 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3D molecular models of whole HIV-1 virions generated with cellPACK
Graham T.
Johnson
*abc,
David S.
Goodsell
c,
Ludovic
Autin
c,
Stefano
Forli
c,
Michel F.
Sanner
c and
Arthur J.
Olson
c
aUniversity of California, San Francisco, CA 94143, USA. E-mail: graham.johnson@ucsf.edu
bCalifornia Institute for Quantitative Biosciences (QB3), QB3 MC: 2522, 1700 4th Street, Byers Hall, Suite 214, San Francisco, CA 94158-2330
cThe Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA
First published on 25th September 2014
Abstract
As knowledge of individual biological processes grows, it becomes increasingly useful to frame new findings within their larger biological contexts in order to generate new systems-scale hypotheses. This report highlights two major iterations of a whole virus model of HIV-1, generated with the cellPACK software. cellPACK integrates structural and systems biology data with packing algorithms to assemble comprehensive 3D models of cell-scale structures in molecular detail. This report describes the biological data, modeling parameters and cellPACK methods used to specify and construct editable models for HIV-1. Anticipating that cellPACK interfaces under development will enable researchers from diverse backgrounds to critique and improve the biological models, we discuss how cellPACK can be used as a framework to unify different types of data across all scales of biology.
Introduction
Biological processes in their mesoscale context
Multiscale biological models that span from atoms to cells can greatly improve our understanding of the mechanisms of health and disease. However, methods do not yet exist to observe, visualize, or model the mesoscale, the intermediate scale of 10−7–10−8 m bridging molecular and cellular biology, in full molecular detail. To address this need, we have developed a software framework called cellPACK, which integrates multiple types of data across scales into comprehensive 3D spatial models. This report describes a cellPACK pipeline used to create increasingly rigorous molecular models of HIV-1 (Human Immunodeficiency Virus) from structural components and systems biology data.Structural modeling of HIV-1 poses a timely challenge. HIV is a membrane-enveloped lentivirus that approaches a cell in its complexity. The genome of HIV is packaged inside a protein capsid, which requires a sophisticated collection of viral and cellular molecules to deliver it to new hosting cells, to initiate replication of the genome, and to repackage the necessary viral components for successful infection.1 NIGMS has recently created a series of Specialized Centers, bringing together dozens of research labs to characterize the structures and mechanisms across the entire lifecycle, and to identify rational targets for drug and vaccine design (http://www.nigms.nih.gov/Research/SpecificAreas/AIDSStructuralBiology/Pages/HIVspecializedcenters.aspx).
The goal of cellPACK is to create rigorous structural models of HIV and its interactions with host cells, which frame our current knowledge on the scale of the entire virion, and in its cellular context in a consistent format. These models will impel hypothesis-driven research by integrating diverse data types, identifying gaps in knowledge, and exploring the ranges of models that are consistent with current knowledge. The level of complexity of HIV is ideally suited to push the current capabilities of cellPACK and to uncover the development needs of the open-source cellPACK project as it evolves to model larger systems.
Methods to bridge from atoms to cells
Experimental techniques for integrative structural biology are now linking molecular structure and supramolecular structure, for instance, to explore the structure of HIV glycoprotein multimers2 and HIV capsid.3,4 However, methods to observe, visualize or model the larger end of the mesoscale in atomic-resolution detail do not yet exist. To make the next step, from atoms to significant portions of cells, many sources of data can be used to synthesize a view of this level. Constraint optimization modelers like IMP,5 procedural and analytical modeling approaches like Molecular Silverware6,7 and relaxation approaches like Brownian Dynamics modeling8,9 are currently used to build large-scale models. However, they are designed to function locally and do not model ultrastructural features like organelle membranes or fibrous molecules like actin. Qualitative manual protocols that use a more artistic approach have typically been required to produce mesoscale models. For example, when a large multidisciplinary collaboration determined the recipe for an average synaptic vesicle, the assembly of a structural model used to visualize the results of this effort took weeks of work using a combination of manual positioning with physics solvers to minimize collision.10,75For the past twenty years, we have taken a semi-quantitative approach for the creation of illustrations of the cellular mesoscale, which integrate data across the nano and microscales. Recipes consisting of structures, concentrations and locations of molecules, macromolecules, and organelles taken from published reports are arranged into mesoscale compositions consistent with ultrastructural data from light and electron microscopy.11,12 The illustrations are created manually with a 2.5D metaphor, typically presenting viruses or cells in cross-section, with simplified representations for the individual molecules. Fig. 1A shows a recent model of HIV-1 created with this method for educational outreach at the Protein Data Bank.13
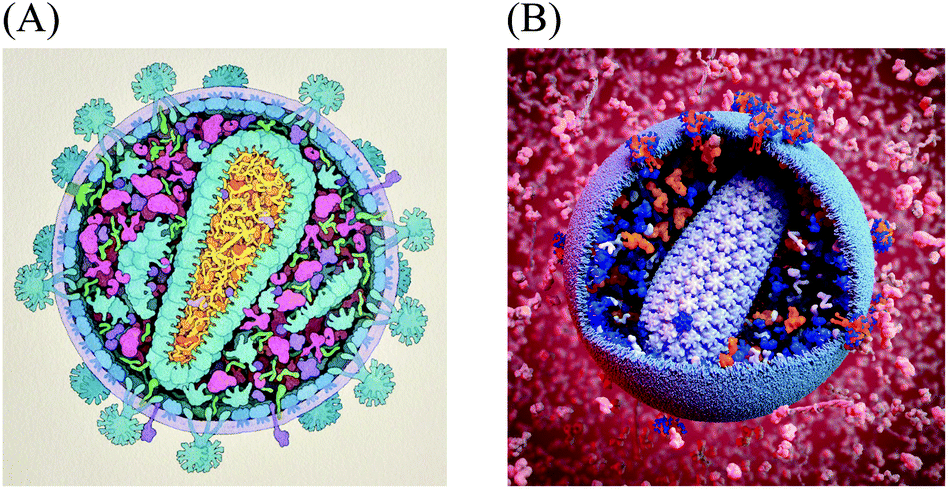 | ||
| Fig. 1 Mesoscale Models of HIV-1. (A) A hand-drawn illustration of HIV-1 integrates data from structural biology, biophysics and microscopy. Data sources from this illustration were later used to create the digitized starting point recipe HIV-1_0.1.0. (B) A 3D model of HIV-1 enables exploration, analysis, animation and simulation, as well as easy variation, modification and updating. This model is based on the cellPACK recipe HIV-1_0.1.4 nested in the larger environment recipe of HIV_in_Blood_Plasma_0.1.2, with a custom lipid bilayer added by the artist. The image was created using the cellPACK viewer plugin by digital artist Andrew McWhae for the autoPACK Visualization Challenge 2013, an outreach project which challenged artists to “convey humanity's complex relationships with this virus, be they emotional, political, or intellectual… to excite general audiences with visuals that will help us spread interest in the search for a cure.” (http://autopack.cgsociety.org) | ||
The data used to specify this illustrated model served as the basis for the HIV-1 cellPACK recipe and 3D model. cellPACK computationally automates the modeling steps of the semi-quantitative artistic approach to extend the results from 2D paintings into 3D models that can be explored, animated, simulated, analyzed, and easily edited and updated (Fig 1B shows a rendering of a 3D model created from the fourth iteration of the digitized recipe called HIV-1_0.1.4 (recipes use a 3 digit semantic versioning system to track them in a database where HIV-1_0.1.4 stands for a recipe named “HIV-1” version 0.1.4).
cellPACK can model the mesoscale at the pace of HIV research
HIV structural biology is currently an area of intense research, with new breakthroughs reported each month. A major goal of cellPACK is to provide a nimble interface for integrating new structural results into the body of available data and evaluating their consequences on the integrative models. For instance, Fig. 4–6 describe two major iterative updates to the HIV-1 recipe, where HIV-1_0.1.6 in Fig. 6 includes structural updates to the envelope glycoprotein, capsid, and other HIV proteins. In addition, cellPACK is designed to allow exploration of ambiguity or imprecision in the various components of the recipe, creating a range of models consistent with the current state of knowledge. Fig. 3, for example, shows 3D structures of HIV that result from varying the parameters affecting surface protein distributions and quantities.Modeling an organism involves two conceptual steps: gathering of data to create a recipe for the model, and use of this recipe to build a virtual model. The goal of this report is to describe the creation of a recipe and model for a mature HIV-1 particle, which will become part of a larger HIV-in-blood-plasma recipe, including details on all of the viral and host macromolecules. This document presents the references used to support the structure, location and interactions of molecules depicted in the rendered models. Several recent review articles provided an overview to the topic and sources for additional information.1,14–18
Methods
HIV-1 cellPACK recipes
Data sources used to create the illustration in Fig. 1A were reused to create the cellPACK recipe HIV-1_0.1.0 as a driving biological project for cellPACK development. Digitization of the recipe involved building 3D representations of molecular components and other needed files, and associating parameters that drive localization, concentration, and interaction into the cellPACK recipe file format. The cellPACK recipe was corrected and updated periodically through four iterations, while developing the cellPACK software over a three-year period, to produce the recipe HIV-1_0.1.4 visualized in Fig 1B. A major update to the recipe, created with the semi-quantitative approach, was published in 2012;19 this recipe was used to update HIV-1_0.1.4 to create HIV-1_0.1.5, which is described in detail in the Results and in Fig. 4 and 5. Three major structural updates to the ingredients have since been published in 2013 and 2014, which were used, as noted in the Results, to update the recipe to HIV-1_0.1.6 (detailed in Fig. 6).cellPACK
A full description of the cellPACK framework has been submitted for publication, meanwhile, details are available on the project website and online documentation at www.cellpack.org. In summary, cellPACK unites existing and novel packing algorithms to generate, visualize and analyze comprehensive 3D models of complex biological environments that integrate data from multiple experimental sources. cellPACK currently generates probabilistic 3D models of large sections of cells that can contain dozens to millions of molecules. It packs molecules and organelles into positions that satisfy constraints consistent with the input data across the full range of scales. cellPACK additionally provides tools to store, visualize, analyze and interact with the results to make mesoscale models and mesoscale modeling accessible to a variety of audiences.As shown in Fig. 2, cellPACK framework defines a desired volume and stochastically packs it with objects, called ingredients, according to a recipe. Ingredients of arbitrary shape are placed into the allowable 3D space with zero or minimal overlap and with random distribution. Constraints and agent behaviors may be associated globally or with selected ingredients to add specific modes of interaction. A grid-based approach is used to generate models quickly while tracking and confirming global parameters. cellPACK's database consists of mesoscale recipes (e.g., cytoplasm, blood plasma, synaptic vesicles, HIV, etc.) and examples of models for each recipe.
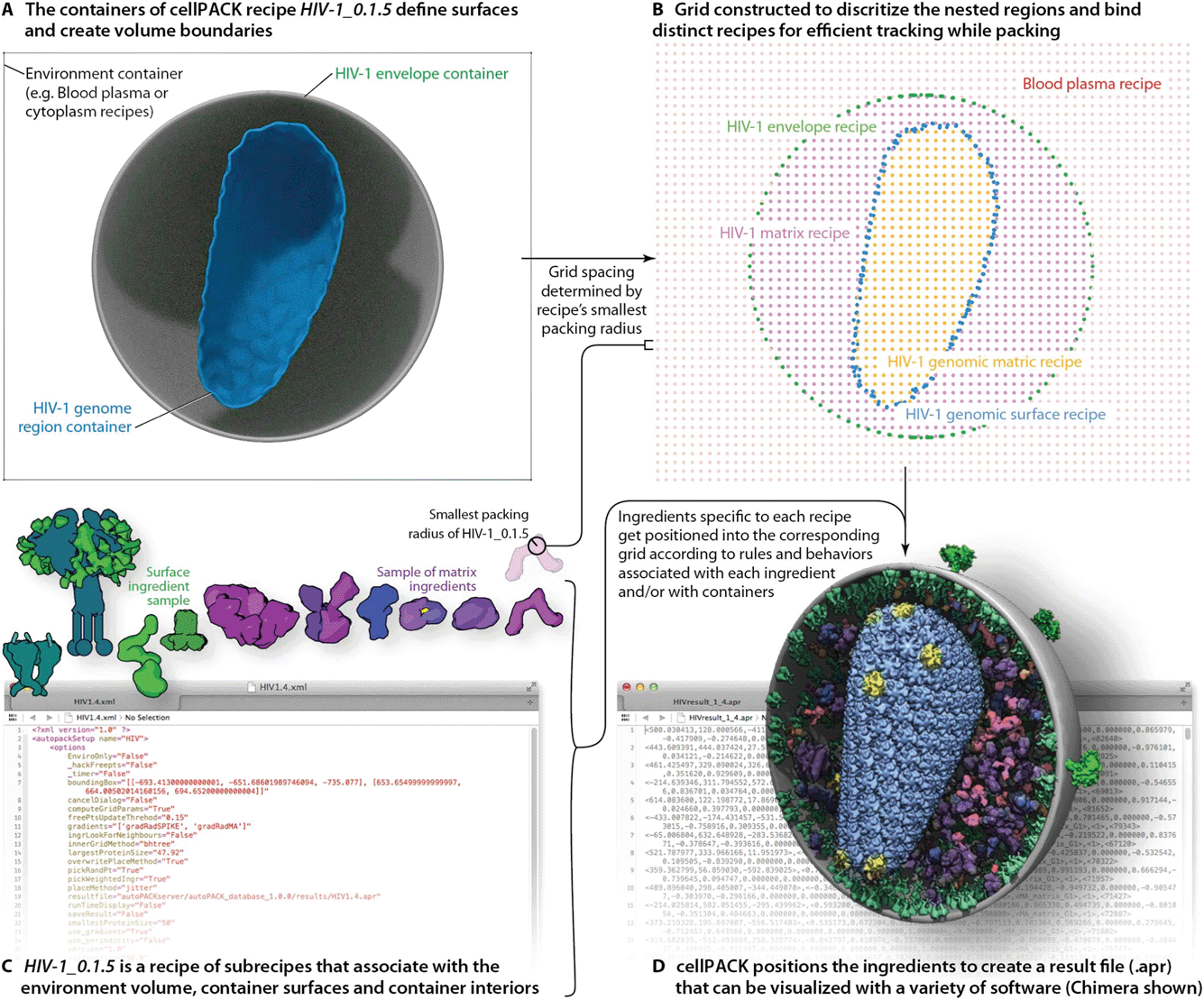 | ||
| Fig. 2 cellPACK uses a grid-based method to track ingredients as they are efficiently packed into recipe-bounding containers. (A) Polyhedral mesh containers create boundaries to restrict the packing of HIV-1 envelope surface ingredients to the surface of the envelope mesh and HIV-1 matrix ingredients to the interior of the envelope and exterior of the capsid (HIV-1 genome region outer container). (B) An omniscient grid parameterizes the space to track ingredients as they pack and to weight regions for preferred packing based on global localization or distance measurements to ingredient interaction partners. The spacing of the grid is based on the radius of gyration of the smallest ingredient to pack. (C) The full recipe of ingredients is packed to (D) generate an .apr file with IDs and matrices for each ingredient packed. The .apr file is used by a variety of visualization (e.g. molecular viewers such as Chimera shown in D) or animation software packages (e.g. Cinema 4D, Maya, Blender, 3D Studio Max, etc.) to rebuild the scene in an interactive viewport where objects can be turned on/off, replaced with more detailed data such as the original PDB file for an ingredient, or analyzed with a collection of cellPACK analysis tools or default analysis tools native to each viewer. | ||
To produce the models and visualizations shown in the figures, HIV-1 recipe ingredients (typically molecules) are stochastically packed into containers (polygonal meshes defining the envelope and capsid ultrastructures as described in the Results), up to densities provided for each ingredient, typically specific to a particular recipe. Each ingredient comes with associated properties, including a structural representation (such as a molecular surface20 or volume occupancy shell generated in this case with the molecular modeling software plugin ePMV21), and behaviors such as a particular collision–detection method or a list of binding partners that will modify the ingredient's global and local packing. For example, in HIV-1_0.1.5 and v0.1.6, the MA proteins are restricted to pack on the inside surface and have a higher probability of packing towards one end of the envelope bilayer as a first-draft global packing constraint that can recapitulate the emergent properties of molecular interactions that have been observed as ENV clustering in fluorescence microscopy.27 The polarization of ENV can be achieved by several methods currently under testing that include simplistic global constraints that force MA or ENV to pack preferentially towards some target location as demonstrated in Fig. 1B, as well as molecular scale interactions that vary association probabilities between MA–MA, MA–ENV, and ENV–ENV. Variations in the ingredient or recipe parameters can produce different results (Fig. 3). In the resultant model for a given recipe, each ingredient retains a connection to various other forms of data to enable deeper analysis, preparation for systems integration or large-scale simulations, or for modifications of the ingredient representations. The representations use coarse molecular surfaces by default, but can be replaced with an interactive molecular modeling session to modify the representation style and to access the atomic details from the structural files, for example, from Protein Data Bank (PDB) files or Electron Microscopy Data Bank (EMDB) files.
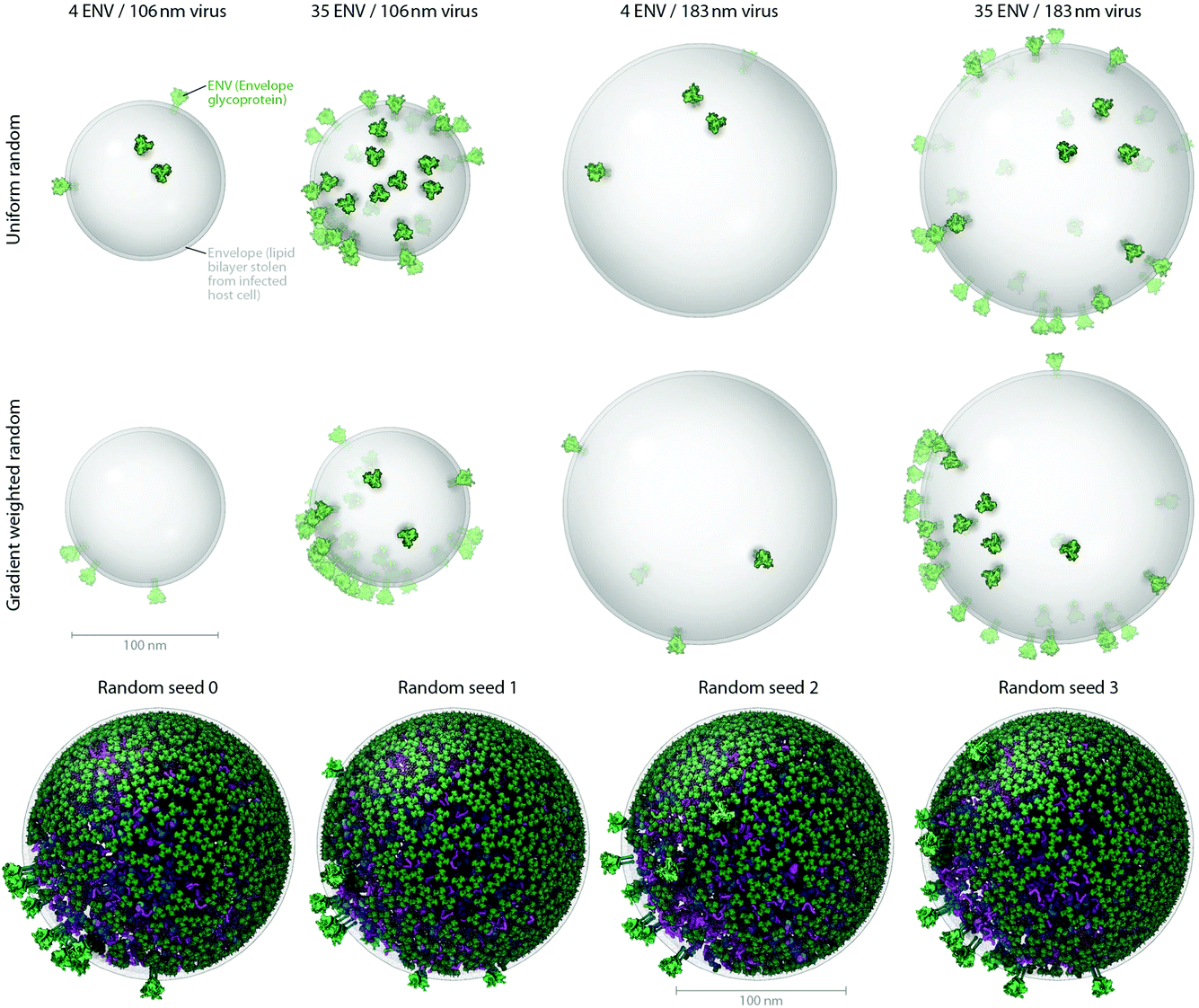 | ||
| Fig. 3 Users can produce representative cellPACK models of HIV-1 by adjusting ingredient or recipe parameters. (A) Using HIV-1_0.1.6 as a starting point, parameters for the ENV ingredient, the MA ingredient, and the envelope container were varied to experimental ranges to create models of the observed structural variability of the virion. The left two columns pack into a 106 nm envelope while the right two columns pack into a 183 nm envelope based on size ranges from electron tomography observations by Briggs et al.25 (compare with the 130 and 145 nm diameters in Fig. 4). The first and third columns pack only 4 ENV per envelope container surface, while the second and fourth columns pack 35 ENV based on ranges from Zhu, et al.26 The top row packs a uniform random distribution of MA, while the bottom row packs with a surface gradient weighted with a preference to deposit MA towards the upper right of the viewport. The dense gradient biased packing of MA excludes much of the upper-right surface of the envelope, forcing ENV to pack in the available space (primarily to the lower left). This simplistic polarization approach is one of several methods of packing and interaction parameter range settings currently being tested that can emulate the polarized clustering of ENV observed by Chojnacki et al.27 (B) Four stochastic variations of HIV-1_0.1.6 result from varying one global parameter– the seed used to generate the random number table. | ||
Unique cellPACK recipes, created for each version of HIV-1, or HIV_in_Blood_Plasma, served as input to the cellPACK software. Packing the recipes with any version of cellPACK (stable release 1.0 and newer) produces the resulting models presented in this paper as autoPACK Result (.apr) files. An .apr file contains a list of ingredient IDs with positions and orientations as described in the online documentation (http://www.autopack.org/documentation/-apr-autopack-result-file-anatomy). As specified in the legends, visualization software including the molecular viewers PMV22 and Chimera23 and a collection of cellPACK-uPy24plugin-enabled 3D animation packages were used to load and visualize the model defined in the HIV-1_0.1.5 and v0.1.6 .apr files. The visualization software all use the same method of downloading the default geometric representation for each unique recipe ingredient followed by generating efficient copies of that representation on the computer's graphics card to enable real-time viewing and fast rendering to create static images, animated sequences, or online viewers.
Community consensus on theoretical mesoscale models
cellPACK provides scripting and GUI interfaces to access viewing, analysis, and modeling modules. A database, scheduled to go online in the Summer of 2014, will soon enable invited users to critique and modify recipes and ingredients while maintaining version histories directly through the project website. Having been through numerous internal edits, upon publication, the HIV-1_0.1.6 recipe will become the first full-release (version 1.0) of HIV in the semantic versioning system (laterally named as HIV-1_1.0.0) and the first official model in the database to be opened for community feedback. Through web browser interfaces and leveraging social-networking technologies, the project website will ask the scientific community to analyze, critique and edit the models in an effort to produce confidence-voted community consensus models and to iteratively improve the recipes.Results
cellPACK models track progress in HIV structural research
The modular and hierarchical architecture of cellPACK is designed to streamline the incorporation of the most current experimental results into representative models. The modules of cellPACK correspond to different experimental techniques with widely different data types. Ultrastructural data from electron microscopy is captured in the containers. Biophysical data on concentrations and localization are captured in the recipes. Structural data is captured in the models for each ingredient. This modularization allows new results to be efficiently added to an existing recipe.We have included several examples of how this modularity can provide tools for exploring the structural biology of a biological system. Fig. 4 shows the models created with our two most recent recipes for HIV-1, HIV-1_0.1.5 and HIV-1_0.1.6. Version 0.1.6 incorporates newly published results for the envelope glycoprotein (ENV), VIF, and capsid that were not available when the previous recipe was created. This figure visualizes the full HIV particles, with sections removed to reveal their internal components. Fig. 5 and 6 chart individual depictions of the molecular ingredient representations where Fig. 5 details the specifics of the v0.1.5 recipe and Fig. 6 highlights the changes between the two versions.
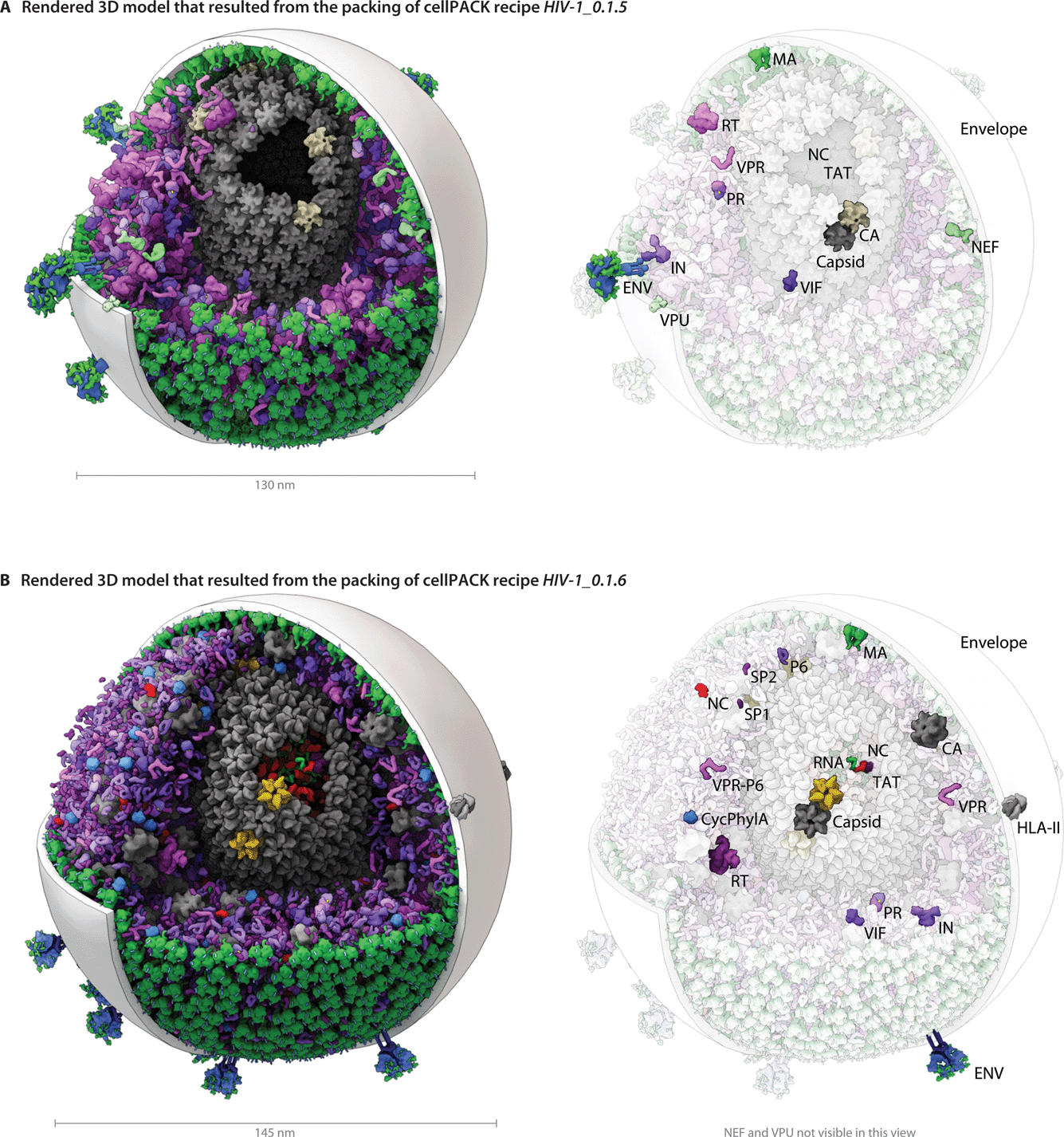 | ||
| Fig. 4 A visual comparison of HIV-1_0.1.5 and HIV-1_0.1.6 reveals consistent, but stochastic packing where ingredients primarily change in their intramolecular structure and quantity rather than in location or localization. (A) The HIV-1_0.1.5 recipe provides a substantial update to the HIV-1_0.1.4 recipe shown in Fig. 1B with numerous ingredient corrections and additions. The model, visualized in a uPy-enabled 3D animation software package shows six different recipes (each a different color coding) unified into a single model. (B) Three major structural updates as well as more subtle quantification and envelope dimension changes were added to update HIV-1_0.1.5 to HIV-1_0.1.6. | ||
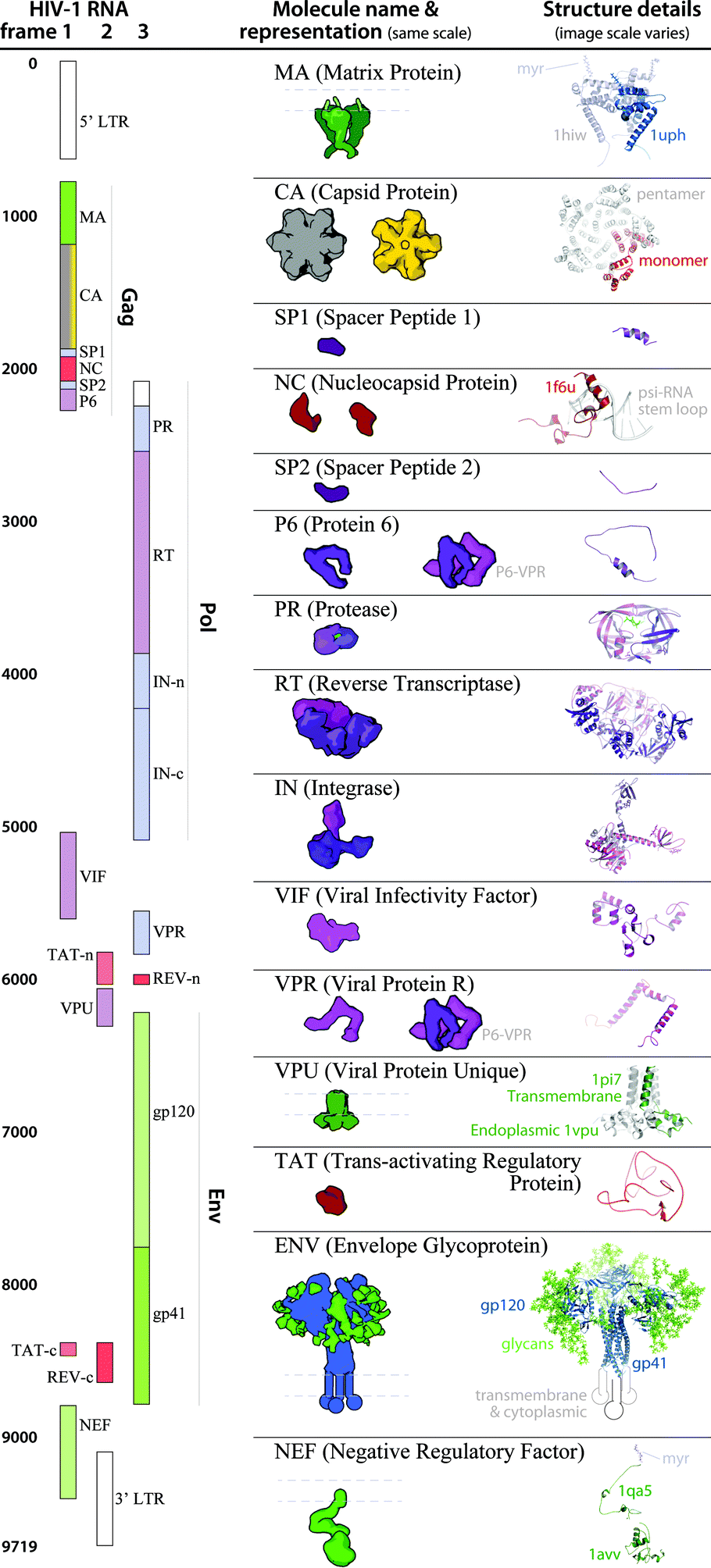 | ||
| Fig. 5 HIV-1_0.1.5 ingredient representations with structural summaries. The HIV genome landmark map on the left side (redrawn from www.hiv.lanl.gov/content/sequence/HIV/MAP/landmark.html) relates to the default coarse molecular surface representations and structural details of the HIV gene products in the center column. Many of the ingredients are updated in the newer iteration of this recipe, HIV-1_0.1.6 as detailed in Fig. 6. | ||
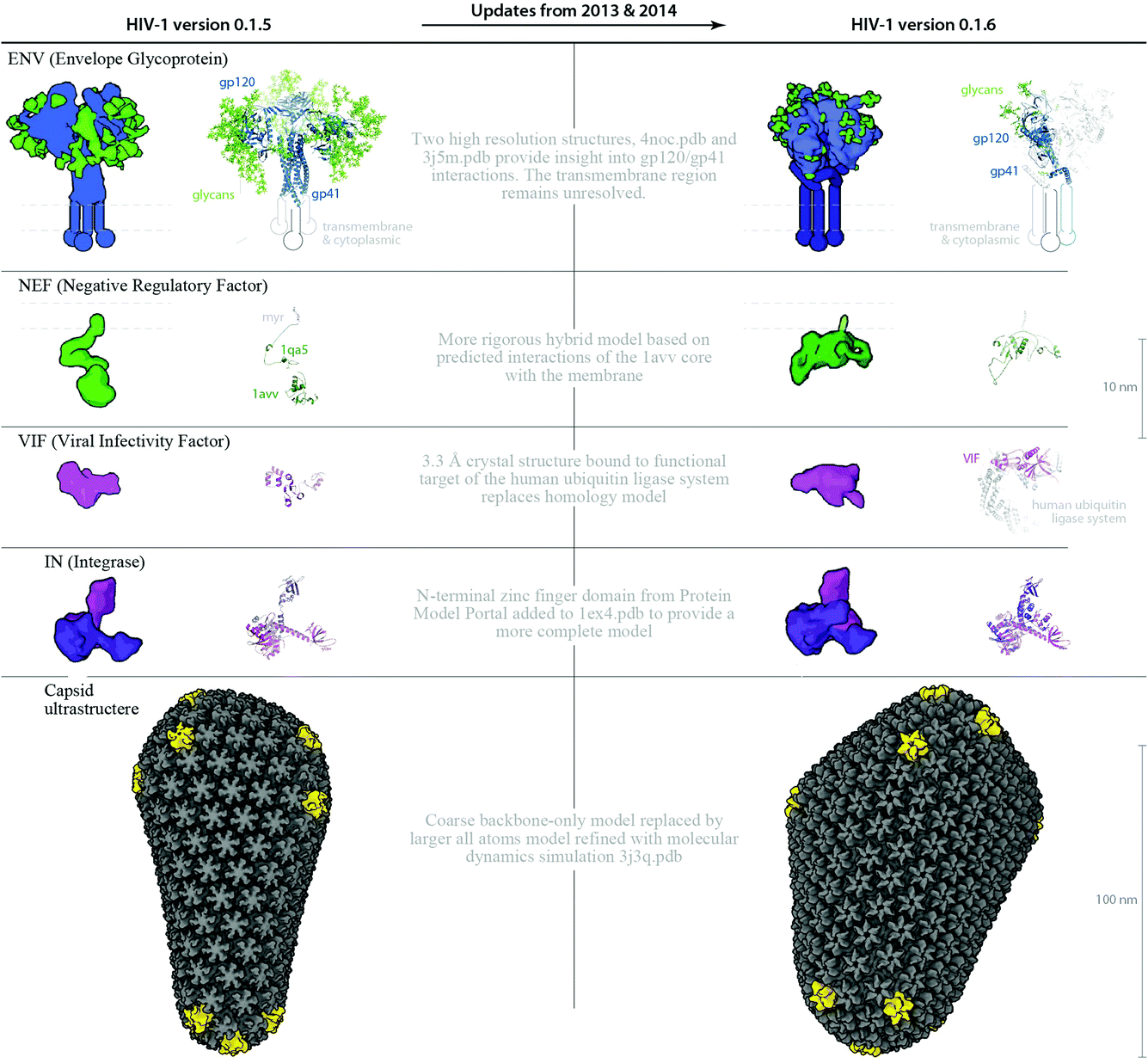 | ||
| Fig. 6 Structure and parameter changes between HIV-1_0.1.5 and HIV-1_0.1.6. Two ingredients ENV and VIF, and the ultrastructural capsid have been updated with new structures published in the past 9 months. The new structures provide important targets for vaccine and drug design, and because there may be steric hindrances imposed by their interactions or localizations, its important to visualize the ingredients in the context of the whole virus. Improved homology models for NEF and IN provide more complete representations for these two ingredients. The left column shows “older” structures and/or parameters from HIV-1_0.1.5 that were changed to iterate the recipe to the “upgraded” HIV-1_0.1.6. | ||
Fig. 3A shows how cellPACK can be used to explore experimental uncertainties or ranges of parameters in experimental data. HIV is a pleomorphic organism, with a range of sizes and numbers of component molecules. cellPACK is able to create models that represent extrema in these ranges or that represent virions with average properties. Finally, Fig. 3B shows that cellPACK is able to create multiple representative models based on a given recipe, stochastically exploring the range of possible structures that are consistent with the given description. Packing times vary widely across recipes depending on the methods specified, the size and detail of the ingredients, the packing volume size, the total ingredient density, the complexity of the interactions, etc. Each of the four models in Fig. 3B pack the default v0.1.6 recipe using a robust collision algorithm (the industry standard Bullet Physics Engine, bulletphysics.org) that allows zero ingredient surface overlaps. The models in Fig. 3B, which vary only in the random seed, each took < 267 seconds to pack on a standard consumer 2012 laptop computer.
The remaining portion of the results will present the sources that were used for creation of the HIV-1_0.1.5 and v0.1.6 recipes, revealing some of the challenges for specifying this type of recipe and identifying several gaps in knowledge where approximations were required. This portion simultaneously includes a brief description of the known functions of each ingredient to provide a comprehensive resource that will be folded into the developing database.
Recipe biology, structure, and parameter details
Ultrastructure: Envelope container and matrix
HIV envelopes are lipid bilayers, captured from the host cell during release, that encapsulate the virus, display the viral ENV proteins, and shield most of the viral machinery from the host immune system. Based on electron micrographs, HIV particle envelopes are primarily spherical, but pleomorphic with occasional subtle lumps in the flexible lipid bilayer surface, and the envelope diameters range from 100 nm to 207 nm (Table 1). Early cellPACK recipes for HIV used distorted spheres (see Fig. 1B) to capture the observed variations in shape, but to simplify analysis (such as the angular distribution variance of ENV) and to describe more average HIV particles, the recipes for HIV-1_0.1.5 and v0.1.6 use perfect spheres as containers for the envelope packing surfaces. HIV-1_0.1.5 envelope has a diameter of 130 nm, measuring to the center of the lipid bilayer. HIV-1_0.1.6 has a diameter of 145 nm. The bilayer, modeled after MD simulated patches, ranges from 2–5 nm in thickness across the hydrophobic region (http://nbcr.ucsd.edu/data/sw/hosted/lipidwrapper). Most of the figures show the bilayer as a 3 nm thick spherical shell for visual clarity.| 133 ± 17 nm (n = 118) (authentic immature virions)28 |
| 145 ± 25 nm (n = 255) (mature virions)29 |
| 139 ± 16 nm (n = 197) (immature virions)29 |
| 135.0 ± 17.4, 116.9 ± 8.5 (n = 10–15, mature virions, two cell lines)30 |
| 125 ± 14 nm (106–183) (mature virions)25 |
| 119 ± 207 nm (95% of mature virions)31 |
| 110 ± 8 nm (mature virions)26 |
| 126 ± 10 nm (n = 30 tomogram)32 |
| 130 ± 34 nm (n = 150 cryoEM)32 |
| 128 ± 32 nm (n = 84 STEM)32 |
| 100–150 nm (review article)14 |
The envelope bilayer in both recipes is populated by both viral and host molecules. The region inside the envelope but excluded by the capsid is termed the matrix. In both of the models, the matrix is populated with a collection of soluble viral and host molecules.
Ultrastructure: Capsid container and genomic region
A distinctive cone-shaped capsid protects the HIV genome (2 strands of RNA visible in Fig. 4B) and delivers the genome to new host cells upon infection. The entire capsid is built from repeating units of a single capsid protein. The large N-terminal domain of a capsid protein binds to other copies of capsid proteins to form stable hexameric rings33 and occasional pentameric rings.34 The C-terminal domain links rings to neighboring rings to form the conic HIV capsid superstructure. HIV capsids have been studied by electron microscopy (EMDB entry 5639) and tomography25 and have been reconstructed as atomic models using hybrid methods in two major publications. HIV-1_0.1.5 uses a smaller coarse, backbone-only model provided by Pornillos and Yeager34 that stands ∼115 nm tall with 1056 monomers (166 hexamers and 12 pentamers, coordinates provided by the authors). HIV-1_0.1.6 uses the larger of two all-atoms models that were refined with molecular dynamics, which stands ∼118 nm tall, but is wider and contains 1356 capsid monomers (216 hexamers and 12 pentamers) comprised of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 440800 atoms3 (PDB entry 3j3q).
440800 atoms3 (PDB entry 3j3q).
The interior of the capsid holds the genome and associated proteins. In our models, we have termed this space the genomic region. Each capsid monomer has a unique conformation. To provide a coarser level of detail useful for interactive visualization, the HIV-1_0.1.6 model uses a single representative monomer (chain A of 3j3q) aligned to each of the 1356 positions. The full detail PDB model with the unique conformers can be overlaid since the coarse model uses the same coordinate system.
Recipes: Molecules and interactions restricted to container boundaries
As new HIV particles bud from the infected host cell, the majority of the viral proteins are found in Gag (p55) and GagPol (p160) polyproteins (a note on nomenclature: the “p” and “gap” names are based on experimental molecular weights, and have largely been replaced by the more descriptive molecular names). Upon maturation, HIV protease (PR) cleaves each Gag and GagPol into 6 and 9 functional proteins respectively. Reports on Gag and GagPol quantification provide the most reliable abundances for proteins derived from Gag or GagPol in both mature and immature virus models. A wide range of values for the number and distribution of Gag and GagPol have been reported. Most current review articles have settled on a value of 2500 Gag polyproteins and a ratio of 20 Gag:1 GagPol, with some primary sources listed in Table 2. HIV-1_0.1.5 assumes 1710 Gag with 90 GagPol for a total of 1800 polyproteins. HIV-1_0.1.6 uses 2375 Gag and 125 GagPol for a total of 2500 polyproteins.
:GagPol ratios
| 20 Gag:1 GagPol (quantified on gels35) |
| 89% Gag:11% GagPol (shorter construct, quantified on gels)36 |
| 2000 Gag:200 GagPol28 |
| Number of Gag |
| 2000 Gag37 |
| 1008, 1512 (calculated from experimental Gag:env ratio and counting of ENV on tomograms)30 |
| 4900 Gag (8 nm lattice at 60 nm radius in 145 nm particle, no gaps)31 |
| 2400 ± 700 (70% coverage, tomography)32 |
| 2400 (61% coverage, STEM)32 |
| 2500 Gag (review article)14 |
| Distribution of Gag |
| 40% coverage (28%–60%) (estimated visually from tomograms)38 |
| 60% ± 18% coverage (n = 64 cryoTEM, sucrose purified virion)39 |
| 82% ± 16% coverage (n = 64 cryoTEM, OptiPrep purified virion)39 |
| 2/3 coverage, continuous, with one gap (reviewed)15 |
Recipes from N to C: Molecules and interactions from Gag proteins
The Gag region of the HIV genome includes the proteins MA, CA, SP1, NC, SP2, and P6. HIV-1_0.1.5 packs 1800 copies of each Gag region protein. HIV-1_0.1.6 packs 2500 copies of each Gag region protein.Recipes: Molecules and interactions from the pol region of GagPol
Roughly 5–11% of the Gag polyproteins are extended by a translational frameshift to create the GagPol polyproteins36 which contain three additional functional subunits (PR, RT, and IN) that are processed by HIV-1 protease to create the mature enzymes. HIV-1_0.1.5 packs 90 copies of each pol region protein. HIV-1_0.1.6 packs 125 copies of each pol region protein.Recipes: Molecules and interactions from the envelope region
Three identical subunits trimerize to form each ENV spike and each trimer has ∼81 glycosylation sites that cover ENV with a shielding layer of carbohydrates that comprises up to 50% of the mass of each spike. HIV-1_0.1.5 uses an older chimeric model templated from 2x7r, 1g9m, 3dnn and glycosylated with glycprot server (http://www.glycosciences.de/modeling/glyprot/php/main.php). HIV-1_0.1.6 uses a mix of two structures of the ectodomain, a crystal structure (PDB entry 4nco), and a single-particle electron microscope structure (PDB structure 3j5m). The new structures contain part of the gp41 stalk in the prefusion state, which provides insights into trimer assembly and the quaternary interaction between gp120 and gp41. The structures also contain bound broadly neutralizing antibodies as a focus for vaccine design. Some of the representations in the model include these antibody (Fab) fragments. The transmembrane region has not been resolved experimentally and is represented by geometric shapes of approximate size and shape based on molecular mass and positioned in close proximity to one another, based on electron tomography.54
The number and structure of ENV projecting from the virion surface has been difficult to quantify (see Table 3 for reported variations). Current reviews state that there are about 10 trimers on the surface.15HIV-1_0.1.5 packs 21 ENV while HIV-1_0.1.6 reduces the number to 12 for the 145nm virus. Fig. 3 shows models that result from varying this number across the published extremes (from 4 to 35).
The arrangement of ENV on the surface is a topic of current research. Fluorescence studies found a single cluster in mature virions, but not in immature virions.27HIV-1_0.1.5 and v0.1.6 pack MA along a preferential gradient that polarizes the distribution of ENV stochastically toward one side by stochastically excluding much of the surface otherwise available to ENV.
Interactions with molecules inside the virion also remain controversial. One study found that cellular protein TIP47 bridges MA and ENV and is required for packaging,57 but a later paper found that MA and TIP47 interact, but is not needed for ENV incorporation.58 Based on a current report, the “mechanism of incorporation still remains unclear”: they present genetic evidence for ENV and MA interaction, but also state “it would seem plausible that passive incorporation could be sufficient to account for the ∼10 copies”,59 therefore the recipes and resulting models do not yet include any specific interactions.
Recipes: Molecules and interactions of HIV-1 accessory proteins
:7 Gag61 equaling 257 VPR in HIV-1_0.1.5 and 357 VPR in HIV-1_0.1.6. 200 of the VPR in the matrix, remain bound to P6.37
NEF appears to be bound to the membrane in the immature virion, but the “majority”64 or 50–70%35 is cleaved to soluble form in mature virion, as quantified on gels. HIV-1_0.1.5 packs 35 envelope bound NEF and 30 soluble NEF. HIV-1_0.1.6 reduces this to 5 envelope bound NEF and 5 soluble.
:1 with the HIV genome. HIV-1_0.1.5 and v0.1.6 do not include cellular RNA.
| Cyclophilin A 1:10 Gag,70 shown in the illustration based on the PDB entry 1ak4 (cyclophilin A and N-terminal domain of CA) |
| Hsp70 1:20 Gag,69 bound to Gag |
| INI1 1:2 integrase, bound to integrase69 |
| Ubiquitin 1:10 Gag69 |
| Monoubiquinated proteins 1:50 Gag69 |
| Lys tRNA synthetase, bound to Gag |
| TIP47 bound to MA57,58 |
| ABCE1 bound to NC71 |
| Staufen1 (ds RNA binding protein) bound to NC72 |
| Annexin2 bound Gag (in macrophages)73 |
The HIV-1_0.1.6 recipe includes these examples, but is not yet comprehensive. We plan a detailed literature search and modeling effort in the next iteration to include the host proteins into the recipe and model. This is an area of intensive research, as the community is uncovering the roles of host proteins in the viral lifecycle.
Conclusions
cellPACK is designed to form a bridge between the approaches of reductionist biology and systems biology. As computational and experimental techniques probe into the mesoscale from smaller and larger scale levels, frameworks like cellPACK, that will unify and contextualize the diverse data, are needed to serve as platforms for consistent communication to facilitate interdisciplinary research. cellPACK models can serve as hypothesis-generating tools, as starting points for mesoscale simulation techniques like Brownian Dynamics,9 and have already been applied extensively for education and outreach.Modeling a virus is a relatively simple task compared to modeling an entire eukaryotic cell in molecular detail. A complex virus like HIV serves as an ideal short-term goal, which now provides a much needed framework for the HIV research community to assemble their data and to better understand lentivirus mechanics. HIV simultaneously serves as a stepping-stone to modeling organisms of increasing complexity. The up-to-date release of HIV-1_0.1.6 demonstrates that cellPACK can keep up with the fast pace of HIV research.
Hierarchical cellPACK models
The HIV-1 models presented here seek to characterize a single representative virion with full molecular detail. The hierarchical architecture of cellPACK allows this model to be used directly as an ingredient inside a larger environment recipe. Fig. 1B for example shows a rendering of HIV_in_Blood_Plasma_0.1.2, which incorporates HIV-1_0.1.4 as a complete ingredient packed amongst other plasma ingredients. Recipes can nest inside of other recipes in this manner, where a whole HIV particle is deposited as a single “large” ingredient according to its target density, or nested recipes can mix their ingredients to pack the HIV molecules at the same time as the blood plasma molecules, which allows depositing ingredients to weight and affect one another as they interact during packing. This specification of models with hierarchical recipes, rather than hierarchical models, offers great potential: In the process of building the model, nested recipes allow the ingredients in the different entities to interact and affect the final form of the model. For instance, antibodies in the blood plasma model can form interactions with the HIV model during creation of a larger model, which can restrict or induce other specific interactions based on densities and kinetics.Future plans
We have initiated several projects to experiment with the HIV-cellPACK system, for example, to explore the vast ranges of interacting parameter space across all of the ingredients. An integrated fluorescence microscopy simulation tool, similar to the approach described by Gardner et al.,76 was recently integrated into cellPACK to enable cellPACK models to be compared more directly to microscope images and to begin to test competing hypotheses for distribution of the Env glycoprotein in immature HIV-1 virions. Upcoming experiments will incorporate probabilities for specific interactions between known binding partners, and will fully explore the structures and interactions of host proteins as the research community identifies them. To further improve the HIV-1 mature virion model and to explore other aspects of the lifecycle, we have initiated a project to model the immature virion. Will exploring the vast parameter space of a cellPACK recipe in an automated fashion with results clustered to higher-order structural scoring functions enable cellPACK to reliably recapitulate the less well-described interaction details from better-understood neighboring molecular building blocks alone? These new approaches have begun to address this question.Having refined the cellPACK software with the driving biological project of modeling HIV-1, it becomes a relatively short step to model a slightly larger and more complex organism such as Mycoplasma genitalium. A systems biology project called WholeCellViz has provided a computational recipe of the hypothetical quantities, localizations, and interactions of every gene product of M. genitalium, dynamically over a full life cycle.74 The WholeCellViz parameters are being transposed into cellPACK recipes that are completed by generating structural representations for each gene product. When complete, the recipe will be packed to generate the first 3D structural model of a whole cell and the first structural snapshots of a whole cell dynamically changing over an entire cell cycle. On a broader scale, because it collects Systems Biology and Structural Biology results into a unified framework, cellPACK can support big data projects like the 3D Virtual Cell project (http://3dvcell.org), where teams of biologists will merge efforts to construct virtual cells that can be probed experimentally to predict outcomes ranging from molecular mechanisms to cell-wide effects.
Acknowledgements
We would like to thank Megan Riel-Mehan for writing suggestions; Andrew McWhae for his award-winning image of HIV_in_Blood_Plasma_0.1.2; Thomas Goddard for efficient/coarse models of 3j3q and HIV RNA; Barbie K. Ganser-Pornillos, Owen Pornillos, Mark Yeager, Juan R. Perilla and Klaus Schulten for additional unpublished versions of their respective HIV capsid models. This work was supported in part by a predoctoral fellowship from NSF (NSF 07576), a grant from the NIH (P41 RR08605), a QB3 Fellowship grant from the California Institute for Quantitative Biosciences, qb3@UCSF, and a UCSF School of Pharmacy, 2013 Mary Anne Koda-Kimble Seed Award for Innovation. This is manuscript 27000 from the Scripps Research Institute.References
- B. K. Ganser-Pornillos, M. Yeager and W. I. Sundquist, Curr. Opin. Struct. Biol., 2008, 18, 203–217 CrossRef CAS PubMed.
- D. Lyumkis, J. P. Julien, N. de Val, A. Cupo, C. S. Potter, P. J. Klasse, D. R. Burton, R. W. Sanders, J. P. Moore, B. Carragher, I. A. Wilson and A. B. Ward, Science, 2013, 342(6165), 1484 CrossRef CAS PubMed.
- G. Zhao, J. R. Perilla, E. L. Yufenyuy, X. Meng, B. Chen, J. Ning, J. Ahn, A. M. Gronenborn, K. Schulten, C. Aiken and P. Zhang, Nature, 2013, 497, 643–646 CrossRef CAS PubMed.
- J. M. Grime and G. A. Voth, Biophys. J., 2012, 103, 1774–1783 CrossRef CAS PubMed.
- D. Russel, K. Lasker, B. Webb, J. Velazquez-Muriel, E. Tjioe, D. Schneidman-Duhovny, B. Peterson and A. Sali, PLoS Biol., 2012, 10, e1001244 CAS.
- M. Blanco, J. Comput. Chem., 1991, 12, 237–247 CrossRef CAS.
- T. Byholm, M. Toivakka and J. Westerholm, Powder Technol., 2009, 196, 139–146 CrossRef CAS PubMed.
- T. Ando J. Skolnick, Proceedings of the International Conference of the Quantum Bio-Informatics IV., ed. L. Accardi, W. Freundenberg and M. Ohya, World Scientific Publishing Co., 2011, 28, pp. 413–426 Search PubMed.
- S. R. McGuffee and A. H. Elcock, PLoS Comput. Biol., 2010, 6, e1000694 Search PubMed.
- S. Takamori, M. Holt, K. Stenius, E. A. Lemke, M. Gronborg, D. Riedel, H. Urlaub, S. Schenck, B. Brugger, P. Ringler, S. A. Muller, B. Rammner, F. Grater, J. S. Hub, B. L. De Groot, G. Mieskes, Y. Moriyama, J. Klingauf, H. Grubmuller, J. Heuser, F. Wieland and R. Jahn, Cell, 2006, 127, 831–846 CrossRef CAS PubMed.
- D. S. Goodsell, Biochem. Mol. Biol. Educ., 2009, 37, 325–332 CrossRef CAS PubMed.
- D. S. Goodsell, Biochem. Mol. Biol. Educ., 2011, 39, 91–101 CrossRef CAS PubMed.
- D. S. Goodsell, http://www.pdb.org/pdb/education_discussion/educational_resources/hiv-animation.html, RCSB Protein Data Bank, 2011.
- B. K. Ganser-Pornillos, M. Yeager and O. Pornillos, Adv. Exp. Med. Biol., 2012, 726, 441–465 CrossRef CAS.
- J. A. Briggs and H. G. Krausslich, J. Mol. Biol., 2011, 410, 491–500 CrossRef CAS PubMed.
- W. I. Sundquist and H. G. Krausslich, Cold Spring Harbor Perspect. Med., 2012, 2, a006924 Search PubMed.
- A. A. Waheed and E. O. Freed, AIDS Res. Hum. Retroviruses, 2012, 28, 54–75 CrossRef CAS PubMed.
- M. Balasubramaniam and E. O. Freed, Physiology (Bethesda), 2011, 26, 236–251 CrossRef CAS PubMed.
- D. S. Goodsell, Biochem Mol Biol Educ, 2012, 40, 291–296 CrossRef CAS PubMed.
- M. F. Sanner, A. J. Olson and J. C. Spehner, Biopolymers, 1996, 38, 305–320 CrossRef CAS.
- G. T. Johnson, L. Autin, D. S. Goodsell, M. F. Sanner and A. J. Olson, Structure, 2011, 19, 293–303 CrossRef CAS PubMed.
- M. F. Sanner, J. Mol. Graph. Model., 1999, 17, 57–61 CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- L. Autin, G. Johnson, J. Hake, A. Olson and M. Sanner, IEEE Comput. Graphics Appl., 2012, 32, 50–61 CrossRef PubMed.
- J. A. Briggs, K. Grunewald, B. Glass, F. Forster, H. G. Krausslich and S. D. Fuller, Structure, 2006, 14, 15–20 CrossRef CAS PubMed.
- P. Zhu, J. Liu, J. Bess, Jr., E. Chertova, J. D. Lifson, H. Grise, G. A. Ofek, K. A. Taylor and K. H. Roux, Nature, 2006, 441, 847–852 CrossRef CAS PubMed.
- J. Chojnacki, T. Staudt, B. Glass, P. Bingen, J. Engelhardt, M. Anders, J. Schneider, B. Muller, S. W. Hell and H. G. Krausslich, Science, 2012, 338, 524–528 CrossRef CAS PubMed.
- T. Wilk, I. Gross, B. E. Gowen, T. Rutten, F. de Haas, R. Welker, H. G. Krausslich, P. Boulanger and S. D. Fuller, J. Virol., 2001, 75, 759–771 CrossRef CAS PubMed.
- J. A. Briggs, T. Wilk, R. Welker, H. G. Krausslich and S. D. Fuller, EMBO J., 2003, 22, 1707–1715 CrossRef CAS PubMed.
- P. Zhu, E. Chertova, J. Bess, Jr., J. D. Lifson, L. O. Arthur, J. Liu, K. A. Taylor and K. H. Roux, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15812–15817 CrossRef CAS PubMed.
- J. A. Briggs, M. N. Simon, I. Gross, H. G. Krausslich, S. D. Fuller, V. M. Vogt and M. C. Johnson, Nat. Struct. Mol. Biol., 2004, 11, 672–675 CAS.
- L. A. Carlson, J. A. Briggs, B. Glass, J. D. Riches, M. N. Simon, M. C. Johnson, B. Muller, K. Grunewald and H. G. Krausslich, Cell Host Microbe, 2008, 4, 592–599 CAS.
- O. Pornillos, B. K. Ganser-Pornillos, B. N. Kelly, Y. Hua, F. G. Whitby, C. D. Stout, W. I. Sundquist, C. P. Hill and M. Yeager, Cell, 2009, 137, 1282–1292 CrossRef PubMed.
- O. Pornillos, B. K. Ganser-Pornillos and M. Yeager, Nature, 2011, 469, 424–427 CrossRef CAS PubMed.
- R. Welker, H. Kottler, H. R. Kalbitzer and H. G. Krausslich, Virology, 1996, 219, 228–236 CrossRef CAS PubMed.
- T. Jacks, M. D. Power, F. R. Masiarz, P. A. Luciw, P. J. Barr and H. E. Varmus, Nature, 1988, 331, 280–283 CrossRef CAS PubMed.
- A. D. Frankel and J. A. Young, Annu. Rev. Biochem., 1998, 67, 1–25 CrossRef CAS PubMed.
- E. R. Wright, J. B. Schooler, H. J. Ding, C. Kieffer, C. Fillmore, W. I. Sundquist and G. J. Jensen, EMBO J., 2007, 26, 2218–2226 CrossRef CAS PubMed.
- N. Kol, M. Tsvitov, L. Hevroni, S. G. Wolf, H. B. Pang, M. S. Kay and I. Rousso, J. Virol. Methods, 2010, 169, 244–247 CrossRef CAS PubMed.
- J. S. Saad, J. Miller, J. Tai, A. Kim, R. H. Ghanam and M. F. Summers, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11364–11369 CrossRef CAS PubMed.
- A. Alfadhli, R. L. Barklis and E. Barklis, Virology, 2009, 387, 466–472 CrossRef CAS PubMed.
- A. Alfadhli, D. Huseby, E. Kapit, D. Colman and E. Barklis, J. Virol., 2007, 81, 1472–1478 CrossRef CAS PubMed.
- J. A. Briggs, M. C. Johnson, M. N. Simon, S. D. Fuller and V. M. Vogt, J. Mol. Biol., 2006, 355, 157–168 CrossRef CAS PubMed.
- S. C. Pettit, M. D. Moody, R. S. Wehbie, A. H. Kaplan, P. V. Nantermet, C. A. Klein and R. Swanstrom, J. Virol., 1994, 68, 8017–8027 CAS.
- H. G. Krausslich, M. Facke, A. M. Heuser, J. Konvalinka and H. Zentgraf, J. Virol., 1995, 69, 3407–3419 CAS.
- J. L. Newman, E. W. Butcher, D. T. Patel, Y. Mikhaylenko and M. F. Summers, Protein Sci., 2004, 13, 2101–2107 CrossRef CAS PubMed.
- M. A. Accola, S. Hoglund and H. G. Gottlinger, J. Virol., 1998, 72, 2072–2078 CAS.
- I. P. O'Carroll, F. Soheilian, A. Kamata, K. Nagashima and A. Rein, Virus Res., 2013, 171, 341–345 CrossRef PubMed.
- R. J. Fisher, A. Rein, M. Fivash, M. A. Urbaneja, J. R. Casas-Finet, M. Medaglia and L. E. Henderson, J. Virol., 1998, 72, 1902–1909 CAS.
- A. de Marco, A. M. Heuser, B. Glass, H. G. Krausslich, B. Muller and J. A. Briggs, J. Virol., 2012, 86, 13708–13716 CrossRef CAS PubMed.
- S. M. Solbak, T. R. Reksten, F. Hahn, V. Wray, P. Henklein, P. Henklein, O. Halskau, U. Schubert and T. Fossen, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 816–823 CrossRef CAS PubMed.
- T. Schwede, J. Kopp, N. Guex and M. C. Peitsch, Nucleic Acids Res., 2003, 31, 3381–3385 CrossRef CAS PubMed.
- D. Lyumkis, J. P. Julien, N. de Val, A. Cupo, C. S. Potter, P. J. Klasse, D. R. Burton, R. W. Sanders, J. P. Moore, B. Carragher, I. A. Wilson and A. B. Ward, Science, 2013, 342, 1484–1490 CrossRef CAS PubMed.
- J. Liu, A. Bartesaghi, M. J. Borgnia, G. Sapiro and S. Subramaniam, Nature, 2008, 455, 109–113 CrossRef CAS PubMed.
- H. R. Gelderblom, E. H. Hausmann, M. Ozel, G. Pauli and M. A. Koch, Virology, 1987, 156, 171–176 CrossRef CAS.
- E. Chertova, J. W. Bess, Jr., B. J. Crise, I. R. Sowder, T. M. Schaden, J. M. Hilburn, J. A. Hoxie, R. E. Benveniste, J. D. Lifson, L. E. Henderson and L. O. Arthur, J. Virol., 2002, 76, 5315–5325 CrossRef CAS.
- S. Lopez-Verges, G. Camus, G. Blot, R. Beauvoir, R. Benarous and C. Berlioz-Torrent, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14947–14952 CrossRef CAS PubMed.
- M. A. Checkley, B. G. Luttge, P. Y. Mercredi, S. K. Kyere, J. Donlan, T. Murakami, M. F. Summers, S. Cocklin and E. O. Freed, J. Virol., 2013, 87, 3561–3570 CrossRef CAS PubMed.
- M. A. Checkley, B. G. Luttge and E. O. Freed, J. Mol. Biol., 2011, 410, 582–608 CrossRef CAS PubMed.
- M. Kogan and J. Rappaport, Retrovirology, 2011, 8, 25 CrossRef CAS PubMed.
- B. Muller, U. Tessmer, U. Schubert and H. G. Krausslich, J. Virol., 2000, 74, 9727–9731 CrossRef CAS.
- X. Ren, S. Y. Park, J. S. Bonifacino and J. H. Hurley, eLife, 2014, 3, e01754 Search PubMed.
- N. Eswar, B. Webb, M. A. Marti-Renom, M. S. Madhusudhan, D. Eramian, M. Y. Shen, U. Pieper and A. Sali, Current protocols in bioinformatics / editoral board, Andreas D. Baxevanis… [et al.], 2006, Chapter 5, Unit 5 6 Search PubMed.
- M. W. Pandori, N. J. Fitch, H. M. Craig, D. D. Richman, C. A. Spina and J. C. Guatelli, J. Virol., 1996, 70, 4283–4290 CAS.
- Y. Guo, L. Dong, X. Qiu, Y. Wang, B. Zhang, H. Liu, Y. Yu, Y. Zang, M. Yang and Z. Huang, Nature, 2014, 505, 229–233 CrossRef CAS PubMed.
- L. Ratner, W. Haseltine, R. Patarca, K. J. Livak, B. Starcich, S. F. Josephs, E. R. Doran, J. A. Rafalski, E. A. Whitehorn and K. Baumeister, et al. , Nature, 1985, 313, 277–284 CrossRef CAS.
- N. Jouvenet, S. Laine, L. Pessel-Vivares and M. Mougel, RNA Biol., 2011, 8, 572–580 CrossRef CAS PubMed.
- J. M. Watts, K. K. Dang, R. J. Gorelick, C. W. Leonard, J. W. Bess, Jr., R. Swanstrom, C. L. Burch and K. M. Weeks, Nature, 2009, 460, 711–716 CrossRef CAS PubMed.
- D. E. Ott, Rev. Med. Virol., 2008, 18, 159–175 CrossRef CAS PubMed.
- E. K. Franke, H. E. Yuan and J. Luban, Nature, 1994, 372, 359–362 CrossRef CAS PubMed.
- J. E. Dooher, B. L. Schneider, J. C. Reed and J. R. Lingappa, Traffic, 2007, 8, 195–211 CrossRef CAS PubMed.
- L. Chatel-Chaix, K. Boulay, A. J. Mouland and L. Desgroseillers, Retrovirology, 2008, 5, 41 CrossRef PubMed.
- E. V. Ryzhova, R. M. Vos, A. V. Albright, A. V. Harrist, T. Harvey and F. Gonzalez-Scarano, J. Virol., 2006, 80, 2694–2704 CrossRef CAS PubMed.
- J. R. Karr, J. C. Sanghvi, D. N. Macklin, M. V. Gutschow, J. M. Jacobs, B. Bolival, Jr., N. Assad-Garcia, J. I. Glass and M. W. Covert, Cell, 2012, 150, 389–401 CrossRef CAS PubMed.
- B. G. Wilhelm, S. Mandad, S. Truckenbrodt, K. Krohnert, C. Schafer, B. Rammner, S. J. Koo, G. A. Classen, M. Krauss, V. Haucke, H. Urlaub and S. O. Rizzoli, Science, 2014, 344, 1023–1028 CrossRef CAS PubMed.
- M. K. Gardner, D. J. Odde and K. Bloom, Methods, 2007, 41, 232–237 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |