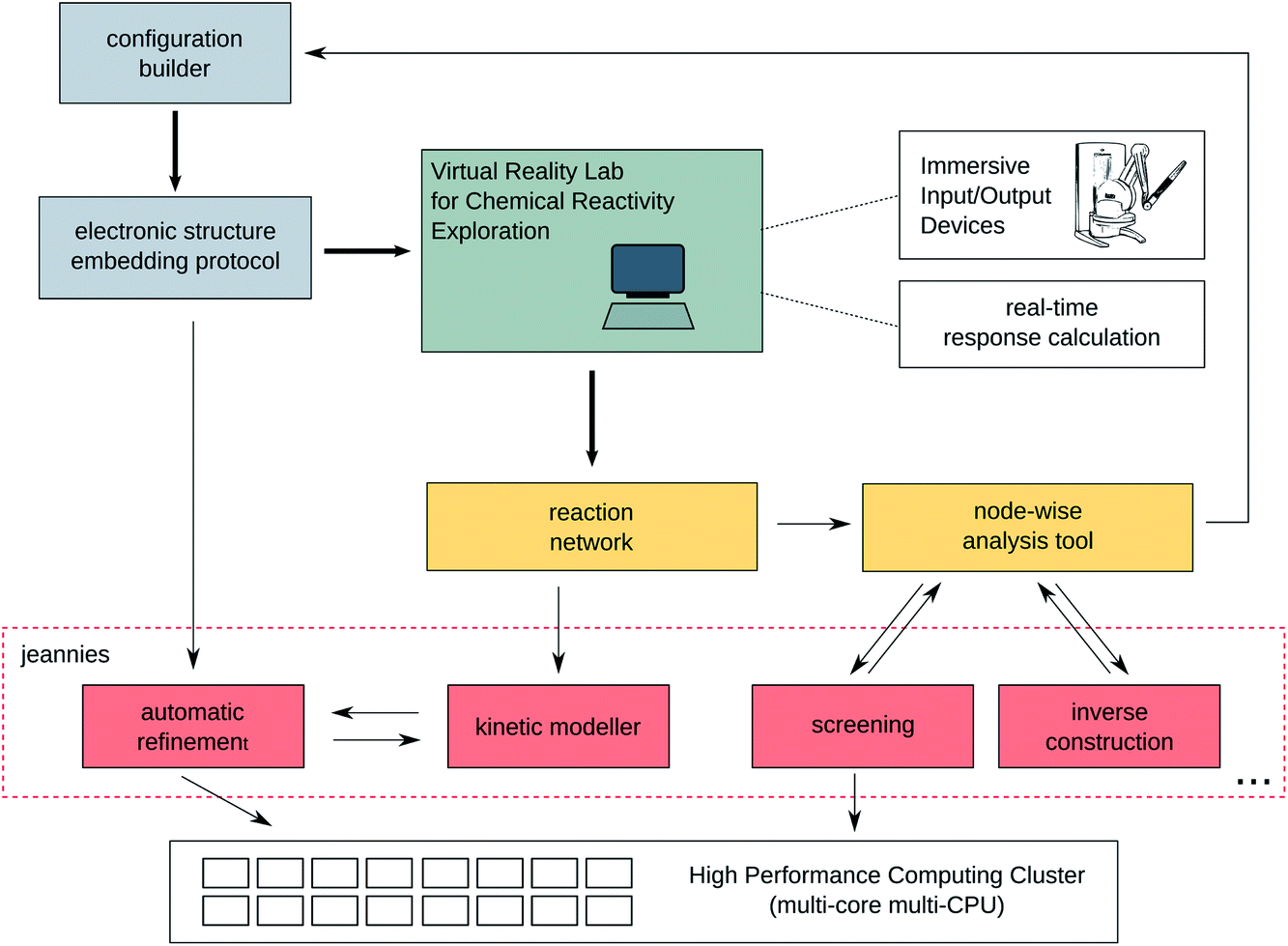 | ||
| Fig. 1 Design of a virtual environment for the investigation of reaction mechanisms and networks. | ||
The components of this virtual reactivity laboratory are as follows: The terminal in green colour is a symbol for the visual interface at which the relevant data can be displayed to the operator. The display may be a simple screen that could be placed right next to a bench in a wet lab or it can be as advanced as a virtual reality cave. The ‘relevant data’ will be the reaction network consisting of nodes and links, which is undergoing a rolling improvement that does not depend on whether the operator is in front of the terminal or not. Instead, service programs take care of as many tedious tasks and decisions as possible. We call these service programs ‘jeannies’. They are supposed to run constantly in the background and independently take as many decisions as possible. The decisions taken are displayed to the user in order to give him or her the opportunity to steer these tasks when deemed necessary.
Information on the nodes and links of the reaction network are graphically displayed upon clicking on these entities. They will then open in a new frame or window on the terminal. The most important frame is the molecule editor which allows the investigation and manipulation of molecular structures to be taken from or eventually integrated into the reaction network. For the manipulation of molecular configurations, new jeannies are set up (denoted in grey and red in Fig. 1), which may fulfil highly system-dependent tasks — from supplementing heavy-atom only PDB structures with hydrogen atoms to the automated construction of reactive complexes, that might lead to new nodes in the reaction network, to inverse and rational design tools13,14 for the construction of totally new reactants and structures.
Many other advanced jeannies can be envisaged. For instance, a kinetic modeller that is able to solve large systems of coupled kinetic differential equations — analogously to algorithms developed in systems biology and bio-informatics — is decisive for an automatic evaluation of the reaction network. Crucial regions in the network can be identified in terms of their kinetic relevance and can then be refined with accurate quantum chemical methods. Also these refinement calculations can be automatically launched on a high-performance compute-cluster (though the operator may actively suppress them if he or she considers them not useful for the exploration).
3 Real-time force calculation from first principles
A key for an interactive exploration of chemical reactivity is the calculation of the quantum mechanical response of the molecular system upon a modification of its structure in real time. In the following we will refer to such modifications as structure manipulations. They include all manipulations that change the relative position of atoms in the system. The primary quantum mechanical response to such changes is a changed electronic structure of the molecular system leading to a different potential energy (electronic energy in the Born–Oppenheimer approximation). From the electronic structure energies and forces can be obtained. The secondary response is the change in the arrangement of the atoms not manipulated, if structural relaxation is allowed. It is important to emphasise that only electronic-structure methods based on the first principles of quantum mechanics are suitable for reactivity studies because bonds are broken and formed, which cannot be reliably predicted by the pre-defined potentials of a (classical) force-field.To obtain the quantum mechanical response, the electronic structure has to be calculated first. The forces can then be obtained analytically from the results of this optimisation procedure. The response of the atomic structure is calculated by minimising the forces on the unmanipulated atoms. To provide the result in real-time there is an upper time limit for all steps. However, in most cases the electronic structure optimisation is the limiting factor. Depending on external factors — such as the details of the optional structure optimisation procedure, the user-defined speed of structure manipulation or the required adiabaticity of the manipulation — the limit can vary considerably. However, an electronic structure (and force) calculation requiring on the order of one hundred milliseconds will be sufficiently fast.
In order to make the electronic structure optimisation sufficiently fast, usually additional approximations have to be introduced. A general rule is: The faster the optimisation is, the more severe are the approximations. Thus, in addition to the time limit it is necessary to specify a lower limit for the quality. To do so we recall the goals of an instantaneous exploration of chemical reactivity in a virtual environment, namely to explore the potential energy surface and identify the structures corresponding to the reactants, products, and transition states of elementary reactions, keeping in mind that they are only a starting point for further refinement using the standard methodology developed in quantum chemistry. Hence, the potential energy surface, i.e., the electronic structure method employed to obtain it, should reproduce all main features of the exact surface. Thus, local minima and transition states should be detectable, while their absolute position and depths or heights are not required with high accuracy.8 Clearly, not in every situation are all of the features needed. A first exploration to familiarise with the molecular system can be of low quality and only later, parts of the surface are examined more accurately. More information on this iterative character of a haptic exploration of potential energy surfaces to continuously refine them can be found in ref. 8.
3.1 Pre-calculated ab initio surfaces and interpolation
The interpolation of pre-calculated potential energies is a fast option to obtain the forces. We found7 that the interpolating moving least squares (IMLS) method15–18 is very well suited for haptic rendering. Other possible schemes are based on splines,19 the modified Shepard method20 or neural networks.21 IMLS is a local method which performs the fitting at each required position giving configurations in the proximity higher weights. This feature enables the introduction of a cut-off procedure to reduce the number of data points needed for the interpolation, which is beneficial if the data set for the whole system is very large. Another advantage is that, because of the analytical expressions obtained at each point, the calculation of the forces is trivial as it involves only derivatives of a polynomial. However, an interpolation solely based on gradients is also possible. Furthermore, IMLS provides a good starting point for an automated refinement procedure of the surfaces.22 To refine the surfaces of contiguous haptic explorations an efficient procedure is available that is guided by the findings of previous explorations.8No matter what technique is chosen, interpolation allows one to achieve a very high accuracy of the potential energy surface and the forces because the raw data can be obtained with high accuracy ab initio methods. However, the application of interpolation is also limited. If the dimensionality of the hyper-surface increases, too many points have to be calculated and the interpolation becomes slow. In addition, if relaxation is allowed, the configuration space spanned by the manipulation is no longer unique but depends on the history of the exploration. Usually, addition or abstraction reactions involving only atoms or diatomic molecules in systems where relaxation does not play a major role are good candidates for the haptic exploration based on interpolation of pre-calculated surfaces.8
3.2 Classical reactive force fields
For cases which require a direct calculation of the forces due to large relaxation effects (introducing significant exploration history effects) during the reaction, reactive classical force fields are the fastest methods available. The bond-order potentials like the Tersoff potential,23 the Brenner potential24 or the Finnis–Sinclair potentials25 are examples for such methods. Other general reactive force fields are the empirical valence bond potential by Warshel and co-workers26 and the ReaxFF27,28 force field. For a recent overview see ref. 29. All of these methods have in common that their simple formalism permits a very fast force calculation for a given atom arrangement. However, the reactive force fields, like all classical force fields, are highly parametrised and of limited transferability.3.3 Standard semi-empirical methods
The next step towards a first-principles treatment of the electronic structure are semi-empirical methods. Since very many different variants have been developed in the past few decades, we focus on the ones suitable for the interactive study of reactive systems.The simplest method of this family is the atom superposition and electron delocalization molecular orbital (ASED-MO) method,30 which is based on the extended Hückel method31 to treat the valence electrons of atoms. It applies atomic-repulsion corrections32 in order to obtain correct structures, which is the main deficiency of the extended Hückel method. This method has already been shown to be feasible for the interactive study of structures containing only carbon and hydrogen.11,12 It has the advantage of being non-iterative as the Fock operator does not depend on the molecular orbital coefficients owing to the severe approximation involved. Moreover, only a few simple integrals have to be evaluated.
