 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Is the equilibrium composition of mechanochemical reactions predictable using computational chemistry?
Peter J.
Bygrave
,
David H.
Case
and
Graeme M.
Day
*
School of Chemistry, University of Southampton, Southampton, United Kingdom. E-mail: g.m.day@soton.ac.uk
First published on 11th June 2014
Abstract
The ability of computational methods to predict the structures and energetics that determine the equilibrium of solid state mechanochemical reactions has been assessed. Two previously characterised base-catalysed metathesis reactions between aromatic disulfides are studied using crystal structure prediction methods and lattice energy calculations that combine molecular electronic structure methods with anisotropic atom–atom potentials. We find that lattice energy searches locate three of the six crystal structures as global minima on their respective crystal energy landscapes. The remaining structures are less successfully predicted, due to problems modelling relative conformational energies due to limitations of the density functional theory method for calculating intramolecular energies. Prediction of the overall reaction energies proves challenging for current methods, but the results show promise as a base on which to build more accurate and reliable approaches.
1 Introduction
The use of mechanochemical reaction conditions for synthesis has gained significant recent interest. These reactions are promoted by the input of mechanical energy and have been employed for the synthesis of a range of materials, such as metal–organic frameworks and organic molecular crystals,1,2 whose construction involves the formation of reversible intermolecular or coordination bonds. Solid state reaction conditions have also been applied to the reversible formation of covalent bonds, providing solvent-free or, in the case of liquid assisted grinding, minimal solvent conditions for organic synthesis.Recently, Belenguer et al.3 demonstrated the thermodynamic equilibration of reversible covalent bond formation in mechanochemical base-catalysed disulfide metathesis reactions; the reactions involve the exchange of substituted aromatic groups between two symmetrical homodimeric disulfides, producing a non-symmetrical heterodimer (Scheme 1). Solution equilibration in the presence of the base catalyst 1,8-diazabicyclo[5.4.0]undec-7-ene (dbu) of all reactions investigated leads to a statistical 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 mixture of the two homodimers and the heterodimer.
:1:2 mixture of the two homodimers and the heterodimer.
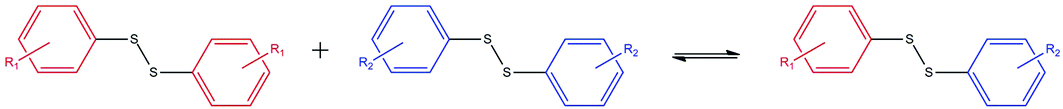 | ||
| Scheme 1 The disulfide homodimer-to-heterodimer reaction scheme. | ||
Ball mill grinding of the crystalline homodimer reactants with a small amount of dbu as catalyst leads fairly rapidly to a steady final composition. However, in contrast to the solution mixture, the final reaction mixture varies depending on the aromatic substituents. The solid-state reaction between bis(4-chlorophenyl)disulfide (4Cl4Cl) and bis(2-nitrophenyl)disulfide (2NO22NO2) leads to a final mixture with a 98% mole fraction of the heterodimer (4Cl2NO2). The analogous reaction between bis(4-methylphenyl)disulfide (4Me4Me) and 2NO22NO2 produces only 20% of the heterodimer (4Me2NO2), with 80% remaining as homodimers. The same final mixtures are obtained from different starting ratios of the homo- and heterodimers, indicating that the final mechanochemical reaction mixtures are a result of thermodynamic equilibration. The chemical diagrams of these systems can be found in Fig. 1.
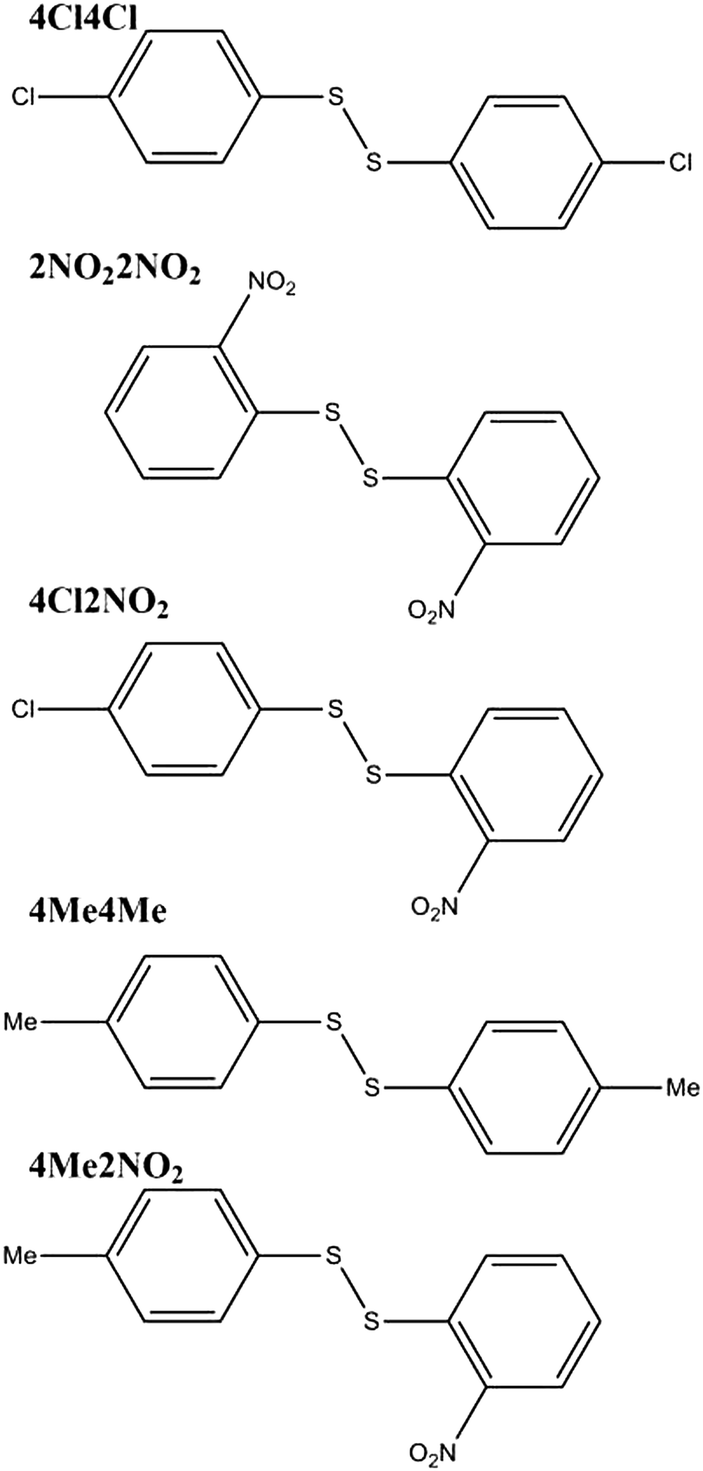 | ||
| Fig. 1 Molecular diagrams of the disulfide homodimers and heterodimers. | ||
The finding that the product mixture differs from that found in solution, and differs from reaction to reaction, demonstrates the importance of understanding the total energetics of the solid-state systems. Given that the reactants and products are crystalline, our hypothesis is that the equilibrium product distribution should be predictable from the lattice energies of the crystal structures of the molecules involved in the reaction. Here, we present an exploratory study of the challenges associated with computational prediction of the thermodynamic equilibrium of such solid state organic molecular reactions, taking the two previously characterised disulfide metathesis reactions described above as model systems.
We set the ultimate goal for computational chemistry in this area as the complete ab initio prediction of the outcome of mechanochemical reactions between molecular crystals. Such techniques would allow predictions that could be made without any experimental input regarding the structure or stability of the materials involved. While the combination of computational methods with experimental data is expected to be a powerful approach to understanding the energetics controlling these reactions, the ambition of completely ab initio predictions is necessary if computational work is to be used to screen possible reactions and decide which to pursue in the lab.
The computational studies performed in ref. 3 demonstrated that intermolecular interactions in the solid state have a key role in determining the lattice energy change across the reaction. Therefore, the crystal packing of reactant and product molecules must be known or predicted. Crystal structure prediction (CSP) has been a challenge for computational chemistry that has received significant attention, particularly for applications in polymorph screening of pharmaceutical molecules.4,5
The two main challenges involved in CSP, as applied to molecular crystals, are: (i) the dimensionality and complexity of the search space that must be explored to identify all possible crystal packing configurations that might be adopted by the molecule; and (ii) the observation that, for most molecules, there exist a plurality of low energy possible crystal structures separated by small lattice energy differences, typically on the order of 1 kJ mol−1 or less. Therefore, practical methods for CSP must involve a rapid evaluation of the stability of a large number of trial structures at the early stages, followed by refinement of the most promising structures. The refinement techniques are able to assess relative energies with high accuracy but come with an increased computational cost and so can only be applied to a small number of structures. The development of CSP methods in the past few years has been promising, with reports of successful applications to a range of challenging, flexible organic molecules.6–11
In this study, we first apply CSP to the reactant and product molecules of both model reactions to assess the predictability of their structures. We then perform total energy calculations on their known crystal structures to evaluate whether, with knowledge of their structures, the thermodynamic preference for homodimer versus heterodimer is predictable.
Our expectation in starting this study is not that these reaction energies can be calculated using existing methods; this would involve both the correct prediction of crystal structures of the reactants and products, and the relative formation energies of the crystal structures. Instead, the study is to assess the challenges involved in these predictions and highlight the areas where further development of predictive computational methods is most urgently required.
Methods
Crystal structure prediction
CSP is approached by global exploration of the lattice energy surface to identify the most stable low energy minima,12 corresponding to the most likely observable crystal structures. The computational methods therefore involve a search for promising trial structures, followed by their refinement and ranking by the calculated lattice energy.Conformation searches
In this study, we separate the search for trial crystal structures into a conformational search for the stable geometries of the isolated molecule, followed by crystal structure generation, where the molecules are initially kept fixed at their idealised gas phase geometries.The first step is a search for all energetically relevant gas phase conformations. Initially, conformations were generated using a force field description of intramolecular interactions; we employed the OPLS2005 force field,13 using the low-mode conformational search,14,15 as implemented in Macromodel.16 The low-mode search explores conformational space by perturbing an initial molecular geometry along the molecule's low energy normal modes, followed by local energy minimisation. For each molecule, the search was continued until 10000 energy minimisations had been performed and all unique conformers within 50 kJ mol−1 of the lowest energy conformers were kept. These conformers were subsequently re-optimised using density functional theory (DFT) to obtain more accurate molecular geometries for CSP, and for a more reliable ranking of the conformational energies. All DFT calculations were performed with Gaussian09 (rev. D01),17 using the B3LYP functional,18 6-31G** basis set19 and GD3BJ empirical correction20 for dispersion interactions. The resulting conformers were clustered to remove duplicates and those within 30 kJ mol−1 of the lowest energy geometry for each molecule were kept as starting points for CSP. This search resulted in 2, 6, 2, 4, and 4 conformers being found for molecules 4Cl4Cl, 2NO22NO2, 4Me4Me, 4Cl2NO2 and 4Me2NO2, respectively.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , P21, Pbca, C2/c, Pna21, Cc, P1, Pbcn, P41212, P43212, P21212, Pc, P31, P32, P41, P43, Fdd2, Pccn, P2/c, P61, P65, I41/a, R
, P21, Pbca, C2/c, Pna21, Cc, P1, Pbcn, P41212, P43212, P21212, Pc, P31, P32, P41, P43, Fdd2, Pccn, P2/c, P61, P65, I41/a, R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ) all with one molecule in the asymmetric unit. The searches were performed using our in-house Global Lattice Energy Explorer (GLEE) software,22 by means of quasi-random sampling of the position and orientations of the molecule in the asymmetric unit, and of unit cell angles and dimensions. The dimensions of the molecule are estimated by projecting the atomic coordinates onto the (orthogonal) vectors which define the principal moments of inertia. These dimensions, in conjunction with the number of symmetry operations, yield a target volume and a set of bounds which are involved in preparing the trial unit cell lengths. Structures with overlapping molecules were rejected and the search was continued until 2000 physically realistic trial crystal structures were generated for each conformer in each space group, yielding 900000 crystal structures in total.
) all with one molecule in the asymmetric unit. The searches were performed using our in-house Global Lattice Energy Explorer (GLEE) software,22 by means of quasi-random sampling of the position and orientations of the molecule in the asymmetric unit, and of unit cell angles and dimensions. The dimensions of the molecule are estimated by projecting the atomic coordinates onto the (orthogonal) vectors which define the principal moments of inertia. These dimensions, in conjunction with the number of symmetry operations, yield a target volume and a set of bounds which are involved in preparing the trial unit cell lengths. Structures with overlapping molecules were rejected and the search was continued until 2000 physically realistic trial crystal structures were generated for each conformer in each space group, yielding 900000 crystal structures in total.
The DMACRYS30 crystal structure modelling software was used for all lattice energy calculations, with a 15 Å cutoff on repulsion–dispersion interactions, an Ewald summation for charge–charge, charge–dipole and dipole–dipole interactions, and all higher order electrostatics summed to a 15 Å cutoff. All clustering of crystal structures was performed using the COMPACK algorithm,31 comparing interatomic distances within a cluster of 30 molecules.
At this stage, the total energy of each crystal structure was calculated as the sum of the DMACRYS intermolecular energy and the DFT energy of the relevant molecular conformation. For each molecule, structures within 20 kJ mol−1 of the global lowest total energy were retained for further structure refinement. This refinement allows for the distortion of the molecular geometry under the influence of intermolecular forces.
CrystalOptimizer calculations that are used during the CSP used the same atomic multipole-based model for intermolecular interactions as described above, with intramolecular forces and energies calculated at the B3LYP/6-31G** level of theory, with the GD3BJ empirical dispersion correction. Further CrystalOptimizer calculations were also performed on the observed crystal structures for the assessment of mechanochemical reaction energies. Here both B3LYP-GD3BJ/6-31G** and MP2/6-31G** electronic structure methods were independently used for both the molecular energy calculations and as a source of atomic multipoles. This allows us to assess the accuracy of the DFT approach and sensitivity to the description of intramolecular energies and charge density.
Results and discussion
Crystal structure prediction
We first assess the results of CSP on the five molecules by comparing the computer-generated structures to the known crystal structures of the reactants and products of both reactions. One crystal structure is known for each of 4Cl4Cl (space group Pbcn, Z′ = 0.5, Cambridge Structural Database code DCPHDS),354Me4Me (space group P21, Z′ = 1, CSD code IPIXUB),32NO22NO2 (space group P21/c, Z′ = 1, CSD code ODNPDS02)36 and 4Me2NO2 (space group P21/c, Z′ = 1, CSD code FUQLEI).37 The 4Cl4Cl + 2NO22NO2 reaction leads to a different polymorph of 4Cl2NO2, depending on whether the reaction is performed under neat grinding conditions or liquid assisted grinding (LAG), with a small amount of acetonitrile added to the grinding jar. The 4Cl2NO2LAG polymorph crystallises in the space group P (CSD code FUQLIM),37 while the 4Cl2NO2Neat polymorph crystallises in P21/n (CSD code FUQLIM01).3
Molecular conformation
The approach used here to deal with conformational flexibility during CSP relies on the molecular geometry in the crystal structure being close enough to one of the optimised conformers of the isolated molecule. After geometry optimisation with molecular flexibility, one of the trial structures generated should lead to the observed crystal packing.We find that all of the molecular geometries found in the observed crystal structures have a fairly good match among the generated gas-phase conformers. It is also useful to observe where each conformer lies on the energy landscape of the generated conformers. In all cases except 4Cl4Cl and 4Me4Me, the geometry adopted in the crystal structure corresponds to the global minimum energy conformer according the dispersion-corrected DFT calculations. For 4Cl4Cl and 4Me4Me, the molecule adopts a conformation more than 5 kJ mol−1 above the most stable gas phase conformer (Fig. 2), highlighting the need to consider multiple conformers during CSP – in these structures, the energy penalty associated with adopting a higher energy conformation must be compensated by more favourable intermolecular interactions.
 | ||
| Fig. 2 Overlays of the molecular geometries in each experimentally determined crystal structure (blue) with the closest match among the computer-generated (B3LYP-GD3BJ/6-31G**) gas phase conformers (red). Also shown are the RMS deviations in atomic position (RMSD1) and the calculated energy of the matching conformer relative to the lowest-energy calculated conformer (0.0 kJ mol−1 indicates that the global minimum conformer is adopted in the crystal structure). | ||
The overlays in Fig. 2 show the extent to which intermolecular forces due to crystal packing distort the molecules; the largest deviations from the nearest gas phase molecular geometry are seen in the orientation of the para-substituted ring for 4Me2NO2 and the Neat polymorph of 4Cl2NO2, which are rotated 33.4° and 61.6° degrees away from the gas phase geometry in the crystal structure, respectively. We show in the following section that, for 4Cl2NO2, this leads to a failure of the structure generation to locate the structure of the Neat polymorph. The overlay for 4Cl4Cl also shows a large RMSD1, but this is due to small rotations of both rings, leading to a smaller change in the overall molecular shape.
Crystal structures
The multiple lattice energy minimisation steps described above are required for computational efficiency of the entire CSP procedure. Here we only show our analysis of the results after the final energy calculation, once the molecular geometry has been optimised within each crystal structure and polarisation of the electron density has been modelled. In analysing the results, we first concern ourselves with whether the experimentally determined crystal structures are found among the predictions and the quality of this match in terms of geometry. Secondly, we consider where this structure lies on the energy landscape of the prediction.| Molecule | Rank (ΔE) | a/Å | b/Å | c/Å | α/° | β/° | γ/° | V/Å3 | RMSD30/Å |
|---|---|---|---|---|---|---|---|---|---|
| a The Neat polymorph of 4Cl2NO2 was not generated by the crystal structure search with the initial set of molecular conformers, but was generated in a search with the molecular geometry constrained to the observed conformation. The rank and relative energy reported for this polymorph refer to where this structure would be ranked within the original set of predicted structures. | |||||||||
| 2NO22NO2 | |||||||||
| Expt. | 7.103 | 22.925 | 7.777 | 90.00 | 95.54 | 90.00 | 1260.42 | ||
| Pred. | 1 (0.0) | 7.056 | 23.106 | 7.836 | 90.00 | 95.88 | 90.00 | 1275.05 | 0.190 |
| Expt opt. | 7.043 | 23.068 | 7.864 | 90.00 | 95.20 | 90.00 | 1272.21 | 0.215 | |
| 4Me4Me | |||||||||
| Expt. | 7.616 | 5.726 | 14.751 | 90.00 | 94.88 | 90.00 | 640.93 | ||
| Pred. | 58 (6.3) | 7.652 | 5.677 | 14.831 | 90.00 | 97.12 | 90.00 | 640.01 | 0.250 |
| Expt opt. | 7.606 | 5.662 | 14.846 | 90.00 | 94.40 | 90.00 | 637.39 | 0.129 | |
| 4Me2NO2 | |||||||||
| Expt. | 6.638 | 24.439 | 7.948 | 90.00 | 93.63 | 90.00 | 1286.82 | ||
| Pred. | 1 (0.0) | 6.891 | 23.847 | 7.610 | 90.00 | 93.30 | 90.00 | 1248.39 | 0.975 |
| Expt opt. | 6.898 | 23.988 | 7.525 | 90.00 | 93.39 | 90.00 | 1242.87 | 1.002 | |
| 4Cl4Cl | |||||||||
| expt. | 7.659 | 5.973 | 27.175 | 90.00 | 90.00 | 90.00 | 1252.08 | ||
| pred. | 245 (13.8) | 7.833 | 5.816 | 27.046 | 90.00 | 90.00 | 90.00 | 1233.54 | 0.268 |
| expt opt. | 7.685 | 5.863 | 27.460 | 90.20 | 90.00 | 90.00 | 1237.17 | 0.150 | |
| 4Cl2NO2 LAG polymorph | |||||||||
| Expt. | 7.043 | 7.832 | 11.371 | 82.58 | 80.69 | 83.19 | 610.71 | ||
| Pred. | 1 (0.0) | 6.807 | 7.902 | 11.620 | 82.53 | 82.26 | 84.15 | 611.83 | 0.287 |
| Expt opt. | 6.776 | 7.901 | 11.645 | 82.81 | 82.03 | 86.00 | 611.71 | 0.357 | |
| 4Cl2NO2 Neat polymorph | |||||||||
| Expt. | 13.456 | 7.103 | 15.707 | 90.00 | 124.00 | 90.00 | 1244.67 | ||
| Pred.a | 18 (4.8)a | 13.540 | 7.014 | 16.202 | 90.00 | 125.92 | 90.00 | 1271.40 | 0.370 |
| Expt opt. | 13.543 | 7.008 | 16.394 | 90.00 | 126.57 | 90.00 | 1249.33 | 0.464 | |
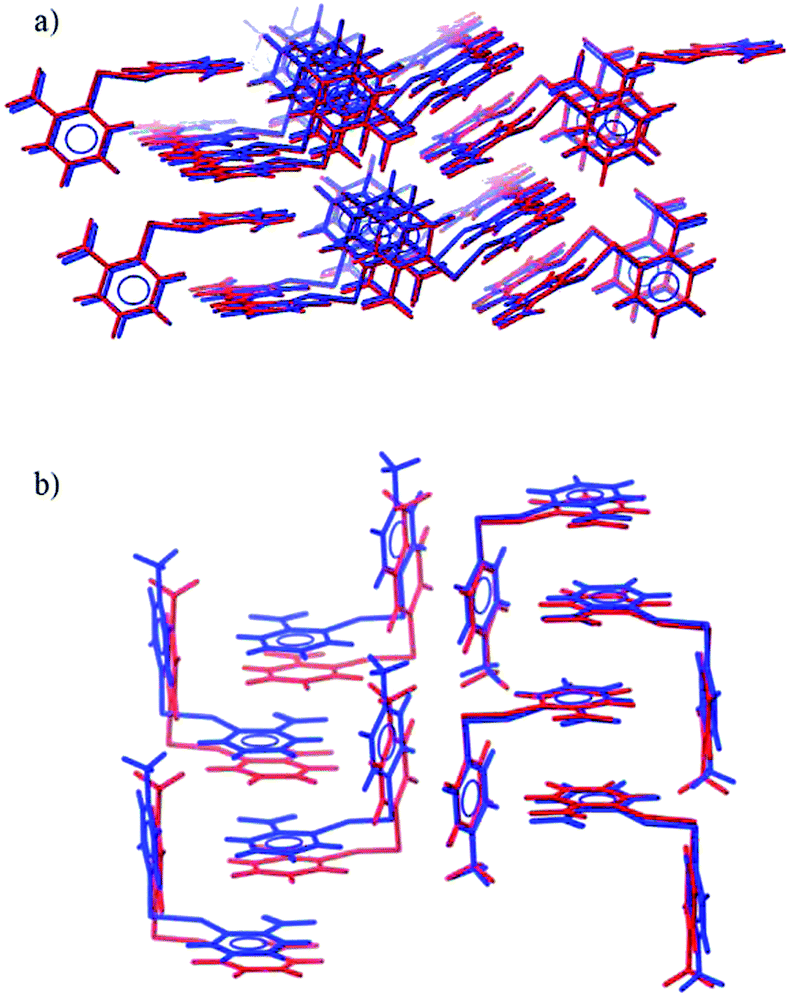 | ||
| Fig. 3 Overlays of the observed (blue) and predicted (red) crystal structures of (a) 2NO22NO2 and (b) 4Me2NO2. | ||
The one failure of the structure generation occurs for the crystal structure (Neat4Cl2NO2) whose molecular geometry was farthest from one of the starting gas phase geometries. The approach of generating crystal structures starting with only gas phase molecular geometries has been successful for many conformationally flexible molecules.7,8,39,40 However, this approach relies on the geometry adopted in the crystal being sufficiently close to one of the gas phase geometries that any molecular distortion due to crystal packing can be recovered during refinement of the trial structures. While previous CSP studies have shown that the crystal structure optimisation phase can allow for large changes in soft intramolecular degrees of freedom,41 it is clear in this case that rotation of the para-substituted ring allows a crystal packing that is not available to the molecule in its gas phase geometry.
To satisfy ourselves that this polymorph would have been found had we started with the distorted molecular geometry, an additional crystal structure generation was performed using a molecular conformation where the S–S–C–C and C–S–S–C torsion angles were fixed to the values seen in the experimentally determined crystal structure. The intramolecular energy penalty for these constraints is only 2.3 kJ mol−1, which must be balanced by improved crystal packing and intermolecular interactions. While the entire set of crystal structures from this additional prediction was not processed through the whole optimisation procedure, we found that the lowest energy structure after rigid-molecule lattice energy minimisation with the atomic multipole electrostatic model matches the observed structure of the Neat polymorph. This confirms the cause of the failure as being due to the distortion of the molecular geometry; molecular flexibility has been introduced at too late a stage in the CSP methodology. We processed the matching structure through the CrystalOptimizer step to find its energy with respect to the rest of the predictions (Table 1).
Matches to the experimentally determined crystal structures were found for all remaining systems. The observed structures of 4Cl4Cl, 4Me4Me, 2NO22NO2 (Fig. 3a) and 4Cl2NO2 are all reproduced very accurately, with an RMSD30 of less than 0.3 Å and the worst lattice parameter being 3.4% different from experiment (most are reproduced to within 1% of observed values).
The structure of 4Me2NO2 is reproduced less well (Table 1). The predicted and experimental structures differ by a ‘slipping’ of the planes of face-to-face stacked arene rings (Fig. 3b). The energy associated with this type of distortion is small and the calculated structure is likely to be very sensitive to small changes in the intermolecular potential. The difference might also be partly attributed to the lack of temperature in the calculated structures; slipping of these weak shear planes might accompany thermal expansion from the temperature-free predictions to the temperature of the observed structure.
The calculated crystal energy landscapes are very crowded, showing that many nearly-isoenergetic crystal structures are available for each molecule (Fig. 4). The observed crystal structures of 2NO22NO2 (Fig. 4a), 4Me2NO2 and the LAG polymorph of 4Cl2NO2 all correspond to the global minimum structure from their respective CSP. While the Neat polymorph of 4Cl2NO2 was not generated in the initial search, its energy would have ranked it 18th among the predicted crystal structures, 4.8 kJ mol−1 above the lowest energy crystal structure. For each of these molecules, the lowest energy gas phase conformer was the closest matching conformer to the molecular geometry in the crystal.
 | ||
| Fig. 4 Calculated crystal energy landscapes of (a) 2NO22NO2 and (b) 4Me4Me. Each point corresponds to a distinct crystal structure, whose total energy is reported relative to the lowest energy predicted structure. The open blue circles are structures generated by the molecular conformer that is closest to the observed conformer, and open red triangles are from the other conformers. The prediction that matches the observed structure is indicated by an open square. | ||
The CSP results are less successful for the two molecules 4Cl4Cl and 4Me4Me, whose observed crystal structure contains a higher energy conformer. The fine balance required between intramolecular and intermolecular energies is highlighted in the crystal energy landscape of 4Me4Me (Fig. 4b); the observed crystal structure is the 4th lowest in energy among the crystal structures generated with that molecular conformation, just 1.6 kJ mol−1 above the best calculated packing of that conformation. The good in-conformation ranking, but relatively high global ranking, indicates errors with our chosen level of theory for treating intramolecular contributions to the total crystal energies. It seems likely that limitations of the DFT functional (B3LYP with empirical dispersion correction) or medium-sized basis set (6-31G**) affect this result. We see a similar distribution of the predicted crystal structures of 4Cl4Cl: all of the lowest energy structures are generated from an alternative conformation to that found in the observed crystal structure. However, in this case, the true crystal structure is also poorly ranked within the structures generated with only the observed conformation, appearing at rank 23. This result causes most concern and is symptomatic of inaccuracies in both the intermolecular energy model and the method used for the intramolecular energies. In Fig. 5 we show the unit cells of the prediction which matches the observed structure, the lowest energy computer-generated packing of the observed conformer and the overall global energy minimum crystal structure. We note that the predicted and observed structures exhibit very different intermolecular interactions. The arene rings form T-shaped interactions in the experimentally observed packing (Fig. 5a), whereas the predicted structures with the lowest calculated energies have rings packed in a face-to-face arrangement (Fig. 5b and c). Furthermore, the observed crystal packing contains planes of close Cl![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) Cl intermolecular interactions, which are absent in the lowest energy predicted structures. We conclude that the intermolecular model potential does not describe these interactions accurately enough to rank correctly the predicted crystal structures. In particular, the chlorine atom parameters, which were added to the W99 intermolecular potential in an ad hoc manner, could be improved to provide a better balance between ClCl interactions and Cl interactions with other atoms.
Cl intermolecular interactions, which are absent in the lowest energy predicted structures. We conclude that the intermolecular model potential does not describe these interactions accurately enough to rank correctly the predicted crystal structures. In particular, the chlorine atom parameters, which were added to the W99 intermolecular potential in an ad hoc manner, could be improved to provide a better balance between ClCl interactions and Cl interactions with other atoms.
 | ||
| Fig. 5 Unit cells of (a) the predicted crystal structure that matches the observed structure, (b) the lowest energy computed structure of the observed conformer and (c) the global minimum predicted crystal structure of 4Cl4Cl. | ||
Reaction energies
From a synthetic point of view, the most important outcome is not the crystal structure, but the position of the equilibrium: does the solid-state reaction favour the heterodimers, homodimers, or are they finely balanced? In this work, we seek to understand how well the computational methods, which have been developed for the application of CSP, perform for determining the relative stabilities of the homodimer and heterodimer crystal structures. We set our target as successfully predicting whether the equilibrium should favour the homodimers or heterodimer in each reaction. Until we can achieve this goal reliably, we make no attempt at predicting the precise equilibrium constant. For the two reactions under investigation, the experimental observations show clearly that, for the reaction of 4Cl4Cl with 2NO22NO2, heterodimers are favoured strongly enough that the grinding experiment leads to 98% yield of heterodimers. When 4Me4Me is reacted with 2NO22NO2, only 20% heterodimer is formed, demonstrating that the heterodimers are less stable than homodimers.Fig. 6 shows the computed reaction energies using the same B3LYP-GD3BJ/6-31G** molecular calculations as used in the CSP studies, before and after applying the final PCM correction for polarisation contributions to the energies. All calculations are performed starting from the experimentally determined crystal structures, after applying the same optimisation procedure that was used in CSP (see Table 1 for the final structures). For comparison with DFT, we have performed the same CrystalOptimizer optimisation of the experimental structures with intramolecular energies and the intermolecular electrostatic model derived from MP2/6-31G** calculations. Because Neat and LAG grinding conditions lead to different polymorphs of 4Cl2NO2, we calculate the reaction energy separately for the two heterodimer polymorphs.
 | ||
| Fig. 6 Calculated reaction energies (in kJ mol−1) for the reactions of 4Cl4Cl + 2NO22NO2 to the LAG and Neat polymorphs, and the 4Me4Me + 2NO22NO2 to 4Me2NO2 reaction. Energies are shown both with and without the PCM corrected model described above. Contributions are separated into intermolecular, intramolecular and total energies and using both B3LYP-GD3BJ/6-31G** (DFT) and MP2/6-31G** (MP2). | ||
Overall, the results for the 4Cl4Cl with 2NO22NO2LAG reaction agree with experimental observations, irrespective of the details of the method used (DFT vs. MP2 molecular energies, with or without PCM estimate of polarisation contributions). This reaction is entirely dominated by the intramolecular energy change according to the DFT-based calculations, while the MP2 results give a description that is more evenly balanced between inter- and intramolecular contributions. Furthermore, it is important to note that the influence of crystal packing on the molecular geometry is crucial. At the geometries of the gas phase molecules, the B3LYP-GD3BJ/6-31G** molecular energies favour the heterodimers by only 0.9 kJ mol−1. The packing forces in the individual crystal structures increase the intramolecular energy difference by an additional 3.7 kJ mol−1 by introducing more molecular strain in the homodimers than the heterodimer. Introducing the effects of polarisation using the PCM model enhances the relative stability of the heterodimers even further.
Although a different polymorph is produced under Neat grinding conditions, the heterodimer is still found to be favoured in the Neat reaction of 4Cl4Cl with 2NO22NO2. Again, the sign of the energy change agrees with experimental observations and, unsurprisingly, there is a similar intramolecular energy change as found for the LAG polymorph of 4Cl2NO2. However, unlike the LAG reaction, the total reaction energies are quite sensitive to the change from DFT to MP2. Interestingly, this sensitivity is found in the intermolecular contribution to the reaction energy, suggesting that these differences result from differences in the description of intermolecular electrostatics between DFT and MP2. One consistent finding between DFT and MP2 results is that the inclusion of polarisation into the calculation shifts the balance of intermolecular energies towards the homodimers. The intermolecular energy change cancels much of the favourable intramolecular energy change in the DFT results and allows the intramolecular energy to dominate the MP2 result. With polarisation included, the reaction energy drops to −0.3 kJ mol−1 in the DFT results (−2.6 kJ mol−1 with MP2).
Our results for the reaction of 4Me4Me with 2NO22NO2 are less satisfactory. When DFT or MP2 is used to for the intramolecular contribution, our calculations would predict that this reaction also favours the heterodimers. Both the intra- and intermolecular energies are calculated to be lower in the heterodimer than the homodimers. This result contradicts what is observed in the grinding experiments. We note that this is the reaction for which the crystal structure of the heterodimer is poorly reproduced (Table 1 and Fig. 3b) and this is also true in the MP2 results (RMSD30 = 1.340 Å). These errors in reproducing the heterodimer crystal structure may be a source of the inconsistency between the observed and predicted behaviour for this reaction.
Using DFT molecular energies and electrostatics, the three reactions each show quite a different balance between inter- and intramolecular energy changes, making these very challenging systems for computational prediction. It is a strength of the hybrid method used for the energy calculations here that the quality of the intermolecular and intramolecular energy model can be separately improved. From our comparison of DFT with MP2, the differences in the overall reaction energies are not dramatic, although the two methods yield quite different individual inter- and intramolecular contributions in the reaction of 4Cl4Cl with 2NO22NO2. Therefore, we suggest that the sensitivity of the results to the method used for molecular calculations is one area that needs further exploration.
With the small magnitude of the reaction energies, and the delicate balance of inter- and intramolecular energies, the inclusion of polarisation should be important in getting the energies correct. In the approach taken here, the polarisation of the molecules in their crystal structures has been modelled with a very approximate model. The crystalline environment is described by a structureless dielectric continuum with a single dielectric constant used to model all of the crystal structures. The model originates from the need to introduce polarisation in a computationally efficient manner for the large numbers of crystal structures considered in CSP studies.33 The success of this polarisation model in CSP probably relies on fortuitous cancellation of error, because all of the crystal structures considered have the same chemical composition. It is inconclusive whether this method of including polarisation is useful in calculating reaction energies when the lattice energies are computed for different molecules. These types of reactions will be a challenging test for the development of more detailed polarisation models.
A relevant question is whether predictions of the thermodynamic balance between reactant and product crystal structures in solid state reactions can accommodate any error in the prediction of the crystal structures themselves. The magnitudes of the calculated reaction energies are similar to the calculated energy differences between the predicted crystal structures of each molecule. Therefore, at least for these reactions, mis-predictions of the crystal structure can lead to energetic errors that are significant. Certainly, the energy differences between the global minimum prediction and the observed structure for 4Cl4Cl and 4Me4Me are so large that using the global minimum predicted structure rather than the observed structure would have introduced substantial errors to the reaction energies.
Conclusions
The results of this assessment of the application of computational methods to solid-state aromatic disulfide metathesis reactions are a mix of successful results with some failures of the applied methodology that serve to highlight areas for development. We cannot conclude from these results that current theoretical methods are predictive of the solid state structures and energetics that govern the equilibrium in mechanochemical reactions. Nevertheless, there are positive results to build on and we find nothing to suggest that the goal of predictive computational methods in this area cannot be achieved.The outcomes of the CSP part of this work are consistent with recent results for other moderately flexible organic molecules. Of the six target crystal structures, three are predicted as the global minimum structure on the molecule's crystal energy landscape; these very encouraging outcomes result from a successful conformational search, crystal structure generation and an accurate energy model for lattice energy minimisation. The structure prediction successes are tempered by the results obtained for the remaining three crystal structures, two being found at fairly high energies on the crystal energy landscape, and one of the known crystal structures being missed by the computational search. The three unsuccessful predictions are each due, at least in part, to problems related to conformational flexibility in the molecules, illustrating the two types of problems that molecular flexibility introduces in CSP. Failure to generate the monoclinic (Neat grinding) polymorph of 4Cl2NO2 highlights the need for a more rigorous sampling of conformational space during the crystal structure search. The preference for unobserved conformations in the predicted crystal structures of 4Cl4Cl and 4Me4Me indicate the requirement for quantum chemical methods that provide more reliable relative intramolecular energies between conformers than the dispersion-corrected DFT that we have used here.
The later requirement is brought to the fore in our calculations of the overall reaction energies. The inter- versus intramolecular balance of the total lattice energy change from homodimer crystals to the heterodimer crystal varies between reactions and, especially for the reaction of 4Cl4Cl with 2NO22NO2, is quite sensitive to the level of theory used for the molecular energy calculation. While the affordable computational cost of DFT is attractive, particularly when considering the large numbers of structures in CSP, the systematic improvability of wavefunction based methods may be required to give reaction energies in which we have confidence. The results also highlight the need for reliable structure prediction: the intermolecular interactions play a key role in determining the relative energies of the products and reactants, such that the crystal structure must be known to predict their equilibrium.
Overall, solid-state organic reactions are an attractive target for the application of computational methods, where we can build on the continuing development of quantum chemical methods used to study gas phase reactions and recent developments in predictive methods for molecular crystal structures. We believe that the results obtained in this study demonstrate that we have a solid base of methods on which to build.
Acknowledgements
P. J. B. and G. M. D. thank the EPSRC for financial support (grant EP/J01110X/1). D. H. C. and G. M. D. acknowledge the European Research Council for funding under grant ERC-StG-2012-307358-ANGLE. The authors acknowledge the use of the IRIDIS High Performance Computing Facility, and associated support services at the University of Southampton, in the completion of this work.References
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- T. Friscic, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC.
- A. M. Belenguer, T. Friscic, G. M. Day and J. K. M. Sanders, Chem. Sci., 2011, 2, 696–700 RSC.
- S. L. Price, Adv. Drug Delivery Rev., 2004, 56, 301–319 CrossRef CAS PubMed.
- A. J. Cruz Cabeza, G. M. Day, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2007, 7, 100–107 CAS.
- G. M. Day and T. G. Cooper, CrystEngComm, 2010, 12, 2443–2453 RSC.
- A. V. Kazantsev, P. G. Karamertzanis, C. S. Adjiman, C. C. Pantelides, S. L. Price, P. T. A. Galek, G. M. Day and A. J. Cruz-Cabeza, Int. J. Pharm., 2011, 418, 168–178 CrossRef CAS PubMed.
- M. Baias, C. M. Widdifield, J.-N. Dumez, H. P. G. Thompson, T. G. Cooper, E. Salager, S. Bassil, R. S. Stein, A. Lesage, G. M. Day and L. Emsley, Phys. Chem. Chem. Phys., 2013, 15, 8069–8080 RSC.
- R. M. Bhardwaj, L. S. Price, S. L. Price, S. M. Reutzel-Edens, G. J. Miller, I. D. H. Oswald, B. F. Johnston and A. J. Florence, Cryst. Growth Des., 2013, 13, 1602–1617 CAS.
- H. Wu, M. Habgood, J. E. Parker, N. Reeves-McLaren, J. K. Cockcroft, M. Vickers, A. R. West and A. G. Jones, CrystEngComm, 2013, 15, 1853–1859 RSC.
- J. Kendrick, G. A. Stephenson, M. A. Neumann and F. J. J. Leusen, Cryst. Growth Des., 2013, 13, 581–589 CAS.
- G. M. Day, Crystallogr. Rev., 2011, 17, 3–52 CrossRef.
- J. L. Banks, H. S. Beard, Y. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 CrossRef CAS PubMed.
- I. Kolossváry and W. C. Guida, J. Am. Chem. Soc., 1996, 118, 5011–5019 CrossRef.
- I. Kolossváry and W. C. Guida, J. Comput. Chem., 1999, 20, 1671–1684 CrossRef.
- MacroModel, version 10.1, Schrödinger, LLC, New York, NY, 2013 Search PubMed.
- Gaussian 09 rev D.01, M. J. T. FrischG. W.; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; B. Mennucci; G. A. Petersson; H. Nakatsuji; M. Caricato; X. Li; H. P. Hratchian; A. F. Izmaylov; J. Bloino; G. Zheng; J. L. Sonnenberg; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; J. A. Montgomery, Jr.; J. E. Peralta; F. Ogliaro; M. Bearpark; J. J. Heyd; E. Brothers; K. N. Kudin; V. N. Staroverov; R. Kobayashi; J. Normand; K. Raghavachari; A. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; N. Rega; N. J. Millam; M. Klene; J. E. Knox; J. B. Cross; V. Bakken; C. Adamo; J. Jaramillo; R. Gomperts; R. E. Stratmann; O. Yazyev; A. J. Austin; R. Cammi; C. Pomelli; J. W. Ochterski; R. L. Martin; K. Morokuma; V. G. Zakrzewski; G. A. Voth; P. Salvador; J. J. Dannenberg; S. Dapprich; A. D. Daniels; Ö. Farkas; J. B. Foresman; J. V. Ortiz; J. Cioslowski; D. J. Fox, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- J. S. Binkley, J. A. Pople and W. J. Hehre, J. Am. Chem. Soc., 1980, 102, 939–947 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- C. P. Brock and J. D. Dunitz, Chem. Mater., 1994, 6, 1118–1127 CrossRef CAS.
- G. M. Day, 2013.
- D. E. Williams, J. Mol. Struct., 1999, 485–486, 321–347 CrossRef CAS.
- D. E. Williams, J. Comput. Chem., 2001, 22, 1154–1166 CrossRef CAS.
- D. E. Williams, J. Comput. Chem., 2001, 22, 1–20 CrossRef CAS.
- A. Abraha and D. E. Williams, Inorg. Chem., 1999, 38, 4224–4228 CrossRef CAS.
- G. M. Day and S. L. Price, J. Am. Chem. Soc., 2003, 125, 16434–16443 CrossRef CAS PubMed.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- A. J. Stone, Chem. Phys. Lett., 1981, 83, 233–239 CrossRef CAS.
- S. L. Price, M. Leslie, G. W. A. Welch, M. Habgood, L. S. Price, P. G. Karamertzanis and G. M. Day, Phys. Chem. Chem. Phys., 2010, 12, 8478–8490 RSC.
- J. A. Chisholm and S. Motherwell, J. Appl. Crystallogr., 2005, 38, 228–231 CrossRef.
- A. V. Kazantsev, P. G. Karamertzanis, C. C. Pantelides and C. S. Adjiman, in Process Systems Engineering, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 1–42 Search PubMed.
- T. G. Cooper, K. E. Hejczyk, W. Jones and G. M. Day, J. Chem. Theory Comput., 2008, 4, 1795–1805 CrossRef CAS.
- Handbook of Chemistry and Physics, CRC, 1992 Search PubMed.
- J. K. Vandavasi, W.-P. Hu, C.-Y. Chen and J.-J. Wang, Tetrahedron, 2011, 67, 8895–8901 CrossRef CAS PubMed.
- M. Song and C. Fan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o2835 CAS.
- C. Glidewell, J. N. Low and J. L. Wardell, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 893–905 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41 Search PubMed.
- D. E. Braun, M. Ardid-Candel, E. D'Oria, P. G. Karamertzanis, J.-B. Arlin, A. J. Florence, A. G. Jones and S. L. Price, Cryst. Growth Des., 2011, 11, 5659–5669 CAS.
- M. Baias, J.-N. Dumez, P. H. Svensson, S. Schantz, G. M. Day and L. Emsley, J. Am. Chem. Soc., 2013, 135, 17501–17507 CrossRef CAS PubMed.
- G. M. Day, W. D. S. Motherwell and W. Jones, Phys. Chem. Chem. Phys., 2007, 9, 1693–1704 RSC.
- B. P. van Eijck, J. Comput. Chem., 2001, 22, 816–826 CrossRef CAS.
- A. Gavezzotti and G. Filippini, J. Am. Chem. Soc., 1995, 117, 12299–12305 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |