
Capabilities of asymmetric flow field-flow fractionation coupled to multi-angle light scattering to detect carbon nanotubes in soot and soil†
Alexander
Gogos
ac,
Ralf
Kaegi
b,
Renato
Zenobi
c and
Thomas D.
Bucheli
*a
aAgroscope, Institute for Sustainability Sciences ISS, 8046 Zurich, Switzerland. E-mail: thomas.bucheli@agroscope.admin.ch; Fax: +41 58 468 72 01; Tel: +41 58 468 73 42
bEawag, Swiss Federal Institute of Aquatic Science and Technology, 8600 Duebendorf, Switzerland
cDepartment of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland
First published on 16th September 2014
Abstract
Analytical detection and quantification of multi-walled carbon nanotubes (MWCNTs) in complex matrices such as soils is very challenging. In an initial approach to this task, we identify MWCNTs by making use of their different (e.g., rod-like) shape compared to other (native) soil particles and in particular soot, which is ubiquitously present in soils. A shape factor ρ, determined using asymmetric flow field-flow fractionation coupled to multi-angle light scattering (aF4-MALS), was used to discriminate MWCNTs of different aspect ratios, as well as mixtures of soot and MWCNTs, in pure suspensions. MALS results were additionally confirmed using automated electron microscopy image analysis. We then analyzed different soil types which consistently showed ρ-values that differed from pure MWCNTs. To test the performance of the method for MWCNT detection in such complex matrices, we conducted standard additions of a MWCNT as well as soot to an agricultural soil. Extracts from these MWCNT-spiked soils showed increased ρ-values compared to soot-spiked or native soil. The method detection limit for the MWCNT was 1.6 to 4.0 mg g−1 soil and lies within the range of commonly used black carbon quantification methods, but is much higher than any currently predicted environmental concentration. Additionally, the method is currently limited by a relatively narrow dynamic range of ρ. Despite these limitations, our results suggest that aF4-MALS provides specific shape information that may be linked to an actual MWCNT presence in soils. Further method improvement potential is outlined along different steps of the workflow.
Nano impactWe developed and evaluated a novel approach based on aF4-MALS to differentiate high aspect ratio particles (MWCNTs) from differently shaped background. This approach may in the future help to identify these emerging NPs in complex matrices such as soils, if current detection limits and the dynamic range can be further improved. |
Introduction
Since Iijima first described tube-like structures of sp2-hybridized carbon1 – now known as carbon nanotubes (CNTs) – this new material type has undergone an enormous development, which is reflected by a production volume that already exceeds several thousand tons per year.2 CNTs are increasingly used in consumer products,2,3 that include, e.g., lightweight composites in cars and aircrafts, antifouling paints, batteries and water filters. As the prices of multi-walled carbon nanotubes (MWCNTs) are still orders of magnitude lower than for the single-wall types (SWCNTs),2 they prevail in terms of production volumes. During their life cycle, these materials could potentially be released into the environment via different routes.4–6 They might even be intentionally applied in agriculture, as several beneficial effects of CNTs on plants were postulated, such as enhanced water uptake, seed germination, and cell growth.7,8 Such direct applications would induce much higher fluxes into soils than predicted to date.9 In contrast, some studies suggest that CNTs negatively affect soil microbial communities as well as plants,10–12 after being released into the environment. This highlights the need to efficiently assess the environmental distribution of CNTs.However, detection and quantification methods for CNTs in soils and sediments are still not well established. One of the main challenges in CNT quantification in soils and sediments is the ubiquitous presence of black carbon (BC) particles. These originate from wildfires or incomplete combustion of fossil fuels,13 and have chemical and physical properties similar to CNTs. First attempts to quantify CNTs in soils were made by Sobek and Bucheli in 2009,14 using standard additions and a chemothermal oxidation method (CTO-375). Unfortunately, this method alone is not able to differentiate between soot-BC and CNT-BC. Alternative methods, such as thermal/optical transmittance/reflectance15 or thermogravimetry (coupled to mass spectrometry16), make use of the lower thermal stability of some SWCNT compared to soot. However, the thermal stability may vary depending on the length and the thickness of the CNTs, and even be comparable to soot,14 resulting in potential co-isolation. Also, quantification of CNTs may be influenced by matrix effects in more complex samples, such as urban dust and sediments.15,16 Further promising techniques include extraction of CNTs with suitable surfactants and near-infrared fluorescence (NIRF),17 or microwave-induced heating.18 However, the former is only applicable to SWCNT, and the latter lacks specificity. Given the limitations of these different methods, it is evident that quantification of CNTs in real-world samples will probably require applying a suite of complementary methods in one single workflow.
A parameter that is very special for CNTs is shape: CNTs are rod-like to randomly coiled structures with high aspect ratios, whereas soot particles consist of spherical primary particles19 aggregated to fractal-like structures.20 A technique that has already been employed for shape description of environmental colloids21 is asymmetric flow field-flow fractionation (aF4) coupled to multi-angle light scattering (MALS). Asymmetric flow field-flow fractionation in combination with UV-vis or electron microscopy (EM) has previously been used to separate CNTs in suspension,22–24 and coupled with MALS for measuring their length and dispersion state.25,26 Thus, aF4-MALS may be able to detect changes in the overall shape of a natural sample once CNTs are present.
Here, we describe the development of an aF4-MALS method to differentiate between a MWCNT and soot in synthetic mixtures of these materials. We used automated EM in combination with image analysis tools to confirm the aF4-MALS results, and tested the robustness and performance of aF4-MALS to identify a MWCNT in natural soil samples spiked with this analyte. The potential and limitations of this approach are discussed in terms of methodological figures of merit, and its applicability to actual environmental samples.
Theory
The principles and theory of aF4 and MALS have been presented in detail elsewhere.27,28 Here we will briefly describe the main concepts of MALS, because our data is mainly based on this technique. All MALS measurements are commonly evaluated using eqn (1): | (1) |
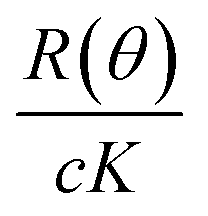 vs. sin2(θ/2) at zero angle. The rg can be interpreted as the weighted average of all possible radii of a particle from its center of mass. Therefore, rg is susceptible to changes in particle shape.
vs. sin2(θ/2) at zero angle. The rg can be interpreted as the weighted average of all possible radii of a particle from its center of mass. Therefore, rg is susceptible to changes in particle shape.