The next more complex group of semi-empirical methods invokes the neglect of differential diatomic overlap (NDDO)33,34 and the modified neglect of diatomic overlap (MNDO)35,36 approximations. The PM637 and OMx38 family of methods are recent members of this group. They are based on the Hartree–Fock theory but apply approximations such as neglecting electron–electron interaction integrals and replacing many of the remaining ones by empirically adjusted parameters.
Also density functional tight-binding (DFTB) methods like the most recent DFTB339–41 approach are suitable semi-empirical methods for a fast and quite reliable force calculation. Although DFTB methods involve fit parameters they are not truly semi-empirical in a strict sense since all these parameters are determined from full Kohn–Sham density functional theory (DFT) calculations. As the name suggests DFTB methods can be understood as a DFT analogue to NDDO/MNDO-based methods.42
All these semi-empirical methods are electronic structure methods (in contrast to reactive force field approaches). From the electronic structure (eventually, from the orbitals) quantum chemical properties can be calculated. Furthermore, all apply a finite basis set expansion of the orbitals. Hence, the optimised molecular orbital coefficients provide a reasonable estimate for more elaborate electronic structure methods to refine the results of the exploration. The connection to more accurate methods is therefore straightforward.
3.4 Minimal Hartree–Fock and density functional methods
The main idea behind this group of methods is to reduce the computational cost of standard quantum chemical calculations based on Hartree–Fock or density functional theory to a justifiable minimum. Since this group of methods does not introduce system-dependent parameters, they are generally applicable and, with increasing computational effort, gradually improvable towards the reference method (i.e., towards the Hartree–Fock limit and the Kohn–Sham DFT result at the basis-set limit free of numerical inaccuracies). It is possible to apply algorithms developed in the field of linear-scaling electronic structure methods. However, to achieve very fast calculations mainly the size of the basis set is decreased, since this provides an easy to apply and very efficient way9 to reduce the computational cost with a built-in natural strategy for systematic improvement. Moreover, effective core potentials (ECPs) reduce the number of explicitly treated electrons and thus also the number of basis functions.We have explored this methodology taking DFT with a minimal basis set and ECPs and applying the usual integral screening and density-fitting techniques to accelerate the calculations. We have applied this minimal set-up first to calculate data points for surface interpolation8 and then to show that reasonable energies and forces can be calculated for small systems even in real time.9 Interestingly, such minimal approaches are experiencing a renaissance in quantum chemistry. The corrected small basis set Hartree–Fock method proposed by Sure and Grimme43 for the fast calculation of mass spectroscopy data takes Hartree–Fock theory as a starting point with a minimal basis set called MINIX. To minimise the errors electronic core potentials and three different atom pair-wise correction terms with pre-fitted parameters have been applied.
The recent development in the field of quantum chemistry to utilise the computer power offered by graphical processing units or other specially tailored hardware, will make first-principles force calculations possible with increasing accuracy. Elaborated screening techniques to avoid unnecessary integral evaluations and sophisticated algorithms to evaluate the remaining integrals will also contribute to this development.9
4 Analysis of contemporary semi-empirical methods
In the following we discuss timings for state-of-the-art implementations and accuracy studies of semi-empirical and density functional tight-binding methods. These results complement our previous work in which we demonstrated the general applicability of interpolation approaches7,8 and minimal one-determinant Hartree–Fock and DFT methods.9 With semi-empirical and density functional tight-binding methods the size of molecules that can be considered in real-time calculations is clearly much larger than the size of the reactive systems studied in ref. 9 (Br2 approaching ethene and a chlorine anion approaching H3CF).4.1 Runtime study
In order to study contemporary semi-empirical methods and density-functional tight-binding methods for their potential to provide real-time energies and forces, we performed single-point energy calculations for different test systems for a variety of NDDO-based semi-empirical Hamiltonians (AM1,44 RM1,45 PM3,46 PM6,37 PM6-D3,37,47 PM748) and the third-order self-consistent-charge density-functional tight-binding method DFTB3.39–41 Since the possibilities to obtain timings from the available implementations are limited and are not comparable among different programs, the timings were taken by measuring the overall execution time. To average out the influence of varying background tasks performed by the operating system 100 consecutive runs were carried out and averaged.Our set of test molecules of different sizes and different elemental composition is shown in Fig. 2. The two alanine oligopeptides represent typical biological molecules. The system denoted as FeGP (iron-guanylylpyridinol) is a model for the active site of [Fe] hydrogenase, where the methylenetetrahydromethanopterin (HMPT) is a cofactor which we chose to model by structure e in Fig. 2. Both structures have been taken from ref. 52. The so-called compound A (cf. structure A in Fig. 3, of ref. 53) is a large organic molecule. The Schrock complex with an approaching N2 molecule54–60 is an example of a large transition metal complex employed in homogeneous nitrogen-fixation catalysis.
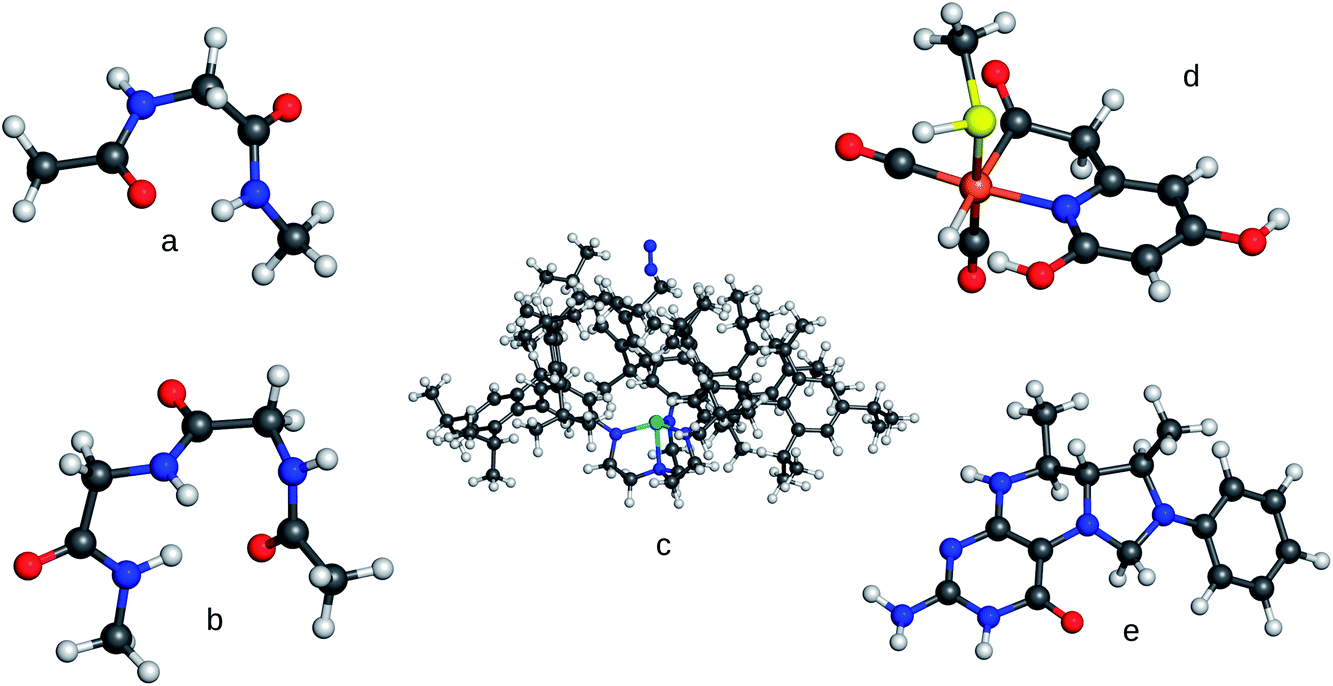 | ||
| Fig. 2 Molecular systems used for the timings. (a) Alanine dimer, (b) Alanine trimer, (c) Schrock trisamidoamine Mo complex with N2, (d) FeGP, (e) model for HMPT. Structures a, b, e have been obtained from a DFTBA49,50 optimisation in Gaussian 0951 Rev. d.01. Structure d has been taken from ref. 52, structure c from ref. 8. | ||
All calculations started from the default molecular orbital (MO) coefficients guess provided by the respective program. The timings obtained are compiled in Table 1. Those molecules containing only C, N, O and H atoms required a similar number of iterations to reach self-consistency. However, a comparison of the required iterations for Ala3 and the FeGP model system shows that the iteration number has only a small influence on the execution time. Although the times measured here contain the overhead of reading and writing to disk, printing information to output files and setting up the SCF iterations, they are all within the time range of what we would require for real-time calculations. Only the very large Schrock complex requires several seconds to optimise an electronic structure, which is mainly due to its size.
| AM1 | RM1 | PM3 | PM6 | PM6-D3 | PM7 | DFTB3 | |
|---|---|---|---|---|---|---|---|
| Ala2 | 248 ± 5 | 249 ± 9 | 249 ± 7 | 248 ± 9 | 251 ± 6 | 249 ± 10 | 344 ± 37 |
| 19 atoms | (14) | (14) | (14) | (14) | (14) | (15) | (11) |
| C, H, N, O | |||||||
| Ala3 | 249 ± 5 | 249 ± 6 | 250 ± 6 | 252 ± 5 | 252 ± 6 | 252 ± 5 | 342 ± 32 |
| 26 atoms | (14) | (14) | (14) | (14) | (14) | (14) | (13) |
| C, H, N, O | |||||||
| FeGP | – | – | – | 258 ± 6 | 259 ± 6 | 259 ± 5 | – |
| 29 atoms | (–) | (–) | (–) | (236) | (236) | (61) | (–) |
| Fe, S, C, O, N, H | |||||||
| HMPT | 255 ± 6 | 255 ± 4 | 255 ± 7 | 256 ± 4 | 257 ± 4 | 257 ± 5 | 354 ± 40 |
| 43 atoms | (14) | (14) | (17) | (20) | (20) | (15) | (12) |
| C, H, N, O | |||||||
| Compound A | 275 ± 21 | 276 ± 29 | 273 ± 6 | 289 ± 54 | 295 ± 63 | 279 ± 34 | 562 ± 32 |
| 84 atoms | (13) | (13) | (17) | (25) | (26) | (14) | (10) |
| C, H, N, O | |||||||
| Schrock-N2 | – | – | – | 9197 ± 498 | 9414 ± 574 | 5719 ± 103 | – |
| 280 atoms | (–) | (–) | (–) | (142) | (142) | (95) | (–) |
| C, H, N, Mo | |||||||
The timings given here are only upper limits. In an implementation tailored to the needs of real-time quantum chemistry execution time can be reduced by restructuring the algorithms considering the following points.
