 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical synthesis and characterisation of cocrystals and metal organic compounds
L.
Tröbs
and
F.
Emmerling
*
BAM Federal Institute for Materials Research and Testing, Richard-Willstätter-Str. 11, 12489 Berlin, Germany. E-mail: franziska.emmerling@bam.de; Fax: +49-30-8104-1139; Tel: +49-30-8104-1133
First published on 26th February 2014
Abstract
The mechanochemical synthesis of two model compounds, a metal organic framework (H2Im)[Bi(1,4-bdc)2] (bdc = benzene dicarboxylate, H2Im = imidazole cation)) and a cocrystal (carbamazepine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :indometacin 1:1) were followed ex situ using a combination of two analytical methods. Powder X-ray diffraction (XRD) and Raman spectroscopy data were evaluated for the synthesis of the metal organic framework. The XRD measurements and REM images were analysed for the synthesis of the cocrystal. The measurements revealed that both model compounds were synthesised within minutes. The metal organic framework (H2Im)[Bi(1,4-bdc)2] is synthesised via an intermediate structure. The cocrystal carbamazepine:indometacin 1:1 is formed within a few seconds. The crystallite size decreases during the further milling treatment.
:indometacin 1:1) were followed ex situ using a combination of two analytical methods. Powder X-ray diffraction (XRD) and Raman spectroscopy data were evaluated for the synthesis of the metal organic framework. The XRD measurements and REM images were analysed for the synthesis of the cocrystal. The measurements revealed that both model compounds were synthesised within minutes. The metal organic framework (H2Im)[Bi(1,4-bdc)2] is synthesised via an intermediate structure. The cocrystal carbamazepine:indometacin 1:1 is formed within a few seconds. The crystallite size decreases during the further milling treatment.
Introduction
Mechanochemistry has gained increasing importance for the synthesis of novel compounds in recent years.1–3 Mechanochemical reactions provide many advantages over their liquid-phase counterparts in terms of shorter reaction times, higher product yields and the elimination of (harmful) organic solvents. Among other methods, mechanochemistry is now an established method for the synthesis of metal organic frameworks (MOFs) and cocrystals.4–8MOFs are compounds consisting of metal or metal-cluster centres connected via organic molecules resulting in a porous coordination system. MOFs offer a wide range of interesting properties and applications.9,10 In particular, they are characterized by large inner surfaces and pronounced porous systems. Based on these properties, MOFs can be utilized in different sectors, such as drug-carrier processes, catalysis, gas storage and gas purification.9–14 Because of the enormous number of available organic ligands and metal centers, MOFs are characterized by a great structural diversity. The use of metal cations with higher and flexible coordination numbers such as bismuth(III) can additionally increase the flexibility of the resulting structure.15,16
Cocrystals are organic compounds consisting of at least two neutral organic molecules, which are connected via intermolecular interactions such as π–π stacking, van der Waals forces and hydrogen bonds. Pharmaceutical cocrystals consist of an active pharmaceutical ingredient (API) and a coformer, typically a small organic molecule.17–19 Cocrystallisation is used to enhance the physicochemical properties of the APIs, including the stability, solubility, compressibility and bioavailability.19,20
For both material classes, mechanochemical reactions offer an effective and environmentally-friendly synthesis pathway, preventing the use of solvents and achieving high material yields. Furthermore, the products are of high purity and the reactions periods are typically completed within minutes. Although, mechanochemistry is applied successfully for the synthesis of these materials, the underlying mechanisms of the milling processes have not been clarified yet. Theories explaining the milling process range from intermediate melts or eutectics to predictions that reactions are occurring via plasma or hot-spots which are induced by the collision of the grinding balls and the grinding jar surfaces.2,21–24
In an elegant experiment, Friščić, and co-workers succeeded in directly monitoring milling reactions using synchrotron XRD.25,26 The authors used a modified ball mill equipped with a transparent Perspex jar, which allows them to follow the synthesis of the MOFs and cocrystals in situ. Further studies have been conducted in terms of an ex situ analysis of grinding reactions using Raman spectroscopy or solid-state fluorescence measurements.27,28
In a different approach, we evaluate the possibility of lab scale XRD measurements combined with further methods to elucidate the milling synthesis of two example compounds. The synthesis of the metal organic framework (H2Im)[Bi(1,4-bdc)2]29 (bdc = benzene dicarboxylate, H2Im = imidazole cation) was investigated based on ex situ XRD measurements and Raman spectroscopy. The synthesis of the cocrystal carbamazepine:indometacin 1:130 was followed via ex situ XRD and REM measurements at different steps during the grinding process. Based on the obtained data it was possible to derive a mechanistic understanding of the formation processes, occurring intermediates and reaction periods.