The hydrodynamic radius (rh) is approximated for non-spherical particles as the radius of a sphere with the same diffusion behavior. It can be obtained either from aF4 retention time calibrated with latex spheres with a known size or by online dynamic light scattering (DLS) measurements. The ratio of these two radii gives a shape factor
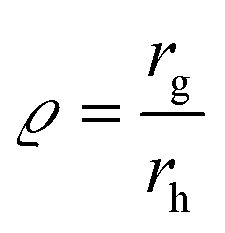 | (2) |
Materials and methods
Chemicals and analytes
Ammonium nitrate (NH4NO3, ≥99%), sodium azide (NaN3, ≥99.5%) and sodium deoxycholate (SDC, ≥98%) were purchased from Sigma-Aldrich (Buchs, Switzerland). Nanosphere™ size standards were purchased from Thermo Scientific (Fremont, CA). Carrier solutions for aF4 were prepared in Milli-Q water (Millipore, Zug, Switzerland). Before use, all carrier solutions were filtered through 0.1 μm Durapore filters (Millipore). Long pristine (MW1), long carboxylized (MW2), short pristine (MW3) and short carboxylized (MW4) MWCNTs were purchased from Cheap Tubes Inc. (Brattleboro, VT). All MWCNTs were declared to be of 90–95% purity and to have an elemental carbon content of >96%. The MWCNTs were used as received without further purification. However, characterization of the very same batch of MWCNTs has been carried out over several years in our group,14,30,31 and detailed data are provided in the ESI† (Table S1). Here, we additionally imaged the suspensions of the MWCNTs using scanning electron microscopy (SEM) (see Fig. S1†). Forklift Diesel soot (Standard Reference Material (SRM) 2975, National Institute of Standards and Technology (NIST), Gaithersburg, MD) with spherical primary particles in the range of 35 nm (ref. 19) served as a BC standard. For reasons of simplicity, we here refer to this material as “soot”.Soils and sediments
A series of well-characterized soils and sediments served as natural samples for our method, i.e., a loamy sand agricultural soil from Germany (LUFA Standard soil 2.2., Landwirtschaftliche Untersuchungs- und Forschungsanstalt, Speyer, Germany), a clay (vertisol) soil from Australia, several soils from the Swiss Soil Monitoring Network (NABO) and a marine sediment from Baltimore Harbor, USA (NIST SRM 1941b). The clay soil as well as the sediment were part of an earlier BC inter-laboratory ring trial, and thus well characterized.32 The NABO soils had been analyzed for BC content by Agarwal and Bucheli.33 For detailed parameters and description of the used soils see Table S2.†Analyte suspensions and sample extracts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500g for 10 min (Sorvall Superspeed, DuPont Instruments, Newton, CT) to remove agglomerates. The overlaying half of the volume then served as a working suspension. This methodology has been shown before to yield very stable and well dispersed suspensions.17 Suspensions were free of agglomerates (see Fig. S1† for SEM images of the suspensions) and very stable, with ζ-potentials around −50 mV.
500g for 10 min (Sorvall Superspeed, DuPont Instruments, Newton, CT) to remove agglomerates. The overlaying half of the volume then served as a working suspension. This methodology has been shown before to yield very stable and well dispersed suspensions.17 Suspensions were free of agglomerates (see Fig. S1† for SEM images of the suspensions) and very stable, with ζ-potentials around −50 mV.
Residual MWCNT and soot concentrations in the supernatants were determined by UV-vis (at λ = 289 nm), calibrated with different concentrations (1–15 μg mL−1) of the analytes in non-centrifuged dispersions. Appropriate volumes of MW1 and soot suspensions were then mixed to yield mass ratios of 0.8:1, 1:1, 1.3:1, 2:1 and 4:1 (corresponding to MW1 mass fractions of 44.5, 50.0, 56.5, 66.7 and 80%).
Separation (aF4) and online detection (UV-vis, MALS)
For aF4, we used a commercial apparatus (AF2000, Postnova Analytics GmbH, Landsberg, Germany). This instrument was connected online to a UV-vis diode array detector (PN3241, Postnova Analytics), a 21-angle MALS detector (PN3621, Postnova Analytics), and a Malvern Zetasizer (Infors, Bottmingen, Switzerland). The aF4 channel was trapezoidal-shaped, 350 μm thick (defined by a spacer), 29.8 cm long (inlet to outlet), and had a 2 cm maximum width. All runs were done with a 10−5 M NH4NO3/0.02% NaN3 mobile phase (pH 6.5), which has previously been shown to be suitable for CNT fractionation in aF4.26 Particle recoveries over aF4 were tested with several commonly used membranes (see ESI† Table S3), and were highly variable for both CNTs and soot. Since the objective of this paper is a differentiation of particles based on shape, we selected regenerated cellulose membranes (RC; 10 kDa). This membrane type was the only one that provided equal recoveries (~50%) for both types of particles. Operating conditions of the aF4 were optimized to separate both size standards, as well as the samples with the same method. Membranes were changed at the latest after 20 to 25 injections. All injections were performed in triplicate with an autosampler (PN5300, Postnova Analytics). Further fractionation conditions were as follows: injection volume adjusted to 5 μg absolute analyte mass (280–410 μL, pure suspensions) or 500 μL (natural samples and standard addition experiments), detector flow 0.5 ml min−1, cross flow 1 ml min−1 with a power gradient of 0.2, injection flow 0.2 ml min−1, injection time 10 min and fractionation time 40 min. At the end of the fractionation process, a 7 min rinse step without cross-flow but full detector flow was applied, to minimize the risk of cross-contamination between injections.Fraction collection, automated electron microscopy and image analysis