• Separate all system specific initialisation and perform it at the beginning.
• Take MO coefficients from previous optimisations as a starting point for the next.
• Eliminate every output that is written to disc or to the console.
• Avoid any reading from or writing to disc.
• Perform as many steps as possible in-core.
• Choose convergence thresholds according to the resolution of the output device(s).
Since all methods investigated compare similarly in terms of providing real-time energies, they may be selected according to their accuracy, their ability of being systematically improvable, and to the possibility of including them in a subsystem approach. Because of its close connection to DFT the DFTB approach is well suited for subsystem approaches using DFT.
4.2 Quality assessment
The ability of an electronic-structure method to yield qualitatively correct potential energy surfaces is of paramount importance for the study of chemical reactivity. As already stated, absolute barrier heights, depths, and accurate positions of local minima and first-order saddle points are not ultimately important as long as these features are present and can be detected.To investigate the semi-empirical and density-functional tight-binding methods for their ability to yield such qualitatively correct surfaces, we chose a rotor-like molecule with three anthracene blades that undergoes a rearrangement with a challenging plateau-like barrier and that has recently been studied in our group with more accurate first-principles methods.53 The energy profile from ref. 53 serves as a reference. The reference potential energy surface features two local minima (A and B) which are connected by two transition states (TS1 and TS2) with a shallow minimum in between (MinAB). For the corresponding structures see Fig. 3.
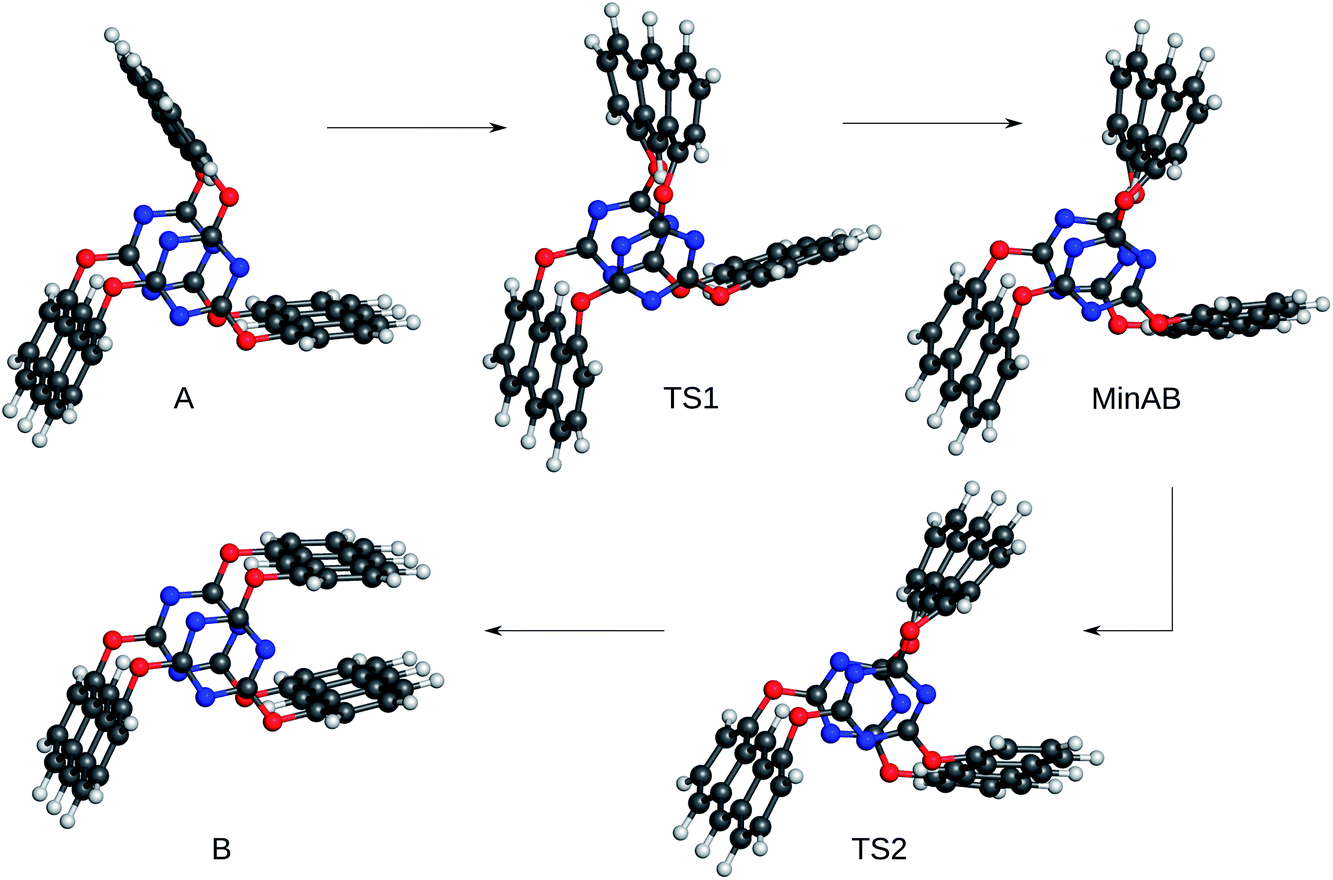 | ||
| Fig. 3 Reference (starting) structures taken from ref. 53 for the reaction profiles in Fig. 4 and Table 2. | ||
Since every semi-empirical Hamiltonian produces a different (approximate) potential energy surface the energies have been calculated after a structure optimisation employing the respective electronic structure method (cf.Table 2). The NDDO-based calculations were performed with MOPAC2012.61,64 The DFTB3 structures were optimised in Gaussian09 d.0151 with the DFTBA49 method and the DFTB339–41 values were obtained from a single-point energy calculation on these structures with the DFTB+62 (v.1.2.2) program. The results are collected in Table 2 and selected energy profiles are shown in Fig. 4.
| A | TS1 | MinAB | TS2 | B | ||
|---|---|---|---|---|---|---|
| BP86-D3/def2-TZVP53 | E | 0.0 | 20.1 | 19.6 | 20.5 | −3.7 |
| AM1 | E | 0.0 | 17.3 | 0.4 | 22.0 | 0.4 |
| RMSD | 0.118 | 0.354 | 1.334 | 0.265 | 0.506 | |
| RM1 | E | 0.0 | 23.4 | 15.1 | 32.0 | 0.6 |
| RMSD | 0.099 | 0.405 | 0.276 | 0.268 | 0.359 | |
| PM3 | E | 0.0 | 16.2 | 13.7 | 18.3 | 0.4 |
| RMSD | 0.135 | 0.314 | 0.392 | 0.194 | 0.612 | |
| PM6 | E | 0.0 | 13.2 | 12.6 | 13.8 | 0.5 |
| RMSD | 0.123 | 0.196 | 0.128 | 0.123 | 0.488 | |
| PM6-D3 | E | 0.0 | 13.0 | 12.3 | 13.3 | −1.4 |
| RMSD | 0.115 | 0.196 | 0.123 | 0.125 | 0.361 | |
| PM7 | E | 0.0 | 18.3 | 12.1 | 30.9 | −8.3 |
| RMSD | 0.085 | 0.405 | 0.237 | 0.273 | 0.090 | |
| DFTBA | E | 0.0 | 21.8 | 0.0 | 18.6 | 0.3 |
| RMSD | 0.122 | 0.731 | 1.199 | 0.194 | 0.353 | |
| DFTB3 | E | 0.0 | 19.2 | 0.0 | 17.9 | 0.2 |
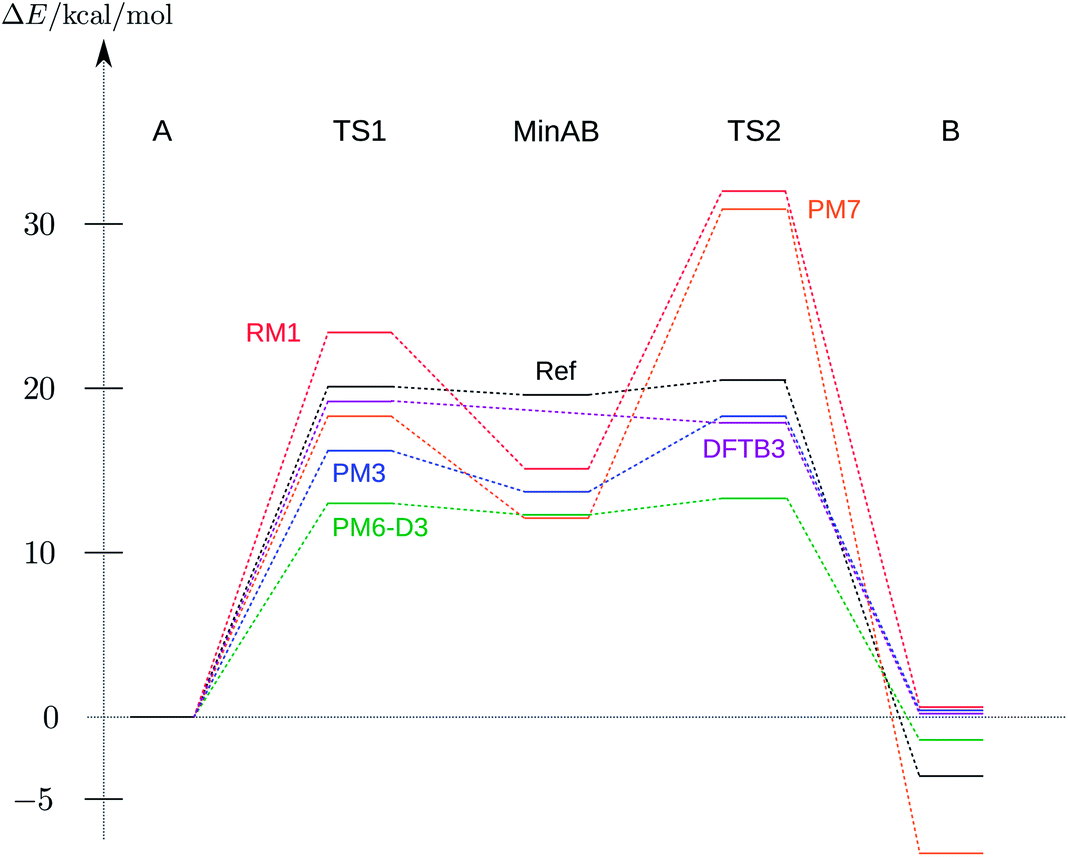 | ||
| Fig. 4 Energy profile of the reaction A → TS1 → MinAB → TS2 → B relative to the energy of A of the corresponding method. Only a selection of the methods from Table 2 is shown. The reference BP86, DFT-D3, def2-TZVP profile has been taken from ref. 53. | ||
Except for AM1 all semi-empirical methods show the local minima and saddle points of the reference surface (cf.Table 2). As expected, the heights or depths vary from method to method but the overall structure of the energy profile is conserved throughout. The transition state structures deviate more from the reference structures than the local minima, as is expected for semi-empirical methods.48 The correct sign of the energy difference between product and reactant is only reproduced by PM6-D337,47 and PM7.48 This is caused by a strong dispersion interaction effect, which is explicitly treated in a post-SCF manner in PM6-D3 and is directly incorporated in the PM7 Hamiltonian.37,47,48 All NDDO-based methods have the first transition state energetically below the second one, as in the reference calculation.