Results and discussion
The crystal structures of both model compounds, (H2Im)[Bi(1,4-bdc)2] and carbamazepine:indometacin 1:1, are shown in Fig. 1. These compounds were chosen to meet the following criteria: i) a typical example for the respective material class, ii) known crystalline structure, and iii) clearly distinguishable scattering areas for educts and products in a limited 2θ range. The mechanochemical syntheses of the compounds were carried out in a conventional ball mill. The reactions were investigated ex situ by a combination of different analytical methods. For both syntheses XRD measurements were performed and backed by either Raman measurements ((H2Im)[Bi(1,4-bdc)2]) or REM investigations (carbamazepine:indometacin 1:1). In a typical experiment, the reactions were stopped at different time intervals and a small amount of sample was retrieved for analysis.
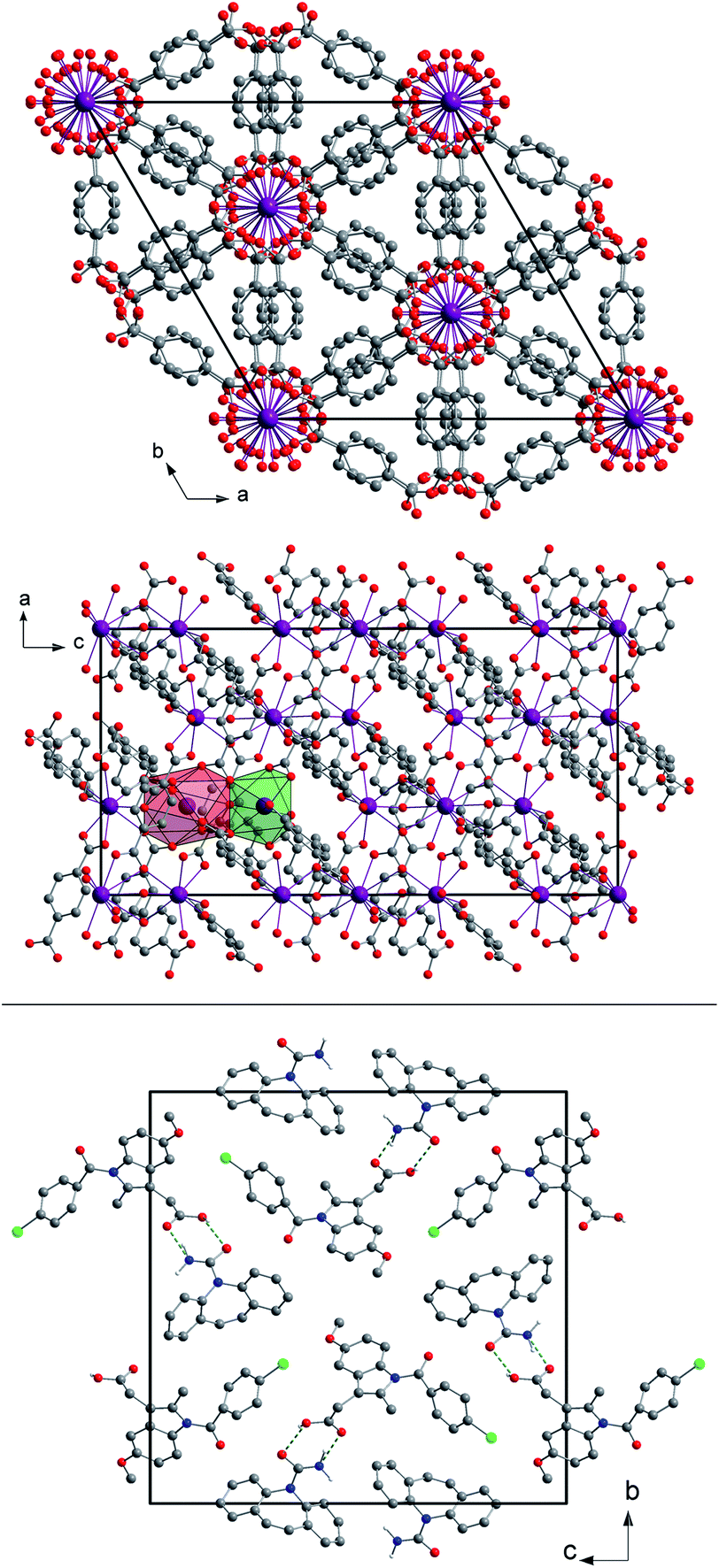 | ||
| Fig. 1 Top: a view of the unit cell of (H2Im)[Bi(1,4-bdc)2] seen along the c- and b-axes with the coordination polyhedra of both Bi3+ cations in the asymmetric unit shown in green (Bi1) and red (Bi2). The hydrogen atoms were omitted for clarity. A detailed description of the crystal structure can be found elsewhere.29 Bottom: a view of the unit cell of the cocrystal CBZ:indometacin 1:1 seen along the a-axis. The green dotted lines indicate the hydrogen bonds. The hydrogen atoms not participating in the hydrogen bonds were omitted for clarity. A detailed description of the crystal structure can be found elsewhere.30 | ||
