DOI:
10.1039/C3FD00125C
(Paper)
Faraday Discuss., 2014,
169, 477-499
Rapid decomposition and visualisation of protein–ligand binding free energies by residue and by water†
Received
6th December 2013
, Accepted 31st January 2014
First published on 31st January 2014
Abstract
Recent advances in computational hardware, software and algorithms enable simulations of protein–ligand complexes to achieve timescales during which complete ligand binding and unbinding pathways can be observed. While observation of such events can promote understanding of binding and unbinding pathways, it does not alone provide information about the molecular drivers for protein–ligand association, nor guidance on how a ligand could be optimised to better bind to the protein. We have developed the waterswap (C. J. Woods et al., J. Chem. Phys., 2011, 134, 054114) absolute binding free energy method that calculates binding affinities by exchanging the ligand with an equivalent volume of water. A significant advantage of this method is that the binding free energy is calculated using a single reaction coordinate from a single simulation. This has enabled the development of new visualisations of binding affinities based on free energy decompositions to per-residue and per-water molecule components. These provide a clear picture of which protein–ligand interactions are strong, and which active site water molecules are stabilised or destabilised upon binding. Optimisation of the algorithms underlying the decomposition enables near-real-time visualisation, allowing these calculations to be used either to provide interactive feedback to a ligand designer, or to provide run-time analysis of protein–ligand molecular dynamics simulations.
1 Introduction
Recent advances in hardware and software enable the running of long timescale (microsecond plus) atomistic, condensed phase molecular dynamics simulations of protein and protein–ligand systems.2–5 This is sufficient to allow biochemical events such as protein folding4 and ligand binding and unbinding5,6 to be observed directly. This is a great advance, but watching events as they occur during a dynamics trajectory provides limited quantitative information on the molecular driving forces of these events. In particular, examining a trajectory where a ligand binds or unbinds once or twice from a protein provides little insight into the binding affinity of the ligand, nor much guidance on how the ligand could be modified to increase binding and so design a better drug.
Binding free energy methods calculate the free energy change associated with ligand binding. There is a direct relationship between binding free energies and equilibrium binding constants. Negative (favourable) binding free energies show that the equilibrium is tilted in favour of the bound complex, while positive values indicate that the equilibrium is tilted towards the unbound protein and ligand. While knowledge of binding free energies is useful, it is difficult to relate changes in structure to changes in binding affinity. One reason is that protein–ligand binding can be viewed as a competition between the ligand and water for the protein active site. This means that, in some cases, differences in binding may be caused by differences in solvation of the ligand in the protein active site.7,8 New methods need to be developed that can link molecular dynamics and binding free energy calculations. This would allow changes in structure and solvation observed during a dynamics trajectory to be related directly to their contribution to the binding affinity of the ligand. In particular, new ways of visualising this data also need to be developed, so that the link between structure, solvation and binding can be easily recognised and understood by drug designers. Such visualisations would ensure that changes in structure and solvation upon ligand binding are readily available and relatable to binding affinities, thereby providing inspiration to drug designers to suggest modifications to the ligand that could improve binding. Here, we derive and then demonstrate how the waterswap1 absolute binding free energy method can be used to analyse snapshots from molecular dynamics trajectories. We show how these binding free energies can be decomposed into components that can be used to annotate the output of a molecular dynamics trajectory. These annotations provide detailed graphical visualisations that reveal the molecular drivers for binding, providing clear suggestions for how stronger-binding ligands could be developed.
2 Background
Many methods have been developed to estimate binding free energies of protein–ligand complexes. Many rely on calculating differences in binding free energy between different ligands to the same protein.9 These relative binding methods, in which one ligand is alchemically morphed onto another, have been used successfully to investigate structure–activity relationships (SARs).10,11 However, these methods struggle to accurately predict differences in binding free energy when the binding modes of the two ligands are different, or when the two ligands vary significantly in size. An alternative is to use an absolute binding method. These methods attempt to calculate the binding free energy of a ligand to a protein directly. Double-decoupling12–14 (or double annihilation15,16) uses two simulations; one that calculates the free energy of decoupling the ligand from the protein binding site and transfers it to vacuum, and another that calculates the free energy of similarly decoupling the ligand from water. The decoupling free energies must be calculated with a high accuracy. This is because, for large or polar ligands, they are typically on the order of 5–20 times the magnitude of the binding affinity. This can lead to large errors because the binding free energy is calculated as the difference between the free energy to decouple fully the ligand from bulk water and the free energy to decouple fully the ligand from the protein. Another problem is that, as the ligand is transferred to vacuum, it leaves behind a cavity in the solvent. This cavity has to be filled by bulk solvent, which is a slow process. One way to solve the cavity problem is to use implicit solvent methods, such as Poisson–Boltzmann Surface Area (PBSA)17,18 or Generalized Born Surface Area (GBSA).19,20 These use an implicit, continuum model of water, with the binding free energy calculated as a combination of the interaction energy between the protein and ligand, and the difference in hydration free energies of the protein–ligand complex, and the individual protein and ligand molecules. PBSA or GBSA methods can be used to analyse the binding observed in molecular dynamics simulations, e.g. via MM-PBSA or MM-GBSA.19–23 Snapshots are taken from the trajectory, and PBSA or GBSA used to calculate the binding free energy for each. The average from all snapshots is taken as the absolute binding free energy, with individual snapshot values giving insight into how changes in binding mode during the trajectory affect the binding. While popular, these techniques have drawbacks. The hydration free energies of the protein and protein–ligand complex are large, and since the difference between them needs to be evaluated, errors or small variations in their value can greatly affect the calculated binding free energy. In addition, the entropy of binding is neglected (or included via an approximation24). Most significantly, the use of an implicit model of water means that the molecular detail of protein–water, and ligand–water interactions is lost. This is particularly important where individual water molecules may form important bridging interactions between the ligand and the protein.25
2.1 Waterswap
Waterswap1 is an absolute binding free energy method that we developed to solve the cavitation and difference-of-large-values problems of double-decoupling. The method uses an explicit model of water, so includes the molecular detail of protein–water, ligand–water and protein–water–ligand interactions that are missing in continuum solvent methods. Waterswap is a comparatively new method, and has seen successful application in the prediction of mutants of influenza neuraminidase that show reduced drug binding.26 The method has been described in detail elsewhere,1 but in summary, it works by using a single simulation that swaps the ligand bound to the protein with an equivalent shape and volume of bulk water molecules. The effect is to push the ligand into bulk water, while filling the resulting cavity in the protein with a cluster of water molecules. This removes the cavitation problem of double-decoupling. In addition, the use of a single reaction coordinate means that the free energy is not calculated as the difference of two large numbers, and so highly accurate values are not required.
The method works by using a pair of coupled simulation boxes: (a) the protein box, containing the protein–ligand complex solvated in a periodic-boundaries box of explicit water molecules, and (b) the water box, containing just a periodic box of water. Both boxes are coupled to the same thermostat, enabling energy transfer between boxes through the heat bath in which both are theoretically placed.
Waterswap uses an identity constraint1,27 to identify a cluster of water molecules in the water box that has approximately the same shape and size as the ligand. It works by changing how water molecules are labelled during a simulation. Rather than labelling water molecules based on their index in an input coordinate file, water molecules are instead labelled by their location in space. This is achieved by placing identity points in space. For example, let us consider a blue and a green identity point (Fig. 1(a)). These are used to label the blue and green water molecules, with the labels assigned so as to minimise the sum of the distances between the centre of the blue water molecule and the blue point, and the centre of the green water molecule and the green point (Fig. 1(b)). As the simulation progresses, and the water molecules move, this constraint is continually re-evaluated. If another water molecule moves such that, for example, it becomes closer to the green point (Fig. 1(c)), then the identities of the water molecules are swapped, and it then becomes the green water molecule (Fig. 1(d)). In this way, the identity of the cluster is maintained, as water molecules that diffuse away from the cluster are replaced by water molecules that diffuse into it. Note that the identity constraint only affects the labelling of the water molecules. The molecular coordinates are unchanged, no forces or restraints are required, and the thermodynamics of the system is unaffected.
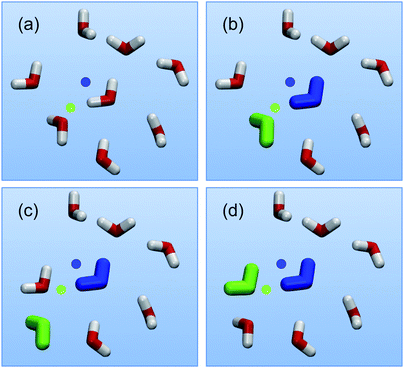 |
| | Fig. 1 Illustration of the identity constraint. Water molecules are labelled by their location in space by using identity points. A green and blue identity point (a) identify a green and blue water molecule (b). This assignment is based on minimising the sum of the distances between each identified water molecule and its corresponding point. As the water molecules diffuse during simulation (c), this constraint is updated to switch the identity to a better matching water molecule (d). | |
Waterswap uses the identity constraint to pick out a cluster of water molecules from the water box that are in the same position, and are approximately the same shape and volume as the ligand in the protein box. Identity points are placed onto selected atoms of the ligand, and these points are copied into the water box to identify a cluster of water molecules. With the cluster identified, the total energy of the system is evaluated using
| |  | (1) |
where
Eproteinbox is the energy of all molecules except the ligand in the protein box,
Ewaterbox is the energy of all molecules except the identified water cluster in the water box,
Eligand is the intramolecular energy of the ligand, and
Ecluster is the intermolecular energy between all of the water molecules in the water cluster. The interaction energy between the ligand and all other atoms in the protein box,
Eligand:proteinbox, and the interaction energy between the water cluster and all other water molecules in the water box,
Ecluster:waterbox, is scaled by l −
λ, where
λ is an alchemical reaction coordinate. The effect is to decouple the ligand from the protein box while simultaneously decoupling the water cluster from the water box. At the same time, the ligand is coupled to the water box, and the water cluster is coupled to the protein box. This is achieved by calculating the energy between the ligand and all water molecules in the water box,
Eligand:waterbox and the water cluster and the molecules in the protein box,
Ecluster:proteinbox, and scaling this by
λ. The result is that
λ is a single reaction coordinate that alchemically morphs the system from
λ = 0, where the ligand is bound to the protein in the protein box, to
λ = 1, where the ligand has unbound and is in bulk water, and a corresponding cluster of water has been transferred to the protein box to fill the resulting cavity.
The absolute binding free energy can be calculated using thermodynamic integration.28–30 This involves calculating the gradient of the energy with respect to λ,
| | 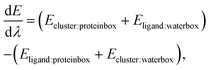 | (2) |
and then calculating the ensemble average of this at different values of
λ to obtain the free energy gradient across
λ,
| | 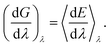 | (3) |
Because the identity constraint introduces discontinuities in the energy surface (associated with identity reassignment events), the free energy gradients must be averaged using Monte Carlo sampling31 at each value of λ. The binding free energy is then obtained by integrating the gradients across λ,
| | 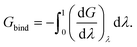 | (4) |
Note that the negative of the integral is used as the waterswap reaction coordinate models unbinding (pulling the ligand out of the protein). The quality of the result depends on the number of Monte Carlo simulations used to obtain free energy gradients across λ. We have found that sixteen Monte Carlo simulations, generating sixteen free energy gradients spaced across λ to be sufficient. The gradients can be integrated either numerically (e.g. via quadrature) or analytically by fitting the gradients to a polynomial expansion and integrating the resulting expression.32
2.2 Free energy decomposition
Waterswap calculates absolute binding free energies using a single reaction coordinate. The free energy is calculated by averaging the gradient of the total waterswap energy with respect to λ to form the free energy gradient (eqn (3)), and then integrating this over the entire waterswap reaction coordinate to obtain the absolute binding free energy (eqn (4)). An advantage of waterswap that will be explored and developed in this paper is that it is equally valid to average the gradient of components of the total waterswap energy with respect to λ. This would result in an approximation of the gradient of the corresponding free energy component. For example, eqn (5)–(8) show how the free energy change associated with only the protein box, Gproteinbox, could be obtained by integrating the gradient of the energy components that involve only interactions between the ligand and water cluster and the molecules of the protein box (Eproteinbox(λ)).| | | Eproteinbox(λ) = (1 − λ)Eligand:proteinbox + λEcluster:proteinbox | (5) |
| |  | (6) |
| | 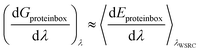 | (7) |
| | 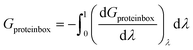 | (8) |
The result, Gproteinbox, provides an estimate of the contribution to the total binding free energy resulting from only the molecules in the protein box. A similar decomposition can be used to estimate the contribution to the total binding free energy from only the molecules in the water box, Gwaterbox.
| | | Ewaterbox(λ) = (1 − λ)Ecluster:waterbox + λEligand:waterbox | (9) |
| |  | (10) |
| | 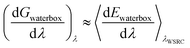 | (11) |
| | 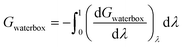 | (12) |
Note that these decompositions are approximate because the free energy is formed as an average over configurations sampled using the total waterswap energy function (as indicated by λWSRC in the equations). Because of this, the sum of Gproteinbox and Gwaterbox will not equal Gbind exactly. In this work, we will explore whether or not this error is small, and will examine if the resulting decomposition provides useful information. Our expectation is that this decomposition will reveal whether a favourable binding free energy is a result of the ligand having a strong affinity for the protein (Gproteinbox is large and favourable), or because the ligand has a low affinity for water (i.e. has a low solubility, with Gwaterbox being the dominant term).
2.3 Thrombin
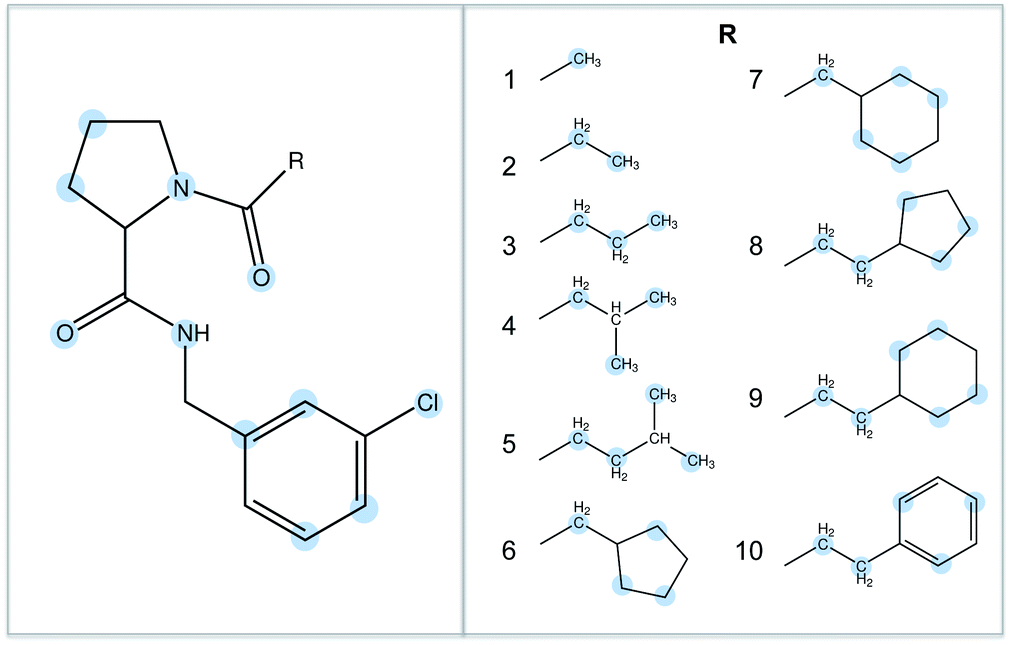
The binding of ten ligands to thrombin was chosen as the test system for this work.33,34 The ligands are shown in Fig. 2, and represent a congeneric series with increasing size and increasing experimentally measured binding affinity. These ligands were chosen because it is known that their binding affinity to thrombin increases in proportion to the hydrophobic contact area with the protein, suggesting that selectivity is driven by hydrophobic (van der Waals) interactions.34 This would provide a strong difference to the binding of the water cluster, where binding should be driven by hydrophilic (hydrogen bonding) interactions.
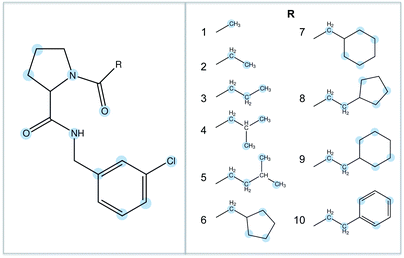 |
| | Fig. 2 The ten thrombin ligands studied in this work. They share a common core, and differ only in the R-group. The atoms used to anchor the identity points during the waterswap calculations are highlighted in blue. | |
3 Results
3.1 Waterswap binding free energies
The first step was to discover whether waterswap could correctly rank the binding affinities of the ten ligands to thrombin. Models of each of the ten thrombin–ligand complexes were built using the Amber FF99SB35 and GAFF36 forcefields. These were solvated in a periodic box of TIP3P37 water, and minimised and equilibrated for 1 nanosecond (ns). Each solvated complex was subjected to a further 50 ns of molecular dynamics using the GPU-accelerated version of the PMEMD.cuda module from Amber 12.2,38,39 Snapshots were taken every 10 ns from each of the 50 ns molecular dynamics trajectories for analysis via waterswap. Full technical details for all of these simulations are given in the Simulation protocols section. The result of all this work was five waterswap binding free energies per ligand, which were combined together to yield a time-averaged value. Fig. 3 shows the resulting time-averaged waterswap versus experimental binding free energies for each of the 10 ligands. It is satisfying that there is a strong degree of correlation between the simulation and the expected values (R2 of 0.8). There is a large difference between the absolute values predicted by waterswap and as measured by experiment. This can in part be explained by waterswap not measuring important contributions to binding, e.g. the effect of protein conformational change, or the loss of translational or rotational entropy for the ligand. This is because waterswap calculates the free energy change for swapping the ligand and water cluster for the conformation and binding mode of the complex used as input to the calculation. While the ligand and protein are flexible, Monte Carlo sampling allows only limited sampling of protein backbone motion. This means that the conformational change between the bound and unbound form of the protein is unlikely to be sampled. Waterswap also does not model concentration effects, as the protein and ligand are both effectively alone in an infinitely-sized heat bath (i.e. they both have an effective concentration of zero).1 Waterswap also overestimates the absolute binding affinity because of the large difference in ionic strength between the protein box (high, due to charged protein residues) and water box (zero). However, these missing contributions should all be roughly equal for the thrombin ligands. The observation of strong correlation, together with a correct prediction of the strongest and weakest binding ligand, suggests that time-averaged waterswap calculations capture the important drivers for binding in this system.
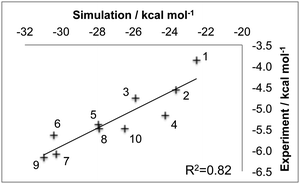 |
| | Fig. 3 Absolute binding free energies of ten ligands to thrombin as calculated using time-averaged waterswap calculations versus the experimental value. Points are labelled with the ligand number, and the line of best fit with corresponding R2 correlation coefficient are shown. | |
3.2 Free energy decomposition
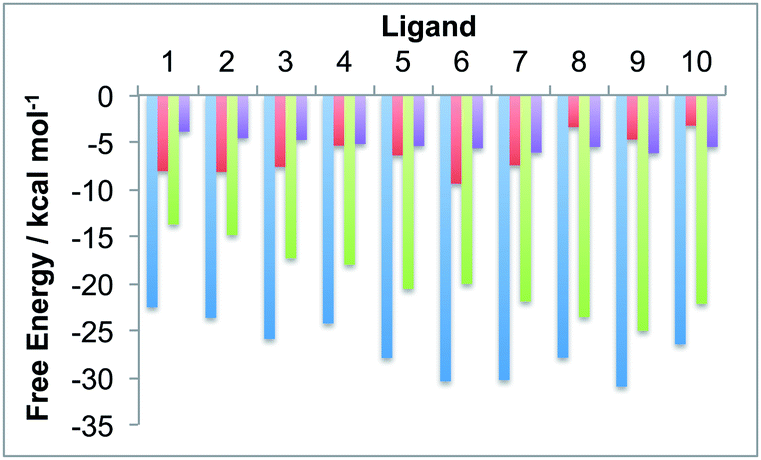
The protein box and water box energy components were separately averaged and integrated to obtain estimates for Gproteinbox and Gwaterbox for each protein–ligand complex, for each snapshot from the dynamics simulations. The error in the decomposition was approximated as the difference between the sum of these components (Gproteinbox + Gwaterbox) and the total predicted binding free energy (Gbind) for each snapshot. The maximum error was 1.0 kcal mol−1 with a mean unsigned error of 0.9 kcal mol−1. The low error, of less than 5% of the binding free energy, suggests that the decomposition should be meaningful. Fig. 4 compares the time averaged, decomposed free energies against the experimental and waterswap binding affinities of each ligand. It is clear from this figure that the waterswap binding free energy is dominated by the contribution from the water box. This accounts for between 60–85% of the total binding free energy. This is unsurprising as the ligands are predominantly hydrophobic, and so it would be expected that there would be a penalty for swapping the ligand into bulk water. In contrast, the protein has several charged and hydrophilic residues in the active site, so it is reassuring that there is little penalty in exchanging the hydrophobic ligand with (obviously) hydrophilic water. What is interesting is the correlation between the water box and protein box components with the experimental binding affinity. This is shown in Fig. 5. It was hoped that there would be a correlation for Gproteinbox, indicating that selectivity of the ligands was driven by specific interactions with the protein. Instead, little to no correlation is seen, and instead strong correlation is found between Gwaterbox and experiment (R2 > 0.8). This suggests that increases in binding affinity of the ligands are not driven by increased affinity with the protein, but are instead driven by decreased affinity with bulk water, i.e. lower solubility.
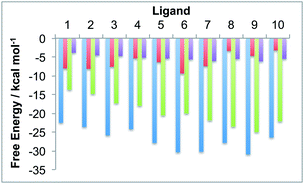 |
| | Fig. 4 Comparison of the time-averaged waterswap (ΔGbind, blue) binding free energy of the ten ligands to thrombin against the protein box (ΔGproteinbox, red) and water box (ΔGwaterbox, green) free energy components. The experimental binding affinities are shown in purple. | |
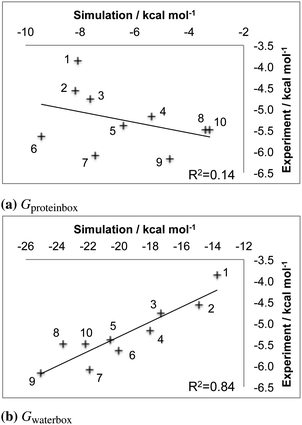 |
| | Fig. 5 (a) Protein box and (b) water box contributions to the binding free energy of ten ligands to thrombin as calculated using a decomposition of the waterswap free energy versus the experimental binding affinity. Points are labelled with the ligand number, and the line of best fit and corresponding R2 correlation coefficients are shown. | |
3.3 Residue-based decomposition
The method used to decompose Gbind to Gproteinbox can be used to decompose the waterswap binding free energy into even smaller components. One possibility is to decompose the free energy into per-residue components (Gresidue). To do this, the interaction energy between each residue and the ligand (Eligand:residue) and each residue in the water cluster (Ecluster:residue) can be averaged and integrated according to| | | Eresidue(λ) = (1 − λ)Eligand:residue + λEcluster:residue | (13) |
| |  | (14) |
| | 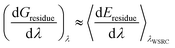 | (15) |
| | 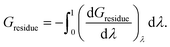 | (16) |
This is likely to be an even greater approximation than for Gproteinbox or Gwaterbox, and the result should perhaps not be called a “free energy”. Instead, this decomposition should be viewed as indicating whether the residue either (a) helps stabilise the protein–ligand complex in preference to the protein–water complex, (b) helps stabilise the protein–water complex over the protein–ligand complex, or (c) provides no support either way. The decomposition for each residue can in turn be broken down into its electrostatic (Gresidue[coulomb]) and van der Waals (Gresidue[LJ]) contributions. This is achieved by averaging and integrating only the coulombic or Lennard Jones (LJ) parts of Eligand:residue and Ecluster:residue, respectively. The values for all residues within 15 Å of each ligand were calculated and time-averaged. As the sum of Gresidue[coulomb] and Gresidue[LJ] should equal Gresidue, the error on the approximation was estimated by examining the difference. Across all snapshots of all ligands and all residues, the maximum error was 0.016 kcal mol−1 with a mean unsigned error of 0.0008 kcal mol−1.
The time-averaged values of the total (Gresidue) components for the twelve residues that show the strongest preferences for the any of the ten ligands or their corresponding swapped water clusters are shown in Fig. 6. This shows that charged residues (Lys60, Arg173 and Asp189) stabilise the water cluster (indicated by positive values), while the ligand is stabilised by its large hydrophobic contact with the neighbouring residues Ser214, Trp215 and Gly216. Ile174 sits at the bottom of the binding site, and can only come into direct contact with the large ligands. This is seen in its increasing contribution to the binding free energy with ligand size, although it is worth noting that this contribution alternates between supporting the water cluster and supporting the ligand.
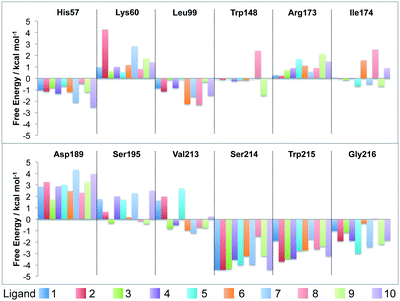 |
| | Fig. 6 Time-averaged decomposition of the waterswap binding free energy into selected per-residue components. The values for each of the ten ligands are shown, with positive values indicating that the residue stabilises the complex with the water cluster, while negative values indicate stabilisation of the protein–ligand complex. Residues are numbered to match their identification in the PDB (2ZC933). | |
To gain further insight, Fig. 7 shows the further decomposition of these values into their corresponding electrostatic and van der Waals terms. This breakdown reveals that the protein residues that are in direct contact with the ligand (His57, Leu99, Ser195, Ser214, Trp215 and Gly216) show a strong van der Waals preference for the ligand. For residues that have partial contact (Val213 and Ile174 for larger ligands), there is some van der Waals stabilisation, but it is weaker and occasionally in favour of the water cluster. This is unsurprising as these ligands are hydrophobic, with binding affinities that increase with increasing hydrophobic contact area with the protein.34 This is a good result, showing that this decomposition is revealing what is known experimentally about the ligands. What is more interesting is that this decomposition shows that the increased van der Waals stabilisation carries an electrostatic penalty. The contact residues almost all show strong electrostatic stabilisation in favour of the swapped water cluster. Examination of the structures shows that this is because the water cluster is able to form hydrogen bonds with the backbone of many of these residues. In addition, the decomposition shows that the stabilisation of the water cluster from the non-contact and charged residues is almost entirely electrostatic. The polar water molecules in the cluster are seen to exploit long range electrostatic interactions with the protein more readily than the hydrophobic ligands.
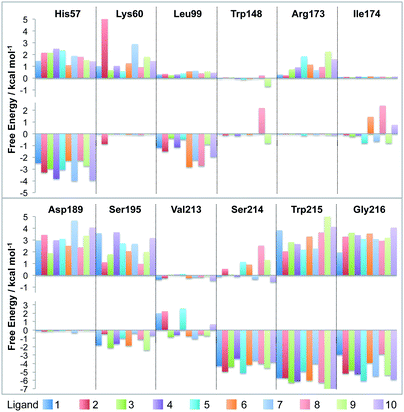 |
| | Fig. 7 Time-averaged decomposition of the waterswap binding free energy into selected per-residue electrostatic (top) and van der Waals (bottom) components for each of the ten thrombin ligands. The twelve residues selected are the same as shown in Fig. 6. | |
These decompositions are useful, but are formed as time averages over each of the molecular dynamics trajectories. To gain more insight, the components can be visualised by converting them into a score that is used to colour each residue in a molecular viewer. The components for each residue from each snapshot were converted into a score that was used to colour the corresponding residue in that snapshot to form an annotated movie of the trajectory. Snapshots from the resulting movie of ligand 9 (the strongest binding ligand) are shown in Fig. 8. The movie shows that the per-residue free energy components are fairly stable across the trajectory, with changes that mirror the motion of the ligand or orientation of the residues. For example, at 20 ns, the ligand sits slightly lower in the binding site, and the contact residues below the ligand show a correspondingly stronger stabilisation (indicated by a stronger blue colour). At 30 ns the m-chlorobenzyl substituent on the right of the ligand rotates up, increasing its contact and stabilisation with Trp148 above. As the ligand moves back down at 50 ns, this stabilisation is weakened. The motion of the residues also appears to be important, with Lys60 showing greater stabilisation for the water cluster (brighter red colour) in snapshots when it points towards the ligand than when it points away. Fig. 6 shows that Lys60 can provide a lot of electrostatic stabilisation to the water cluster, revealing the potential to design a better ligand by adding on hydrophilic substituents that target hydrogen bonding with that residue. This visualisation provides new insight by providing a direct link between changes in structure and their impact on binding affinity.
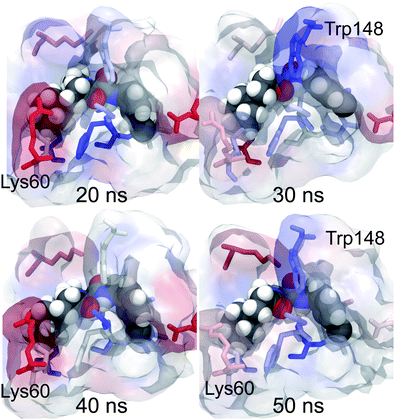 |
| | Fig. 8 Snapshots taken at selected times from the trajectory of ligand 9 complexed with thrombin. The residues are coloured by their component of the waterswap binding free energy calculated for that timestep. Red values indicate that the residue stabilises the water cluster, while blue values indicate that the residue stabilises the protein–ligand complex. Residues discussed in the text are highlighted. | |
Movies can be made looking at the total stabilisation, or at the electrostatic or van der Waals components. Of equal interest is the conformations adopted by the ligand and water cluster during the Monte Carlo trajectories used to calculate the waterswap free energy. Fig. 9 shows the annotated structure at the end of the Monte Carlo trajectories at λ = 0.005 (ligand) and λ = 0.995 (water cluster) for the 20 ns snapshot for ligand 9. The residues are coloured by their total, electrostatic and van der Waals free energy components. This shows how the swapped water cluster packs into the volume originally occupied by the ligand. Features such as the cluster-equivalent of the cyclohexyl and m-chlorobenzyl rings are clearly apparent. The figure shows that the water cluster is able to form hydrogen bonds with the oxygens of the contact residues along the backbone of the protein. This provides the cluster with strong electrostatic stabilisation (indicated by red) compared to the ligand. In compensation, the ligand receives strong van der Waals stabilisation (indicated by blue) compared to the cluster from those residues. This decomposition reflects the experimental observation that the ligands bind via hydrophobic interactions. The decomposition also shows that a path to strong-binding ligands may be to add hydrogen bond donor groups to pick up the same hydrogen bonding interactions that are observed to stabilise the swapped water cluster. In particular, it may be worth replacing the chlorine on the m-chlorobenzyl group with a hydroxyl or other group that can pick up the hydrogen bond to the oxygen on the backbone of Phe227. In addition to providing insight into how changes in structure affect the electrostatic and van der Waals components of the binding affinity, this visualisation also reveals the molecular detail of how water solvates the binding site. To bind, the ligand must displace the water molecules of the swapped water cluster, and make interactions with the protein that are stronger than those made by the water. The visualisation in Fig. 9 reveals the detail of the interactions between the protein and water. This should provide inspiration for the design of ligands that can replicate these interactions, thereby leading to the development of compounds with improved binding affinity.
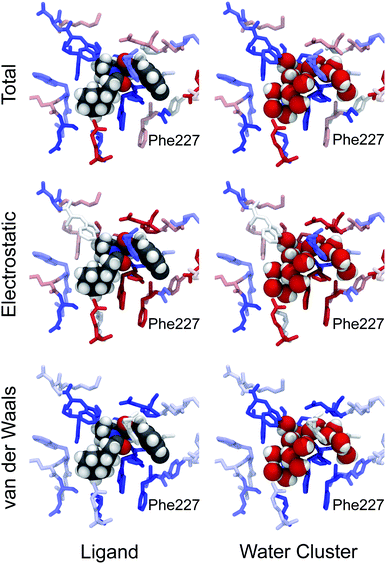 |
| | Fig. 9 Final configurations at λ = 0.005 (ligand, left) and λ = 0.995 (swapped water cluster, right) from the Monte Carlo simulations used to calculate the waterswap free energy for the 20 ns snapshot of ligand 9 bound to thrombin. The coordinates of the ligand and swapped water cluster are shown. The residues are coloured by their total (top), electrostatic (middle) and van der Waals (bottom) components of the waterswap free energy, with red values indicating stabilisation of the water cluster, and blue values indicating stabilisation of the ligand. Phe227, discussed in the text, is highlighted. | |
3.4 Water-based decomposition
In addition to decomposing the waterswap binding free energy per-residue, it is also possible to decompose it to per-water molecule components. To do this, the identity of water molecules in the binding site has to be constrained. This is to ensure that the free energy gradient is averaged over energy components calculated using a consistently identified water molecule, and so that free energy gradients calculated across λ can be correctly matched up and integrated. The identity constraint was used to hold onto the identity of each of the water molecules in the binding site around the ligand. Identity points were placed on all of the oxygen atoms of water molecules in the protein box within 5 Å of the ligand. These points were fixed in space, with a water molecule identified by each point. As each Monte Carlo trajectory across λ started from the same configuration, it was possible to identify a single water molecule across λ and to match and integrate its corresponding free energy gradients. Gradients of the component of the waterswap free energy for each water molecule were calculated in a similar manner to those for each residue. These components were themselves decomposed into electrostatic and van der Waals terms. These were all collected and integrated for each snapshot for each ligand to obtain Gwater, Gwater[coulomb] and Gwater[LJ] for each identified water molecule. Fig. 10 shows the annotated structure at the end of the Monte Carlo trajectories at λ = 0.005 (ligand) and λ = 0.995 (water cluster) for the same 20 ns snapshot for ligand 9 as seen in Fig. 9. The residues and water molecules are coloured by their total, electrostatic and van der Waals free energy components. This shows that water molecules near the cyclohexyl substituent on the solvent-exposed face of the ligand provide net stabilisation to the water cluster. This is to be expected, as the cyclohexyl substituent is hydrophobic and will likely disrupt the bulk water that is in contact with the surface of the protein–ligand complex. This suggests that one way to improve the binding affinity of the ligand would be to add hydrophilic groups that point out towards the surface of the complex. These would stabilise the water molecules, or at least reduce the preferential stabilisation provided to the water cluster. More interestingly, this figure also reveals a bridging water molecule that aids in the electrostatic stabilisation of the water cluster by residue Asp189. When the ligand is bound, this water molecule is trapped in the binding pocket in contact with the m-chlorobenzyl group. Here it provides van der Waals stabilisation to the ligand. However, when the water cluster is bound, the water molecule forms a hydrogen bonding bridge between the water cluster and Asp189. This allows both the water molecule and Asp189 to provide electrostatic stabilisation to the water cluster. The electrostatic stabilisation from this water molecule to the cluster is cancelled out by the van der Waals stabilisation provided to the ligand, so the net stabilisation is near zero. Despite this, the role of this water molecule in bridging to Asp189 is clear. A similar pattern is seen with another water molecule that bridges from the water cluster to Glu192. This helps both it and Glu192 to electrostatically stabilise the cluster over the ligand. This suggests that ligands that are designed to displace these water molecules and that make direct hydrogen bond or salt bridge interactions with Asp189 or Glu192 may have stronger binding affinities. Fortunately, the original experimental study of these ligands included a series where the m-chlorobenzyl group was replaced with benzamidine. Benzamidine analogs of all ten ligands were synthesised and their binding to thrombin measured.34 It was seen that these analogs did indeed displace water and form a direct interaction with Asp189, with each having a binding affinity that is about 2 kcal mol−1 stronger than that for the corresponding m-chlorobenzyl ligand (see ESI†).
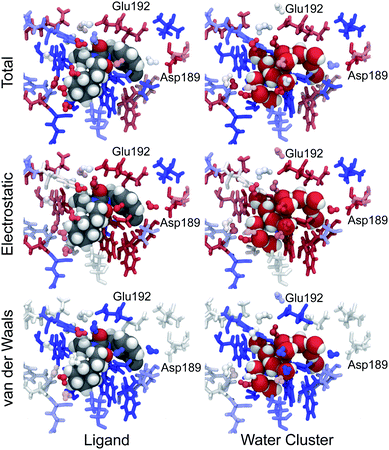 |
| | Fig. 10 Final configurations at λ = 0.005 (ligand, left) and λ = 0.995 (swapped water cluster, right) from the Monte Carlo simulations used to calculate the waterswap free energy for the 20 ns snapshot of ligand 9 bound to thrombin. The coordinates of the ligand and swapped water cluster are shown. The residues and selected binding site water molecules are coloured by their total (top), electrostatic (middle) and van der Waals (bottom) components of the waterswap free energy, with red values indicating stabilisation of the water cluster, and blue values indicating stabilisation of the ligand. Residues discussed in the text are highlighted. | |
These visualisations show directly how the ligand disrupts solvation of the protein. The swapped water cluster is seen to provide a seamless hydrogen-bonding network between all of the water molecules in the active site. In contrast, the ligand breaks this network, with lone water molecules trapped in what are now revealed to be electrostatically unfavourable conformations between the protein and the ligand. This new way of visualising ligand binding, as a competition between the ligand and water, should ensure that the impact of the ligand on solvation in the active site is readily visible and comprehendible throughout the drug design process. This should aid drug designers in their quest for better-binding ligands that do not unwittingly destabilise active site water molecules. In addition, these visualisations should help when rationalising the structure–activity relationships (SARs) of ligands where changes in structure cause differences in solvation. One such example of this is the different specificity of ligands binding to Itk, Src, cKit and Flt-3 kinases.7,8 In this case, the different SARs to different kinases were rationalised by using WATERMAP40 calculations to work out how water would be distributed in the active site of the apoproteins, and then consider how different ligands would displace that water on binding. We believe that waterswap decomposition and visualisation will provide a deeper and more detailed understanding of this system. We are now performing these calculations, the results of which we plan to discuss in a future paper.
4 Discussion
Decompositions of the waterswap binding free energy have been shown to provide useful insight and meaningful annotation of trajectories generated by molecular dynamics simulations of protein–ligand complexes. Performing a waterswap calculation is relatively straightforward. A waterswap executable is available as part of Sire41 and performs the calculations directly from the Amber-format topology and restart files that are produced by PMEMD.cuda during a molecular dynamics simulation. The only user intervention that is suggested is to specify the atoms of the ligand on which the identity points used to identify the swapped water cluster are placed. We have developed an algorithm to do this automatically, but it is not yet sufficiently robust to rely on blindly. The calculations are relatively quick, taking four minutes per iteration (50 thousand Monte Carlo moves per λ value) on a 2 × 8 core Intel Xeon E5-2670 compute node. We performed these calculations using 1000 iterations (50 million Monte Carlo moves per λ value), which took nearly 3 days. This is comparable to the 4 days needed to generate each 50 ns molecular dynamics trajectory (accelerated using nVidia Tesla M2090 GPUs). While the calculations were performed in series, it is not hard to imagine that a compute cluster could run heterogenous jobs involving waterswap calculations running on the CPUs and dynamics calculations on the GPUs. This would allow analysis to be performed on snapshots from a dynamics simulation as it was being generated. In addition, for this work we over-specified the number of iterations so that we could fully converge the free energy averages. Acceptable binding free energies, albeit with larger errors, could be generated with 400–600 iterations. Because the phase space is smaller, the residue- and water-free energy components converge extremely quickly, with initial estimates of their value available within the first 2–5 iterations, and acceptable values available after about 200 iterations (see the ESI†). This means that the colour-coding of the residues and water could be performed in near-real time (10–20 min). Waterswap is currently implemented in a prototyping code, and optimisation is on-going to create a custom application. Our goal is for this optimisation to accelerate the code by at least a factor of 10, which would allow estimates of components in 1–2 min, with converged free energies available within 5 h. This would allow near-interactive applications, e.g. allowing a drug designer to quickly see the effect on residue and water stabilisation of modifications made to the ligand in an interactive viewer. Non-interactive jobs could then be submitted to refine the components and binding free energies of promising designs.
One major challenge posed by these decompositions is that they significantly increase the amount of information that accompanies each molecular dynamics trajectory. Annotation adds several extra dimensions to the data. In addition to viewing a trajectory by “time”, waterswap adds in a λ dimension for swapping in the water cluster, a Monte Carlo “timestep” dimension for each simulation at each value of λ, and a “type” dimension that allows either total, electrostatic or van der Waals components to be visualised. Analysis and easy visualisation of all of this data is a significant challenge. We have written and rely on custom Python and Tcl scripts in VMD42 to manage this data deluge, but this is not ideal and not intuitive for non-experts. We feel that there is an urgent need for molecular visualisation tools that can readily present complex, multidimensional data in a manner that would be easy to navigate and manipulate by drug designers.
5 Conclusions
The utility of free energy decompositions of the waterswap binding free energy for annotating and visualising molecular dynamics trajectories of thrombin–ligand complexes has been demonstrated. These visualisations provide detailed molecular-level information that should be useful to drug designers aiming to create better-binding ligands. While we tested these decompositions on thrombin, we believe that they should be just as useful when applied to other protein–ligand systems. The calculations are robust, computationally inexpensive, almost fully automated, and implemented in software41 that is available for download under an open source license. These results show that waterswap, and its decomposition into free energy components, provides a promising new direction in the search for better tools for analysing and visualising the molecular driving forces in simulations of protein–ligand complexes.
6 Simulation protocols
A crystallographic structure of human thrombin in a complex with a thrombin ligand structurally related to the ligands simulated in this work was downloaded from the PDB databank (PDB code 2ZC933). All subsequent structure modifications were done with the software Maestro.43 The hirugen chain was removed from the structure. The side-chain of Arg75 in chain H was completed in a solvent exposed conformation. The incomplete light chain was capped before Glu1C with an ACE residue and after Ile14L with an NME residue. The incomplete heavy chain was capped after Gly246 with an NME residue. Missing residues Trp148, Thr149, Ala149A, Asn149B, Val149C, Gly149D, Lys149E in chain H were modelled in the structure using the FALC-Loop web server.44 Standard protonation states were assumed for protein side-chains. On the basis of visual inspection of hydrogen bonding patterns, His57 and His71 were modelled in their uncharged, δ-tautomer. His91, His119 and His230 were modelled in the δ-tautomer. Disulfide bridges were modelled between Cys42–Cys58, Cys1–Cys122, Cys168–Cys182 and Cys191–Cys220. Models of the ligands were prepared by editing the structure of the ligand present in the crystal structure. Substituents were positioned in the S3 pocket in plausible starting conformations, on the basis of the available experimental data.33,34 The protein was modelled using the Amber FF99SB forcefield,35 with GAFF36 used for the ligands. Ligand parameters and AM1BCC partial charges were generated using Antechamber.45 Each protein–ligand complex was solvated in a periodic box of TIP3P37 water molecules using the LEaP module of Amber, with the water molecules added to create a buffer of at least 10 Å around the protein. The resulting box had dimensions of approximately 70 × 71 × 74 Å3. Seven chloride ions were added using LEaP to neutralise the system. This provided a starting point for minimisation, equilibration and production using the GPU-accelerated PMEMD.cuda module of Amber 12 (version 12.1, bugfix 9, released August 2012).2,38,39 The mixed single-precision/fixed precision (SPFP) version of pmemd.CUDA was employed. The Particle Mesh Ewald (PME) method was used to account for long-range electrostatics, with a 10 Å real space cutoff, with the same cutoff used for the Lennard Jones potential. SHAKE was used to constrain bonds involving hydrogen. Each complex was subjected to two cycles of 3000 steps of minimisation, after which molecular dynamics (MD) simulations were carried out, also using pmemd.CUDA. A timestep of 2 fs was used, with the simulation divided into thermalisation, equilibration and production phases. In the thermalisation step, the temperature was increased linearly to 300 K over a period of 100 ps using canonical (NVT) dynamics. A Langevin thermostat was used to maintain temperature, using a collision frequency of 5 ps−1. Next, the system was equilibrated for a further 100 ps of NVT dynamics, then a further 800 ps of isothermal–isobaric (NPT) dynamics, with a Langevin thermostat used to maintain a temperature of 300 K, and the isotropic pressure scaling algorithm implemented in pmemd.CUDA used to maintain the pressure at 1 bar, using a pressure relaxation time of 1 ps. Visual inspection of each system together with monitoring of RMSD confirmed that the starting structures closely matched the original crystal structures. Production dynamics was performed for 50 ns at 300 K and 1 bar. Thermalisation, equilibration and production was carried out using a single M2090 nVidia Tesla GPU per protein–ligand complex, with production dynamics of each 33 thousand atom system running at a rate of approximately 14 ns of sampling per day. Waterswap calculations were performed using the waterswap executable in Sire (devel branch version 2169, obtainable via Google Code,46 and released as Sire 2014.1),41 with calculations using the same force field parameters and solvent model as the dynamics simulations. Identity points were placed manually onto the ligands, anchored to the atoms highlighted in Fig. 2. These points were set to move as the ligand moved, thereby ensuring maximum overlap between the ligand and swapped water cluster. The points were spread evenly over the ligand, with care taken to ensure that the identified water clusters had approximately the same shape and size as the ligands. The waterswap binding free energy was calculated using thermodynamic integration28–30 over 16 λ windows (values 0.005, 0.071, 0.137, 0.203, 0.269, 0.335, 0.401, 0.467, 0.533, 0.599, 0.665, 0.731, 0.797, 0.863, 0.929 and 0.995). The free energy gradients were fitted to a polynomial of order 14 and integrated analytically.32 50 million Monte Carlo (MC) moves were performed for each window, with free energy averages collected over the last 30 million steps. The location and orientation of the ligand was fixed, but all internal degrees of freedom were sampled using Z-matrices that were automatically generated by Sire.41 Non-bonded interactions were subjected to a 15 Å coulomb and Lennard Jones cutoff. PME is too expensive to use with Monte Carlo, so instead a shifted-force47 electrostatic cutoff was used. This has been shown to show give good agreement with PME.47 The free energy components of all residues within 15 Å of the ligand, and for all water molecules within 5 Å were evaluated, with interactions evaluated and averaged every 1000 MC steps. To aid convergence of the free energy averages, a soft-core potential was used to evaluate the interaction energies between the ligand, water cluster and the protein and water boxes, using the “Set A” parameters already developed for use with waterswap (shift delta of 1.2, water softening parameter of 1.1 and coulomb power of 0).1 This meant that the interaction energies involving the ligand or swapped water cluster had a dependency on λ, and so the simple differentiation used to obtain the free energy gradients in eqn (3), (7), (11) and (15) was not possible. Instead, a finite-difference approximation was used.48,49 The free energy gradient, 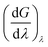 , was approximated using the Zwanzig equation50 to calculate the free energy difference between λ and λ + Δλ,
, was approximated using the Zwanzig equation50 to calculate the free energy difference between λ and λ + Δλ,| | 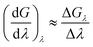 | (17) |
| | 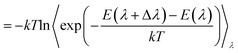 | (19) |
This finite difference approximation was used to calculate all of the free energy gradients, using a value of Δλ of 0.001. To check the approximation, both forwards gradients (using E(λ + Δλ) − E(λ)) and backwards gradients (using E(λ) − E(λ − Δλ)) were evaluated and compared.
To improve the efficiency of the calculation, all residues and all water molecules wholly beyond 15 Å of the ligand were fixed. All residues and water molecules within 15 Å were free to move. Each mobile residue had a full range of motion, with sampling of both the intra-residue degrees of freedom, and a backbone-anchored translation and rotation move that enabled limited backbone sampling. Each mobile water molecule was kept rigid, with sampling only of its translational and rotational degrees of freedom. To prevent water molecules from moving beyond 15 Å of the ligand, a reflection sphere boundary condition was employed. This condition ensured that any Monte Carlo moves that would take the centre of mass of the water molecule out of a sphere of radius 15 Å centred on the ligand, were reflected back into the sphere. This boundary condition was developed and implemented in Sire,41 and has the advantage that no restraints are needed to keep the mobile water molecules inside the sphere. In addition, as all fixed water molecules and protein residues outside the reflection sphere were still included in the energy calculation, it meant that there were no artefacts associated with mobile water molecules encountering a vacuum boundary (as is the case in “droplet” boundary conditions, whereby fixed atoms are deleted and mobile atoms kept in the sphere through use of a half-harmonic boundary potential). A further benefit of the reflection sphere was that as the molecules/residues outside the sphere were fixed, evaluating their contribution to the long range non-bonded interactions could be optimised, e.g. by using pre-allocated arrays and pre-calculated values. While not used for this work, it would also be possible to use an electrostatic grid to pre-compute the electrostatic interactions between the fixed and mobile region, thereby further improving the efficiency of the calculation.
In total, this work generated 510 ns of dynamics trajectories (requiring access to 10 nVidia Tesla M2040 GPUs for four days, and generating 14 GB of data) and required 8 billion Monte Carlo moves (requiring access to 10 16-core Intel Xeon E5-2670 compute nodes for 3 days, and generating 57 GB of data). We are grateful to e-Infrastructure South for providing access to the Emerald GPU cluster to run the dynamics simulations, and to the ACRC at Bristol for access to the newly-opened bluecrystal3 cluster for the waterswap calculations. We are also grateful to the Bristol Research Data Storage Facility for providing backed-up disk space to hold the data for this work.
Acknowledgements
The work was carried out with the kind support and the computational facilities of e-Infrastructure South (http://einfrastructuresouth.ac.uk) and the University of Bristol Advanced Computing Research Centre (http://www.bris.ac.uk/acrc). We thank the EPSRC (EP/I030395/1) for funding the development of Sire and the development of the fundamental research underpinning waterswap and its decomposition, and the BBSRC (BB/K016601/1) for funding this work. AJM is an EPSRC leadership fellow (EP/G007705/1).
References
- C. J. Woods, M. Malaisree, S. Hannongbua and A. J. Mulholland, J. Chem. Phys., 2011, 134, 054114 CrossRef PubMed.
- A. W. Goetz, M. J. Williamson, D. Xu, D. Poole, S. L. Grand and R. C. Walker, J. Chem. Theory Comput., 2012, 8, 1542–1555 CrossRef PubMed.
- R. O. Dror, R. M. Dirks, J. P. Grossman, H. Xu and D. E. Shaw, Annu. Rev. Biophys., 2012, 41, 429–452 CrossRef CAS PubMed.
-
D. E. Shaw, R. O. Dror, J. K. Salmon, J. P. Grossman, K. M. Mackenzie, J. A. Bank, C. Young, M. M. Deneroff, B. Batson, K. J. Bowers, E. Chow, M. P. Eastwood, D. J. Ierardi, J. L. Klepeis, J. S. Kuskin, R. H. Larson, K. L. Larsen, P. Maragakis, M. A. Moraes, S. Piana, Y. Shan and B. Towles, Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis, New York, NY, USA, 2009, pp. 1–11 Search PubMed.
- Y. Wang and E. Tajkhorshid, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9598–9603 CrossRef CAS PubMed.
- C. J. Woods, M. Malaisree, B. Long, S. McIntosh-Smith and A. J. Mulholland, Biochemistry, 2013, 52, 8150–8164 CrossRef CAS PubMed.
- J.-D. Charrier, A. Miller, D. P. Kay, G. Brenchley, H. C. Twin, P. N. Collier, S. Ramaya, S. B. Keily, S. J. Durrant, R. M. Knegtel, A. J. Tanner, K. Brown, A. P. Curnock and J.-M. Jimenez, J. Med. Chem., 2011, 54, 2341–2350 CrossRef CAS PubMed.
- K. R. M. A. and D. Robinson, Mol. Inf., 2011, 30, 950–959 CrossRef.
- B. L. Tembe and J. A. McCammon, Comput. Chem., 1984, 8, 281–283 CrossRef CAS.
- J. Michel, N. Foloppe and J. W. Essex, Mol. Inf., 2010, 29, 570–578 CrossRef CAS.
- J. Michel and J. W. Essex, J. Comput.-Aided Mol. Des., 2010, 24, 639–658 CrossRef CAS PubMed.
- D. Hamelburg and J. A. McCammon, J. Am. Chem. Soc., 2004, 126, 7683–7689 CrossRef PubMed.
- J. Hermans and L. Wang, J. Am. Chem. Soc., 1997, 119, 2707–2714 CrossRef CAS.
- C. Barillari, J. Taylor, R. Viner and J. W. Essex, J. Am. Chem. Soc., 2007, 129, 2577–2587 CrossRef CAS PubMed.
- W. L. Jorgensen, J. K. Buckner, S. Boudon and J. Tirado-Rives, J. Chem. Phys., 1988, 89, 3742–3764 CrossRef CAS PubMed.
- S. B. Singh and P. A. Kollman, J. Am. Chem. Soc., 1999, 121, 3267–3271 CrossRef CAS.
- M. A. L. Eriksson, J. Pitera and P. A. Kollman, J. Med. Chem., 1999, 42, 868–881 CrossRef CAS PubMed.
- B. Honig and A. Nicholls, Science, 1995, 268, 1144–1149 CAS.
- F. Godschalk, S. Genheden, P. Soderhjelma and U. Ryde, Phys. Chem. Chem. Phys., 2013, 15, 7731–7739 RSC.
- J. Michel, M. L. Verdonk and J. W. Essex, J. Med. Chem., 2006, 49, 7427–7439 CrossRef CAS PubMed.
- I. Massova and P. A. Kollman, Perspect. Drug Discovery Des., 2000, 18, 113–135 CrossRef CAS.
- I. Massova and P. A. Kollman, J. Am. Chem. Soc., 1999, 121, 8133–8143 CrossRef CAS.
- L. T. Chong, Y. Duan, L. Wang, I. Massova and P. A. Kollman, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14330–14335 CrossRef CAS.
- S. Genheden, O. Kuhn, P. Mikulskis, D. Hoffmann and U. Ryde, J. Chem. Inf. Model., 2012, 52, 2079–2088 CrossRef CAS PubMed.
- Z. Li and T. Lazaridis, Phys. Chem. Chem. Phys., 2007, 9, 573–581 RSC.
- C. J. Woods, M. Malaisree, B. Long, S. McIntosh-Smith and A. J. Mulholland, Sci. Reports, 2013, 3, 3561 Search PubMed.
- M. D. Tyka, R. B. Sessions and A. R. Clarke, J. Phys. Chem. B, 2007, 111, 9571–9580 CrossRef CAS PubMed.
- D. A. Pearlman and P. S. Charifson, J. Med. Chem., 2001, 44, 3417–3423 CrossRef CAS PubMed.
- B. C. Oostenbrink, J. W. Pitera, M. M. H. Lipzig, J. H. N. Meerman and W. F. van Gunsteren, J. Med. Chem., 2000, 43, 4594–4605 CrossRef CAS PubMed.
- X. Barril, M. Orozco and F. J. Luque, J. Med. Chem., 1999, 42, 5110–5119 CrossRef CAS PubMed.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087–1092 CrossRef CAS PubMed.
- F. M. Y. C. Shyu, J. Comp. Chem., 2009, 30, 2297–2304 Search PubMed.
- B. Baum, L. Muley, A. Heine, S. M., D. Hangauer and G. Klebe, J. Mol. Biol., 2009, 391, 552–564 CrossRef CAS PubMed.
- L. Muley, B. Baum, M. Smolinski, M. Freindorf, A. Heine, G. Klebe and D. G. Hangauer, J. Med. Chem., 2010, 53, 2126–2135 CrossRef CAS PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins: Struct., Funct., Bioinf., 2006, 65, 712–725 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS PubMed.
- R. Salomon-Ferrer, A. W. Goetz, D. Poole, S. L. Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS.
-
D. Case, T. Darden, T. Cheatham, C. S. III, J. Wang, R. Duke, R. Luo, R. Walker, W. Zhang, K. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. Goetz, I. Kolossvry, K. Wong, F. Paesani, J. Vanicek, R. Wolf, J. Liu, X. Wu, S. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M. J. Hsieh, G. Cui, D. Roe, D. Mathews, M. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- R. Abel, T. Young, R. Farid, B. J. Berne and R. A. Friesner, J. Am. Chem. Soc., 2008, 130, 2817–2831 CrossRef CAS PubMed.
-
C. J. Woods and J. Michel, Sire 2014.1, a complete molecular simulation framework, 2014, http://siremol.org Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
-
Suite 2011: Maestro, version 9.2, Schrödinger, LLC, New York, NY, 2011 Search PubMed.
- J. Ko, D. Lee, H. Park, E. A. Coutsias, J. Lee and C. Seok, Nucleic Acids Res., 2011, 39, W210–W214 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
-
C. J. Woods and J. Michel, Sire, Public Source Code Repository, 2013, http://sire.googlecode.com Search PubMed.
- C. J. Fennell and J. D. Gezelter, J. Chem. Phys., 2006, 124, 234104 CrossRef PubMed.
- M. Mezei, J. Chem. Phys., 1987, 86, 7084–7088 CrossRef CAS PubMed.
- C. R. W. Guimaraes and R. B. Alencastro, Int. J. Quantum Chem., 2001, 85, 713–726 CrossRef CAS.
- R. W. Zwanzig, J. Chem. Phys., 1954, 22, 1420–1426 CrossRef CAS PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Graph comparing binding of m-chlorophenyl and benzamidine ligands, graph showing the convergence of the residue components of the waterswap free energy and an animated version of Fig. 8. See DOI: 10.1039/c3fd00125c/ |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 

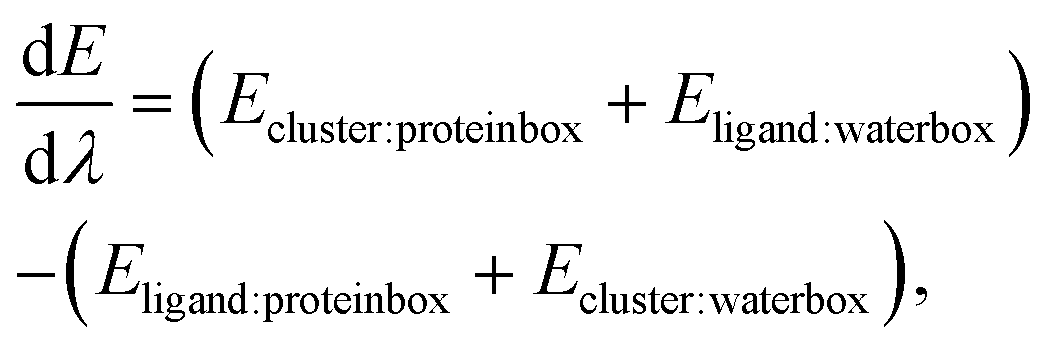

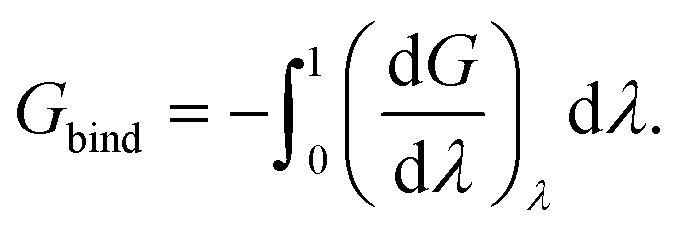
















 , was approximated using the Zwanzig equation50 to calculate the free energy difference between λ and λ + Δλ,
, was approximated using the Zwanzig equation50 to calculate the free energy difference between λ and λ + Δλ,