PM6-D3 shows the best performance of all methods analysed. Although the heights of the TS1, MinAB and TS2 are significantly lower compared to the DFT reference the relative energies of the transition states and the middle minimum as well as the relative energy between product and reactant are reproduced. The best method for obtaining the correct structures of reactant and product (A and B) is PM7 with RMSD values below 0.1Å.
Interestingly, it was not possible to locate the stable minimum MinAB with the DFTB methods, but they reproduced the two transition states and the minima A and B quite well.
The overall performance of all methods studied here is astonishingly good, i.e. they perfectly meet the quality requirements stated in the beginning. The fact that the semi-empirical approaches without dispersion correction overestimate the stability of the MinAB structure is no drawback as the detection of a potential intermediate is feasible. Applied in an interactive chemical reactivity study these methods would show the desired features of the potential energy surface and would guide the user to the correct structures. The relatively small RMSD values with respect to the DFT reference (cf.Table 2) indicate that, if the user is able to correctly locate the minima, they will be perfectly suited as starting structures for a refinement employing a more accurate method.
5 Reactivity exploration in virtual environments
In the following we describe in detail how a virtual reactivity exploration experience should proceed. We work out what is needed from the chemist's perspective in order to successfully study chemical reactivity.5.1 Preparatory steps
The procedure starts with the construction of the molecular structure of interest. This construction requires the definition of the type and position of atoms and the determination of the overall charge and spin multiplicity. To arrive at a stable configuration the energy can be continuously minimised in the background while the molecular structure is constructed.5,6 However, applying a first-principles method can be problematic for placing additional atoms one by one. It may produce unwanted structural distortions upon continuous structure optimisation or may already show a non-converging electronic structure optimisation and artificially large gradients are the result. Classical force fields can be more suitable at this stage.5.2 The exploration procedure
After the build-up of the structure the operator exploits ab initio force calculations to start the exploration. A possible first step is to grab one or more atoms and distort the structure by pulling at them. If the device with which the manipulation is performed features force–feedback functionality, the force acting on the manipulated atoms and hence the reactivity can be directly felt (haptic quantum chemistry7). Without haptic interaction the user has to rely on the visual information to infer the reactivity. In that case the user needs to be informed about how likely the applied distortion is by visual elements like force arrows on the atoms or a panel that indicates high energy configurations, which is neither a direct sensation nor is it practical for large molecular systems (the user would drown in a sea of force vectors).In the case of intermolecular reactions, the starting point is a stable configuration with no intermolecular interactions, e.g., at the dissociation limit. This means that many orientational changes are possible without a change in potential energy and therefore without forces. The reactive fragments are oriented such that a suitable initial position is obtained to start the reactive exploration. From there the scientist may record all configurations visited during the exploration. The recording can be stopped and restarted several times which results in a multitude of different paths. Usually both, the initial and the end configuration, are local minima that are stored separately from the paths. During the exploration the chemist is guided by the forces and by the structural changes occurring as a result of the structure manipulations. The exploration is therefore driven by a combination of the operator's prior knowledge, the information provided through the forces, and the structural changes upon structural manipulations.
Such an exploration can be performed to study the shape of the potential energy surface in a localised region or to directly find possible reaction paths between already known stable configurations (local minima).
5.3 Output for reaction networks
The structures recorded during the exploration together with the associated energies and gradients form the primary (raw) output data. They are what we recently defined as ‘core quantities’ in our definition of Real-time Quantum Chemistry.9 Additional data such as dipole moments, the electron density and other quantum chemical properties can also be part of the output if calculated on the fly.The structures, energies and gradients can be analysed and the distinct points with zero gradients can be extracted. They can be considered as nodes of a reaction network that emerges successively during an exploratory study. The paths between the structures with zero gradients (stationary points) connect the nodes as links. If they correspond to a minimum energy path, the associated transition state structure node can be marked as such. They are usually the main objective of the exploration. If, however, the stationary points are far apart in configuration space, no unique transition structure will be found for any connecting path, which can then be taken as a hint to search for additional local minimum structures in between (new nodes). This then produces a more fine-grained reaction network for which new connecting paths are defined. They are the starting point for further transition state searches—either by haptic exploration or by automatised protocols launched in the background.
5.4 Output for reaction paths
Obtaining all relevant nodes will be mandatory if a complete reaction network is needed. For the study of reaction mechanisms, however, the links connecting the nodes are the prime objective, since they contain kinetic information about how the atoms rearrange during a reaction. The analysis of these paths can be seen as an interactive analogue to the Reaction Path Hamiltonian methods65–67 or the Unified Reaction Valley67,68 methods. A very close connection exists to the Reaction Force approach69 since the operator can follow these paths directly if a haptic device is employed. The output of the exploration can be used as a starting point for a more detailed analysis of the reaction dynamics or kinetics with standard approaches70–72 eventually applying more accurate electronic structure methods.5.5 Experimental realisation of manual force exertion
The virtual exploration of chemical reactivity is in fact connected to experimental realisations for the manual manipulations of molecular matter. To study chemical reactivity the resisting forces rendered to the user are an indirect descriptor of the topology of the potential energy surface. Such forces can be directly measured in special experiments. In these experimental setups mechanical forces can be applied to single molecules to change their structure and even to alter their reactivity. There exists a variety of experimental techniques for the realisation of this mechanochemistry. Examples are optical/magnetic tweezers, the bio-membrane force probe technique, hydrodynamic techniques and atomic force microscopy. The manipulations applied by the user in the virtual environment can thus be directly linked to exerting real forces on parts of the molecular assembly. For a review of these experimental techniques see ref. 73. In fact, such experiments allow the characterisation of chemical bonds in a direct way74,75 and the virtual environment can become a convenient means to steer such experiments.The mechanical manipulation of molecular matter is called mechanochemistry. Theoretical work on mechanochemistry has been performed on ring opening reactions.76,77 In this example it has been shown how additional mechanical forces alter the potential energy profiles of reactions that involve the rupture of chemical bonds. A review on the theoretical concepts and the computational tools has recently been provided by Ribas-Arino and Marx.78
6 Interaction with molecular systems in virtual environments
The core objective of an interactive and real-time treatment of molecular systems is the instantaneous calculation of energy and gradients, i.e. the response of a given atom arrangement.9 Since a plethora of structures, energies and gradients are produced very quickly and in a cumulative manner, they can no longer be mapped out in huge data files as these can hardly be condensed to intuitive insights. Immersion into the virtual environment of the molecular system allows the scientist to perceive the vast amount of data more easily and intuitively.79 Immersion refers to the process of creating a perception in a virtual reality.6.1 Immersive perception
The visual presentation of the structure plays an important role for immersion. Hence, it should be done in the way elaborated in chemistry more than hundred years ago, namely in terms of balls as atoms and sticks as bonds. This is the default way to present the structures as applied in molecular visualisation tools like VMD,80 PyMOL81 or Chimera82 to name only a few. Also conventional molecular structure builders like GaussView,83 the graphical user interface of ADF,84 Avogadro6 and SAMSON5 can serve as examples.Suitable visual output devices for an efficient immersion are devices that are able to give a comprehensive and intuitive picture of the virtual world. The larger the visual devices are, the more they exclude the perception of reality around the user. Panoramic screens or head-mounted displays85–87 are best suited for this purpose. Rendering the atoms, bonds and all volumetric data with ray tracing is very helpful in this regard. Also current three dimensional presentation techniques like polarised or active shutter systems are beneficial, since they allow for a much easier perception of the spatial configuration of the molecular system.
To intensify the immersion by addressing the tactile sense requires the rendering of the occurring forces by haptic devices. Haptic pointer devices, for instance, can render forces in three to six dimensions. They can be employed to probe the forces acting on different sites of the molecular assembly. The scientist picks an atom and feels the resulting force response of the system. Also two or more haptic pointer devices can be utilised simultaneously to understand the interplay of forces. Such devices can be purchased at modest prices, which makes them attractive for virtual environments employed in chemical laboratories.
A more elaborate way to render forces and to address the human tactile sense are external skeletons. Since they render the force directly for each finger, they provide an even more natural experience for the tactile sense. However, these devices are still very expensive and currently not useful for a large scale deployment in chemical laboratories.
Additional data like force vectors on atoms not probed by a haptic device, the total energy, partial charges, isosurfaces of electron densities or molecular orbitals need to be seamlessly integrated as well. The most intuitive form to do so is to visualise them in three dimensions together with the molecular structure. Since these quantities as well as the structures change very fast during the manual exploration efficient implementations using graphical processing units and multi-core central processing units are needed.88,89
Global scalar quantities like the electronic energy could be rendered by addressing even more senses. For example, the auditory sense could be utilised for this purpose. High-frequency sounds could signal a configuration with very high energy and thus regions in configuration space that are not accessible in a given thermal setting. However, a change of background colour achieves the same and is certainly less annoying. Accordingly, the tactile sense remains the most important one besides human vision.
6.2 Immersive interaction
Human–computer interaction is a key ingredient in immersive technologies as it allows the user to participate in the virtual environment. For the study of chemical reactivity, interaction is mandatory because it drives the reactivity exploration.Input devices facilitate the human–computer interaction and therefore play an important role. In an immersive virtual environment they allow for a manipulation of the virtual objects. Therefore, the real-world position of the device is transferred into virtual-world coordinates of a virtual representation of the device. With that the virtual objects, in our case, atoms or atom groups, can be moved. A general requirement for devices to be suitable for immersion is that a physical movement of the operator is transferred directly into a movement in the virtual world. Transformations applied in between, e.g. to avoid range limitations of the physical device or to stabilise motions, need to be transparent for the operator, since the positions of virtual objects are controlled with them.
Since the forces resulting from such manipulations can be very sensitive with respect to position, a precise position control is most important. A lack of precision renders the control over the manipulation nontransparent which means that physical movements of the device are not consistent with the virtual movement. This also implies that the devices should not loose accuracy if the user quickly moves the devices. Fluctuations in the position due to instabilities caused by the user is also a reason for a less precise position. Especially freely movable input devices require attention to avoid unintentional small amplitude motions. The stabilisation procedure can take place already in the hardware, but also on the software side. Stabilisation can be efficiently achieved by scaling down the input movement.
The interaction with large molecular assemblies in a virtual environment is challenging because they are complex and dense (many objects in a small space). Moving in and interacting with such an environment should be done with devices that allow a manipulation directly in three dimensions. Input devices with less dimensions, like an ordinary computer mouse, allow only a two dimensional position control and are therefore less intuitive to handle. Such input devices acting in two dimensions require additional visual cues and sometimes also additional input with another device to be able to manipulate three dimensional objects.
Three dimensional input devices can be divided into two categories. To the first category belong devices in which a physical object like a ball or a pen is moved. The haptic pointer device, which we have been using in the framework of haptic quantum chemistry, features a pen connected via joints to a machine which calculates position and orientation and sends them to the computer. Such devices allow a relatively precise control over position in three dimensions (below 0.01mm according to manufacturer specifications90,91).
The second category contains devices which do not require to move a physical object. They directly capture the motion of the user's hand. Such motion sensing devices provide an even more natural form of three-dimensional input. Modern motion sensing devices can have an accuracy down to the sub-millimetre range.92 A problem with such devices is the detection of two fingers very close to each other. Hence, grabbing atoms with such devices is difficult, since it involves two fingers coming close to each other. Pointing at the atoms and moving them by just one finger is easier to implement, but is also less intuitive. The accuracy of the finger tip positions can be improved by wearing passive or active markers that make the detection easier.
Currently haptic pointer devices are more suitable since input and output can be combined in one such device.
6.3 Rendering forces in reactive potentials
The rendering of the forces is a very important component of the virtual environment proposed in this work, since they convey significant information. One has to distinguish between the force rendered by the device, which is felt by the user, and the force calculated by the chosen electronic structure method. The former we may call ‘haptic force’ and the latter ‘molecular force’.In a straightforward implementation both forces would be exactly the same. However, the molecular forces are in the range of 1 nN, whereas the forces which can be felt by the user are on the order of 1 N. It is immediately clear that both forces cannot be the same but have to be transformed. This transformation needs to fulfil two requirements in order to be suitable for studying chemical reactivity. As a guide through the configuration space avoiding unphysical configurations the rendered forces need to be intuitive and smooth. Yet, the transformation between the molecular and the haptic force needs to be as transparent as possible, since from the force direction and magnitude the user infers the shape of the potential energy surface. The most transparent transformation is to multiply the molecular force by a constant factor to obtain the haptic force. This is how we transform the nanoscale molecular forces to a haptic force that is experienced by the user.
The shape of potential energy surfaces occurring in reactive chemical systems requires additional transformations. Shallow and deep minima, steep walls and flat hills may lie in close proximity on a reactive potential energy surface. This results in a wide range of forces that need to be rendered. Since the range of forces which can be rendered by the devices is limited and also the tactile sense has a limited resolution, the forces need to be scaled. For instance, in strongly repulsive regions huge forces quickly exceed the limits of the devices. Hence, the forces need to be scaled down. In almost flat regions, however, where shallow minima need to be detectable the force requires to be scaled up. A constant force scaling93 as mentioned before is therefore not suited. A variable gain scheme94 can account for both situations without the need to change the scaling manually.
The third reason why one needs to transform forces is more technical. To avoid instabilities and to compensate for slow simulation loops often frequency filters are applied. However, a too dominant filter can lead to an in-transparent force rendering. To obtain a stable control scheme usually the position is not directly controlled but the user applies an additional force that is incorporated into the simulation.95,96 This, however, hides the true forces acting on the manipulated atom from the user, which makes it unsuitable for studying chemical reactivity, where the forces transport a substantial part of the desired information.
6.4 Moving objects vs. probing reactivity
For the manipulation of molecular assemblies one may distinguish between two different intentions. The first intention is to move parts of the assembly around to either see them from a different perspective or to arrange them in a way better suited to start the reactivity exploration. During such a rearrangement the internal structure of the molecules stays intact. The second intention is to probe the chemical reactivity, which involves the breaking and/or formation of chemical bonds. The virtual environment has to accommodate both scenarios.A seamless transition from moving a molecule to probing reactivity is needed when atoms come closer and the forming or breaking of bonds becomes possible. The problem can be illustrated by considering the abstraction of an atom from a molecule versus just pulling at an atom to move the molecule when no further constraints are applied. Whether pulling at an atom results in an abstraction or just in a movement of the whole molecule depends on how fast the system responds in terms of the relaxation described at the beginning of this section. A manipulation within an infinitely fast relaxing system would correspond to a reversible adiabatic process. If the virtual environment is designed to mimic this scenario there would be no net change in energy and therefore also no reactivity. A non-adiabatic process is emulated when the relaxation rate is slower than the manipulation by the user. In this scenario moving a molecule by just pulling at an atom would not be possible without breaking a chemical bond.
This dilemma can be resolved in different ways depending on the limits imposed by the underlying force calculations: either by adjusting the relaxation rate or by adding additional constraints. The first is only possible if the relaxation (structure optimisation) is always faster than the manipulation. Then one can adjust the rate such that the users can switch between adiabatic and non-adiabatic manipulations by adjusting their manipulation speed. Pulling fast at an atom induces the abstraction, while pulling slowly just moves the molecule. Complementary to that would be to change the relaxation rate instead.
If an adjustment of the relaxation rate is not possible or unwanted, additional constraints can be introduced. Possible constraints are to fix atoms in space, preserve internal structures or pin the orientation and centre of mass of a molecule. The user can realise them employing additional input devices (e.g., one hand holds a metal complex in position and the other probes the reactivity of a possible ligand) or by keeping the corresponding degrees of freedom fixed during the relaxation procedure.
In both solutions employing additional input devices is the preferred way since then the constraints can easily be altered or removed if necessary. By contrast, changing the parameters of the relaxation procedure, e.g., changing the rate or the constraints, requires interruption of the exploration. Accordingly, this should only be done if the constraints are expected to remain constant during the exploration, e.g., when certain parts of the molecular assembly are known to be non-reactive.
6.5 Restricting the manipulation to avoid high energy configurations
If in a molecular structure every atom is freely movable, then the manipulation can easily lead to configurations with very high energy. For instance, if bond lengths are decreased below the equilibrium distance of two fragments, very high electronic energies result. Since high energy configurations may not be accessible by the system under real conditions at finite temperature such manipulations have to be avoided or at least made hard to perform.A natural way of restriction is provided by force–feedback devices. The rendered force guides the user and allows only reasonable manipulations, since a rapid increase in energy is associated with a steep gradient pushing the user in the opposite direction. By this an artificial blocking of certain manipulations can be avoided.
Another possibility is to perform a very fast relaxation of the remaining degrees of freedom. The energy is thereby removed from the system so that the system always maintains a configuration with a reasonable energy. For the physical implications of such an energy removal see next section.
7 Physical laws in virtual environments
Clearly, any useful virtual environment designed to immerse the chemist into the reactive molecular assembly has to obey certain physical laws. They are a trade-off between an intuitive presentation and the ‘true’ physics. For the former most of the requirements have already been outlined in the sections above. Here, we will show which consequences for the design of the virtual environment follow from the physical theory.7.1 Potential energy and force field of the molecular systems
Although the atoms are visually represented as hard spheres they are described as classical point-like objects in the calculations. Also the interaction of the operator with the molecular system necessitates a classical treatment of the atoms, since at all times they have a completely determined position in space, i.e., their movement corresponds to classical trajectories. Accordingly, the virtual environment implies a ‘macroscopic’ picture of the molecular entities which is classical by definition. Hence, quantum mechanical effects of the atoms, such as tunnelling, are not directly representable. One may account for them by discontinuous atom position dislocations when induced by a tunnelling probability measure in separate studies if the potential energy surface is sufficiently well known. However, this will only be necessary for potential proton transfer steps.The dynamics of the electrons are not explicitly represented in the virtual environment, but they largely define the interactions between the atoms. This separated treatment of the electron and atom dynamics is based on the Born–Oppenheimer approximation.97 In fact, the electrons give rise to the potential in which the atoms move. The electronic energy is then defined by the electronic Schrödinger equation
| ĤelΨel = EelΨel | (1) |
[![[T with combining circumflex]](https://www.rsc.org/images/entities/i_char_0054_0302.gif) nuc + Eel]Ψnuc = EnucΨnuc. nuc + Eel]Ψnuc = EnucΨnuc. | (2) |
The electronic energy Eel ({RI}) is a parametric function of the nuclear coordinates {RI}. A first-principles treatment of the electrons requires that Eel ({RI}) has to be calculated by solving equation (1). This has to be done at discrete configurations of the atoms.
Despite its calculation as a discrete function, Eel can be considered as a continuous function of all atom coordinates. We may define a continuous variable x in 3M dimensional space as
| x = (R1, R2,…, RM)T with RI = (xI, yI, zI)T. | (3) |
Accordingly, Eel(x) is a hyper-surface in a 3M-dimensional Cartesian configuration space spanned by the coordinates of all atoms. There is a conservative force field connected to this surface. It describes the forces acting on each atom A and is given by the negative gradient gA of the potential energy Eel(x)
| fA (x) = −gA with gA = ∇AEel(x). | (4) |
The gradients (or forces) can be calculated directly from the electronic structure. Only this analytic differentiation is sufficiently fast for immersive techniques.
From this setting it follows naturally that the molecular system has to be described classically with respect to the atom movements and quantum mechanically with respect to the electronic structure and derived properties—a standard and well-investigated approximation for mechanistic studies in complex molecular systems. Interestingly, this partition into a classical and a quantum mechanical description reveals another way to determine which information has to be presented visually and which information needs to be rendered by addressing other senses. The part described by classical mechanics is presented visually and the highly non-intuitive quantum behaviour of the electrons is expressed and presented as forces rendered to the tactile sense.
A fully quantum mechanical treatment of the atom movement would also be possible, but the visualisation of such a situation cannot be easily realised in a virtual environment where molecules are represented by ball-and-stick arrangements. Instead, nuclear probability distributions need to be considered, which are related to the molecular structures in the Born–Oppenheimer approximation.98,99 However, the tremendous computational demands for such a full quantum treatment are prohibitive, apart from the fact that the force on an atom is then no longer straightforwardly defined.
7.2 Coordinates of the configuration space
The operator manipulates the molecular structures by changing the Cartesian coordinates of one or a few atoms at a time. Hence, the Cartesian coordinate system employed above is the most natural way to define the configuration space. Alternatively, coordinates based on the internal degrees of freedom of the molecular system can be chosen. But for the discussions to follow, the coordinate system does not need to be specified.We assume that the collection of coordinates can be separated into different sets (subsystems). The first separation follows naturally from the way the user interacts with the system. For every manipulation we can define a set of coordinates s which are strictly defined by the manipulation (system coordinates) and the set of all remaining (environment) coordinates e,
| (R1, R2,…, RM) → (s, e). | (5) |
Since the set of environment coordinates is not constrained by the manipulation, different ways to treat them are possible. They may be completely frozen, they may be subdivided into subsystems with reduced degrees of freedom or they may be optimised. Completely frozen inactive coordinates are only reasonable if they are not very much affected by the manipulation (detected by forces defined as gradient components with respect to these coordinates). Although the environment is frozen the active (system) part movement is not just ballistic, since interactions can still occur. Partly frozen coordinates (frozen subsystems) are useful, when some groups of atoms are expected to be internally rigid. The most general treatment is to optimise the inactive coordinates according to a certain protocol, which is discussed in the following section.
7.3 Energy deposition, redistribution and dissipation
Most of the manipulations performed to study reactivity involve a change in electronic energy. If a low temperature is to be maintained any excess energy has to be removed from the system. The energy deposited by the structure manipulation of the operator may be restricted according to a single measure proportional to room temperature. Any excess energy can result in a relaxation of all unconstrained coordinates. That is to minimise the energy by varying the unconstrained coordinates,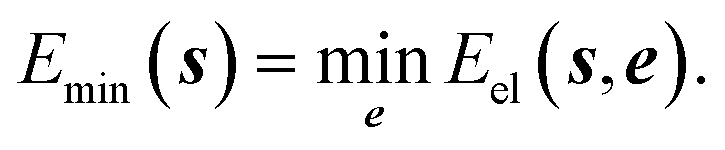 | (6) |
Removing the excess energy from the free coordinates is an artificial dissipation of energy. If always the direction with minimum resistance is chosen, the path is equal to the so called minimum energy path between reactants and products.
The redistributed energy gives rise to a motion of the atoms along the unconstrained degrees of freedom. Apart from structure relaxation, this motion can also be treated by the usual methods of molecular dynamics (MD) and corresponds then to a steered BO-MD100,101 scheme similar to steered classical MD.102 Under certain conditions, the work performed by the operator is equal to the free energy change of an equilibrium process according to the Jarzynski equality.103
7.4 Reaction path and reaction force
The structures x recorded during a virtual exploration correspond to points in the configuration space for a pseudo-one-dimensional reaction coordinate ξ(x), the arc length of the reaction path. With each point in configuration space along the path ξ a set of forces is associated| x(ξ) → f (ξ). | (7) |
If ξ describes a minimum energy path (MEP), there are only forces along the constrained coordinates and the forces in the (orthogonal) environment coordinates are reduced or zero by structure minimisation. Therefore, the work W along the MEP can be obtained from
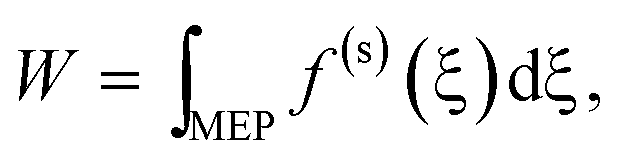 | (8) |
7.5 Virtual inertia and friction
The correct physical dynamics of an object in vacuo requires that a force exerted on it accelerates it and the object moves then with constant velocity when the force is switched off. In the case of molecules, any structure manipulation is performed by exerting forces which would lead to a continuous linear movement after the force vanished at the end of the manipulation. The operator would be constantly occupied with stopping unintentionally accelerated molecules. The energy minimisation discussed before can remove excess energy from the inactive coordinates and therefore also from the overall translation. In any case, a molecule which had been pulled at should not move after it has been released by the operator (provided that no other forces are present).The technical implementation of the energy dissipation process, however, gives rise to a sensation of viscosity when a molecule is grabbed and pulled around in the virtual environment. The removal of energy by relaxing all unconstrained degrees of freedom requires a certain amount of time. This delays the relaxation and, hence, pulling at a molecule shows a resisting force. This resisting force leads to a sensation of friction as if the molecule would be dragged through a viscous liquid. It appears that this artifact is in fact useful, since the user naturally expects inertia when accelerating atoms of fragments, although the haptic exploration does not consider any mass-dependent reaction to the exerted force (action).
7.6 Adiabatic and non-adiabatic manipulations
Depending on the rate of the relaxation process relative to the speed of manipulation, the manipulations are either adiabatic or non-adiabatic. In the adiabatic case the relaxation is faster than the manipulation and hence the path described by the manipulation including relaxation is a minimum energy path. If the relaxation is slower than the manipulation, a non-adiabatic manipulation is performed and no minimum energy path results.7.7 Implications of modified forces
As has already been discussed in section 6 an intuitive interaction with molecular systems in virtual environments often requires the manipulation of the calculated forces before they are rendered to the user. In a more formal way the forces felt by the operator fh are a modified version of the molecular force fm as| fh = C (|fm|)fm, | (9) |
As we have already discussed in our work on the haptic exploration of potential energy surfaces8 the requirement for the scaled surface is that it preserves the main features like the positions of minima and first-order saddle points. The steepness of walls or valleys is not that important if their relative depth or height is approximately preserved. Especially, schemes with a constant scaling factor93 for the forces (C = const) need to meet this requirement. Clearly, the reaction network explored in such a way can be refined by accurate quantum chemical methods in the background.
In variable gaining schemes94 the scaling factor is a function of the absolute value of the haptic force. To preserve the existence of all features allowing only a modification of their relative heights or depths, the function C is subject to some conditions. (1) C = 0 only at |fm| = 0 and nowhere else and (2) C must be strictly positive. Only then are no artificial saddle points created. With such a function, for instance, a shallow minimum is detectable but its depth may be exaggerated.
It is important to give the operator the freedom to change the scaling during an exploration. Since during an exploration all structures, gradients and energies can be recorded the true potential energy surface can be reconstructed afterwards.
7.8 Influence of interactive manipulations
Every input device implicitly introduces a constraint by keeping certain atoms at fixed positions. Additional constraints may be imposed by the parameters of the optimisation procedures. Hence, in the course of an exploration many constraints can be simultaneously active. In fact every constraint that is introduced restricts the part of configuration space that can be explored.However, since every constraint introduces additional forces, the force on a certain atom, can be significantly different with and without a constraint. If, for instance, some atom positions are fixed either by an additional input device or by just excluding them from the relaxation procedure, new constraints are introduced. These constraints in turn alter the behaviour of the system and can be regarded as additional forces. It is clear that the design of such constraints is a crucial ingredient for the manual exploration of reaction mechanisms.
7.9 Local reactivity vs. conformational entropy and large amplitude motions
We envisage a virtual exploration of chemical reactivity that involves the making and breaking of chemical bonds featuring a bond energy that is at least one order of magnitude larger than the thermal energy. Only under these conditions can a manual exploration of individual molecular structures be meaningful. A typical example is the formation of coordinative bonds to a transition metal ion. The thermodynamics and kinetics of such a process in solution is usually dominated by the changes in electronic energy, while temperature and entropic effects may be neglected in a first step. This is the reason why computational studies on transition-metal catalysed reactions can be successfully carried out by stationary quantum chemical methods. We may call such situations dominated by ‘local reactivity’. Temperature and entropic effects are then considered within simplified models for the degrees of freedom (e.g., by harmonic potentials). Usually, they lead to small modifications of the electronic energy profile. Significant departures of the free energy surface from the Born–Oppenheimer potential energy surface can be observed if the number of reactants changes in an elementary reaction step in a gas-phase process because of non-negligible contributions from the translational entropy change. This situation changes when many weak contacts among conformationally flexible reactants and surrounding molecules become important such that entropy changes upon chemical transformations can pile up. Then, resorting to statistical methods as provided by molecular dynamics or stochastic simulation schemes becomes mandatory. Other chemical processes that may require such techniques are the occurrence of structural rearrangements on long time scales (such as large amplitude motions that may induce massive overall structural changes in a macromolecular assembly). However, sampling approaches that may account for these processes can be called upon within the virtual reactivity environment described here. In fact, the hardware demands are currently just exorbitantly larger. As a consequence, the first implementation of a virtual reactivity lab may only deal with typical local-reactivity problems.8 General discussion and conclusions
The remarkable theoretical and algorithmic achievements in quantum chemistry in the past decades made it possible to assign an electronic energy and molecular properties to a molecular structure in a reasonable time. Traditional research along these lines seeks to improve on accuracy as well as on increasing the size of molecules for which such calculations are still feasible. We have argued that a different focus on algorithmic developments will lead to a paradigm shift in this field. This produces a virtual environment for the intuitive and direct study of chemical reaction mechanisms. Its ingredients are (i) the possibility to calculate quantum-mechanical information about a reactive system of at most a few hundred atoms in real time9 and (ii) the possibility to explore large amounts of data by physically experiencing their contents through the tactile human sense.7 The latter can be realised directly9 or through an intermediate interpolation layer.7,8 It is important to understand that such a reactivity exploration tool requires new concepts for the step-by-step exploration of a reaction path.10 For reactivity exploration, algorithmic tools need to be developed10 that are significantly advanced compared to interactive structure optimisations, which aim to aid the set-up of configurations in molecule editors. For the latter, the final result, i.e., a minimum structure on the Born–Oppenheimer surface counts, while considering physical principles for the way in which such a minimum structure is found is not decisive.In this work, we undertook the next step and elaborated on a new route of development in quantum chemistry towards ubiquitous computing that deeply immerses a chemist into his/her molecular target. This virtual environment poses new challenges for the organisation and presentation of tons of quantum chemical raw data. Generating alpha-numerical input files for complicated electronic structure calculations and processing the alpha-numerical output is one of the main obstacles prohibiting ubiquitous interactive computing in chemistry. This traditional, but pedestrian way of reactivity exploration is likely to die out, when ubiquitous computing, big-data handling, and immersive technology becomes available within a single implementation. Before such a virtual reactivity lab can be made available, a thorough analysis of its components is mandatory.
A central part is certainly how the quantum mechanical response is calculated in such an environment. The three main requirements for a suitable electronic structure method are: (1) it needs to be fast enough for a real-time execution, (2) it has to be based on the first principles of quantum mechanics for bond breaking and making and (3) the quality has to be such that the main features of the potential energy surface are detectable. As a first step one has to resort to approximate electronic structure methods. Hence, we discussed and evaluated contemporary semi-empirical methods suitable to study chemical reactivity in real time. With respect to both, time and accuracy, the semi-empirical methods investigated here permit an interactive study of chemical reactivity within a virtual environment. Combining the results of this work with the results we obtained in our previous studies7–9 we now have a range of methods at hand, which allow the calculation of real-time quantum mechanical responses. These methods range from interpolation techniques, semi-empirical methods up to minimal DFT and HF calculations.
Furthermore, we describe in detail here the design of a virtual environment tailored for the exploration of chemical reaction mechanisms and the subsequent study of reaction networks. We show how the quantities presented in the proposed virtual environment are connected to those manifest in a quantum mechanical description. The introduced approximations, which are necessary to allow for an interactive study of chemical reactivity have been analysed and the inherent limits have been worked out. We also discuss routes on how these limits can be partly alleviated for the incorporation of, e.g., tunnelling or entropic effects. The scaling of the forces (and therefore also of the potential energy surface) in a virtual environment has been analysed, too. Carefully chosen, they do not impair the physics of the system but can even enhance the experience. In fact, an interactive exploration of the Born–Oppenheimer potential energy surface given by the electronic energy can be carried out with a minimal quantum chemical model that recovers the rough position of minima and first-order saddle points and thus the specific structure of the surface (i.e., without introducing artifacts like spurious or missing stable intermediates).
The operator does not need to analyse any intermediate results. Instead, good candidates for stable structures are stored as nodes of a reaction network, which the operator sees emerging on the screen. Connecting lines of these nodes can be supplemented with a barrier height from an automated transition state search, which can be launched in the background by one of many service programs, which we have called ‘jeannies’ here. If the jeannie realises that there is no unique transition state, the connecting line will most likely not represent an elementary reaction step and this fact can be visualised. The jeannie may hand this information to another service program responsible for the optimisation of local minimum structures (within the structural interval defined by the two nodes in the reaction network). Since real-time quantum chemical data is a prerequisite for the whole virtual environment, the local-minimum and the transition-state searches do not need to be very efficient, but they need to be very stable. This poses a whole new set of constraints on such algorithms.
While the operator observes the expanding reaction network on the screen and can investigate structures on nodes as well as the connecting transition-state structure by a simple mouse click, he/she may choose to steer the construction of the emerging network. Certain branches of the network may thus be shut down and excluded from further exploration at will when deemed necessary. Accepted nodes may be refined by more accurate quantum chemical calculations, automatically launched in the background by another jeannie. While the accurate optimisation of a minimum structure at a node is certainly valuable for obtaining reliable structural information, the associated electronic energy is only of secondary importance. Instead, the energetical position of the node within the reaction network is of primary importance. In this systems-chemical view of complex chemical processes, a rolling kinetic modelling of all elementary reaction steps emerging in the network will determine, which nodes and links are most important. These are the ones for which more accurate quantum chemical calculations are then launched by the jeannie.
While we have already realised some of the components necessary for the chemical virtual reality lab, many ideas described here await their implementation and we will continue to finally provide such a tool to the chemistry and materials science communities. Currently, the chemical reactivity of hydrocarbons can already be explored in real time and reaction networks for these compounds can be mapped out.10 If the fully-fledged tool is completed, it will represent a new type of predictive and creative tool for chemical research in silico. Imagine a catalytic reaction mediated by some catalyst. Modelling a catalytic cycle is currently a very laborious and time-consuming undertaking. Because of the huge effort required, side reactions, of which there are orders of magnitude more than there are reaction steps in the catalytic cycle, are usually omitted. The virtual reality lab would be able to deal with side reactions, too. The data can be naturally absorbed into the components of the reaction network introducing new configurations to be constructed and finally added to the existing nodes. Natural reaction partners of the intermediates in the catalytic cycle are all molecules occurring in the cycle (including the intermediates themselves) as well as standard molecules from the environment like solvent molecules (especially water) and dioxygen. The plethora of structures needed for such a screening of potentially degradative reactions can again be set up and screened automatically by jeannies. Clearly, another useful ingredient for this endeavour is a jeannie that aids designing structures for reactive systems with specific functionality.13,14
Hence, the virtual environment envisaged here will be much more than a simple tool to control and manage quantum chemical calculation (as by standard graphical user interfaces to quantum chemistry program packages). Instead, it will be a new tool for truly creative work in chemistry that can challenge the work of experimental chemists.
Acknowledgements
This work has been supported by ETH Zurich (grant number: ETH-08 11-2).References
- J. F. Prins, J. Hermans, G. Mann, L. S. Nyland and M. Simons, Future Gener. Comput. Syst., 1999, 15, 485–495 CrossRef.
- A. Bolopion, B. Cagneau, S. Redon and S. Régnier, Intelligent Robots and Systems, 2009. IROS 2009. IEEE/RSJ International Conference on, 2009, pp. 237–242 Search PubMed.
- N. Férey, J. Nelson, C. Martin, L. Picinali, G. Bouyer, A. Tek, P. Bourdot, J. Burkhardt, B. Katz, M. Ammi, C. Etchebest and L. Autin, Virtual Reality, 2009, 13, 273–293 CrossRef.
- J. Heyd and S. Birmanns, Virtual Reality, 2009, 13, 245–255 CrossRef.
- M. Bosson, S. Grudinin, X. Bouju and S. Redon, J. Comput. Phys., 2012, 231, 2581–2598 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- K. H. Marti and M. Reiher, J. Comput. Chem., 2009, 30, 2010–2020 CrossRef CAS PubMed.
- M. P. Haag, K. H. Marti and M. Reiher, ChemPhysChem, 2011, 12, 3204–3213 CrossRef CAS PubMed.
- M. P. Haag and M. Reiher, Int. J. Quantum Chem., 2013, 113, 8–20 CrossRef CAS.
- M. P. Haag, A. C. Vaucher, M. Bosson, S. Redon and M. Reiher, ChemPhysChem Search PubMed , submitted [arXiv: 1405.4036].
- M. Bosson, C. Richard, A. Plet, S. Grudinin and S. Redon, J. Comput. Chem., 2012, 33, 779–790 CrossRef CAS PubMed.
- M. Bosson, S. Grudinin and S. Redon, J. Comput. Chem., 2013, 34, 492–504 CrossRef CAS PubMed.
- T. Weymuth and M. Reiher, Int. J. Quantum Chem., 2014, 114, 838–850 CrossRef CAS.
- T. Weymuth and M. Reiher, Int. J. Quantum Chem., 2014, 114, 823–837 CrossRef CAS.
- R. Dawes, D. L. Thompson, Y. Guo, A. F. Wagner and M. Minkoff, J. Chem. Phys., 2007, 126, 184108 CrossRef PubMed.
- Y. Guo, L. B. Harding, A. F. Wagner, M. Minkoff and D. L. Thompson, J. Chem. Phys., 2007, 126, 104105 CrossRef PubMed.
- G. G. Maisuradze and D. L. Thompson, J. Phys. Chem. A, 2003, 107, 7118–7124 CrossRef CAS.
- G. G. Maisuradze, D. L. Thompson, A. F. Wagner and M. Minkoff, J. Chem. Phys., 2003, 119, 10002–10014 CrossRef CAS PubMed.
- J. M. Bowman, J. S. Bittman and L. B. Harding, J. Chem. Phys., 1986, 85, 911–921 CrossRef CAS PubMed.
- M. A. Collins, Theor. Chem. Acc., 2002, 108, 313–324 CrossRef CAS PubMed.
- L. M. Raff, M. Malshe, M. Hagan, D. I. Doughan, M. G. Rockley and R. Komanduri, J. Chem. Phys., 2005, 122, 084104 CrossRef CAS PubMed.
- R. Dawes, D. L. Thompson, A. F. Wagner and M. Minkoff, J. Chem. Phys., 2008, 128, 084107 CrossRef PubMed.
- J. Tersoff, Phys. Rev. B, 1988, 37, 6991–7000 CrossRef.
- D. W. Brenner, Phys. Rev. B: Condens. Matter, 1990, 42, 9458–9471 CrossRef CAS.
- M. W. Finnis and J. E. Sinclair, Philos. Mag. A, 1984, 50, 45–55 CrossRef CAS.
- A. Warshel and R. M. Weiss, J. Am. Chem. Soc., 1980, 102, 6218–6226 CrossRef CAS.