The XRD patterns obtained during the synthesis of (H2Im)[Bi(1,4-bdc)2] are shown in Fig. 2 (reaction 1). Until a reaction period of 3 min was reached, samples were extracted every 10 s. Afterwards, the samples were taken every 30 s. After a reaction period of 20 s, the XRD measurements indicate the presence of an intermediate which is characterized by an intensive reflection at 6.5° 2θ (intermediate - reaction 1). The corresponding XRD pattern revealed, that the benzene-1,4-dicarboxylic acid is still present in the reaction mixture, whereas the reflections of bismuth(III) nitrate pentahydrate and imidazole vanished (Fig. 2, top). It can be assumed, that an intermediate was formed at the beginning of the reaction. The diffractogram corresponds rather well with the diffraction pattern of the basic bismuth nitrate Bi6O5(OH)3(NO3)5(H2O)3.31,32 Based on this assignment, it is reasonable that the first step in the formation of the framework consists of a release of H2O and NOx from bismuth(III) nitrate pentahydrate along with a protonation of imidazole. The resulting coordination sphere around the bismuth atoms consists solely of water molecules. Afterwards, this intermediate reacts with benzene-1,4-dicarboxylic acid to form the metal organic framework (H2Im)[Bi(1,4-bdc)2]. The product is formed after a reaction period of 2 min. The corresponding crystallite size amounts to 108 nm. The crystallite size of the product at different time periods was calculated based on the Scherrer equation.33,34 The product remains stable until the end of the grinding process at 15 min, indicating no significant changes in the crystallite size. Obviously, a longer grinding process is not necessary for the completeness of the reaction and the crystallite size is not influenced significantly by a prolonged reaction period.
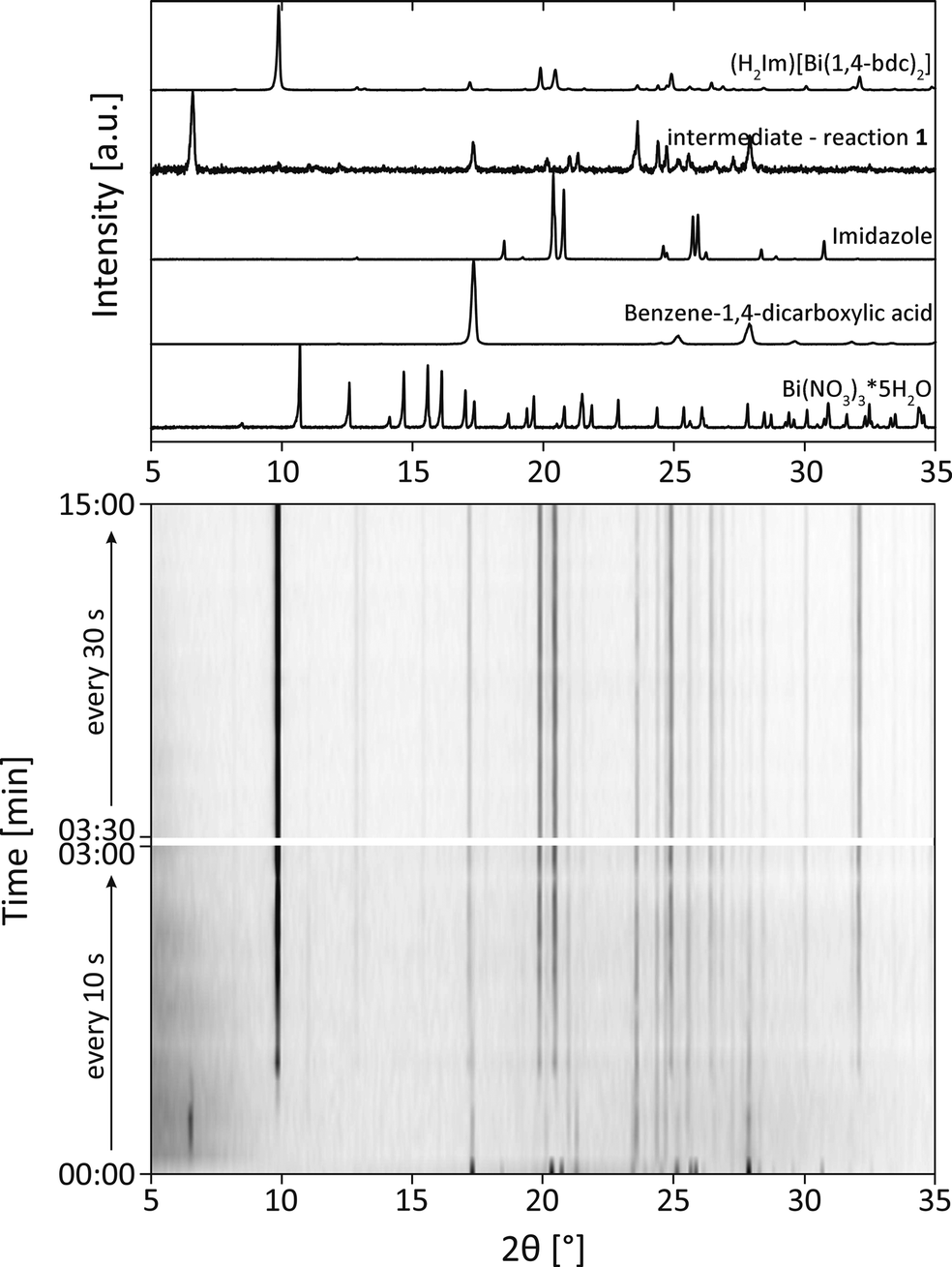 | ||
| Fig. 2 The diffractograms at milling times between 0–15 min during the mechanochemical synthesis of (H2Im)[Bi(1,4-bdc)2]. For comparison the XRD patterns of (H2Im)[Bi(1,4-bdc)2], the intermediate of reaction 1, bismuth(III) nitrate pentahydrate (Bi(NO3)3*5H2O), benzene-1,4-dicarboxylic acid and imidazole are depicted above. | ||
These findings are supported by the simultaneously collected Raman spectra (Fig. 3). The Raman spectra show that the formation of (H2Im)[Bi(1,4-bdc)2] is completed after a milling time of 2 min. The protonation of the imidazole molecule can be verified based on the Raman spectra. Two intensive bands at 1220 cm−1 and 1445 cm−1 characterize the spectrum of the imidazolium ion (blue bars in Fig. 3).35 These bands are detectable after 30 s, indicating that the protonation of imidazole proceeds in the first seconds of the milling reaction. The imidazolium molecule might act as a template, triggering the formation of the MOF. The Raman spectra of (H2Im)[Bi(1,4-bdc)2] show similar Raman bands compared to the Raman spectra of the educts benzene-1,4-dicarboxylic acid and imidazole. They are clearly shifted in the Raman spectrum of the product due to the different coordination spheres of the participating molecules.
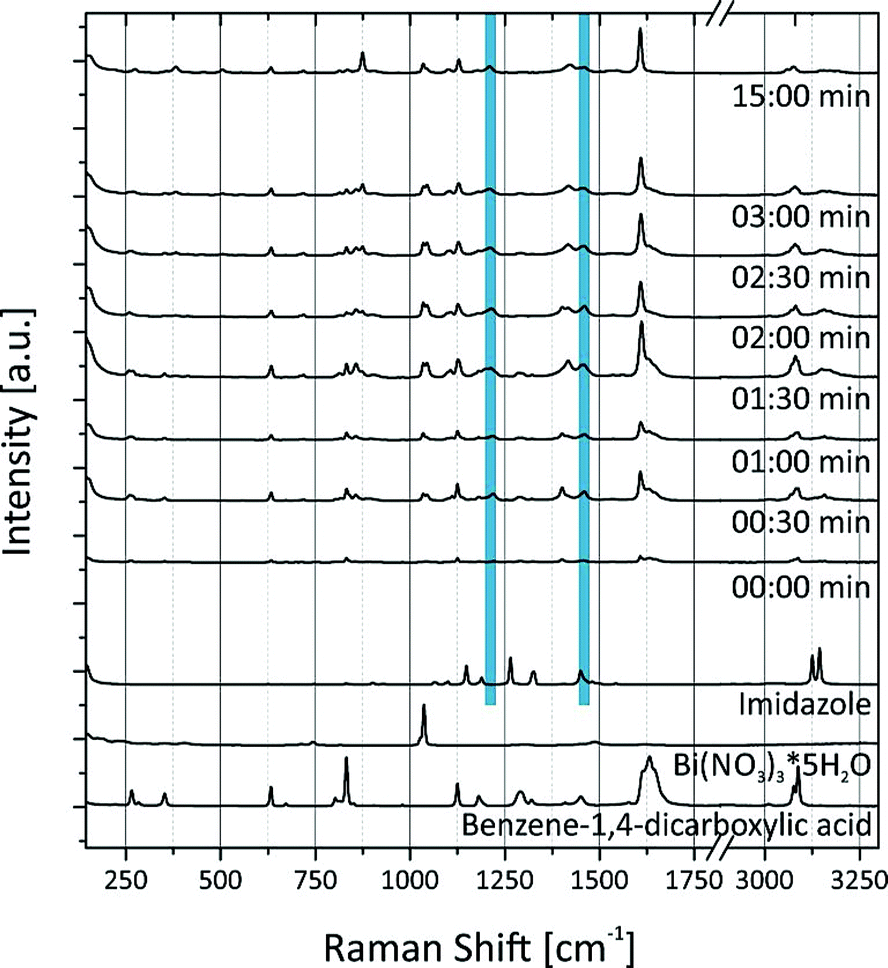 | ||
| Fig. 3 The Raman spectra recorded during the mechanochemical synthesis of (H2Im)[Bi(1,4-bdc)2] in comparison to the Raman spectra of the educts bismuth(III) nitrate pentahydrate (Bi(NO3)3*5H2O), benzene-1,4-dicarboxylic acid and imidazole. The blue bars indicate the characteristic bands for the imidazolium molecule. | ||
Similar experiments were conducted for the mechanochemical synthesis of the cocrystal carbamazepine:indometacin 1:1 (reaction 2a). The diffractograms recorded during the milling process are shown in Fig. 4. At the beginning of the reaction, the XRD measurements were performed every 10 s, after 3 minutes every 30 s, and after 10 minutes every 60 s. The diffractograms indicate that the formation of the cocrystal is completed after 1 min. Obviously, the crystal structure of both educts is destroyed and the new structure is formed within these first seconds of the reaction. Crystalline intermediates could not be detected. A closer investigation of the reflections indicates an increasing width of the reflections with a prolonged milling time. After a milling time of 1 min, the crystallite size amounts to 61 nm. After 5 min a crystallite size of 44 nm was determined, and after 15 min a crystallite size of 22 nm was determined. After 25 min, the crystallite size was below 5 nm. These findings indicate that prolonged milling can destroy the intermediately formed crystallites, whereas large crystals are formed within the first seconds of the experiment.
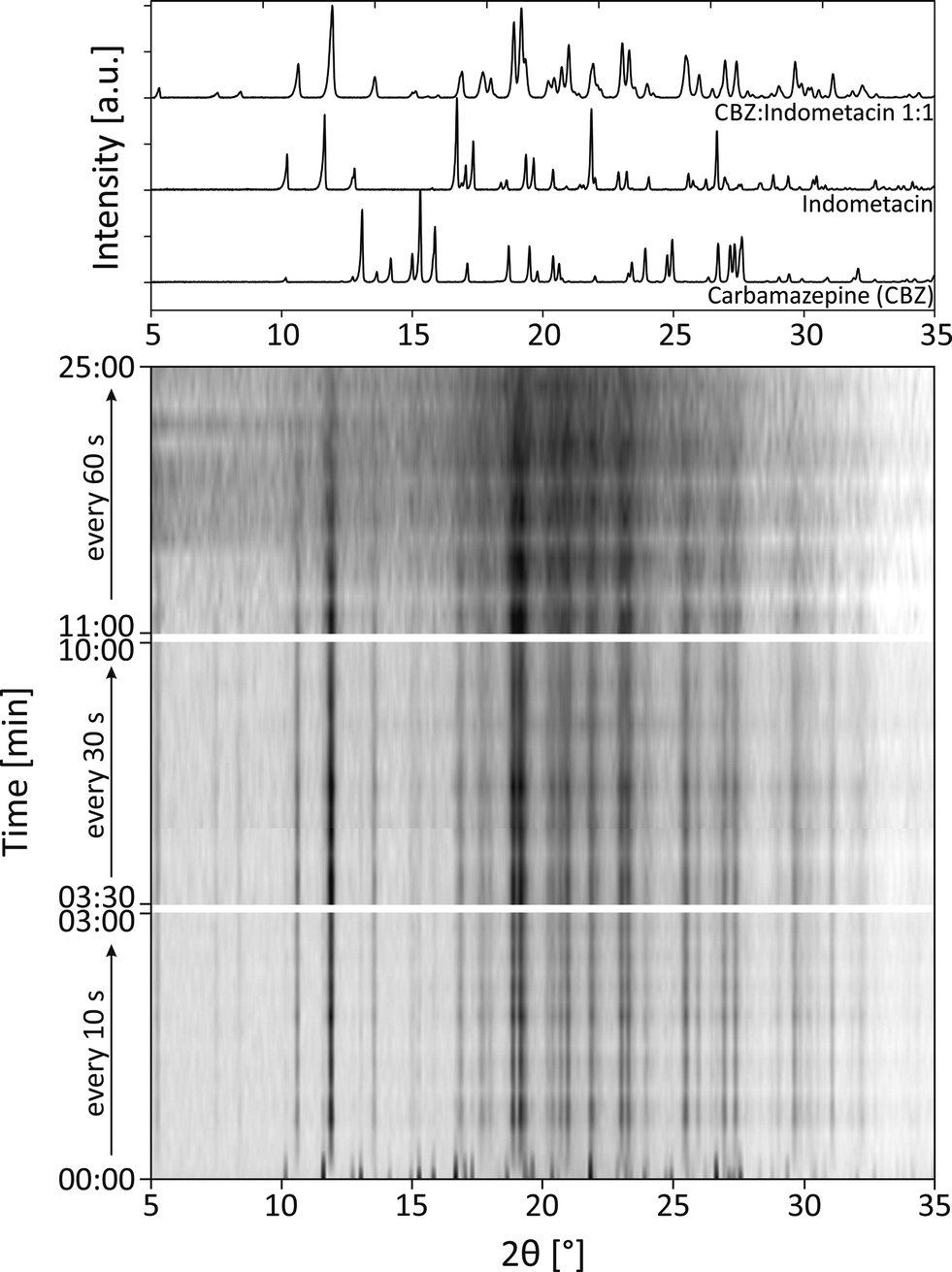 | ||
| Fig. 4 The diffractograms at milling times between 0–25 min during the mechanochemical synthesis of the cocrystal carbamazepine:indometacin 1:1. For comparison the XRD patterns of carbamazepine:indometacin 1:1, carbamazepine and indometacin are depicted above. | ||
REM images were taken at different steps of the mechanochemical synthesis, to monitor the development of the crystal morphologies (Fig. 5). Before the start of the reaction (0 s), the two cocrystal components can be clearly separated from each other within the reaction mixture. The indometacin crystals are plate-like whereas the carbamazepine crystals appear powder-like with no distinct morphology. After a reaction time of 30 s, the REM images clearly indicate the presence of needle-shaped product crystallites next to educt crystallites, mainly indometacin plates. After 60 s only needle-shaped crystals can be identified in the REM image. This observation is in accordance with the XRD experiment. Here, the formation of the cocrystal is completed after 60 s. No traces of the educt crystallites could be detected. The length of the needles is in the range of 2000–4000 nm. After 120 s the length of the needles decreases (1500–3000 nm). Prolonged milling leads to a further decrease in the length of the needles (700–1000 nm after 5 min, and 600–700 nm after 10 min). After a milling reaction of 15 min and 25 min, only a few needles are detectable, whereas the majority of the sample consists of small powder-like crystallites. These findings are in accordance with the decreasing crystallite size determined by the XRD measurements. The combination of both analytical methods reveals the formation of needle shaped crystals after a reaction period of 60 s and the destruction of the crystals during the further milling process.
 | ||
| Fig. 5 REM images of different time periods of the mechanochemical synthesis of the cocrystal carbamazepine:indometacin 1:1. | ||
Keeping in mind that a mechanochemical process conducted in a stepwise manner might yield a different result compared to an uninterrupted one,36 an additional experiment was performed. Here, the reactants carbamazepine and indometacin were ground together for 1 min. Afterwards, the reaction mixture was stored for four days in the closed grinding jar at ambient conditions to allow an uninfluenced crystallisation of the cocrystal (reaction 2b). An REM image of the final product of reaction 2b is shown in Fig. 6 in comparison to an REM image of the cocrystal synthesised in reaction 2a after a milling time of 60 s. The product from reaction 2b consists of long needles, which are interwoven into each other. The thickness of the needles obtained in both reactions is comparable whereas the length of the needles crystallised without interruption (reaction 2b) is significantly larger. The diffractogram of the product derived from reaction 2b indicates the completeness of the reaction. The crystallite size was determined to be 51 nm. These findings suggest that a short initial milling period followed by an uninfluenced crystallisation could lead to the same but more crystalline product.
 | ||
| Fig. 6 REM images for comparison of the crystalline products (cocrystal carbamazepine:indometacin 1:1) from reaction 2a after 60 s grinding time and from reaction 2b. In the latter reaction, the cocrystal was synthesised by 60 s grinding and four days storage at ambient conditions in the closed grinding jar. | ||
The analysis of the synthesis of both model compounds revealed that mechanochemistry is a very fast synthesis method. Both products were gained after milling times of 2 min ((H2Im)[Bi(1,4-bdc)2]) or 1 min (CBZ:indometacin 1:1). Especially the synthesis of the cocrystal proceeded within a few seconds. The speed of the cocrystallisation can be attributed to the fact that the intermolecular interactions between the organic molecules are more a coordination of the cocrystal components to each other.
Based on the data obtained from the combination of different analytical methods, it was possible to derive a mechanistic understanding of the formation processes, intermediates and reaction periods. In particular for the synthesis of the cocrystal carbamazepine:indometacin 1:1 the knowledge of the optimal reaction periods is essential to receive a product of high crystallinity. Amorphous, liquid, or eutectic intermediates could not be detected for both syntheses. The propensity to form a product could be associated to an increase in the contact areas with decreasing particle size during the grinding process.
Experimental
Materials
Bismuth(III) nitrate pentahydrate Bi(NO3)3·5H2O (98%), benzene-1,4-dicarboxylic acid (terephthalic acid) C6H4(COOH)2 (98+%) and imidazole C3H4N2 (99%) were purchased from Alfa Aesar, Karlsruhe, Germany and used without further purification. Carbamazepine C15H12N2O2 (≥99.5%, Fluka, Steinheim, Germany), indometacin C19H16ClNO4 (≥99%, Sigma, Steinheim, Germany) and methanol (99.9%, Roth, Karlsruhe, Germany) were also purchased commercially and used without further purification.Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :indometacin 1:1 (reaction 2).
Carbamazepine (0.1989 g, 1 eq.) and indometacin (0.3011 g, 1 eq.) were ground together after adding 250 µL methanol. The reaction was performed in a conventional ball mill (Pulverisette 23, FRITSCH GmbH, Idar-Oberstein, Germany) at 30 Hz using a 10 mL steel grinding jar and two steel balls (10 mm diameter). Two types of experiment were conducted to analyse this reaction:
:indometacin 1:1 (reaction 2).
Carbamazepine (0.1989 g, 1 eq.) and indometacin (0.3011 g, 1 eq.) were ground together after adding 250 µL methanol. The reaction was performed in a conventional ball mill (Pulverisette 23, FRITSCH GmbH, Idar-Oberstein, Germany) at 30 Hz using a 10 mL steel grinding jar and two steel balls (10 mm diameter). Two types of experiment were conducted to analyse this reaction:
(a) To follow the reaction by X-ray analysis and REM measurements, the following sampling scheme was applied: 00:00 min–03:00 min every 10 s; 03:30 min–10:00 min every 30 s; 11:00 min–25:00 min every 60 s. At these steps, the reaction was interrupted and a small amount of the sample was taken from the reaction mixture.
(b) The educts were ground for 60 s before the grinding jar was placed under ambient conditions (20 °C) without opening the jar for four days. The product was obtained as a white powder and analysed by XRD and REM measurements.
Methods
Conclusions
The presented ex situ approach proved to be completely suitable to determine the formation processes during mechanochemical reactions. The combination of two analytical techniques provides further information for a thorough understanding of the processes. Surprisingly, both model compounds are synthesised very fast within the first minutes of the milling process. The synthesis of the metal organic framework (H2Im)[Bi(1,4-bdc)2] proceeds via the formation of an intermediate. The analysis of the synthesis of the cocrystal carbamazepine:indometacin 1:1 revealed that the initially formed crystallites are destroyed during the prolonged milling times. The in situ monitoring analysis has the potential to elucidate the mechanisms of the milling reactions and provide optimized synthesis parameters.
Acknowledgements
We are grateful to Mrs Feldmann for providing the REM measurements.Notes and references
- D. Braga, S. L. Giaffreda, F. Grepioni, A. Pettersen, L. Maini, M. Curzi and M. Polito, Dalton Trans., 2006, 1249–1263 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- T. Friščić, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC.
- A. Delori, T. Friščić and W. Jones, CrystEngComm, 2012, 14, 2350–2362 RSC.
- D. R. Weyna, T. Shattock, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2009, 9, 1106–1123 CAS.
- S. Heiden, L. Tröbs, K. J. Wenzel and F. Emmerling, CrystEngComm, 2012, 14, 5128–5129 RSC.
- M. Klimakow, P. Klobes, A. F. Thunemann, K. Rademann and F. Emmerling, Chem. Mater., 2010, 22, 5216–5221 CrossRef CAS.
- E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS PubMed.
- J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef CAS PubMed.
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Ferey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, Angew. Chem., Int. Ed., 2006, 45, 5974–5978 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- A. Thirumurugan and A. K. Cheetham, Eur. J. Inorg. Chem., 2010, 3823–3828 CrossRef CAS.
- A. C. Wibowo, M. D. Smith and H. C. zur Loye, Cryst. Growth Des., 2011, 11, 4449–4457 CAS.
- N. Qiao, M. Li, W. Schlindwein, N. Malek, A. Davies and G. Trappitt, Int. J. Pharm., 2011, 419, 1–11 CrossRef CAS PubMed.
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950–2967 CAS.
- T. Friščić and W. Jones, J. Pharm. Pharmacol., 2010, 62, 1547–1559 CrossRef PubMed.
- S. Karki, T. Friščić, L. Fabian, P. R. Laity, G. M. Day and W. Jones, Adv. Mater., 2009, 21, 3905–3909 CrossRef CAS.
- P. Balaz, Mechanochemistry in Nanoscience and Minerals Engineering, Springer-Verlag, Berlin, Heidelberg, 2008 Search PubMed.
- J. F. Fernandez-Bertran, Pure Appl. Chem., 1999, 71, 581–586 CrossRef CAS.
- F. K. Urakaev and V. V. Boldyrev, Powder Technol., 2000, 107, 93–107 CrossRef CAS.
- D. Braga, L. Brammer and N. R. Champness, CrystEngComm, 2005, 7, 1–19 RSC.
- T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimaki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66–73 CrossRef PubMed.
- I. Halasz, A. Puskaric, S. A. J. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimaki, R. E. Dinnebier, B. Patel, W. Jones, V. Strukil and T. Friščić, Angew. Chem., Int. Ed., 2013, 52, 11538–11541 CrossRef CAS PubMed.
- M. Frenette, G. Cosa and T. Friščić, CrystEngComm, 2013, 15, 5100–5106 RSC.
- S. Rehder, M. Klukkert, K. A. M. Löbmann, C. J. Strachan, A. Sakmann, K. Gordon, T. Rades and C. S. Leopold, Pharmaceutics, 2011, 3, 706–722 CrossRef CAS PubMed.
- L. Tröbs, M. Wilke, W. Szczerba, U. Reinholz and F. Emmerling, CrystEngComm, 2014, 16, 5560 RSC.
- L. Tröbs, N. Zientek, D. Rump, M. Maiwald and F. Emmerling, 2014, in progress.
- F. Lazarini, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3169–3173 CrossRef.
- B. Liu, W. W. Zhou, Z. Q. Zhou and X. Y. Zhang, Inorg. Chem. Commun., 2007, 10, 1145–1148 CrossRef CAS PubMed.
- P. Scherrer, in Nachrichten von der Gesellschaft der Wissenschaften, Akademie der Wissenschaften, Göttingen, 1918, vol. 2, pp. 98–100 Search PubMed.
- H. P. Klug and L. E. Alexander, X-Ray Diffraction Procedures: For Polycrystalline and Amorphous Materials, 2nd edn, Wiley-Interscience, New York, 1974 Search PubMed.
- L. M. Markham, L. C. Mayne, B. S. Hudson and M. Z. Zgierski, J. Phys. Chem., 1993, 97, 10319–10325 CrossRef.
- V. Strukil, L. Fabian, D. G. Reid, M. J. Duer, G. J. Jackson, M. Eckert-Maksic and T. Friščić, Chem. Commun., 2010, 46, 9191–9193 RSC.
| This journal is © The Royal Society of Chemistry 2014 |