Suspensions of pure MW1, soot, and mixtures thereof were fractionated after aF4 using a fraction collector (PN8050, Postnova Analytics). For their analysis by SEM, 375 μL of the collected fractions were filled into a conical Eppendorf tube with a carbon-coated TEM grid on a plastic stopper. This setup resulted in a water column of 5 mm height above the TEM grid. The particles were deposited on the TEM grids by centrifugation (1 h at 16000g) using a swinging bucket rotor. Images were recorded on a SEM (Nova Nano-SEM 230, FEI, USA) with a transmission electron detector. The bright field signal was used for image formation. In total, 40 (soot), 30 (MW1) and 20 (mixture) images were automatically recorded at a fixed magnification (20000) using INCA suite 4.15 (Oxford Instruments, Oxford, UK). All images were automatically processed in Fiji (background removal and thresholding) to obtain binary images and then skeletonized using the ‘Skeletonize 2D/3D’ and ‘Analyze Skeleton 2D/3D’ plugins.34–36 The “longest–shortest” branch was then used as the skeleton length. Distributions of this skeleton length from the pure materials then served to calculate percentages of MW1 and soot in mixed suspensions by applying the following linear model:37| Xmix = α·XMWCNT + β·Xsoot + ε | (3) |
MALS data analysis
Data analysis was always performed on the last of three replicate injections (if not stated otherwise), to ensure data acquisition at stable run conditions. MALS data were analyzed with NovaMALS 1.0.0.9. (Postnova Analytics). Since the actual geometry of the particles (MWCNT and soot) was unknown, a 5th grade polynomial Debye fit for P(θ) that implies an arbitrary shape21 was used for rg calculations (for examples, see Fig. S2†). The rh was determined by calibrating the retention time (UV-vis) with two different mixtures of Nanosphere™ size standards (10, 50, 100, 175 nm and 30, 75, 150, 200 nm, Fig. S3†). The resulting calibrated values for rh were in good agreement with values from online DLS measurements (7–19% deviation). According to eqn (2), ρ was then calculated over the aF4 retention time from rg and rh values, and represented in a fractogram. For further evaluation, a shape factor difference | (4) |
Quality control, method validation and statistics
A correct normalization of the light scattering detector was assured by repeated measurements of size standards and subsequent verification of obtained rg values. Blank measurements were run regularly to assure artefact free analyses. Additionally, alternate injections of MW1 and soot on the same membrane were performed to ensure that no carry-over effects occurred.The recovery from aF4 runs was determined as follows:
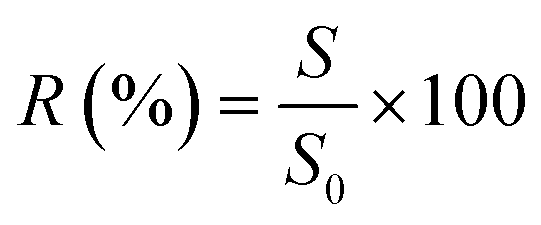 | (5) |
To determine the instrumental limit of detection (LOD), we measured ρ distributions of decreasing MW1 amounts (from 5 down to 0.05 μg, constant injection volume) in pure suspensions. Here, we define the LOD to be the lowest injected MW1 mass, until which the distribution of ρ remains stable.
Method detection limits were calculated for the matrices soot and soil. Three different cases were evaluated (for the description of the samples see sections before): (i) mixtures of MW1 and soot (MDLsoot), (ii) standard additions of MW1 to soil extracts (nominal MDLsoil) and (iii) standard additions of MW1 to soil (effective MDLsoil). For the first one, a t-test was applied on the ρ-values obtained over 50% of the MALS 92° peak width to compare each mixture (one replicate, n = 1) with pure soot. For the second and third, the area under each ρ-distribution (three independent measurements, n = 3) was calculated as “area under curve”, evaluated using one-way analysis of variance (ANOVA) and finally compared by pairwise comparison with a Bonferroni test. To compare these results with a more conventional way of determining MDL, we used the well-established concept by Keith et al.,38 according to which the MDL is defined as three standard deviations of a blank sample (3σ), the detection criterion being (St − Sb) > 3σ (with St representing the average signal for the sample and Sb the average signal for the blank). All statistical analyses were done with the software R (version 3.01, the R Foundation for Statistical Computing) integrated in RStudio (version 0.97.551, RStudio, Boston, MA).
Results and discussion
General features of aF4-MALS fractograms and consequences for their interpretation
During all measurements, rg values decreased with increasing time in the beginning of the aF4 fractograms (Fig. S2†). This was caused by void/steric elution of particles (≤160 nm rg) that were not retained after focusing. After this void/steric elution, particles always showed increasing rg values with increasing time (normal mode) throughout the remaining elution. Because calibrated rh values referred to normal mode elution (see also Fig. S3†), meaningful ρ-values could only be obtained beyond this inversion point. This also explains the high ρ-values in the beginning of the fractograms (Fig. 1) which were therefore disregarded. Toward the end of the peak (e.g., after 40 to 45 min, Fig. S2†), rg values sometimes showed a slower increase compared to rh, which led to a steep decrease of ρ, depending on the particle or respective mixture. This was due to a more pronounced curvature of at larger particle sizes that could not be fitted correctly anymore using the Debye algorithm. Consequently, for further data evaluation, we chose 50% of the MALS 92° peak width (full width at half maximum), where both effects were negligible.
at larger particle sizes that could not be fitted correctly anymore using the Debye algorithm. Consequently, for further data evaluation, we chose 50% of the MALS 92° peak width (full width at half maximum), where both effects were negligible.
 | ||
Fig. 1 (A) Fractograms obtained by aF4-MALS with shape factor ρ (filled circles) for equal mass injections (5 μg) of different types of pure MWCNT suspensions: MW1 (pristine, medium aspect ratio (1500),  ), MW2 (–COOH, high aspect ratio (3750), ), MW2 (–COOH, high aspect ratio (3750),  ), MW3 (pristine, low aspect ratio (200), ), MW3 (pristine, low aspect ratio (200),  ) and MW4 (–COOH, low aspect ratio (200), ) and MW4 (–COOH, low aspect ratio (200),  ). Vertical lines indicate the transition points between void/steric and normal mode elution and solid lines show the 92° MALS signal for each type of MWCNT in the respective color. The shaded area corresponds to particle sizes too large for appropriate fitting using the Debye algorithm. (B) Average shape factor ρ obtained from 50% of the MALS 92° peak width vs. nominal aspect ratio of the different MWCNTs. Bars represent the standard deviation of the ρ-values over the selected retention time window. ). Vertical lines indicate the transition points between void/steric and normal mode elution and solid lines show the 92° MALS signal for each type of MWCNT in the respective color. The shaded area corresponds to particle sizes too large for appropriate fitting using the Debye algorithm. (B) Average shape factor ρ obtained from 50% of the MALS 92° peak width vs. nominal aspect ratio of the different MWCNTs. Bars represent the standard deviation of the ρ-values over the selected retention time window. | ||
An injected mass of 0.5 μg pure MW1 was the lowest amount until which the ρ distribution remained stable (Fig. S4†), and was therefore defined as LOD. While the MALS 92° signal-to-noise ratio (S/N) was still above 10 (Fig. S4†), we generally preferred ρ over the more conventional S/N ratio at a given (shape dependent) MALS angle as a measure of detection limit, because it provides the more integral analyte signal information. Moreover, we chose injection amounts >2.5 μg as a starting point for all subsequent experiments to measure in a range well above the LOD.
aF4-MALS analysis of MWCNTs and MW1-soot mixtures
To assess the relation between ρ and nominal aspect ratios, we measured various MWCNTs that differed in this parameter (Fig. 1). Shape factors for these MWCNTs were found to correlate with their nominal aspect ratios (R2 = 0.877, y = 6 × 10−5x + 1.052, p = 0.058, Fig. 1B). The dynamic range, however, was less pronounced than expected. This may be due to selective losses of longer MWCNT to the aF4 membrane (as described in detail in the following section).Consequently, recoveries for MWCNTs (and soot) in aF4 were highly variable, depending also on the nature of the membrane material (see Table S3†), as well as on carrier composition26 and cross-flow selection.39 After preconditioning a 10 kDa RC membrane with two injections, we obtained stable recoveries of 50 ± 9% for soot and 50 ± 5% for the pristine long MWCNT (MW1, also see Tables S1 and S3†) determined by aF4-UV/Vis analysis. These recoveries were favorable for our type of analysis, as they were similar for both kinds of particles, avoiding a bias on the shape factors from the start. These values were, however, significantly lower compared to those reported by Gigault et al.26 for short carboxylized MWCNTs (94 ± 2%) under similar separation conditions. We measured such shorter, functionalized MWCNTs as well (see Fig. 1), and achieved recoveries of around 75% in the first injections, which then stabilized between 41 and 49%, possibly indicating interactions between mobile phase-dispersed and membrane-bound MWCNTs.
Fig. 2A shows the ρ-value fractograms for soot, MW1, and mixtures of both. Soot exhibited rather uniform ρ-values in the retention time window between 23 and 37 min. With an increasing fraction of MW1 in the mixtures, the elution profile of the ρ-values developed an increasing maximum at about 31 min, similar to pure MW1. The increase of this maximum was concentration dependent (Fig. 2B). In addition, the increase in ρ was different at different time points (Fig. 2A). Such an increase in ρ points to a separation by length, which has been described before using EM.22,23 However, fractions of MW1 in the MW1/soot mixtures remained rather constant throughout the fractogram (see Fig. S5†). Thus, it was not possible to physically separate MW1 and soot using aF4 under the selected conditions.
 | ||
Fig. 2 (A) Fractograms obtained by aF4-MALS with shape factor ρ (symbols) for equal mass injections (5 μg) of a pristine long MWCNT (MW1,  ), soot (●) and different mixtures (w/w) of both: 44.5% ( ), soot (●) and different mixtures (w/w) of both: 44.5% ( ), 50.0% ( ), 50.0% ( ), 56.5% ( ), 56.5% ( ), 66.7% ( ), 66.7% ( ), and 80.0% ( ), and 80.0% ( ) MW1 in soot. The vertical line indicates the average transition point between void/steric and normal mode elution and solid lines show the 92° MALS signal of pure soot (black) and pure MW1 (red). The shaded area corresponds to particle sizes too large for appropriate fitting using the Debye algorithm. (B) Average shape factor difference Δρ obtained from 50% of the MALS 92° peak width vs. MWCNT content in the mixtures. Bars represent the standard deviation of the Δρ-values over the selected retention time window. ) MW1 in soot. The vertical line indicates the average transition point between void/steric and normal mode elution and solid lines show the 92° MALS signal of pure soot (black) and pure MW1 (red). The shaded area corresponds to particle sizes too large for appropriate fitting using the Debye algorithm. (B) Average shape factor difference Δρ obtained from 50% of the MALS 92° peak width vs. MWCNT content in the mixtures. Bars represent the standard deviation of the Δρ-values over the selected retention time window. | ||
The increase in the MW1 fraction in the mixtures correlated well with the average Δρ of each concentration (R2 = 0.997, y = 0.0041 x − 0.146, p = 0.0025, Fig. 2B). Statistical analysis (t-test, p < 0.001) resulted in a MDLsoot of 44.4% (w/w, corresponding to 2.2 μg injected MW1 mass). Conversely, a soot content of 20% was significantly (t-test, p < 0.001) discriminated from MW1. Amorphous carbon/soot contents in commercial CNT products can range up to approx. 50%,40,41 and would thus influence the shape properties of the overall product. Thus, the method presented here may contribute to identifying the soot content of CNT powders after production.
Orthogonal confirmation of aF4-MALS results using automated EM in combination with image analysis
Fractions of MW1, soot and a mixture of both (1:1 w/w), collected from aF4 analyses similar to those displayed in Fig. 2, were imaged and analyzed using automated EM in combination with image analysis tools (Fig. 3). The skeleton length distribution of MW1 and soot was different and allowed a quantification of their fractions in a mixed sample after aF4. This was accomplished by a linear combination fit using the skeleton length distributions of pure MW1 and soot as references. Results indicated that the mixture consisted of 34% MW1 and 58% soot. The linear model explained 92% of the observed skeleton length distribution of the mixture, while 8% remained unexplained. The fraction of MW1 derived from the EM analysis was somewhat lower than the corresponding fraction determined from aF4-MALS generated ρ-values averaged over the retention time collected for EM analysis (52% MW1, 48% soot). Deviations between the two methods may be explained by artifacts related to the sample preparation protocol for EM analysis, resulting in different image qualities that affected the results from the image analysis procedure to various degrees. Common EM artifacts, such as particle recognition, overlapping particles or contaminations on the EM grid may affect the length distributions of pure MWCNT and soot source profiles and thus the particle quantification. The observed contamination effects were mostly related to carbonaceous materials condensing at the location of the electron beam which resulted in a darkening and loss of contrast of the images. An additional cleanup, such as a thermal treatment under vacuum, may reduce these contamination effects and thus help to increase image qualities and the figures of merit of the EM technique. If number based losses in the aF4 can additionally be quantified, the combination of aF4 – as a technique able to gently reduce sample complexity – with EM analysis holds the potential to obtain number based concentrations of MWCNTs and soot from complex samples.
 | ||
| Fig. 3 EM image analysis workflow (from left to right: original, thresholded and skeletonized images with longest–shortest branch through the particle (red) and side branches (grey)) and skeleton length distributions obtained from skeletonized images of MW1, soot and a 1:1 (w/w) mixture of both. Images were taken and analyzed from aF4 fractions collected during 28–32 min retention time. The distributions of MW1 and soot (solid/dashed, red: MW1; black: soot) are displayed together with the 1:1 (w/w) mixture (solid, yellow). | ||
Furthermore, EM analysis of aF4 fractions revealed additional information on the behavior of MW1 in the aF4 channel. Nanotube-shaped particles were modeled to elute in normal mode up to 500 nm rod length (1 nm diameter).42 Nominal lengths of MW1, however, were much larger (see Table S1†). Image analysis of EM images indicated that eluting MW1 ranged from 0.1 to 1 μm in length, with a maximum number at around 300 nm (Fig. 3). These findings support the hypothesis that longer MWCNTs initially present (see Fig. S6†) were lost, most likely on the aF4 membrane, and that skeleton lengths of up to 1 μm eluted in normal mode. As discussed in greater detail by Phelan and Bauer,42 the loss of longer CNTs may be explained by decreased rotation with increasing length and alignment of the particles at the accumulation wall.
aF4-MALS analysis of native soils
In a next step, we measured ρ-values of a diverse range of native soil and sediment samples to assess the range of possible background situations (Fig. 4, for raw data, see Fig. S7†). Recoveries of their extracts over aF4 ranged between 43% (NIST SRM 1941b) and 66% (BC Vertisol), determined using the MALS 92° peak areas (n = 3). Values for ρ of all analyzed soils/sediments were different from pure MWCNTs (Fig 1B) and varied between approx. 0.65 (NABO 89) and 1.0 (BC Vertisol) (Fig. 4). They should thus provide enough contrast for detection of spiked MW1, with a Δρmin of ~0.26 (Lufa 2.2) and a Δρmax of ~0.61 (NABO 89). Colloids extracted from soil have been measured by von der Kammer et al.,21 who obtained higher ρ-values (ρaverage = 1.12) compared to extracts of the native soils in this study. This difference may stem from different sample preparation (e.g., lower centrifugation forces and different extraction conditions used by von der Kammer et al.21) as well as chemical/physical differences between the analyzed soils. For example, organic soils such as NABO 67 and 89 showed lower ρ-values than some of the clay soils (NABO 1 and 46) or sandy soils such as the Lufa 2.2. | ||
Fig. 4 Compilation of average shape factor ρ-values obtained from 50% of the MALS 92° peak width of suspensions of (i) pure MW1 and soot (■), (ii) native soils ( ): Lufa 2.2 (agricultural soil), BC Vertisol (Clay vertisol from Hammes et al.32 ring trial), NABO 46 (agricultural soil), NABO 1 (grassland soil), NABO 89 (turf) and NABO 67 (organic soil from a vegetable garden) and (iii) a marine sediment ( ): Lufa 2.2 (agricultural soil), BC Vertisol (Clay vertisol from Hammes et al.32 ring trial), NABO 46 (agricultural soil), NABO 1 (grassland soil), NABO 89 (turf) and NABO 67 (organic soil from a vegetable garden) and (iii) a marine sediment ( ): NIST SRM 1941b. Bars represent the standard deviation of the ρ-values over the selected retention time window. ): NIST SRM 1941b. Bars represent the standard deviation of the ρ-values over the selected retention time window. | ||
aF4-MALS analysis of MW1 and soot spiked to soil extracts
While differences in ρ between pure MW1 and soil reached values up to Δρ ~ 0.61 (Fig. 4), we tested whether such differences also apply to mixtures of both. To do so, we selected the Lufa 2.2 agricultural soil for standard addition experiments. This soil had several advantages; (i) it is a possible CNT recipient matrix due to its agricultural origin, (ii) it is well characterized and commercially available to other researchers in its function as a reference soil and (iii) it provides a “worst case” scenario in terms of ρ, as it showed one of the highest values among the soils we studied, similar to pure soot (Fig. 4).Asymmetric flow field-flow fractionation served to eliminate some of the matrix constituents that would otherwise disturb MALS detection and ρ determination. Compared to pure MW1 suspensions (Fig. 2A), higher ratios of the void peak relative to the analytically accessible part of the MALS 92° peak were observed (see Fig. S8†). Thus, some soil constituents that were not retained after focusing were thereby separated from the peak of interest.
Spiking of MW1 to soil extracts (Fig. 5A, for raw data see Fig. S8†) led to a concentration dependent increase in Δρ up to an apparent plateau above 25 μg mL−1 (corresponding to 12.5 μg injected mass). The highest concentration showed ρ-values comparable to pure MW1 (up to approx. 1.2; see Fig. 2A, 4 and S8†). Obviously, the soil extract did not influence the aF4-MALS analysis negatively and provided a contrasting background for the MWCNTs, as all Δρ-values were above the soil baseline (Δρ = 0). Conversely, spiking of soot yielded negative Δρ-values (Fig. 5B). Absolute ρ-values were comparable to pure soot (approximately 0.9; see comparatively Fig. 2A and S6†). Also, ρ-values for soot were again more uniform compared to MWCNTs over the main peak region (Fig. S7†). This behavior could already be observed in pure suspensions (Fig. 2A). Interestingly, the lowest soot concentration showed the highest difference in ρ to the blank soil, while the highest soot concentration was identical to it. Currently, we cannot provide a satisfying explanation for this observation.
 | ||
| Fig. 5 Average shape factor differences Δρ obtained from 50% of the MALS 92° peak width in relation to spiked concentrations and injected masses of (A) MW1 and (B) soot, both spiked to soil extracts of the Lufa 2.2 soil that is represented by the dashed red line (Δρ = 0). Bars indicate the standard deviation of the ρ-values over the selected retention time window. | ||
aF4-MALS analysis of MW1 and soot spiked to soils
When MW1 was added directly to soil and then extracted (Fig. 6A, for raw data see Fig. S9†), Δρ increased with increasing MW1 concentration, similar to Fig. 5A, but with a lower standard deviation. Shape factor differences also plateaued between 8.4 and 16.4 mg g−1. The highest MW1 concentrations again showed ρ-values comparable to pure MW1 (see comparatively Fig. 2A and S9†). No concentration-dependent increase in Δρ was observed upon addition of soot (Fig. 6B), and Δρ-values for the different concentrations were consistently below the soil baseline (Δρ = 0). This is surprising, as from Fig. 4 and 5B, it could have been expected that Δρ for soot mixed to the Lufa 2.2 soil should be closer to, or even overlapping, the baseline (Δρ = 0). Although this is again not easily explained, the different Δρ behavior of MW1 and soot in soil nicely shows the different reaction of the measurement system to the presence of differently shaped particles. | ||
| Fig. 6 Average shape factor differences Δρ obtained from 50% of the MALS 92° peak width in relation to spiked concentrations of (A) MW1 and (B) soot, both spiked to the Lufa 2.2 soil that is represented by the dashed red line (Δρ = 0). Bars indicate the standard deviation of the ρ-values over the selected retention time window. | ||
Quality control and method validation
Sodium deoxycholate was employed far above the critical micelle concentration (2.4 mM (ref. 43) or 0.09%, respectively) to efficiently extract MWCNTs. In this concentration it did not influence the measurements (see blank analyses in Fig. S10B†). Alternating injections of pure MW1 and soot suspensions showed that no detectable carry-over effects occurred between injections in terms of ρ-development (Fig. S10A†). Between these injections, ρ-values showed relative standard deviations (RSDs) of approx. 7% (soot) to 8% (MW1). Reproducibility of aF4-MALS analyses of MW1 spiked to soil extracts and soils was highest in the peak center and end; approx. 2–10% RSD in case of soil extracts (Fig. S11A†) and 1–6% in case of soils (Fig. S11B†). The higher reproducibility of the latter may be attributed to the centrifugation step, which removes agglomerates.When applied to the soil, the dynamic range of the measured signal ρ spread between 0.7 and 1.2. This range is rather narrow and limits the approach in general. It could potentially be extended if losses of longer MWCNTs during aF4 separation are reduced. For example, Gigault et al. achieved recoveries of 89% for SWCNTs and measured ρ-values of up to 3.6, corresponding to SWCNT lengths up to 2 μm.39 Also, depending on the type of the CNT, ρ may be higher (Fig. 1B, MW2).
The nominal MDLsoil obtained from the standard additions to soil extracts was 5 μg mL−1 (corresponding to 2.5 μg injected mass or 1.6 mg g−1 of soil, respectively) based on statistical analysis. This MDL was comparable to the MDLsoot in terms of injected mass, which nicely illustrates the selectivity of the method in these different matrices. Applying the approach by Keith et al.,38 we calculated the nominal MDLsoil according to the criterion (St − Sb) > 3σ, with St = 0.956 (average ρ-value of three replicate measurements of the lowest concentration within 50% of the MALS 92° peak width), Sb = 0.868 (average ρ-value of the blank) and 3σ = 0.051 (three times the standard deviation of the blank). Thus, the lowest MWCNT concentration fulfilled the criterion with 0.088 > 0.051 and both methods resulted in the same nominal MDLsoil. This also held true for the effective MDLsoil, that was higher (4.0 mg g−1) though. This increase can be explained by limited analyte extraction efficiencies of 62 ± 10% (n = 4), which were somewhat lower than those of Schierz et al.17 (approx. 75%). Differences may be explained by differences in sonication (bath vs. horn) and extraction procedure (single vs. sequential).
The nominal MDLsoil is in the range of traditional MWCNT/BC-determination methods such as CTO-375 (0.23 mg g−1)33 or more recently developed methods such as TGA-MS (0.1 mg g−1).16 However all of these are still far above any currently predicted environmental concentration (e.g., 0.01–0.1 μg g−1 for sediments and 0.01 μg g−1 in soils treated with biosolids44).
Further optimization of aF4-MALS
Several improvements of the analytical workflow that were beyond the scope of this conceptual paper may alleviate the current shortcomings of the method and lead to better recoveries. Analyte losses of 30–40% during extraction are problematic considering the expected low environmental concentrations. They may be minimized using, e.g., other surfactants or solvents, and improved extraction procedures. Additional (selective) analyte loss that occurs during aF4 could be reduced by exploring new carrier compositions, separation conditions or membrane materials. For example, identifying membranes that are unfavorable for soot but well suited for CNTs (e.g. the polyether sulfone type shown in Table S3†), may be an elegant way to increase aF4 recoveries and gain CNT selectivity.The measures suggested above may also lead to lower MDLs. Additionally, extracted analyte amounts may be increased through introduction of selective enrichment steps (e.g., density gradient ultracentrifugation45). Analyte amounts at the detector may be increased through the use of preparative aF4 injection volumes or use of smart stream splitting.46
Another shortcoming is related to the aF4-MALS technique itself: as every environmental sample – even the one that is analyte-free – will provide a signal (i.e., ρ), there will be a need for some kind of a “baseline” measurement of the uncontaminated matrix. The best way to deal with this problem may be the establishment of a aF4-MALS database of MWCNT-free soils, spanning over as many representative soils/soil types as possible.
aF4-MALS within the larger analytical workflow
We envision the presented method to be part of a larger analytical workflow for detection and quantification of CNTs in natural samples. Already now, the method presented is considered useful to accompany exposure studies in soils and sediments, in which CNTs are present in the range of native BC concentrations. Ideally, the method may be preceded, e.g., by extraction and isolation of BC by different means (e.g., CTO-375,14 see also ESI,† and other thermo-analytical methods47), and followed by fraction collection, elemental (e.g., by monitoring embedded trace catalytic metals48) and spectroscopic analyses (such as NIRS,17 Raman49). Automated EM may then deliver orthogonal shape information, provide additional particle number concentrations, and may, coupled to X-ray analysis, also be useful for BC identification.50Conclusions
We used aF4-MALS to differentiate between particles of different shapes (MWCNTs, soot and native soil particles). Different MWCNT aspect ratios as well as mixtures of soot and MWCNTs were efficiently discriminated. Fractions of MWCNTs in MWCNT-soot mixtures calculated based on ρ obtained by aF4-MALS were in reasonable agreement with results from automated EM analysis. Compared with native soil, addition of MWCNT led to increased ρ-values, while addition of soot decreased them. The current MDLs for MWCNT are within the range of other BC quantification methods, but far above any currently predicted environmental concentration. To overcome this limitation, further development of suitable enrichment techniques will be necessary. While the method is currently limited by a rather narrow dynamic range, natural soils exhibited ρ-values that were consistently at the lower end, allowing for specific identification of high aspect ratio particles, such as MWCNTs. Electron microscopic analysis could support aF4-MALS by providing orthogonal confirmation as well as quantification capabilities, if contamination effects and losses over aF4 can be reduced. Overall, aF4-MALS in combination with EM and image analysis could be a valuable technique to be integrated into future analytical workflows to confirm MWCNT contents in soils.Acknowledgements
This work is part of the project “Effects of NANOparticles on beneficial soil MIcrobes and CROPS (NANOMICROPS)”, within the Swiss National Research Programme NRP 64 “Opportunities and Risks of Nanomaterials”. We thank the Swiss National Science Foundation for financial support. We also would like to thank the Swiss Federal Office for Environment for instrument funding and Andres Kaech and the Center for Microscopy and Image Analysis of the University of Zurich for EM Support. D. Xanat Flores-Cervantes and Felix Wettstein are acknowledged for fruitful CNT/aF4-discussions, and Isabel Hilber for help with the statistical analysis. We also thank the company Postnova Analytics GmbH for their technical support.References
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- Nanotechnology Consumer Products Inventory, http://www.nanotechproject.org/inventories/consumer/search/, Accessed 28.1., 2014.
- F. Gottschalk and B. Nowack, J. Environ. Monit., 2011, 13, 1145–1155 RSC.
- F. Gottschalk, T. Sun and B. Nowack, Environ. Pollut., 2013, 181, 287–300 CrossRef CAS PubMed.
- E. J. Petersen, L. Zhang, N. T. Mattison, D. M. O'Carroll, A. J. Whelton, N. Uddin, T. Nguyen, Q. Huang, T. B. Henry, R. D. Holbrook and K. L. Chen, Environ. Sci. Technol., 2011, 45, 9837–9856 CrossRef CAS PubMed.
- M. V. Khodakovskaya, B.-S. Kim, J. N. Kim, M. Alimohammadi, E. Dervishi, T. Mustafa and C. E. Cernigla, Small, 2013, 9, 115–123 CrossRef CAS PubMed.
- S. Tripathi, S. K. Sonkar and S. Sarkar, Nanoscale, 2011, 3, 1176–1181 RSC.
- A. Gogos, K. Knauer and T. D. Bucheli, J. Agric. Food Chem., 2012, 60, 9781–9792 CrossRef CAS PubMed.
- Z. Tong, M. Bischoff, L. F. Nies, P. Myer, B. Applegate and R. F. Turco, Environ. Sci. Technol., 2012, 46, 13471–13479 CrossRef CAS PubMed.
- J. E. Canas, M. Q. Long, S. Nations, R. Vadan, L. Dai, M. X. Luo, R. Ambikapathi, E. H. Lee and D. Olszyk, Environ. Toxicol. Chem., 2008, 27, 1922–1931 CrossRef CAS.
- C. M. Rico, S. Majumdar, M. Duarte-Gardea, J. R. Peralta-Videa and J. L. Gardea-Torresdey, J. Agric. Food Chem., 2011, 59, 3485–3498 CrossRef CAS PubMed.
- M. W. I. Schmidt and A. G. Noack, Global Biogeochem. Cycles, 2000, 14, 777–793 CrossRef CAS.
- A. Sobek and T. D. Bucheli, Environ. Pollut., 2009, 157, 1065–1071 CrossRef CAS PubMed.
- K. Doudrick, P. Herckes and P. Westerhoff, Environ. Sci. Technol., 2012, 46, 12246–12253 CrossRef CAS PubMed.
- D. L. Plata, C. M. Reddy and P. M. Gschwend, Environ. Sci. Technol., 2012, 46, 12254–12261 CrossRef CAS PubMed.
- A. Schierz, A. N. Parks, K. M. Washburn, G. T. Chandler and P. L. Ferguson, Environ. Sci. Technol., 2012, 46, 12262–12271 CrossRef CAS PubMed.
- F. Irin, B. Shrestha, J. E. Canas, M. A. Saed and M. J. Green, Carbon, 2012, 50, 4441–4449 CrossRef CAS PubMed.
- O. Gustafsson, T. D. Bucheli, Z. Kukulska, M. Andersson, C. Largeau, J. N. Rouzaud, C. M. Reddy and T. I. Eglinton, Global Biogeochem. Cycles, 2001, 15, 881–890 CrossRef CAS.
- I. Colbeck, B. Atkinson and Y. Johar, J. Aerosol Sci., 1997, 28, 715–723 CrossRef CAS.
- F. von der Kammer, M. Baborowski and K. Friese, Anal. Chim. Acta, 2005, 552, 166–174 CrossRef CAS PubMed.
- B. Chen and J. P. Selegue, Anal. Chem., 2002, 74, 4774–4780 CrossRef CAS.
- N. Tagmatarchis, A. Zattoni, P. Reschiglian and M. Prato, Carbon, 2005, 43, 1984–1989 CrossRef CAS PubMed.
- J. Chun, J. A. Fagan, E. K. Hobbie and B. J. Bauer, Anal. Chem., 2008, 80, 2514–2523 CrossRef CAS PubMed.
- J. Gigault, B. Grassl, I. Le Hecho and G. Lespes, Microchim. Acta, 2011, 175, 265–271 CrossRef CAS.
- J. Gigault, B. Grassl and G. Lespes, Anal. Bioanal. Chem., 2011, 401, 3345–3353 CrossRef CAS PubMed.
- M. Martin and P. S. Williams, in Theoretical Advancement in Chromatography and Related Separation Techniques, Springer, 1992, pp. 513–580 Search PubMed.
- P. J. Wyatt, Anal. Chim. Acta, 1993, 272, 1–40 CrossRef CAS.
- F. von der Kammer, Characterization of Environmental Colloids applying Field-Flow Fractionation – Multi Detection Analysis with Emphasis on Light Scattering Techniques, PhD Thesis, TU Hamburg-Harburg, 2004 Search PubMed.
- F. Schwab, T. D. Bucheli, L. P. Lukhele, A. Magrez, B. Nowack, L. Sigg and K. Knauer, Environ. Sci. Technol., 2011, 45, 6136–6144 CrossRef CAS PubMed.
- F. Schwab, L. Camenzuli, K. Knauer, B. Nowack, A. Magrez, L. Sigg and T. D. Bucheli, Environ. Pollut., 2014, 192, 147–153 CrossRef CAS PubMed.
- K. Hammes, M. W. I. Schmidt, R. J. Smernik, L. A. Currie, W. P. Ball, T. H. Nguyen, P. Louchouarn, S. Houel, Ö. Gustafsson, M. Elmquist, G. Cornelissen, J. O. Skjemstad, C. A. Masiello, J. Song, P. A. Peng, S. Mitra, J. C. Dunn, P. G. Hatcher, W. C. Hockaday, D. M. Smith, C. Hartkopf-Fröder, A. Böhmer, B. Lüer, B. J. Huebert, W. Amelung, S. Brodowski, L. Huang, W. Zhang, P. M. Gschwend, D. X. Flores-Cervantes, C. Largeau, J.-N. Rouzaud, C. Rumpel, G. Guggenberger, K. Kaiser, A. Rodionov, F. J. Gonzalez-Vila, J. A. Gonzalez-Perez, J. M. de la Rosa, D. A. C. Manning, E. López-Capél and L. Ding, Global Biogeochem. Cycles, 2007, 21, GB3016 CrossRef.
- T. Agarwal and T. D. Bucheli, Environ. Pollut., 2011, 159, 532–538 CrossRef CAS PubMed.
- I. Arganda-Carreras, R. Fernández-González, A. Muñoz-Barrutia and C. Ortiz-De-Solorzano, Microsc. Res. Tech., 2010, 73, 1019–1029 CrossRef PubMed.
- M. Doube, M. M. Kłosowski, I. Arganda-Carreras, F. P. Cordelières, R. P. Dougherty, J. S. Jackson, B. Schmid, J. R. Hutchinson and S. J. Shefelbine, Bone, 2010, 47, 1076–1079 CrossRef PubMed.
- G. Polder, H. L. E. Hovens and A. J. Zweers, Proceedings of the ImageJ User and Developer Conference, 2010, vol. 27–29, pp. 172–177 Search PubMed.
- R. Kaegi, J. Aerosol Sci., 2004, 35, 621–632 CrossRef CAS PubMed.
- L. H. Keith, W. Crummett, J. Deegan, R. A. Libby, J. K. Taylor and G. Wentler, Anal. Chem., 1983, 55, 2210–2218 CrossRef CAS.
- J. Gigault, I. Le Hécho, S. Dubascoux, M. Potin-Gautier and G. Lespes, J. Chromatogr. A, 2010, 1217, 7891–7897 CrossRef CAS PubMed.
- D. L. Plata, P. M. Gschwend and C. M. Reddy, Nanotechnology, 2008, 19, 185706 CrossRef CAS PubMed.
- Y. Miyata, K. Mizuno and H. Kataura, J. Nanomater., 2011, 2011, 1–7 CrossRef PubMed.
- F. R. Phelan Jr and B. J. Bauer, Chem. Eng. Sci., 2009, 64, 1747–1758 CrossRef PubMed.
- K. Matsuoka and Y. Moroi, Biochim. Biophys. Acta, 2002, 1580, 189–199 CrossRef CAS.
- F. Gottschalk, T. Sun and B. Nowack, Environ. Pollut., 2013, 181, 287–300 CrossRef CAS PubMed.
- M. S. Arnold, A. A. Green, J. F. Hulvat, S. I. Stupp and M. C. Hersam, Nat. Nanotechnol., 2006, 1, 60–65 CrossRef CAS PubMed.
- M. Hassellöv, F. von der Kammer and R. Beckett, in Environmental Colloids and Particles, John Wiley & Sons, Ltd, 2007, DOI:10.1002/9780470024539.ch5, pp. 223–276.
- A. W. Musumeci, G. G. Silva, W. N. Martens, E. R. Waclawik and R. L. Frost, J. Therm. Anal. Calorim., 2007, 88, 885–891 CrossRef CAS.
- R. B. Reed, D. G. Goodwin, K. L. Marsh, S. S. Capracotta, C. P. Higgins, D. H. Fairbrother and J. F. Ranville, Environ. Sci.: Processes Impacts, 2013, 15, 204–213 CAS.
- A. I. Lopez-Lorente, B. M. Simonet and M. Valcarcel, Talanta, 2012, 105, 75–79 CrossRef PubMed.
- D. T. F. Kuo, J. B. Vander Sande and P. M. Gschwend, Org. Geochem., 2013, 56, 81–93 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Scanning electron micrographs of the particles, soil extracts and CNTs before fractionation. Detailed property description of the used particles and soils, parts of quality assurance (including determination of the instrument detection limit, blank measurement, recoveries of the analytes with different membrane types, alternating injections, relative standard deviations of independent injections, examples of Debye fits and rg distributions, rh calibration), raw data for Fig. 4–6 and particle shape measured after CTO-375. See DOI: 10.1039/c4en00070f |
| This journal is © The Royal Society of Chemistry 2014 |