- A. C. T. van Duin, S. Dasgupta, F. Lorant and W. A. Goddard, J. Phys. Chem. A, 2001, 105, 9396–9409 CrossRef CAS.
- K. D. Nielson, A. C. T. van Duin, J. Oxgaard, W.-Q. Deng and W. A. Goddard, J. Phys. Chem. A, 2005, 109, 493–499 CrossRef CAS PubMed.
- S. Mishra and M. Meuwly, Kinetics and Dynamics, SpringerNetherlands, 2010, vol. 12, pp. 137–155 Search PubMed.
- A. B. Anderson, Int. J. Quantum Chem., 1994, 49, 581–589 CrossRef CAS.
- R. Hoffmann, J. Chem. Phys., 1963, 39, 1397–1412 CrossRef CAS PubMed.
- A. B. Anderson, J. Chem. Phys., 1975, 62, 1187–1188 CrossRef CAS PubMed.
- J. A. Pople, D. P. Santry and G. A. Segal, J. Chem. Phys., 1965, 43, S129–S135 CrossRef CAS PubMed.
- J. A. Pople, D. L. Beveridge and P. A. Dobosh, J. Chem. Phys., 1967, 47, 2026–2033 CrossRef CAS PubMed.
- M. J. S. Dewar and W. Thiel, J. Am. Chem. Soc., 1977, 99, 4907–4917 CrossRef CAS.
- M. J. S. Dewar and W. Thiel, J. Am. Chem. Soc., 1977, 99, 4899–4907 CrossRef CAS.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- W. Weber and W. Thiel, Theor. Chem. Acc., 2000, 103, 495–506 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter, 1998, 58, 7260–7268 CrossRef CAS.
- Yang, H. Yu, D. York, Q. Cui and M. Elstner, J. Phys. Chem. A, 2007, 111, 10861–10873 CrossRef CAS PubMed.
- M. Gaus, Q. Cui and M. Elstner, J. Chem. Theory Comput., 2011, 7, 931–948 CrossRef CAS PubMed.
- N. Otte, M. Scholten and W. Thiel, J. Phys. Chem. A, 2007, 111, 5751–5755 CrossRef CAS PubMed.
- R. Sure and S. Grimme, J. Comput. Chem., 2013, 34, 1672–1685 CrossRef CAS PubMed.
- M. J. Dewar, E. G. Zoebisch, E. F. Healy and J. J. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- G. B. Rocha, R. O. Freire, A. M. Simas and J. J. P. Stewart, J. Comput. Chem., 2006, 27, 1101–1111 CrossRef CAS PubMed.
- J. J. P. Stewart, J. Comput. Chem., 1989, 10, 209–220 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. J. P. Stewart, J. Mol. Model., 2013, 19, 1–32 CrossRef CAS PubMed.
- G. Zheng, H. A. Witek, P. Bobadova-Parvanova, S. Irle, D. G. Musaev, R. Prabhakar, K. Morokuma, M. Lundberg, M. Elstner, C. Köhler and T. Frauenheim, J. Chem. Theory Comput., 2007, 3, 1349–1367 CrossRef.
- G. Zheng, M. Lundberg, J. Jakowski, T. Vreven, M. J. Frisch and K. Morokuma, Int. J. Quantum Chem., 2009, 109, 1841–1854 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Foxls, Gaussian 09, Gaussian Inc.Wallingford CT 2009 Search PubMed.
- A. R. Finkelmann, M. T. Stiebritz and M. Reiher, J. Phys. Chem. B, 2013, 117, 4806–4817 CrossRef CAS PubMed.
- M. J. Kory, M. Bergeler, M. Reiher and A. D. Schlüter, Chem.–Eur. J., 2014 DOI:10.1002/chem.201400364.
- D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed.
- R. Schrock, Angew. Chem., Int. Ed., 2008, 47, 5512–5522 CrossRef CAS PubMed.
- M. Reiher, B. Le Guennic and B. Kirchner, Inorg. Chem., 2005, 44, 9640–9642 CrossRef CAS PubMed.
- B. Le Guennic, B. Kirchner and M. Reiher, Chem.–Eur. J., 2005, 11, 7448–7460 CrossRef CAS PubMed.
- S. Schenk, B. Le Guennic, B. Kirchner and M. Reiher, Inorg. Chem., 2008, 47, 3634–3650 CrossRef CAS PubMed.
- S. Schenk and M. Reiher, Inorg. Chem., 2009, 48, 1638–1648 CrossRef CAS PubMed.
- S. Luber, J. Neugebauer and M. Reiher, J. Chem. Phys., 2009, 130, 064105 CrossRef PubMed.
- J. J. P. Stewart, MOPAC2012, http://openmopac.net, Stewart Computational Chemistry.
- B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678–5684 CrossRef CAS PubMed.
- M. Gaus, A. Goez and M. Elstner, J. Chem. Theory Comput., 2013, 9, 338–354 CrossRef CAS.
- J. D. C. Maia, G. A. Urquiza Carvalho, C. P. Mangueira, S. R. Santana, L. A. F. Cabral and G. B. Rocha, J. Chem. Theory Comput., 2012, 8, 3072–3081 CrossRef CAS.
- W. H. Miller, N. C. Handy and J. E. Adams, J. Chem. Phys., 1980, 72, 99–112 CrossRef CAS PubMed.
- B. A. Ruf and W. H. Miller, J. Chem. Soc., Faraday Trans. 2, 1988, 84, 1523–1534 RSC.
- E. Kraka, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 531–556 CrossRef CAS.
- Z. Konkoli, E. Kraka and D. Cremer, J. Phys. Chem. A, 1997, 101, 1742–1757 CrossRef CAS.
- A. Toro-Labbé, S. Gutiérrez-Oliva, J. S. Murray and P. Politzer, Mol. Phys., 2007, 105, 2619–2625 CrossRef.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- C. Dellago, P. G. Bolhuis, F. S. Csajka and D. Chandler, J. Chem. Phys., 1998, 108, 1964–1977 CrossRef CAS PubMed.
- R. Alves, F. Antunes and A. Salvador, Nat. Biotechnol., 2006, 24, 667–672 CrossRef CAS PubMed.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- M. Grandbois, M. Beyer, M. Rief, H. Clausen-Schaumann and H. E. Gaub, Science, 1999, 283, 1727–1730 CrossRef CAS.
- S. W. Schmidt, A. Kersch, M. K. Beyer and H. Clausen-Schaumann, Phys. Chem. Chem. Phys., 2011, 13, 5994–5999 RSC.
- M. T. Ong, J. Leiding, H. Tao, A. M. Virshup and T. J. Martinez, J. Am. Chem. Soc., 2009, 131, 6377–6379 CrossRef CAS PubMed.
- J. Ribas-Arino, M. Shiga and D. Marx, Angew. Chem., Int. Ed., 2009, 48, 4190–4193 CrossRef CAS PubMed.
- J. Ribas-Arino and D. Marx, Chem. Rev., 2012, 112, 5412–5487 CrossRef CAS PubMed.
- M. Schirski, C. Bischof and T. Kuhlen, Proceedings of the 13th Eurographics conference on Virtual Environments, Aire-la-Ville, Switzerland, Switzerland, 2007, pp. 69–76 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- Schrödinger, LLC, The PyMOL Molecular Graphics System, http://www.pymol.org.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView Version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- J. C. Chung, M. R. Harris, F. P. Brooks, H. Fuchs, M. T. Kelley, J. Hughes, M. Ouh-young, C. Cheung, R. L. Holloway and M. Pique, Proc. SPIE–Int. Soc. Opt. Eng., 1989, 1083, 42–52 CrossRef PubMed.
- E. Patrick, D. Cosgrove, A. Slavkovic, J. A. Rode, T. Verratti and G. Chiselko, Proceedings of the SIGCHI conference on Human Factors in Computing Systems, New York, NY, USA, 2000, pp. 478–485 Search PubMed.
- T. Shibata, Displays, 2002, 23, 57–64 CrossRef.
- J. E. Stone, J. Saam, D. J. Hardy, K. L. Vandivort, W.-m. W. Hwu and K. Schulten, Proceedings of 2nd Workshop on General Purpose Processing on Graphics Processing Units, New York, NY, USA, 2009, pp. 9–18 Search PubMed.
- J. E. Stone, D. J. Hardy, I. S. Ufimtsev and K. Schulten, J. Mol. Graphics Modell., 2010, 29, 116–125 CrossRef CAS PubMed.
- SensAble Technologies, Inc., http://www.sensable.com.
- Force Dimension, http://www.forcedimension.com.
- F. Weichert, D. Bachmann, B. Rudak and D. Fisseler, Sensors, 2013, 13, 6380–6393 CrossRef PubMed.
- Y.-G. Lee and K. W. Lyons, Computer-Aided Design, 2004, 36, 75–90 CrossRef.
- A. Bolopion, B. Cagneau, S. Redon and S. Régnier, World Haptics Conference (WHC), 2011 IEEE, 2011, pp. 469–474 Search PubMed.
- A. Bolopion, B. Cagneau, S. Redon and S. Régnier, J. Mol. Graphics Modell., 2010, 29, 280–289 CrossRef CAS PubMed.
- A. Bolopion, B. Cagneau, S. Redon and S. Régnier, Advanced Intelligent Mechatronics (AIM), 2010 IEEE/ASME International Conference on, 2010, pp. 329–334 Search PubMed.
- M. Born and R. Oppenheimer, Ann. Phys., 1927, 389, 457–484 CrossRef.
- E. Mátyus, J. Hutter, U. Müller-Herold and M. Reiher, Phys. Rev. A: At., Mol., Opt. Phys., 2011, 83, 052512 CrossRef.
- E. Mátyus, J. Hutter, U. Müller-Herold and M. Reiher, J. Chem. Phys., 2011, 135, 204302 CrossRef PubMed.
- D. Marx and J. Hutter, Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, Cambridge University Press, 2009 Search PubMed.
- B. Kirchner, P. J. Dio and J. Hutter, Multiscale Molecular Methods in Applied Chemistry, Springer Berlin Heidelberg, 2012, vol. 307, pp. 109–153 Search PubMed.
- J. E. Stone, J. Gullingsrud and K. Schulten, Proceedings of the 2001 Symposium on Interactive 3D Graphics, New York, NY, USA, 2001, pp. 191–194 Search PubMed.
- C. Jarzynski, Phys. Rev. Lett., 1997, 78, 2690–2693 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |