
Nanoceria: factors affecting its pro- and anti-oxidant properties
Eric
Grulke
*a,
Kenneth
Reed
b,
Matthew
Beck
a,
Xing
Huang
a,
Alastair
Cormack
c and
Sudipta
Seal
d
aChemical and Materials Engineering, University of Kentucky, Lexington, KY, USA. E-mail: eric.grulke@uky.edu
bCerion Enterprises, LLC, 1 Blossom Road, Rochester, NY, USA
cNY State College of Ceramics, Alfred University, Alfred, NY, USA
dAdvanced Materials Processing and Analysis Centre, University of Central Florida, Orlando, FL, USA
First published on 18th August 2014
Abstract
Nanoceria redox properties are affected by particle size, particle shape, surface chemistry, and other factors, such as additives that coat the surface, local pH, and ligands that can participate in redox reactions. Each CeO2 crystal facet has a different chemistry, surface energy, and surface reactivity. Unlike nanoceria's industrial catalytic applications, biological and environment exposures are characterized by high water activity values and relatively high oxygen activity values. Electrochemical data show that oxygen levels, pH, and redox species affect its phase equilibria for solution and dissolution. However, not much is known about how the many and varied redox ligands in environmental and biological systems might affect nanoceria's redox behaviour, the effects of coated surfaces on redox rates and mechanisms, and whether the ceria solid phase undergoes dissolution at physiologically relevant pH and oxygen levels. Research that could answer these questions would improve our understanding of the links between nanoceria's redox performance and its morphology and environmental conditions in the local milieu.
Nano impactNanoceria is a well-known redox catalyst, which has been linked to redox effects in biological and environmental systems. The perspective shows connections between nanoceria morphology, including crystallite growth and surface structure, electrochemical phase equilibria, surface coatings (intentional and unintentional), and surface reactivity (both experimental plus computational modeling) to its apparent redox properties. These connections, or lack of them, are used to identify what is known about nanoceria's redox properties and to generate research gaps that might be addressed by future work. |
1. Introduction
Pro- and anti-oxidant systems
Pro-oxidants and anti-oxidants are involved in a number of important biological and environmental reactions. In biological systems, pro-oxidants induce oxidative stress either by generating reactive oxygen species (ROS), such as hydrogen peroxide (H2O2), the hydroxyl radical (˙OH), and the superoxide anion (O2−), or by inhibiting anti-oxidant systems. The ROS can damage cells and tissues, termed oxidative stress. Anti-oxidants inhibit the oxidation of other molecules from ROS by being oxidized themselves. There are multiple levels of anti-oxidants in living organisms. Anti-oxidants include small molecules, vitamin A, vitamin C, vitamin E, glutathione, and enzymes, catalase, superoxide dismutase and the peroxidase family. Oxidative stress plays a role in many human diseases, such as cancer, stroke, and neurodegenerative diseases. Anti-oxidants are used as food preservatives, stabilizers for cosmetics, and stabilizers for polymers and gasoline.Some compounds act as both pro- and anti-oxidants. Vitamin C is an example of a monosaccharide oxidation–reduction (redox) catalyst. It can reduce reactive oxygen species such as hydrogen peroxide and can act as a substrate for ascorbate peroxidase, a redox enzyme. In redox reactions, atoms have their oxidation states changed: loss of an electron leads to an increase in the oxidation state of an atom, ion or molecule, whilst reduction occurs when an atom, ion, or molecule gains electrons. Redox reactions are matched sets, i.e., one compound is being oxidized while the other is being reduced. Redox reactions often, but not always, involve an electron transfer between the species.
Nanoceria
Ceria is a well-known redox catalyst: surface cerium atoms can cycle between Ce3+ and Ce4+ and the surface can store or release oxygen. Nanoceria is of particular interest because, with its high surface area to volume ratio, there are many surface cerium atoms that can participate in various redox reactions. The physical properties of the two bulk cerium oxides, CeO2 (with Ce4+) and Ce2O3 (with Ce3+), have significant differences. CeO2 has the cubic fluorite crystal structure, with a density of 7215 kg m−3, while Ce2O3 has the hexagonal A-type rare earth sesquioxide crystal structure with a density of 6869 kg m−3. Nano-sized metal oxides, including nanoceria, have large surface areas and ‘defect’ sites (closely located cations and anions) linked to the faces, edges, and corners of their polyhedral shapes.1 Changes either in the oxidation state of the cerium or in oxygen levels can lead to significant local stresses in the crystal structure and changes in lattice size.2 Both hydrogen and oxygen can be transported readily through nanoceria so its structure can be dynamic as its surfaces groups participate in redox reactions.Nanoceria applications
For decades, ceria has been under study as a three way catalyst3 and as a base material for electrolytes and electrodes in solid oxide fuel cell systems.4 Ceria can be a catalyst support, a catalyst, and/or a co-catalyst based on its ability to provide local oxygen ‘storage’. Doping of ceria with other metals has also been used to improve its catalytic properties. Many commercial gas-phase processes using nanoceria operate at temperatures above 200 °C with either oxidizing or reducing atmospheres and no liquid water-containing phase (ref. 108 has more detail on such issues). As a corollary, much of the computational and experimental studies of ceria reactivity are linked to similar conditions. These applications have moderate oxygen activities and low water activities.Other nanoceria applications involve a liquid water phase, such as the polishing aids, anti-corrosion systems (the paint or coating would be in contact with water or sea water), oxidants for electrode sensors,5 and redox agents for treatment of disease (ref. 109 has more detail on such issues). Environmental problems might be associated with the release of nanoceria or nanoceria products into soil and water milieu. Human health applications can include intentional injection or exposure for diseases, or the effects of long-term exposure in the workplace (ref. 110 has more detail on such issues). Ceria nanoparticles have been proposed for a wide variety of oxidative stress conditions, including: radiation,6 acting as ROS scavengers,7–9 cancer,10 neurological oxidative stress diseases,11 ischemic stroke,12 and cosmoceutical applications.13 In these milieux, the water activity at the nanoceria surface is high, and the oxygen activity may be high or low. For such cases, there is less experimental and computational data on nanoceria's surface reactivity, redox reaction mechanisms, and ligand adsorption. There are a number of sources for nanoceria release, including paints, batteries, non-battery metals, catalytic converters, polishing slurries, glass additives, fuel additives and phosphors.
With respect to biological and environmental system, nanoceria will act as a colloid in aqueous, body fluid, and soil environments. The dispersion stability of colloidal material is affected by temperature, surface atomic arrangements, inorganic or organic ligands adsorbed on its surface, ions in solution and their levels, and pH among other factors.
Factors affecting nanoceria's redox reactions
Nanoceria's surface atoms and groups, such as cerium, oxygen and hydroxyl, can be involved in various reaction mechanisms. Catalytic properties of nanoparticles usually depend on their crystal structures, size distributions and exposed surfaces, edges, and corners. The initial morphology is established during the synthesis process. By controlling the growth rates of specific crystal faces, different morphologies can be produced (for example, gold14 or ceria15). Toxicity can be affected by nanoparticle size and shape, making this an important metric for risk assessment.16,17 In liquid phases, ions, organic acids and polymers, pH, oxygen levels, and redox agents can affect nanoceria's reactivity, even leading to crystallite dissolution and reprecipitation.The term, coating, is used broadly in this perspective. Metal oxides (MOx) have a variety of surface chemistries; the metal–oxygen composition of the near-surface layer can often be quite different from that in the bulk material. On ‘neat’ samples, in which there are no surface adducts, there can be a variety of surface-terminated oxygens, such as hydroxyls, M–O–M, and M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties. In environmental systems, there can be a number of species adsorbed to the nanoparticle surface, including organic acids and bases, organic ligands, herbicides, pesticides, surfactants, dispersants, proteins and phosphates, to name a few. Adsorbed species are assumed to be in dynamic equilibrium with their levels in aqueous solution, i.e., adsorption and desorption are constantly underway. Therefore, adsorbed species might block nanoparticle reactive surface sites sterically, but only during their adsorption phase. Additional coatings types include ligands covalently bound to the nanoparticle surface (such as coupling agents) or materials form a shell covering the surface. Covalently bound agents may not interact with all reactive sites on the nanoparticle surface. Shell-type coatings also may not react with surface sites, but could block larger molecules from diffusing to the metal oxide surface.
O moieties. In environmental systems, there can be a number of species adsorbed to the nanoparticle surface, including organic acids and bases, organic ligands, herbicides, pesticides, surfactants, dispersants, proteins and phosphates, to name a few. Adsorbed species are assumed to be in dynamic equilibrium with their levels in aqueous solution, i.e., adsorption and desorption are constantly underway. Therefore, adsorbed species might block nanoparticle reactive surface sites sterically, but only during their adsorption phase. Additional coatings types include ligands covalently bound to the nanoparticle surface (such as coupling agents) or materials form a shell covering the surface. Covalently bound agents may not interact with all reactive sites on the nanoparticle surface. Shell-type coatings also may not react with surface sites, but could block larger molecules from diffusing to the metal oxide surface.
Finally, nanoceria dispersions in water are often unstable, which can lead to agglomeration and can affect its transport and biodistribution. The local activities of oxygen and water directly affect nanoceria's surface chemistry. Table 1 categorizes the oxygen and water activities of specific nanoceria applications. The activity of a pure liquid is unity (ai = 1), and the activity of a gas component near 1 atmosphere pressure is its partial pressure divided by its saturated vapour pressure at the temperature of interest (ai = pi/pisat). The activity of a species characterizes its chemical availability. Differences in the activity of, e.g., water or oxygen can alter the stable structure and/or oxidation state of ceria surfaces. For example, the water activity of biological fluids at physiological conditions is often between 0.95 and 1. The water activity of air is similar to its relative humidity (on a fractional basis, not a percent). The oxygen activity in air at room temperature is ~0.20.
| Application area | aoxygen | awater | Ligands in the application environment |
|---|---|---|---|
| a Oxygen activity can be high if the water is near saturation with oxygen. However, the local oxygen ‘capacity’ may be low as it is sparingly soluble. | |||
| Gas phase catalysis | Moderate | Very low | Combustion products |
| Solid oxide fuel cells | Moderate | Very low | Fuels |
| Dispersed diesel fuel additives | Moderate | Very low | Diesel fuels |
| Polishing aids | Higha | High | Chemical reaction with Si–O species |
| Oxygen sensor | Moderate | Varied | Varied |
| Polymer or ceramic nanocomposites | Low | Low | |
| Anti-corrosion coatings | Higha (salt water) | High | Ions in aqueous solution |
| Environmental systems: soil and water | Higha | High | Salts in aqueous dispersions; adsorbing organic acids/polymers; oxidizing/reducing agents and systems |
| Biological systems: redox agents for treatment of disease | Higha | High | Salts in aqueous solution; metabolic organic acids; adsorbing proteins; oxidizing/reducing agents and systems |
An objective of this perspective is to illustrate how nanoceria's structure, composition, surface groups, and its local environment affect oxidation/reduction reactions, particularly in aqueous systems. Nanoceria morphology established, in part, by crystallite growth during synthesis. Morphology characterization is relevant to the surface structure of nanoceria at dosing conditions, its dissolution and re-precipitation in biological and environmental systems, and specific surface groups on different crystal facets that might participate in the redox reactions. Electrochemical reactions of soluble cerium salts and cerium solid phases are reviewed; these are relevant both to synthesis conditions and morphologies, as well as dissolution/reprecipitation conditions. Nanoceria particle surfaces may be coated during the manufacturing process for commercial materials, or may be coated by organic or inorganic ligands in local environments. Material on nanoceria surface reactivity is divided into four sections: experimental systems with low water activities, experimental systems with high water activities, computational models for nanoceria reactivity, and acellular reactions of nanoceria. After each section, there is a summary of what is known and research gaps relevant to nanoceria surface reactivity and redox properties in biological and environmental milieu.
2. Nanoceria morphology
Nanoceria can be synthesized by a number of pathways,18 including hydrolysis,19 precipitation,20–23 thermal deposition,24 combustion or flame synthesis,25,26 sol–gel,27,28 hydrothermal or solvothermal,29,30 microemulsion,31,32 gas condensation,33 sonochemical,34 or electrochemical.35 Nanoceria produced in the liquid phase are often based on redox reactions of inorganic salts of Ce3+. In these systems, cerium is oxidized from the Ce3+ to the Ce4+ state. For example, a precursor such as Ce(OH)3(s) would be converted to CeO2. Because of our interest in high water activity environments, this section focuses on synthesis in liquid systems.Control of crystallite growth and morphology
Different particle sizes and shapes of nanoceria are synthesized by controlling the growth of specific crystal facets. Control of the shape of colloidal inorganic nanocrystals has been reviewed by Lee,36 covering a range of shapes from isotropic (similar in each orthogonal direction), such as cubes and spheres, to anisotropic morphologies, such as nanorods and more complex structures. Shape control begins with the crystallization of the nucleating seeds; this sets a ‘template’ for the unit cell structure of the nanocrystal. Temperature appears to have a significant effect on the thermodynamic stability of nuclei. Additional control can be accomplished by directing preferential growth of specific crystallographic faces kinetically. Once the crystal starts to grow, several factors can control the growth rates and the final structure of the product. If the process is controlled thermodynamically, i.e., there is a low flux of molecules adding to the facet, the most stable form of the nanocrystal is preferred. When kinetics drives the process via either a high flux of adding molecules or blocking of specific crystallographic faces, anisotropic shapes are possible. The interfacial energy of a growing crystallographic face impacts its growth rate: the kinetic energy barrier, i.e., the free energy of the process, is inversely proportional to the surface energy. Often, growth rate can be controlled with organic “capping” molecules that adsorb preferentially to specific faces, leading to anisotropic forms, such as nanorods. When the desired size and shape has been attained, the synthesis process needs to be arrested by separating the nanoparticles from their reaction medium. Otherwise, the crystallites would re-precipitate to their thermodynamically preferred shapes and crystal faces over long times.Organic acids can be used to complex with ceria nanoparticles during synthesis or with cerium ions in solution, thereby improving their solubility. For example, cerium(III) ions (Ce3+) are generated during ore digestion using strong acids. Oxalic acid, a multidentate carboxylic acid, is added at this stage to form a soluble salt that can be recovered from the mixture. Fig. 1 shows the structure of cerium oxalate; this is typical of a bidentate organic ion complexing with metal atoms. In the case of ceria dissolution in environmental or biological milieu, organic acid complexes with cerium ions could generate soluble salts.
 | ||
| Fig. 1 Cerium oxalate structure. | ||
Carboxylic acids are commonly used to control crystallite growth. Taguchi and coworkers15 reported the effects of a series of dicarboxylic acids with varied chain lengths for this purpose. The synthesis was carried out in supercritical water systems (400 °C, 38 MPa) with Ce(OH)4 as the ceria precursor. Assuming that the nanoceria surface consists only of metal and oxygen atoms (no hydroxyls), the carboxylic acid groups could form bidentate, bridging or unidentate configurations on the nanoparticle surfaces (Fig. 2). In this case, all dicarboxylic acids formed bidentate structures, based on FTIR data.
 | ||
| Fig. 2 Three binding states for carboxylate anions and metal atoms on metal oxide surfaces.15 | ||
Table 2 shows the product morphologies as a function of the –CH2– chain length for the dicarboxylic additives. All of the dicarboxylic acid additives caused changes in the nanoparticle morphologies. For ceria crystallites, the surface energies of the lowest index crystal facets are in the order, γ(111) < γ(100) < γ(110). That is, the (111) crystal face is the most stable.
| –(CH2) – chain length | Carboxyl state | Carboxylate coverage of surface, % [TGA data] | Crystallite morphology | Plane that limits growth |
|---|---|---|---|---|
| None | NA | NA | Truncated octahedral | (111) |
| 4 | No free carboxyl | 54.8 | Cubo-octahedral | (111) ~ (100) |
| 5 | No free carboxyl | 33.8 | Cubo-octahedral | (111) ~ (100) |
| 8 | Free carboxyl | 92.4 | Cubic | (100) |
| 10 | Free carboxyl | 89 | Cubic | (100) |
Crystallite faces, edges, and corners all have different reactivities (see the review of Reed). Adsorption of additives to a crystallite face should hinder its growth. The additives in Table 2 have different effects on the morphologies of nanoceria (Fig. 3). The carboxylic acid groups appear to bind solely to the (100) crystallite faces. When no additive was used, this synthesis method produced truncated octahedrons with large (111) faces (pathway a). Short chain dicarboxylic acids gave moderate protection of the (100) surface and growth was relatively balanced between the two faces, resulting in cubo-octahedrons (pathway b). When the additive provided dense packing on the (100) face, this facet became the surface that limited the rate of crystal growth (pathway c). Both carboxylic acids of the additives with shorter methylene chain segments bound to the nanoceria surface, resulting in moderate surface coverages that left no free carboxylate groups. Long chain acids aligned themselves perpendicular (normal) to the surface, resulting in higher surface coverage and free carboxylates on the exterior of the nanoparticles. Cerium atoms on edges and corners have different local coordinations, and will have different reactivities. As the nanoparticle size decreases, the fractions of surface atoms on edges or corners increases. One of the challenges in understanding the surface reactivity of nanoceria is describing the surface reactivity of all surface atoms for a particular nanoparticle size and morphology.
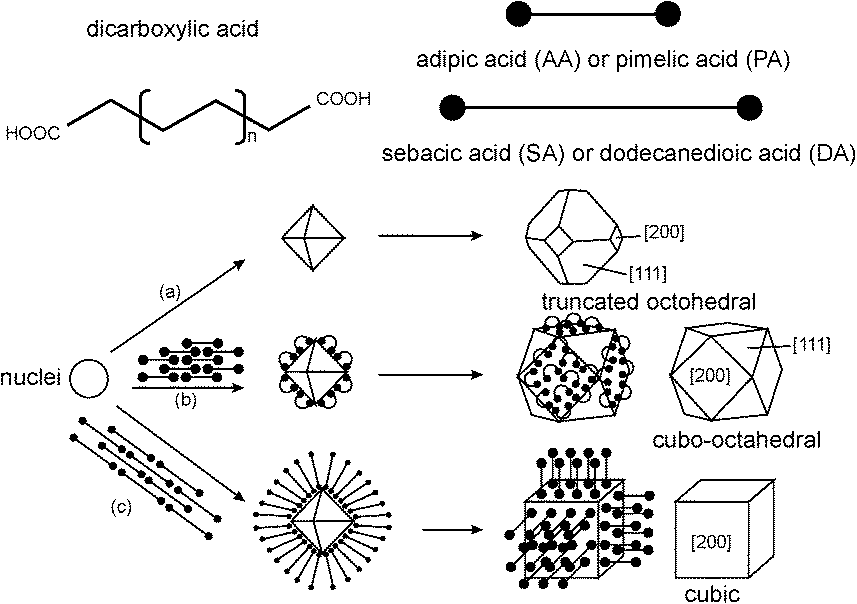 | ||
| Fig. 3 Structural models of truncated octahedral, cubo-octahedral, and cubic morphologies.15 | ||
Nanocrystal growth mechanisms have particular relevance for nanoparticles that may undergo chemical transformations in environmental or biological milieu. Should nanoparticles be exposed to conditions that change their redox environment, i.e., new combinations of temperature, pH, ionic species and/or ionic species concentrations, oxygen, other oxidizing ligands, or reducing ligands, it is possible for them to re-equilibrate their structure (that is, dissolve and/or precipitate).
Realistic surface structures: nanoceria and other metal oxides
The surfaces of metal oxides have different compositions and are often terminated in oxygen (such as M–O–M or MO) or hydroxyls (M–OH). The presence of hydroxyls on the surface of ceria has been known for more than four decades.37 Driven by the importance of ceria for gas phase catalysis, much computational and experimental research on ceria surface reactions has been based on crystallite structures relevant to gas phase systems in which the activity of water may be quite low. Much data on nanoceria surface chemistry is based on analytical tools requiring ultrahigh vacuum, in which the activities of key species, i.e., oxygen and water, will be quite low.38–40 It is not clear how well data and models for nanoceria reactivity in systems with low activities of O2 and H2O will relate to data for systems with high activities of O2 and H2O.
This conundrum has been identified in a recent review on adsorption, diffusion and reaction, and structural sensitivity of heterogeneous catalysts. Sterre and Freund41 point out that adsorption structures and reaction pathways observed under ultrahigh vacuum conditions may not be the same as those under realistic pressures and fluid phase compositions. One of the more important modifications, in practice, can include hydroxylation of the metal oxide surface. One of the remaining challenges is the characterization and modeling of reaction mechanisms for surface hydroxyls at relevant water activities. For several metal oxides, dissociative adsorption of water is thermodynamically unfavorable; however, this process occurs readily on defect sites such as step edges or oxygen vacancies. Ref. 108 describes mechanisms in greater detail.
Table 3 links specific nanoceria morphologies to their typical crystal facets and the surface energy of that facet. The two references are based on gas phase42 and liquid phase15 data. In general, a crystal facet with high surface energy has a higher reactivity.
Ceria abrasive, with aggregate sizes of ~125 nm and constituent particles of ~50 nm, have a ‘shell’ near their surfaces that is enriched in Ce3+.43 Doping impurities, such as lanthanum which is added to aid redox catalysis, also tend to accumulate near the outer surface of nanoceria particles.43 Ceria nanoparticles synthesized by vapour phase condensation have increasing fractions of Ce3+ ions in the particles as the particle diameter decreases.44 For example, Ce3+ levels on ceria surfaces increase as the particle size drops below 11 nm.44 The amount of CeO1.5 seems to depend on the synthesis method and the crystallite product; nanoparticles with the more stable (111) morphology are less likely to have reduce stoichiometry (CeO1.5).44 Specific analysis of (111) and (100) facets by STEM-EELs showed that, for (111) facets and surface islands, the reduction shell exists over the surface plane and extends 1–2 mixed valence planes below the surface.45 For (100) facets, the reduction shells extends to 5–6 levels of oxygen-valency planes below the surface.45
• Each crystal facet of CeO2 will have a different growth rate, surface energy, and surface reactivity. For very small nanoparticles, the different reactivities of cerium atoms on edges and corners can be important.
• At environmentally-relevant conditions (high oxygen and water activities), some metal oxide surfaces are terminated by an oxygen layer and become fully hydroxylated in the presence of water. Therefore, it would be expected that the chemical properties of these hydroxyls and their surface reactivity would be strong functions of the environmental conditions.
• Crystallite morphology and specific crystal facets should be part of the nanoceria's characterization for the best understanding of its redox properties and its function as a pro- or anti-oxidant. For very small nanoparticles, it will be important to include atoms on edges and corners in the reactivity ‘audit’.
• Reaction mechanisms for specific crystal facets are not always known.
3. Phase equilibria: cerium ions, salts, and solids in aqueous solutions
The phase equilibria for cerium ions and solids in aqueous solutions have been modeled thermodynamically using half-cell redox reactions and are often visualized in Pourbaix diagrams. These diagrams map stable equilibrium phases (electrical potential vs. pH) of aqueous electrochemical systems. Ion boundaries are represented by lines, which represent transitions between stable species but not the kinetic rates by which they are achieved. These calculations are based on a series of half-cell potentials for the various chemical species. Insoluble coatings (such as covalently-bound coupling agents or polymeric shells) can block reactive sites on the metal oxide surface; in these cases, the Pourbaix diagram will not be applicable.The original Pourbaix diagram for aqueous cerium46 was revised by Hayes et al.;47 (see Fig. 4) by considering the precipitation of Ce4+ species and the potential reaction between soluble Ce3+ species and insoluble Ce4+ precipitates. In general, this is based on the aqueous solubility of the compounds, Ce(OH)3 and Ce(OH)4. Other cerium compounds, such as those used for nanoceria synthesis or that might form due to redox reactions with surface cerium atoms, will have different solubilities. Experimental data were obtained via a series of titration studies covering a range of pHs under inert (argon) and oxygen atmospheres. For Ce3+ solutions, exposure of aqueous systems to air slowed the pH stabilization during potassium hydroxide titrations, while an argon purge resulted in rapid pH stabilization. Solutions under an air atmosphere developed yellow precipitates, which were consistent with the formation of Ce4+ species. In argon, only white precipitates were noted, which were consistent with no Ce4+ formation. Thus, the presence or absence of dissolved oxygen in aqueous solutions makes a difference in the phase equilibria achieved. Ce3+ and Ce4+ ions in solution tended to hydrolyze and complex. Both Ce(OH)3 and Ce(OH)4/CeO2·2H2O species were soluble to some extent, but were the major components in precipitates. Furthermore, they found that the morphology, agglomeration, and surface chemistry of the solids would be expected to change with the experimental methodology.
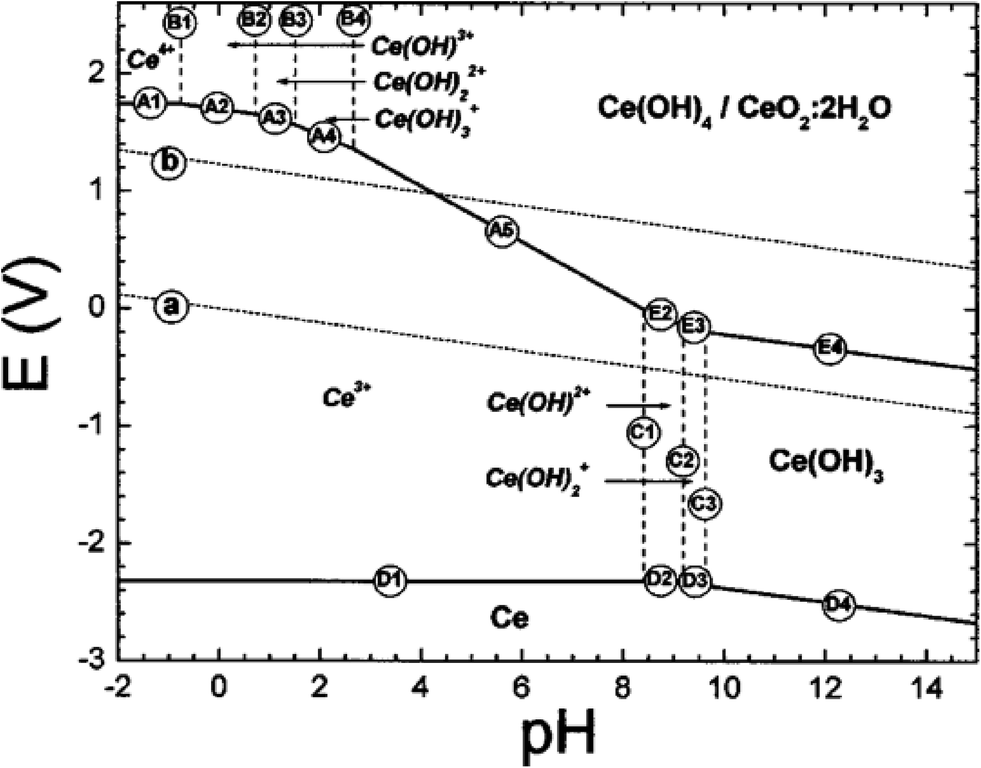 | ||
| Fig. 4 Updated E-pH (Pourbaix) diagram for cerium in aqueous perchlorate solutions.47 Reproduced with permission from J. Electrochem. Soc., 2002, 149, C623. | ||
Yu and coworkers48 developed a model for the cerium–water–hydrogen peroxide system (Fig. 5), varying these species plus pH and oxygen as well. Typical half-cell reactions for the cerium–water system alone included:
| Ce3+ + 4H2O → Ce(OH)4 + 4H+ + e− | (1) |
| O2 + 4H+ + 4e− → 2H2O | (2) |
| 4Ce3+ + O2 + 14H2O → 4Ce(OH)4 + 12H+ | (3) |
 | ||
| Fig. 5 Simplified E-pH diagram of Ce–H2O–H2O2 system showing when the Ce4+ precipitate is CeO2(precip).48 Reproduced with permission from J. Electrochem. Soc., 2006, 153, C74. | ||
Different half-cell reactions apply when hydrogen peroxide is added to the system. Peroxide acts as a reducing agent for Ce4+ at low pH (<4) and as an oxidizing agent for Ce3+ at higher pH (>4). However, the actual states of the cerium precipitates were not assessed. These diagrams should be considered as computations only and based on kinetics that were readily observed during titrations.
Fig. 5 shows that Ce3+ is oxidized to Ce4+ by hydrogen peroxide, i.e., H2O2 is a stronger oxidizing agent than Ce4+. A typical overall reaction for a soluble Ce3+ ion would be:48
| 2Ce3+ + H2O2 + xH2O → 2Ce(OH)3−x/21+x/2 + xH+ | (4) |
The half-cell reaction for hydrogen peroxide is:
| H2O2 + 2H+ + 2e− → 2H2O | (5) |
In the case that a Ce3+ salt is oxidized to a solid precipitate, the overall reaction would be:
| 2Ce3+ + 2H2O2 + 2H2O → 2CeO2(s) + 6H+ | (6) |
In addition, it is possible that Ce4+ could form a complex with hydrogen peroxide. In general, Ce3+ is relatively soluble at pH up to 11 while Ce4+ is soluble at pH less than 4 (shown as a series of species in the upper left hand corner of the E(V) vs. pH plot). Eqn (6) is typical of cerium redox systems in solution. If nanoceria is dissolved in the local milieu, it would be most likely to be as a Ce3+ salt. A local oxidizing agent or system might then oxidize the Ce3+ salt to CeO2, which would precipitate and, presumably, recrystallize to minimize surface energy. Similar chemistries occur in aqueous phase synthesis of nanoceria. For example, even bulk CeCl3 can be transformed to CeO2 under oxygen excess.49 However, CeO2 in biological systems could be reduced to form Ce3+ salts if appropriate reducing agents and conditions are present. For example, lysosomal fluid is acidic (~pH 4.5 due to acid hydrolases).50 The presence of different redox systems (such as ligands H2O2, O2, H+, e−) can cause shifts in the phase equilibria between Ce3+ and CeO2 (Ce4+), as shown in Fig. 5.
There is at least one report of nanoceria dissolving and reprecipitating in biological studies.51 In this case, ~30 nm cubic nanoceria, in which the (100) face predominated, were intravenously administered to rats. Samples of organ tissues were gather for analysis up to 90 days after the dose, which was high and well above the previously determined therapeutic window for ceria in vivo.52 Many of the cubic nanoceria particles had become fragmented and rounded along their edges. There were clouds of small nanoceria, 1–3 nm in size, with dominant faces of (111), (200), and (311), which were not seen in the original dosed nanoceria (primarily (100) faces). In addition, polycrystalline large grains of nanoceria were observed that could have been the result of Ostwald-type ripening. Therefore, these nanoceria appeared to undergo in vivo bioprocessing after prolonged exposure in the liver. In addition to this one report of processing, there are a number of reports showing that stored nanoceria can change its structure and composition. Some of these occurrences may be related to the local redox conditions during storage.
• It seems likely that a number of oxidizing and reducing agents could play roles in ceria dissolution and/or precipitation at storage, in vitro, or in vivo conditions.
• When nanoceria are in aqueous biological or environmental milieu, the following mechanisms may alter their surface morphologies: Ostwald ripening, in which less stable crystal facets lose surface atoms to more stable crystal facets; adsorption of inorganic and organic ligands, such as proteins leading to coronas; and reactions with liquid-phase ligands, which can include redox-type reactions.
• Demonstration of systems in which nanoceria's redox cycles are ‘turned on’ and ‘turned off’. This could lead to better understanding of how nanoceria's redox capabilities, i.e., pro- or anti-oxidant, can be controlled in environmental and biological systems.
4. Coated surface chemistry of nanoceria in aqueous dispersions
Many ceria nanoparticles are dispersed in liquid phases, either during synthesis, functionalization, or in use. As is discussed elsewhere in this perspective, small changes in nanoceria surface chemistry or in the suspending medium can drastically change their colloidal stability. Therefore, many manufacturers modify the surface chemistry of their products by applying coatings. This technology has been known for decades and, often, the coatings are specific to given applications. These coatings will affect the hydrophilicity/hydrophobicity of the nanoceria and the adsorption of ligands from environment exposures (such as natural organic matter) or from biological exposures (such as proteins or natural surfactants). For example, phospholipid-PEG12 and chitosan53 coatings are effective in improving nanoceria stability in dispersions. Two surface coating examples are discussed here: intentional coatings with silanes and unintentional coatings with proteins.Functionalization
Fig. 6 shows a typical functionalization sequence, which assumes that the nanoparticle was created in the gas phase (very low water activity) and would have few hydroxyls on its surface. The first stage develops functional groups on the neat particle (coating 1) and the second stage applies the coating needed for the application (coating 2). A variety of techniques are available to generate surface hydroxyls on metal oxides, including acid etching. Nanoceria produced by hydrothermal or solvothermal processes often have surfactants or other surface-active salts associated with their surface. These would typically be removed prior to coating. | ||
| Fig. 6 Process for nanoparticle coatings. Functionalization + silanation. | ||
Silanization
Mono-, di-, and tri-functional silanes are commonly used coupling agents that are known to react with the hydroxyls on the surfaces of many metal oxides,54–58 including ceria. A key factor in preparing uniform and complete coatings is measuring the surface density of hydroxyl groups (# of hydroxyls per nm2, for example)59 and then tailoring the functionalization recipe to assure uniform surface densities of the silane. As shown in Fig. 6 (shell 2), many coating types are possible. For example, silanes can be found to make metal oxide surfaces hydrophobic,56 to control protein adsorption,58 or permit electrochemical detection of DNA hybridization.60 The electrical charge of the coated nanoparticle will be affected both by unreacted ions on the metal oxide and by chemical groups on the coating material. Not all intentional coating processes are well-controlled (there can be batch-to-batch variation), provide reproducible surface coverage (sub-monolayer, monolayer, or multilayer), or react with all reactive groups on the metal oxide surface. Therefore, while the coating may improve nanoceria dispersion in a specific liquid environment, the coated nanoceria may not necessarily be chemically inert, particularly with respect to small molecules diffusing to and reacting with ‘free’ surface groups.Protein adsorption
Proteins are known to adsorb to many nanoparticles when they contact biological fluids, creating protein ‘coronas’. In mixtures of proteins, the adsorption is competitive and specific proteins may displace others from the surface, often called the Vroman effect. These coronas can be considered coatings and have thicknesses from sub-monolayer to multi-layer, depending on the nanoparticle morphology and the conditions of the medium. Protein coronas appear to form rapidly, approaching initial equilibrium within seconds of the exposure. Over time, the initially adsorbed proteins may be displaced with proteins that generate lower energy surfaces. Ref. 109 have more details on these issues.Rezwan and coworkers published a series of papers illustrating the general principles of protein adsorption on nanoparticles. In the first study,61 adsorption isotherms of bovine serum albumin (negatively charged) and lysozyme (positively charged) were determined for hydrophilic (alumina, silica, and titania) and hydrophobic (zirconia) nanoparticles. In all cases, the adsorbed proteins changed the zeta potentials curves of the metal oxide dispersion. For neat, hydrophilic nanoparticles, the amounts of protein adsorbed correlated with zeta potential. Hydrophobic zirconia nanoparticles adsorbed high amounts of protein even under electrostatically repulsive conditions, suggesting that multiple adsorption sites and mechanisms are possible.
In a second study,62 protein mixtures were adsorbed on hydrophilic nanoparticles (alumina and titania). The amount of protein in solution was sufficient to develop coronas that completely ‘masked’ the nanoparticle surfaces. The researchers were able to model the shift in the isoelectric point of the complex nanoparticle. This included analysis of the amino acid compositions and pK values of the two proteins, estimating their charges, using a weighted average to estimate the isoelectric point of the adsorbed mixture, and using a factor for the accessible surface area to fit the data. In this case, bovine serum albumin had the most effect on the isoelectric point; its accessible surface area was 2.6 times that of lysozyme. Additional discussion of the zeta potential for protein-coated nanoparticles is given by the Das et al. perspective plus other recent references.63–66
Wang and coworkers67 studied the effects of surface charge (positive, neutral, and negative) of various ceria nanoparticles on the adsorption of one protein, lysozyme (net positive charge). The data were modeled using the Toth and Sip isotherms, and compared to monolayer coverage estimates for both ‘side-on’ and ‘end-on’ packing of the lysozyme molecules under the assumption of random packing on the nanoparticle surface. Adsorption of a protein with a net positive charge on negatively charged nanoparticles had a very broad adsorption site energy distribution, which controlled the adsorption process. On the other hand, adsorption levels of this protein onto a positively charged surface were lower, but were influenced by lateral effects from adsorbed protein species.
Aqueous dispersion effects
There is a growing consensus within the risk assessment community of the need for standardized and validated dispersion methods to ensure reliable and reproducible results when doing nanoparticle toxicology studies. A method has been reported for titania dispersion in aqueous media; this method is harmonized with respect to the particle size distribution, the pH, the isoelectric point of the dispersion, the nanoparticle concentration, and the batch variability.68Flocculation and agglomeration of ceria dispersions were studied by Kong and Leong.69 Below the isoelectric point (~pH 7), high concentration (30 wt%) ceria dispersions flocculated but were homogeneous. Above this pH, dispersions phase separated rapidly, forming large agglomerates, consistent with the work of Nabavi,70 which was based on nanoceria dispersions stabilized with nitrate anions. Below the isoelectric point, nitrate anions adsorbed and were bound covalently to the nanoceria, forming a steric layer that prevented particle aggregation. Aggregation was shown to occur when the nitrate groups were displaced by hydroxyls, followed by condensation reactions between contacting nanoparticles (forming Ce–O–Ce bonding bridges). At high pH, agglomeration was prevented by adding a pyrophosphate. However, phosphate ligands may complex with nanoceria particles, changing their stability in water dispersions.32,53
A number of coating types are preferred for drug delivery by nanoparticles, including poly(ethylene glycol), sugar solutions for controlling osmotic pressure, organic acids (mono- to multidentate), polymers, and oligomers. These are intended to improve the dispersion stability at dosing conditions and during the transport and biodistribution of the nanoparticles to their intended target.
• Adsorption of ligands, such as proteins, is dynamic; protein composition within the coating can change over time.
• Does the coating prevent redox reactions or merely retard redox reactions of the nanoceria, i.e., is the coating porous to ligands participating in redox reactions?
• Does the coating help reduce or control dispersion agglomeration or aggregation at environmental or biological exposures?
• Does the coating affect the transport and biodistribution of the nanoparticle?
5. Nanoceria surface reactivity
Many of the applications in which nanoceria is used commercially (Table 1) have well-controlled process conditions, known potential reactants, are well above physiological temperatures and have low water activities. In addition, much of the instrumentation used to assess surface chemistry, surface structure, and morphology operates at very low water activities. This section reviews experimental systems with both low and high water activities. If surface hydroxyls are important for nanoceria reactivity at high water activities, the experimental reactivity and surface characterization results obtained in low water activity environments may not be relevant to the pro- or anti-oxidant properties in biological environments.Experimental systems with low water activities
For gas phase catalysis on metal oxides, it is well known that the chemistry of the surface, the surface structure (including the crystallographic face and the atomic structure of its surface), the degree of dehydration, and the presence of anionic or cationic ligands all affect the surface acidity, hydrophilicity, and reactivity.71 However, even in gas phase reactions, only a small fraction of surface sites may be involved in the catalysis.In a highly cited review, Mogensen and coworkers72 evaluated the physical, chemical and electrochemical properties of pure and doped ceria; the data available at that time (2000) was mostly focused on high temperature, low water conditions relevant to applications such as solid oxide fuel cells. The process of ceria reduction was modeled as generating a defect in the form of Ce3+, rather than linked to oxygen vacancies.
Experimental systems with high water activities
Nanoceria can be synthesized directly in monosaccharide and polysaccharide solutions by oxidizing the precursors (such as cerium(III) nitrate) in either acidic or basic environments.85 The initial complex between the saccharides and the nanoceria surfaces does not appear to affect the redox performance of the nanoceria. However, redox reactions proceed more rapidly in acidic media. The Ce4+ oxidation state was retained for more than 2 months, suggesting good dispersion stability. The pH of the solution must be maintained in order to prevent the precipitation of cerium hydroxide.In 1993, Nabavi and coworkers evaluated the acid–base behavior of surface hydroxyls in aqueous dispersions70 using a model (the “MUSIC” model) developed by Van Riemsdijk.86,87 In bulk ceria, Ce4+ coordinates with eight oxygen atoms while each oxygen atom coordinates with four cerium atoms. The formal valence bond, the number of shared electrons, is v = 0.5. In the bulk ionic crystal, the charges balance. Assuming that a ceria surface is isolated from the bulk, a ceria surface in water would have the coordination of metal atoms completed by the oxygen(s) of adsorbed water. Thus, oxygens on the surface would have a charge. The model was used to evaluate the equilibria for various hydroxyl sites, which could be coordinated with different numbers of cerium atoms. The general equilibria equations were:
| Mn − Onv−2 + H+ ↔ Mn − OHnv−1 + H+ ↔ Mn − OHnv2 |
| Ce2 − OH2+1 ↔ Ce2 − OH0 + H+ ↔ Ce2 − O−1 + 2H+ |
| Ce1 − OH2+0.5 ↔ Ce1 − OH−0.5 + H+ ↔ Ce1 − O−1.5 + 2H+ |
| Ce3 − OH2+1.5 ↔ Ce3 − OH0.5 + H+ ↔ Ce3 − O−0.5 + 2H+ |
Quantum mechanical calculations of, e.g. Mulliken population analyses on ceria nano-particles (A.N. Cormack, personal communication, and see the discussion of Fig. 7, below) suggest that while the surface atoms do have a different effective charge from those in the bulk, they do not carry a net charge, as supposed by the “MUSIC” model: the particle is, overall, electrostatically neutral. Thus, the bond valence analysis must differ from that used by Nabavi et al., and the parameters used in calculating the equilibrium constants are, therefore, suspect.
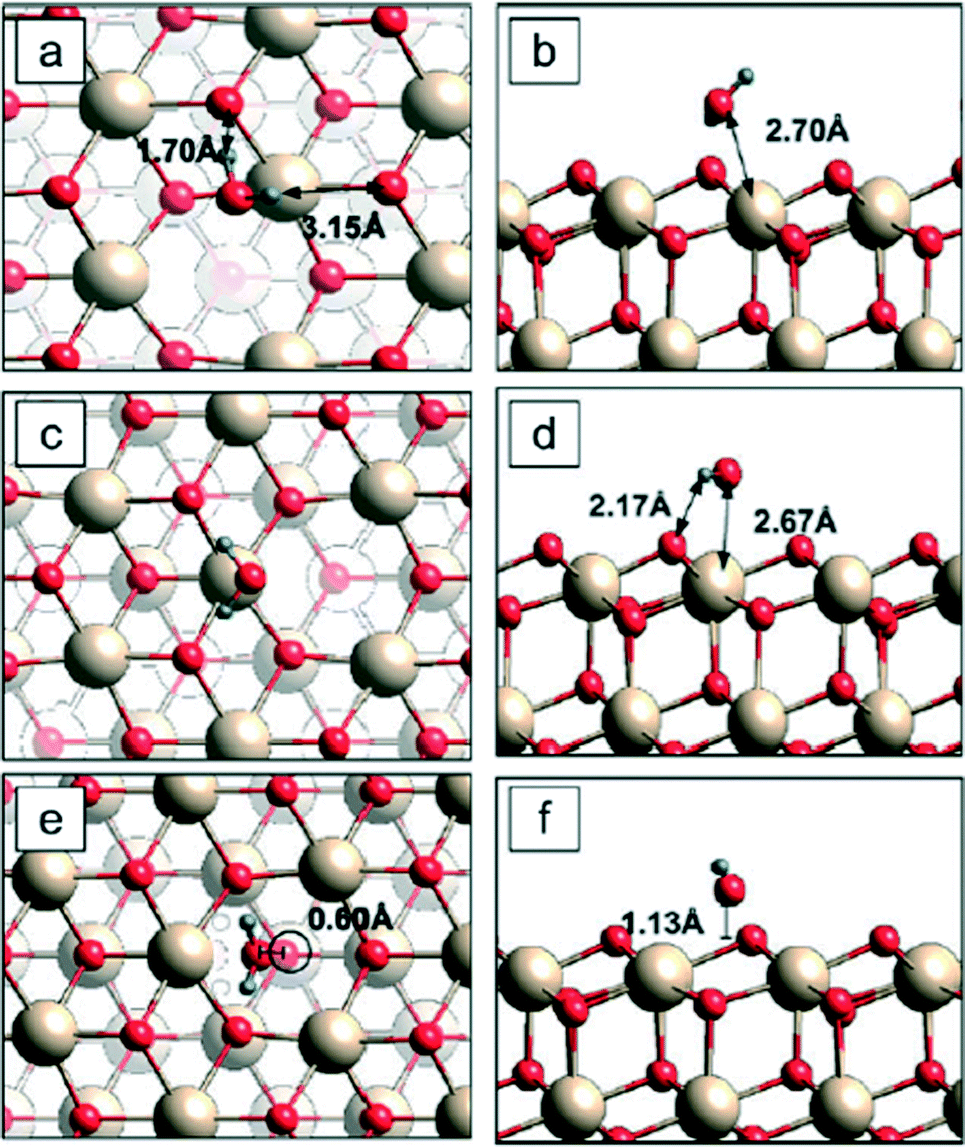 | ||
| Fig. 7 Three adsorption sites for a water molecule on reduced (111) surfaces.100 Left = top view; right = side view. Cerium = beige; oxygen = red; hydrogen = blue. The lowest binding energy configuration is c–d. In the side views (b, d, and f), the transecting plane passes through the oxygen on the water molecule; therefore, the second hydrogen is outside the plane and does not appear in d and f. | ||
Nabavi and coworkers explored the surface chemistry of ceria nanoparticles in aqueous dispersions through a series of titrations.70 Their nanoceria was synthesized using Ce3+ nitrate, by which nitrate ions are associated with the surface. The agglomeration and aggregation properties of colloidal dispersions are usually related to their surface chemistry plus the presence of other ions at their surfaces. This dispersion exhibited three distinct states: 1) between pH 2 to pH 6, nitrates are released and the dispersion aggregated, 2) between pH 6 to pH 10, there were no more transformations, and 3) above pH 10, the surfaces underwent transformation(s) that suppressed the sites for nitrate binding. In particular, when long times occurred prior to using the nanoceria, Ce3+ could be detected in solutions, presumably from the dissolution of Ce4+ ions and their reduction. The rates of these processes will be affected by the nature and size of the nanoparticles.
The monodentate and tridentate sites appear to take part in binding equilibria for protons and hydroxyls, while bidentate sites appear to be inactive over the range of pHs studied. When nitrates begin to be released during titration (~pH 6), covalent nitrates are released and the nanoparticles aggregate. Reactions of active monodentate and tridentate sites were thought to cause this change in surface energy. When the dispersion has additional hydroxyls (titration above pH 10), the ceria surfaces restructure by transforming bidentate sites into tridentate sites. These surfaces have lost the ability to bind nitrates with partly covalent bonds. The links between surface chemistry and colloidal stability for nanoceria appear to be typical of other metal oxide nanoparticle dispersions. Dispersions are typically stable in narrow pH ranges; washing or titrating these systems results in a loss of surface functional groups and unstable performance.
Adhesion between ceria nanoparticles and other surfaces has been used to study the complexation of various ligands on nanoceria.88 In the presence of complexing agents, such as nitrates, ceria surfaces are protected and adhesion is prevented (mica as an adsorbate in this case). The efficiency of the ligand increases in the order, nitrate < acetate < acetyl acetone. In this study, controlling adhesion at nanoparticle contact appeared more important than achieving long-range electrostatic repulsion. In the case of nitrate ions, they also found that nitrate was released at pH > 3, with the dispersion becoming unstable. Some nitrate ions appeared to be more strongly bound to the nanoceria than others. In general, adhesion at contact might be eliminated by the addition of complexing molecules, i.e., by applying coatings that provide steric or electrostatic stabilization.
• Titration experiments of nanoceria demonstrate that specific surface hydroxyls, which have different numbers of coordinating cerium atoms, have different apparent charges. This should lead to differences in adsorption, complexation, and reaction for specific sites.
6. Computational studies of nanoceria systems
Computational studies provide a different perspective on the redox properties of nanoceria surfaces. Computational results on systems linked to commercial processes have been described in ref. 108. A number of these studies have been based on the surface structures of bulk ceria as a starting point. Table 4 shows the crystal facets investigated, the method, and the typical conditions considered. Several research groups have studied water adsorption89–94 and hydroxyl stability95 on nanoceria.| Condition | Crystal facets | Method | Ref. |
|---|---|---|---|
| Low water activity | |||
| CO adsorption | (110), slab | 96 | |
| Reduction of ceria by hydrogen | (111), (110) bulk | Ultra-accelerated QCMD, DFT | 81 |
| Oxygen vacancies | (111) – nanoparticles | DFT | 97 |
| Oxygen defect formation | (110), (111) | QCMD, others | 98 |
| (111) effects of oxidizing environment | DFT | 99 | |
| High water activity | |||
| Chemical polishing | SiO2 surface with hydroxyls, bulk CeO2 | QCMD | 92 |
| Adsorption of water | (111) – CeO2; CeO2−x | DFT, SRPES | 91 |
| (111) – CeO2; CeO2−x | DFT | 100 and 89 | |
| Preferred adsorption on (100), (110), edges, corners | DFT | 93 | |
| Oxygen vacancies increase adsorption energy of water on (111) | DFT | 90 | |
| Configuration of low energy surfaces | 94 | ||
| Stability of surface hydroxyls | Nanoceria supercell | DFT | 95 |
Since water is ubiquitously present in environmental or biological milieu, the role of water on the surface structure and redox chemistry of nanoceria cannot be ignored. To address this issue, Traversa's group100 have investigated the variation of surface structures for (111) ceria faces and Huang et al.95 have focused on the expected surface structural evolution for ceria nanoparticles in response to changing conditions in high water activity environments.
Water adsorption has been used as a probe to evaluate the effects of ceria surface on binding energy, among other factors.89,100 Fronzi and coworkers100 evaluated binding energies for water molecules adsorbing and/or dissociating on CeO2 (111) surfaces. Two different surfaces were considered: clean stoichiometric (111) surfaces and reduced (111) surfaces with oxygen vacancies. An example is shown in Fig. 7, based on 2 × 2 unit cells and considering one molecule adsorbing from the gas phase on different available surface sites of a reduced (111) surface. The most energetically favorable adsorption site for a single water molecule was a surface Ce atom above which the water was in a planar geometry parallel to the ceria surface with its own oxygen atom positioned, and each hydrogen atom bound to a nearest surface oxygen [c & d in Fig. 7]. This site had a lowest binding energy of 0.49 eV, about the same as those reported for similar experimental conditions.89 All three of the dissimilar water adsorption configurations on reduced (111) surfaces had higher binding energies than that of water adsorption configurations on the clean (111) surface. When oxygen vacancies were present at the ceria surface, the binding energy of water molecules to ceria surface (above oxygen vacancy sites) became stronger, suggesting an attraction between the water molecule and the oxygen vacancy. The presence of water appeared to lower the energy cost of creating oxygen vacancies on (111) surfaces. Some simulations and experiments suggest that the reactivity of ceria nanoparticles is environmentally dependent and is influenced by its history in reaching a particular milieu.93
Different assumptions regarding nanoparticle surface energies can lead to different results. For example, it is possible to construct nanoparticles assuming they conserve stoichiometry or mass ratios, or that they conserve net charge and oxidation state. Recent quantum mechanical calculations of the stability of cubic nanoceria examined this issue in detail, considering the density and distribution of bound surface groups, and deriving scaling relationships for the stable surface concentration of these groups.95 While the analysis of acid–base behaviour of surface hydroxyls by Nabavi70 assumed that the surface atoms are separated from the bulk, quantum mechanical calculations permit charge to be distributed throughout the nanocrystal. Conservation of stoichiometry did not lead to minimum energy nanoceria structures, but non-stoichiometric configurations that conserved oxidation state did. These calculations reveal the stable concentration and configuration of species of cubic nanoceria as a function of oxygen and hydrogen chemical potentials, which are dictated by its environment. Nanoceria might be considered as consisting of a bulk “core” decorated with combinations of anionic surface groups (e.g., –Ox and –OH groups). Hence stable nanoceria configurations balance the energy costs of anions from the environment against energy variations within the nanoceria itself, including effects of changes in oxidation state of Ce cations.101
For cubic nanoceria structures considered in ref. 74, DFT calculations predict stable configurations similar to those shown in Fig. 8. Analysis of the local arrangement of various surface groups at nanoceria corners, edges and facets allows the development of analytic scaling relationships that can be used to predict the relative concentration of various surface groups on similarly-shaped nanoceria of arbitrary size. Based on these results, cubic 30 nm nanoceria terminated with only hydroxyl groups is predicted to exhibit hydroxyl coverages that are 33% higher than reported experimentally. Nanoceria structures with multiple surface groups, i.e., –Ox and –OH, have predicted surface areal densities of hydroxyl groups on 30 nm nanoceria of 12.5 per nm2, quite close to the measured surface density of 12.9 per nm2. As environmental conditions control the chemical potential of available surface groups along with their concentration and configuration, these results suggest that changes to nanoceria synthesis conditions can tune nanoceria surface structures, i.e., the type, distribution and density of surface functional groups. This provides an approach to tuning the surface structure, and thus an avenue to manipulate the redox properties of nanoceria.
 | ||
| Fig. 8 Relaxed, ground-state atomic structures of three ceria nanoparticles terminated with mixed –OH and –Ox surface groups. Atomic representations: ceria = yellow, hydrogen = green, O = purple (either O atoms or –OH groups), O atoms in O2− molecules = blue, and O atoms in O3− molecules = pink.95 | ||
• There have been a few other studies that address possible mechanisms occurring at high water activities and temperatures relevant to environmental and biological system exposures. The surface structures of nanoceria at these conditions are still a matter of debate.
• This work could include studying relevant redox couples and ligands, as well as common ions that are known to form complexes with cerium. One example of the latter is phosphate ions, which complex with cerium. For more information, see ref. 110.
7. Acellular redox reactions with nanoceria
Nanoceria catalyst for gas phase redox reactions are often evaluated for redox reactivity and oxygen storage capacity independently from their performance in specific applications.42 Nanoceria have previously been studied in aqueous acellular systems for their redox reactions with hydrogen peroxide,102,103 complex systems such as superoxide via inhibition of ferricytochrome C,102,104 antioxidants105(Fig. 3), dopamine,106 and serotonin.107 As shown in Table 5, a variety of techniques were used to assay the reactions, some of which used the liquid phase only.| Oxidant or chemical | Liquid phase analysis | Other analyses | Reference |
|---|---|---|---|
| Hydrogen peroxide | UV-Vis; difficult to quantify | EPR, XPS | 102 |
| Hydrogen peroxide | UV-Vis, 500 nm | FTIR, XPS | 103 |
| Ferricytochrome C; competitive inhibition by superoxide | UV-Vis | 102 and 104 | |
| Antioxidants: ascorbic acid, gallic acid | UV-Vis | 105 (Fig. 3) | |
| Dopamine (citric acid solution); proposed mechanism is surface attachment | UV-Vis | FTIR, TGA, XPS | 106 |
| Serotonin (5-HT) | UV-Vis; supernatant | FTIR, TGA, DLS | 107 |
Some of these acellular redox reactions could be used to characterize the surface reactivity of nanoceria prior to in vitro or in vivo tests. Surface activity assays would then be part of the characterization of nanoparticle properties, and should provide a better basis for conclusions regarding the effects of specific environmental or biological conditions.
8. Conclusions
Nanoceria morphology and surface chemistry differ depending on synthesis methods, functionalization, intentional or adventitious coatings, and its local environment. Different crystal faces have different arrangements of atoms at the surface, leading to different reactivity. Experimental and computational studies of ceria reactivity in environments with low oxygen and water activities suggest that oxygen vacancies control reaction pathways and mechanisms. However, a few experimental and computational studies at high water activities suggest that hydroxyl groups on the surfaces control redox reaction mechanisms. Since the surface oxygens and surface hydroxyls on different crystal facets have different reactivities, the ‘overall’ redox reactivity of a specific ceria nanoparticle depends on its specific surface structure.A wide variety of ligands, from organic acids, to silanes, to proteins, can adsorb onto nanoceria surfaces. Coverage levels range from sub-monolayer to multilayer. However, surface coatings may only slow, but may not completely prevent, redox reactions.
In environmental and biological milieu, there are a plethora of redox ligand and cycles. Nanoceria may participate in these via two mechanisms: 1) direct activity as an oxidant or reductant for the specific molecule, or 2) indirectly, by provide or receiving oxygen to an existing cycle. Acellular redox reactions (e.g., the nanoceria test kit for glucose in which glucose oxidase provides the catalytic activity and nanoceria provides oxygen storage) may be a way to study these redox cycles more fully.
Finally, a wide variety of ligands could provide half-reactions to drive the dissolution or re-precipitation of nanoceria. Local pH, temperature, and oxygen levels will affect these phase equilibria processes. While initial reaction products will be driven by kinetics (for example, capping molecules can permit a less stable facet to grow quickly), the final crystallite structure will be the more thermodynamically stable. Typically, the most stable crystal face is (111), which has the lowest surface energy, and, generally, the lowest redox activity. However, since adsorption of ligands on the surface of nanoceria can alter the relative surface energies, one needs to consider the equilibrium of the whole system, nanoceria plus adsorbents plus solution conditions (pH, ions, etc.). In addition, there is now evidence for dissolution and re-precipitation of nanoceria for long-term in vivo exposures. The different surface structures of crystal facets, and the wide variety of electrochemical reactions that are possible, seems to be a relatively unexplored area of research with high relevance to the performance of nanoceria in environmental and biological milieu.
Acknowledgements
This article is a product of a workshop on nanoceria held November 2, 2013 at the Fess Parker Doubletree Resort, Santa Barbara, CA, made possible by financial support from the Sustainable Nanotechnology Organization, the Tracy Farmer Institute for Sustainability and the Environment, the Department of Pharmaceutical Sciences, Associate Dean of Research of the College of Pharmacy, and Office of the Vice President for Research, University of Kentucky.Notes and references
- J. Rebellato, M. M. Natile and A. Glisenti, Appl. Catal., A, 2008, 339, 108–120 CrossRef CAS PubMed.
- L. Chen, P. Fleming, V. Morris, J. D. Holmes and M. A. Morris, J. Phys. Chem. C, 2010, 114, 12909–12919 CAS.
- A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439–520 CAS.
- N. Q. Minh and T. Takahashi, Science and technology of ceramic fuel cells, Elsevier, New York, 1995 Search PubMed.
- S. H. Choi, S. D. Lee, J. H. Shin, J. Ha, H. Nam and G. S. Cha, Anal. Chim. Acta, 2002, 461, 251–260 CrossRef CAS.
- J. Colon, N. Hsieh, A. Ferguson, P. Kupelian, S. Seal, D. W. Jenkins and C. H. Baker, Nanomedicine, 2010, 6, 698–705 CrossRef CAS PubMed.
- G. Ciofani, G. G. Genchi, B. Mazzolai and V. Mattoli, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 495–506 CrossRef CAS PubMed.
- S. M. Hirst, A. Karakoti, S. Singh, W. Self, R. Tyler, S. Seal and C. M. Reilly, Environ. Toxicol., 2013, 28, 107–118 CrossRef CAS PubMed.
- M. S. Lord, M. Jung, W. Y. Teoh, C. Gunawan, J. A. Vassie, R. Amal and J. M. Whitelock, Biomaterials, 2012, 33, 7915–7924 CrossRef CAS PubMed.
- S. Giri, A. Karakoti, R. P. Graham, J. L. Maguire, C. M. Reilly, S. Seal, R. Rattan and V. Shridhar, PLoS One, 2013, 8, e54578 CAS.
- A. Y. Estevez and J. S. Erlichman, ACS Symp. Ser., 2011, 1083, 255–288 CrossRef CAS PubMed , 253 plates.
- C. K. Kim, T. Kim, I.-Y. Choi, M. Soh, D. Kim, Y.-J. Kim, H. Jang, H.-S. Yang, J. Y. Kim, H.-K. Park, S. P. Park, S. Park, T. Yu, B.-W. Yoon, S.-H. Lee and T. Hyeon, Angew. Chem., Int. Ed., 2012, 51, 11039–11043 CrossRef CAS PubMed.
- N. M. Zholobak, A. B. Shcherbakov, A. S. Bogorad-Kobelska, O. S. Ivanova, A. Y. Baranchikov, N. Y. Spivak and V. K. Ivanov, J. Photochem. Photobiol., B, 2014, 130, 102–108 CrossRef CAS PubMed.
- T. Morita, E. Tanaka, Y. Inagaki, H. Hotta, R. Shingai, Y. Hatakeyama, K. Nishikawa, H. Murai, H. Nakano and K. Hino, J. Phys. Chem. C, 2010, 114, 3804–3810 CAS.
- M. Taguchi, S. Takami, T. Naka and T. Adschiri, Cryst. Growth Des., 2009, 9, 5297–5303 CAS.
- R. C. Macphail, E. A. Grulke and R. A. Yokel, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 374–387 CrossRef CAS PubMed.
- R. Yokel, E. Grulke and R. MacPhail, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 346–373 CrossRef CAS PubMed.
- C. Sun, H. Li and L. Chen, Energy Environ. Sci., 2012, 5, 8475–8505 CAS.
- M. Hirano and M. Inagaki, J. Mater. Chem., 2000, 10, 437–477 RSC.
- J. E. Spanier, R. D. Robinson, F. Zhang, W. W. Chan and I. P. Herman, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 245407 CrossRef.
- E. N. S. Muccillo, R. A. Rocha, S. K. Tadokoro, J. F. Q. Rey, R. Muccillo and M. C. Steil, J. Electroceram., 2004, 13, 609–612 CrossRef CAS.
- F. Zhang, S. W. Chan, J. E. Spanier, E. Apak, Q. Jin, R. D. Robinson and I. P. Herman, Appl. Phys. Lett., 2002, 80, 127–129 CrossRef CAS PubMed.
- F. Zhang, Q. Jin and S. W. Chan, J. Appl. Phys., 2004, 95, 4319–4326 CrossRef CAS PubMed.
- M. Kamruddin, P. K. Ajikumar, R. Nithya, A. K. Tyagi and B. Raj, Scr. Mater., 2004, 50, 417–422 CrossRef CAS PubMed.
- L. Madler, W. J. Stark and S. E. Pratsinis, J. Mater. Res., 2002, 17, 1356–1362 CrossRef CAS.
- R. D. Purohit, B. P. Sharma, K. T. Pillai and A. K. Tyagi, Mater. Res. Bull., 2001, 36, 2711–2721 CrossRef CAS.
- N. Ozer, Sol. Energy Mater. Sol. Cells, 2011, 68, 391–400 CrossRef.
- C. Laberty-Robert, J. W. Long, E. M. Lucas, K. A. Pettigrew, R. M. Stround, M. S. Doescher and D. R. Rolison, Chem. Mater., 2006, 18, 50–58 CrossRef CAS.
- M. Hirano and E. Kato, J. Am. Ceram. Soc., 1999, 82, 786–788 CrossRef CAS PubMed.
- C. W. Sun and L. Q. Chen, Eur. J. Inorg. Chem., 2009, 3883–3887 CrossRef CAS PubMed.
- T. Matsui, K. Fujiwara, K. Machida, G. Adachi, T. Sakata and H. Mori, Chem. Mater., 1997, 9, 2197–2204 CrossRef.
- Y. J. He, B. L. Yang and G. X. Cheng, Mater. Lett., 2003, 57, 1880–1884 CrossRef CAS.
- N. Guillou, L. C. Nistor, H. Fuess and H. Hahn, Nanostruct. Mater., 1997, 8, 545–557 CrossRef CAS.
- L. Lin, Y. Wang, G. Pang, Y. Koltypin and A. Gedanken, J. Colloid Interface Sci., 2002, 246, 78–84 CrossRef PubMed.
- Y. Zhou, R. J. Phillips and J. A. Switzer, J. Am. Ceram. Soc., 1995, 78, 981–985 CrossRef CAS PubMed.
- S.-M. Lee, S.-N. Cho and J. Cheon, Adv. Mater., 2003, 15, 441–447 CrossRef CAS PubMed.
- R. K. Singh, A. Suryanarayan and G. K. Roy, Indian Chem. Eng., Sect. A, 1995, 37, 17–21 CAS.
- D. R. Baer, J. Surf. Anal., 2011, 17, 163–169 CAS.
- A. S. Karakoti, P. Munusamy, K. Hostetler, V. Kodali, S. Kuchibhatla, G. Orr, J. G. Pounds, J. G. Teeguarden, B. D. Thrall and D. R. Baer, Surf. Interface Anal., 2012, 44, 882–889 CrossRef CAS PubMed.
- S. V. N. T. Kuchibhatla, A. S. Karakoti, D. R. Baer, S. Samudrala, M. H. Engelhard, J. E. Amonette, S. Thevuthasan and S. Seal, J. Phys. Chem. C, 2012, 116, 14108–14114 CAS.
- M. Sterrer and H.-J. Freund, Catal. Lett., 2013, 143, 375–385 CrossRef CAS PubMed.
- E. Aneggi, D. Wiater, C. de Leitenurg, J. Llorca and A. Trovarelli, ACS Catal., 2014, 4, 172–181 CrossRef CAS.
- S. R. Gilliss, J. Bentley and C. B. Carter, Mater. Res. Soc. Symp. Proc., 2004, 818, 9–14 CrossRef CAS PubMed.
- L. Wu, H. J. Wiesmann, A. R. Moodenbaugh, R. F. Klie, Y. Zhu, D. O. Welch and M. Suenaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 125415 CrossRef.
- S. Turner, S. Lazar, B. Freitag, R. Egoavil, J. Verbeeck, S. Put, Y. Strauven and G. Van Tendeloo, Nanoscale, 2011, 3, 3385–3390 RSC.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, NACE, Houston, TX, 1974 Search PubMed.
- S. A. Hayes, P. Yu, T. J. O'Keefe, M. J. O'Keefe and J. O. Stoffer, J. Electrochem. Soc., 2002, 149, C623–C630 CrossRef CAS PubMed.
- P. Yu, S. A. Hayes, T. J. O'Keefe, M. J. O'Keefe and J. O. Stoffer, J. Electrochem. Soc., 2006, 153, C74–C79 CrossRef CAS PubMed.
- R. Farra, M. Eichelbaum, R. Schloegl, L. Szentmiklosi, T. Schmidt, A. P. Amrute, C. Mondelli, J. Perez-Ramirez and D. Teschner, J. Catal., 2013, 297, 119–127 CrossRef CAS PubMed.
- Y. Hedberg, J. Gustafsson, H. L. Karlsson, L. Moller and I. O. Wallinder, Part. Fibre Toxicol., 2010, 7, 23–36 CrossRef PubMed.
- U. M. Graham, M. T. Tseng, J. B. Jasinski, R. A. Yokel, J. M. Unrine, B. H. Davis, A. K. Dozier, S. S. Hardas, R. Sultana, E. A. Grulke and D. A. Butterfield, ChemPlusChem, 2014, 79(8), 1083–1088 CrossRef CAS PubMed.
- T. Pirmohamed, J. M. Dowding, S. Singh, B. Wasserman, E. Heckert, A. S. Karakoti, J. E. S. King, S. Seal and W. T. Self, Chem. Commun., 2010, 46, 2736–2738 RSC.
- Y. Zhai, K. Zhou, Y. Xue, F. Qin, L. Yang and X. Yao, RSC Adv., 2013, 3, 6833–6838 RSC.
- Q. J. Fu, A. M. Steele and S. C. Tsang, Green Chem., 2001, 3, 71–73 RSC.
- M. Green, T. Kramer, M. Parish, J. Fox, R. Lalanandham, W. Rhine, S. Barclay, P. Calvert and H. K. Bowen, Adv. Ceram., 1987, 21, 449–465 CAS.
- A. Hozumi, K. Ushiyama, H. Sugimura and O. Takai, Langmuir, 1999, 15, 7600–7604 CrossRef CAS.
- S. Mishra and J. J. Weimer, J. Adhes. Sci. Technol., 1997, 11, 337–357 CrossRef CAS PubMed.
- M. Advincula, X. Fan, J. Lemons and R. Advincula, Colloids Surf., B, 2005, 42, 29–43 CrossRef CAS PubMed.
- S. Takeda and M. Fukawa, Mater. Sci. Eng., B, 2005, 119, 265–267 CrossRef PubMed.
- A. Zebda, V. Stambouli, M. Labeau, C. Guiducci, J. P. Diard and G. B. Le, Biosens. Bioelectron., 2006, 22, 178–184 CrossRef CAS PubMed.
- K. Rezwan, L. P. Meier and L. J. Gauckler, Langmuir, 2005, 21, 3493–3497 CrossRef CAS PubMed.
- K. Rezwan, L. P. Meier and L. J. Gauckler, Langmuir, 2005, 21, 3494–3497 CrossRef PubMed.
- L. Treuel, S. Brandholt, P. Maffre, S. Wiegele, L. Shang and G. U. Nienhaus, ACS Nano, 2014, 8, 503–513 CrossRef CAS PubMed.
- A. Lesniak, A. Campbell, M. P. Monopoli, I. Lynch, A. Salvati and K. A. Dawson, Biomaterials, 2010, 31, 9511–9518 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Aberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- B. Wang, P. Wu, R. A. Yokel and E. A. Grulke, Appl. Surf. Sci., 2012, 258, 5332–5341 CrossRef CAS PubMed.
- J. S. Taurozzi, V. A. Hackley and M. R. Wiesner, Nanotoxicology, 2013, 7, 389–401 CrossRef CAS PubMed.
- C. Kong and Y.-K. Leong, J. Dispersion Sci. Technol., 2011, 32, 1235–1238 CrossRef CAS.
- M. Nabavi, O. Spalla and B. Cabane, J. Colloid Interface Sci., 1993, 160, 459–471 CrossRef CAS.
- V. Bolis, G. Cerrato, G. Magnacca and C. Morterra, Thermochim. Acta, 1998, 312, 63–77 CrossRef CAS.
- M. Mogensen, N. M. Sammes and G. A. Tompsett, Solid State Ionics, 2000, 129, 63–94 CrossRef CAS.
- D. R. Baer and M. H. Engelhard, J. Electron Spectrosc. Relat. Phenom., 2010, 178–179, 415–432 CrossRef CAS PubMed.
- D. R. Baer, D. J. Gaspar, P. Nachimuthu, S. D. Techane and D. G. Castner, Anal. Bioanal. Chem., 2010, 396, 983–1002 CrossRef CAS PubMed.
- D. M. Lyons, J. P. McGrath and M. A. Morris, J. Phys. Chem. B, 2003, 107, 4607–4617 CrossRef CAS.
- D. R. Mullins, S. H. Overbury and D. R. Huntley, Surf. Sci., 1998, 409, 307–319 CrossRef CAS.
- M. Li, S. Ge, W. Qiao, L. Zhang, Y. Zuo and S. Yan, Appl. Phys. Lett., 2009, 94, 152511 CrossRef PubMed.
- A. Sundaresan, R. Bhargavi, N. Rangarajan, U. Siddesh and C. N. R. Rao, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 161306 CrossRef.
- M. Y. Ge, H. Wang, E. Z. Liu, J. F. Liu, J. Z. Jiang, Y. K. Li, Z. A. Xu and H. Y. Li, Appl. Phys. Lett., 2008, 93, 062505 CrossRef PubMed.
- S. Agarwal, L. Lefferts, B. L. Mojet, D. A. J. M. Ligthart, E. J. M. Hensen, D. R. G. Mitchell, W. J. Erasmus, B. G. Anderson, E. J. Olivier, J. H. Neethling and A. K. Datye, ChemSusChem, 2013, 6, 1898–1906 CrossRef CAS PubMed.
- M. K. Alam, F. Ahmed, K. Nakamura, A. Suzuki, R. Sahnoun, H. Tsuboi, M. Koyama, N. Hatakeyama, A. Endou, H. Takaba, C. C. A. Del, M. Kubo and A. Miyamoto, J. Phys. Chem. C, 2009, 113, 7723–7727 CAS.
- Z. Wu, M. Li, A. K. Mann, D. R. Mullins and S. H. Overbury, 246th ACS National Meeting & Exposition, Indianapolis, IN, ENFL-289, 2013 Search PubMed.
- D. R. Mullins, P. M. Albrecht, T.-L. Chen, F. C. Calaza, M. D. Biegalski, H. M. Christen and S. H. Overbury, J. Phys. Chem. C, 2012, 116, 19419–19428 CAS.
- D. R. Mullins, P. M. Albrecht and F. Calaza, Top. Catal., 2013, 56, 1345–1362 CrossRef CAS PubMed.
- A. S. Karakoti, S. V. N. T. Kuchibhatla, K. S. Babu and S. Seal, J. Phys. Chem. C, 2007, 111, 17232–17240 CAS.
- T. Hiemstra, J. C. M. De Wit and W. H. Van Riemsdijk, J. Colloid Interface Sci., 1989, 133, 105–117 CrossRef CAS.
- T. Hiemstra, W. H. Van Riemsdijk and G. H. Bolt, J. Colloid Interface Sci., 1989, 133, 91–104 CrossRef CAS.
- O. Spalla and P. Kekicheff, J. Colloid Interface Sci., 1997, 192, 43–65 CrossRef CAS.
- M. A. Henderson, C. L. Perkins, M. H. Engelhard, S. Thevuthasan and C. H. F. Peden, Surf. Sci., 2003, 526, 1–18 CrossRef CAS.
- S. Kumar and P. K. Schelling, J. Chem. Phys., 2006, 125, 204704 CrossRef PubMed.
- Y. Lykhach, V. Johanek, H. A. Aleksandrov, S. M. Kozlov, M. Happel, T. Skala, P. S. Petkov, N. Tsud, G. N. Vayssilov, K. C. Prince, K. M. Neyman, V. Matolin and J. Libuda, J. Phys. Chem. C, 2012, 116, 12103–12113 CAS.
- A. Rajendran, Y. Takahashi, M. Koyama, M. Kubo and A. Miyamoto, Appl. Surf. Sci., 2005, 244, 34–38 CrossRef CAS PubMed.
- T. X. T. Sayle, M. Molinari, S. Das, U. M. Bhatta, G. Moebus, S. C. Parker, S. Seal and D. C. Sayle, Nanoscale, 2013, 5, 6063–6073 RSC.
- S. Vyas, R. W. Grimes, D. H. Gay and A. L. Rohl, J. Chem. Soc., Faraday Trans., 1998, 94, 427–434 RSC.
- X. Huang, B. Wang, E. A. Grulke and M. J. Beck, J. Chem. Phys., 2014, 140, 074703 CrossRef PubMed.
- M. Nolan, J. Phys. Chem. C, 2009, 113, 2425–2432 CAS.
- C. T. Campbell and C. H. F. Peden, Science, 2005, 309, 713–714 CrossRef CAS PubMed.
- J. Paier, C. Penschke and J. Sauer, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS PubMed.
- M. Fronzi, A. Soon, B. Delley, E. Traversa and C. Stampfl, J. Chem. Phys., 2009, 131, 104701–104715 CrossRef PubMed.
- M. Fronzi, S. Piccinin, B. Delley, E. Traversa and C. Stampfl, Phys. Chem. Chem. Phys., 2009, 11, 9188–9199 RSC.
- X. Huang and M. J. Beck, Comput. Mater. Sci., 2014, 91, 122–133 CrossRef CAS PubMed.
- E. G. Heckert, A. S. Karakoti, S. Seal and W. T. Self, Biomaterials, 2008, 29, 2705–2709 CrossRef CAS PubMed.
- M. Ornatska, E. Sharpe, D. Andreescu and S. Andreescu, Anal. Chem., 2011, 83, 4273–4280 CrossRef CAS PubMed.
- M. Das, S. Patil, N. NBhargava, J.-F. Kang, L. M. Riedel, S. Seal and J. J. Hickman, Biomaterials, 2007, 28, 1918–1925 CrossRef CAS PubMed.
- E. Sharpe, T. Frasco, D. Andreescu and S. Andreescu, Analyst, 2013, 138, 249–262 RSC.
- A. Hayat, D. Andreescu, G. Bulbul and S. Andreescu, J. Colloid Interface Sci., 2014, 418, 240–245 CrossRef CAS PubMed.
- R. E. Oezel, A. Hayat, K. N. Wallace and S. Andreescu, RSC Adv., 2013, 3, 15298–15309 RSC.
- K. Reed, A. Cormack, A. Kulkarni, M. Mayton, D. Sayle, F. Klaessig and B. Stadler, Exploring the properties and applications of nanoceria: is there still plenty of room at the bottom?, Environ. Sci.: Nano, 2014, 1 10.1039/C4EN00079J.
- A. Kumar, S. Das, P. Munusamy, W. Self, D. R. Baer, D. C. Sayle and S. Seal, Behavior of nanoceria in biologically-relevant environments, Environ. Sci.: Nano, 2014, 1 10.1039/C4EN00052H , in revision.
- R. A. Yokel, S. Hussain, S. Garantziotis, P. Demokritou, V. Castranova and F. R. Cassee, The Yin: an adverse health perspective of nanoceria: uptake, distribution, accumulation, and mechanisms of its toxicity, Environ. Sci.: Nano, 2014, 1 10.1039/C4EN00039K.
| This journal is © The Royal Society of Chemistry 2014 |