
The yin: an adverse health perspective of nanoceria: uptake, distribution, accumulation, and mechanisms of its toxicity
Robert A.
Yokel
*ab,
Salik
Hussain
c,
Stavros
Garantziotis
c,
Philip
Demokritou
d,
Vincent
Castranova
ef and
Flemming R.
Cassee
gh
aPharmaceutical Sciences, University of Kentucky, USA. E-mail: ryokel@uky.edu
bGraduate Center for Toxicology, University of Kentucky, USA
cClinical Research Unit, National Institute of Environmental Health Sciences, National Institutes of Health, USA
dEnvironmental Health, Harvard, USA
eNational Institute for Occupational Safety and Health, USA
fWest Virginia University School of Pharmacy, Morgantown, WV, USA
gCentre for Sustainability, Environmental & Health, National Institute for Public Health and the Environment, Bilthoven, The Netherlands
hInstitute of Risk Assessment Sciences, Utrecht University, Utrecht, The Netherlands
First published on 21st August 2014
Abstract
This critical review evolved from a SNO Special Workshop on Nanoceria panel presentation addressing the toxicological risks of nanoceria: accumulation, target organs, and issues of clearance; how exposure dose/concentration, exposure route, and experimental preparation/model influence the different reported effects of nanoceria; and how can safer by design concepts be applied to nanoceria? It focuses on the most relevant routes of human nanoceria exposure and uptake, disposition, persistence, and resultant adverse effects. The pulmonary, oral, dermal, and topical ocular exposure routes are addressed as well as the intravenous route, as the latter provides a reference for the pharmacokinetic fate of nanoceria once introduced into blood. Nanoceria reaching the blood is primarily distributed to mononuclear phagocytic system organs. Available data suggest nanoceria's distribution is not greatly affected by dose, shape, or dosing schedule. Significant attention has been paid to the inhalation exposure route. Nanoceria distribution from the lung to the rest of the body is less than 1% of the deposited dose, and from the gastrointestinal tract even less. Intracellular nanoceria and organ burdens persist for at least months, suggesting very slow clearance rates. The acute toxicity of nanoceria is very low. However, large/accumulated doses produce granuloma in the lung and liver, and fibrosis in the lung. Toxicity, including genotoxicity, increases with exposure time; the effects disappear slowly, possibly due to nanoceria's biopersistence. Nanoceria may exert toxicity through oxidative stress. Adverse effects seen at sites distal to exposure may be due to nanoceria translocation or released biomolecules. An example is elevated oxidative stress indicators in the brain, in the absence of appreciable brain nanoceria. Nanoceria may change its nature in biological environments and cause changes in biological molecules. Increased toxicity has been related to greater surface Ce3+, which becomes more relevant as particle size decreases and the ratio of surface area to volume increases. Given its biopersistence and resulting increased toxicity with time, there is a risk that long-term exposure to low nanoceria levels may eventually lead to adverse health effects. This critical review provides recommendations for research to resolve some of the many unknowns of nanoceria's fate and adverse effects.
Nano impactThis critical review focuses on nanoceria's pharmacokinetics and adverse effects. It is the product of a workshop panel presentation on nanoceria that included panels focusing on other aspects of nanoceria, including its beneficial effects/applications and environmental impact. Nanoceria's pharmacokinetics and in vitro and in vivo pharmacodynamics following intravenous, pulmonary, oral, dermal, and ocular exposure and its molecular effects are thoroughly reviewed. Data gaps are identified and research recommendations presented to resolve some of the many unknowns of nanoceria's fate and adverse effects, to support the advancement of the many demonstrated and future applications of nanoceria. An example is provided of a safer by design engineering approach to improve nanoceria's benefit risk ratio. |
Introduction
The availability of cerium dioxide (a.k.a.: CeO2, ceria, cerium oxide) as one of the most abundant rare earth oxides has prompted research on the synthesis and development of functional ceria nanoparticles. As a result, nanoceria is increasingly used in a variety of industrial and commercial applications, including catalysis,1,2e.g., as a diesel fuel additive to increase fuel combustion efficiency and decrease soot emissions;3,4 in chemical mechanical planarization/polishing;5 UV-shielding;6 in semiconductors; in toner formulations;7 and as an additive in various nanocomposites, as reviewed.8 The inevitable increase in consumer and occupational exposures raises the need for a comprehensive toxicological characterization of nanoceria.9 Many companies and organizations have already identified nanoceria as a high priority material for toxicological evaluations.10,11This critical review focuses on the potential routes of human exposure to nanoceria and its extent of uptake from those routes, and its distribution, retention, and resultant effects. The mechanisms of its adverse effects described to date are primarily gleaned from in vitro studies. Each major section is concluded by what we know and knowledge gaps. Research recommendations, that will hopefully advance the understanding of nanoceria risk, are offered in the conclusion of this critical review.
In the context of unintended nanoceria exposure, pulmonary exposure has received the most attention. Other exposure routes are discussed, including intravenous, dermal, oral, and ocular.
Systemic nanoceria exposure
The pharmacokinetics of systemically-administered nanoceria
The intravenous (IV) route provides pharmacokinetic and pharmacodynamic insight into the fate of nanoceria that is introduced into blood, or that is in blood after distribution from the site of uptake. Introduction into blood provides 100% bioavailability, against which uptake from other sites can be compared. Intravenous administration is also a potential route of administration for the use of nanoceria as a therapeutic agent, for described conditions,12 and those identified in the future.The distribution and short term persistence of a commercial platelet nanoceria, that had a median primary particle diameter of 31 nm (based on size), was assessed after IV administration of 50, 250, and 750 mg kg−1 bw to rats. It was infused at a rate of 100 mg kg−1 h−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 as a 5% dispersion in water with concurrent IV infusion in a second cannula of 1.8% sodium chloride. Blood cerium concentration initially decreased with a t1/2 of 7.5 min. Blood cerium concentration increased slightly from 120 to 240 min after completion of the nanoceria infusion, an unexpected trend. Cerium concentrations in the spleen, liver, blood, and brain were higher 20 h than 1 h post-infusion, suggesting some nanoceria was in other compartments at the earlier time point and translocated back into blood and to the 3 measured organs at 20 h. This observation was investigated with intra-arterial infusion at 20 ml min−1 of up to 500 μg ml−1 of a 5 nm ceria for 120 seconds, which demonstrated nanoceria association with the brain microvasculature luminal wall.14 The capillary depletion method showed essentially all of the nanoceria associated with the capillary fraction, and electron microscopy showed it located on the endothelial cell luminal surface.14 Given the uptake t1/2 of nanoparticles by several cell types was 1 to >2 h,15 nanoceria associated with the luminal surface of endothelial cells 120 seconds after its intra-carotid artery infusion might eventually be taken up by cells. However, light and electron microscopy have not revealed any appreciable nanoceria in brain parenchyma (neurons or astrocytes), including the median eminence, a brain region lacking a blood–brain barrier (BBB), after IV administration of 5, 15, or 30 nm citrate-coated polyhedral or cubic nanoceria.13,16–18 Nanoceria adsorption to the luminal wall of vascular endothelial cells may be the site of nanoceria distribution after leaving circulating blood, from which it re-entered circulating blood and distributed to peripheral organs.
13 as a 5% dispersion in water with concurrent IV infusion in a second cannula of 1.8% sodium chloride. Blood cerium concentration initially decreased with a t1/2 of 7.5 min. Blood cerium concentration increased slightly from 120 to 240 min after completion of the nanoceria infusion, an unexpected trend. Cerium concentrations in the spleen, liver, blood, and brain were higher 20 h than 1 h post-infusion, suggesting some nanoceria was in other compartments at the earlier time point and translocated back into blood and to the 3 measured organs at 20 h. This observation was investigated with intra-arterial infusion at 20 ml min−1 of up to 500 μg ml−1 of a 5 nm ceria for 120 seconds, which demonstrated nanoceria association with the brain microvasculature luminal wall.14 The capillary depletion method showed essentially all of the nanoceria associated with the capillary fraction, and electron microscopy showed it located on the endothelial cell luminal surface.14 Given the uptake t1/2 of nanoparticles by several cell types was 1 to >2 h,15 nanoceria associated with the luminal surface of endothelial cells 120 seconds after its intra-carotid artery infusion might eventually be taken up by cells. However, light and electron microscopy have not revealed any appreciable nanoceria in brain parenchyma (neurons or astrocytes), including the median eminence, a brain region lacking a blood–brain barrier (BBB), after IV administration of 5, 15, or 30 nm citrate-coated polyhedral or cubic nanoceria.13,16–18 Nanoceria adsorption to the luminal wall of vascular endothelial cells may be the site of nanoceria distribution after leaving circulating blood, from which it re-entered circulating blood and distributed to peripheral organs.
When terminated 1 or 20 h after completion of 50, 250, and 750 mg kg−1 of the IV infusion of the 30 nm platelet nanoceria, a dose-dependent increase of cerium in the spleen, liver, blood, and brain was seen, indicating the lack of organ saturation of nanoceria accumulation within this short time frame. Cerium concentrations were higher in the spleen than liver. The percentages of the injected dose in the liver and spleen are shown in Fig. 1. Less was in the blood (0.2 to 4%) and much less in the brain (<0.1%). This distribution is similar to that reported by others (Fig. 1) and described below.19 Twenty h after its infusion, intracellular nanoceria agglomerations were seen in macrophage-rich spleen red pulp, Kupffer cells, and hepatocytes. Nanoceria accumulations were seen in the afferent arterioles leading into the glomeruli. Much of the cerium in the brain was probably nanoceria in circulating blood within the brain's vasculature, and perhaps associated with the endothelial cells lining the brain's blood vessels as seen after intra-carotid nanoceria infusion, noted above, and as 100 to 200 nm irregular granules within plasmoid material in blood vessels in the tail after IV tail vein nanoceria injection to the mouse.14,20 The very few nanoceria particles seen in brain parenchyma might be an artifact of tissue preparation (sectioning) for electron microscopic visualization. In summary, this study showed rapid clearance from blood circulation of most of the nanoceria into mononuclear phagocyte system (MPS) organs and very little to no nanoceria BBB penetration into brain parenchyma.
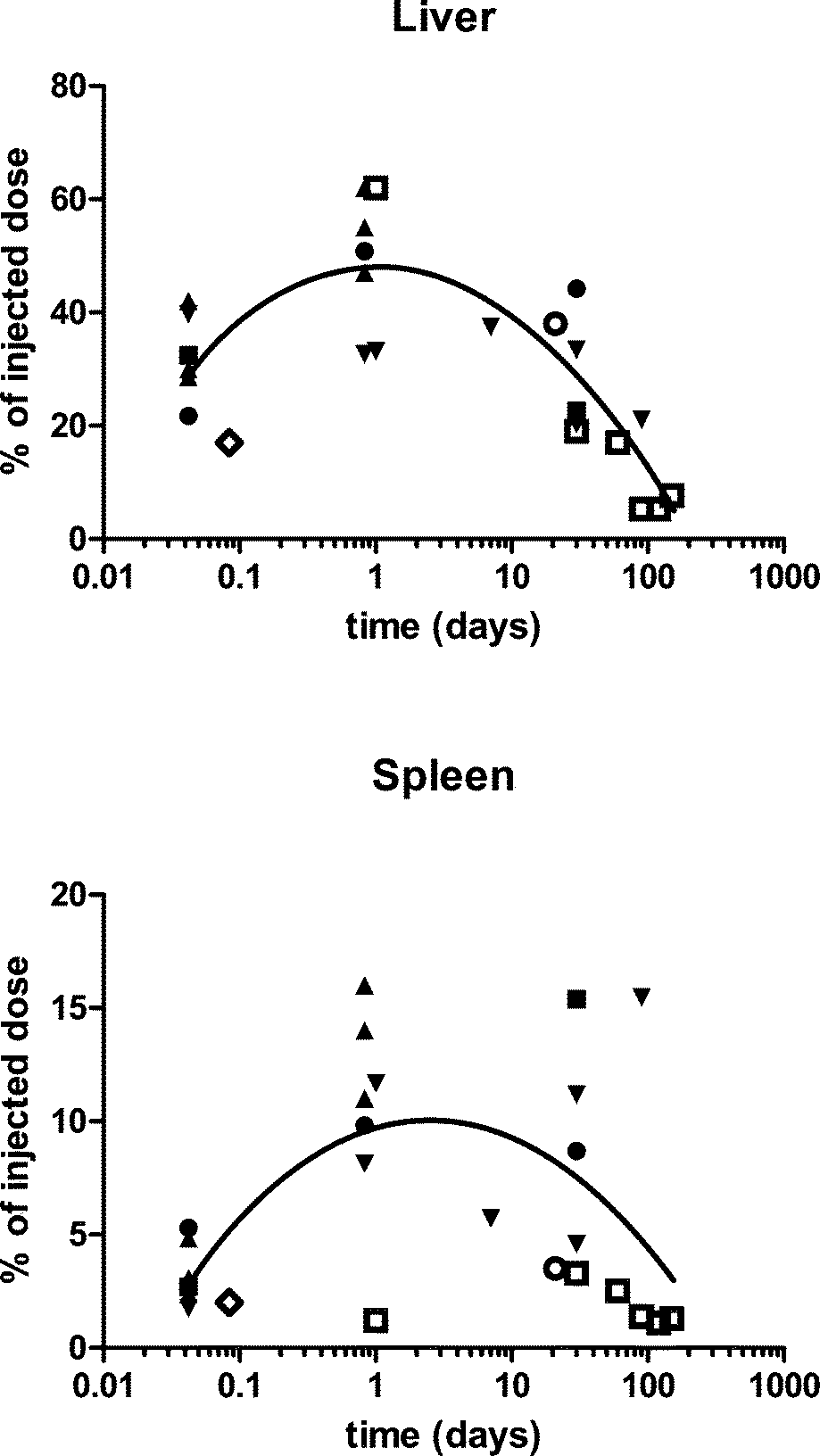 | ||
| Fig. 1 The percentage of the injected dose in the liver and spleen after intravenous nanoceria administration of various sizes, shapes, doses, and surface-coatings. ● = five nm, polyhedral, citrate-coated, 85 mg kg−1 to rats.22 ■ = fifteen nm, polyhedral, citrate-coated, 70 mg kg−1 to rats.22 ▲ = thirty nm, platelets, coated with an unknown carboxylate, 50, 250 and 750 mg kg−1 to rats.13 ▼ = thirty nm, cubic, citrate coated, 85 mg kg−1 to rats. ○ = three to 5 nm, 10 to 50 nm agglomerates, fluorescent-tagged, 0.5 mg kg−1 weekly for 5 weeks to mice, terminated 1 week later.19 ◊ = 5.6 nm, 3-aminopropylsilyl-anchored N-succinimidyl 4-[18F]fluorobenzoate coated, 4.8 MBq to rats.23 □ = 2.9 nm, citrate-EDTA coated, 20 mg kg−1 to mice.24 Line is best polynomial (quadratic) fit. Cerium was quantified by ICP-OES or ICP-MS,13 ICP-MS,18,19,22,24 or positron emission tomography.23 | ||
The biodistribution of a smaller (3 to 5 nm primary particle size, 15 to 20 nm agglomerate21) fluorescent-tagged crystalline ceria was determined in CD-1 mice 7 days after 2 or 5 weekly IV 0.5 mg kg−1 injections.19 The spleen had the highest cerium concentration, followed closely by the liver, consistent with the study presented above. More of the dose was in the liver and spleen (Fig. 1) than the lung (0.15%) or kidney (0.008%). None was detectible in the brain. Organ cerium concentrations were about 3 times higher in the mice that received 5 vs. 2 injections. Cerium clearance into urine was not detectable. The results demonstrate that this nanoceria dose did not saturate the organs and that very little was cleared during the 35 day post exposure period. Assuming the 3-aminopropylsilyl-anchored N-succinimidyl 4-[18F]fluorobenzoate coated nanoceria retained the positive surface charge (+18.5 mV) that it had before N-succinimidyl 4-[18F]fluorobenzoate coating, this is the only positively-charged nanoceria for which biodistribution results have been reported after its IV administration. The results shown in Fig. 1 do not suggest it behaved significantly differently from the other nanoceria that had a negative surface charge. Distribution and retention of a ceria nanorod (diameter and length averages of 10 and 264 nm) was generally not different from that seen after IV infusion of a 5 nm polyhedral or a 30 nm cubic ceria.25 Available results do not suggest nanoceria shape has a profound influence on cerium distribution.
Several studies report the use of functionalized nanoceria. Distribution of a N-succinimidyl 4-[18F]fluorobenzoate covalently anchored 3-(aminopropyl)triethoxysilane functionalized ~5 nm ceria in rats detected by positron emission tomography showed ~6, 2, 2, and 1% of the dose per gram liver, spleen, lung, and kidney, respectively.23Fig. 1 shows the percentage of the dose of this small nanoceria in the liver and spleen was similar to results obtained with other nanoceria. A 2.9 nm citrate/EDTA-coated nanoceria injected IV to mice (20 mg kg−1) in a citrate buffer produced a similar pattern; highest levels in the liver and spleen (Fig. 1), lower in the kidney, and lowest in the brain.24 Levels decreased over 5 months (Fig. 1). The rats were vascularly perfused prior to organ removal so the brain cerium levels should not reflect nanoceria in blood within the brain. After repeated dosing, cerium levels in the cerebellum were higher than in the combined brain and spinal cord. Brain nanoceria was much higher in mice with experimental autoimmune encephalomyelitis, perhaps due to the loss of BBB integrity in this pathology.24 TEM showed intracellular nanoceria in myelinated processes, axons, dendrites, and mitochondria of the cerebellum. This is the only report with good evidence of nanoceria distribution across the BBB into brain parenchyma.
To determine if nanoceria size has a significant effect on its pharmacokinetics after IV administration, citrate-coated (ζ potential −32 to −57 mV), ~5, 15, 30, and 55 nm cubic/polyhedral nanoceria and a mixture of 30 nm cubic and rod nanoceria were prepared and extensively characterized.22 Their pharmacokinetics after a 1 h IV infusion of a single dose (70 or 85 mg kg−1) to rats was determined by repeated blood sampling. Partitioning between blood components, compared to the cerium ion, was also determined.26 The percentage of the nanoceria dose remaining in circulating blood 10 minutes after completion of its IV infusion was <2% for the 15, 30, and 55 nm ceria, indicating very rapid translocation, as seen with the 30 nm commercial nanoceria.13 In contrast, 10 minutes after the 1 h infusion of 5 nm ceria ~35% was still circulating in the blood, perhaps because it was too small to be readily recognized by macrophages and was able to avoid opsonin adsorption or blood cell attachment.12 This was cleared with initial and beta t1/2s of 0.4 and 124 h. Similarly, a citrate-EDTA-coated 2.9 nm ceria was cleared from blood with a plasma t1/2 of 3.7 h.24 An increase of ceria in blood was seen 2 to 4 h after infusion of the 15 and 30, but not 5 or 55 nm, ceria or the cerium ion. This is not typical following IV administration, and suggests nanoceria re-entry into circulating blood, an observation seen with the commercial 30 nm ceria, above,13 or solubilization of nanoceria releasing cerium ions, although this is much less likely within 2 to 4 h.
To ascertain the distribution of these 4 nanoceria within blood after their IV administration, plasma was separated from blood cells by allowing the blood to clot and separating the clot from plasma without using centrifugation that would bring down the nanoceria. This separation showed an increase of nanoceria associated with the clot over 4 h of 15 and 30, but not 5 or 55 nm, ceria. This could be due to protein coating, given this is a very rapid process and known to occur with nanoceria.12,27 Such coating could explain the release of nanoceria from vascular endothelial cell wall adsorption and its re-entry into circulating blood. Nanoceria agglomerations were seen in blood after 1 h incubation of 30 nm ceria13 and 5 nm ceria.16 Some of the agglomerates appeared to be adherent to erythrocytes,16 perhaps a process similar to the adsorption of 7 nm ceria agglomerates to the external cell membrane of fibroblasts.28 Nanoceria may agglomerate or be protein coated quickly once it contacts blood, affecting the ability of macrophages to recognize them as foreign particles to be cleared into MPS organs.
Classical pharmacokinetic analysis, based on blood levels of 5, 15, 30, and 55 nm ceria and a mixture of 30 nm cubic and rod nanoceria after their IV administration, did not describe their fate well.26 This is consistent with observations that the fate of insoluble nanomaterials is different than that of small molecules.29–31 Differences include agglomeration that probably occurs in circulating blood and is seen in MPS organs where they accumulate (below), rapid clearance from circulation presumably by macrophages rather than blood flow (yielding short blood t1/2s that do not reflect the whole organism t1/2), some re-entry into circulating blood resulting in increased concentration hours after IV dosing, movement across cell membranes that is not diffusion-based or as substrates for active transporters, and distribution into MPS sites in which they persist with little elimination from the organism which is inconsistent with partition coefficient distribution between blood and tissues.
To determine the fate of the 5, 15, 30, and 55 nm ceria, liver, spleen, blood, and brain were collected up to 30 days after their single IV infusion over 1 h to rats.22 Blood 5 and 15 nm ceria decreased from 1 to 30 days whereas the 30 nm ceria was higher 20 h, compared to 1 h, after administration, again suggesting mobilization from a site outside of circulating blood. Brain cerium decreased over time, probably reflecting the clearance of ceria from blood, and perhaps mobilization of vascular endothelial cell wall-adsorbed nanoceria. Liver and spleen cerium did not decrease from 1 h to 30 days, and in some cases significantly increased (Fig. 1). These results are consistent with some redistribution within, and the lack of significant elimination from, the rat, as observed up to 14 days by us, noted below18 and 42 days by others.19
To extend the duration of observation and much more extensively characterize nanoceria's fate, rats were evaluated 1, 7, 30, or 90 days after a single 85 mg kg−1 IV infusion of 30 nm ceria.18 Total urine and feces collection for 14 days showed 0.01 and 0.4% of the dose eliminated in these excretia, respectively. Similarly, some cerium was eliminated in feces, but none was detected in urine, after IV administration of 0.5 mg kg−1 of a 3 to 5 nm ceria.19 Cerium concentration in 16 organs showed spleen, liver, bone marrow, and skeletal system with 21 to 37, 5 to 15, 9 to 27, and 2 to 8% of the dose, respectively.18 The percentage of the dose in liver and spleen is shown in Fig. 1, compared to other studies. Distribution among these 16 organs was consistent with prior findings with 30 and 3 to 5 nm primary particle size nanoceria (above) and its distribution in organs distal to the site of administration when given orally or into the lungs (below). Blood, cerebrospinal fluid, and most organs showed an increase of cerium 90 days after nanoceria administration, whereas this was not seen in the liver (Fig. 1), bone marrow, or skeletal muscle, suggesting some redistribution from 30 to 90 days after nanoceria infusion. Light and electron microscopic assessment of MPS organs of these animals showed large (up to 5 μm) nanoceria aggregates predominantly in the phagolysosomes of Kupffer cells, but also in hepatocytes, as early as 1 h after its administration. The nanoceria aggregates persisted to 90 days, consistent with the lack of appreciable change in cerium in the liver. Nanoceria was not seen in bile canaliculi, consistent with the lack of significant fecal excretion.
In summary, the above studies generally show similar nanoceria distribution after IV administration, characterized by rapid translocation from the blood into the MPS with spleen having with the highest concentration and liver the greatest burden. The 2.9 and 5 nm ceria were an exception as these were much more slowly cleared from circulation. In general the clearance t1/2 from organs is long and there is some clearance of nanoceria months after administration that seems to be due to dissolution and some redistribution.
The pharmacokinetics of parenteral non-intravenous nanoceria administration
When 3 to 5 nm primary particle sized-fluorescent-tagged nanoceria was IP injected into CD-1 mice at the same dose (0.5 mg kg−1) and schedule (2 to 5 weekly injections) as given IV (above),19 ~4.2, 1.25, 0.02 and 0.01% of the dose was in the liver, spleen, lung, and kidney, respectively, 7 days after the last dose.19 These results suggest some uptake of nanoceria from the peritoneal cavity, or nanoceria dissolution and cerium uptake.The pharmacokinetics of the cerium ion
The pharmacokinetics and distribution of nanoceria are quite different than cerium, consistent with observations of many nanomaterials that their properties are different from their solute or bulk forms. Approximately 2.5% of IV cerium chloride was excreted in urine during the first day, decreasing to ~0.1% by day 14, resulting in elimination of ~5.5% in the first 15 days, whereas fecal excretion was ~4.5% of the dose 7 days after injection, ~42% in the first 15 days.32 In another study, less than 1% of IV injected cerium was excreted in the urine over 4 days.33 In the first 7 days after IV cerium chloride administration total cerium eliminated in urine and feces of rats was 0.02 and 0.85% of the dose, respectively.18 In contrast, only 0.01% of a 30 nm ceria was eliminated in urine during the first 14 days after its dosing, and only 0.4% appeared in the feces.18 One h after IV injection of cerium chloride 10% remained in serum, which was then cleared with a t1/2 of ~10 h33 and initial and beta t1/2s of 0.6 and 16 h.26 In contrast, >98% of 15, 30, and 55 nm ceria was cleared from blood within minutes and 10 minutes after a 1 h IV infusion of 5 nm ceria ~35% was in blood, which was cleared with an initial and beta t1/2 of 0.4 and 124 h,26 as noted above. Fifteen days after an IV injection of cerium chloride, 20, 16, 2, and 2% was in the skeleton, liver, kidneys, and gastrointestinal (GI) tract, whereas 0.7, 0.5, 0.08, 0.08, and 0.05% was in the muscle, spleen, lung, testes, and heart.32,34 In contrast, nanoceria is predominantly cleared into MPS organs, the liver, spleen, and bone marrow.13,18,19,22,23 During the first 8 days after inhalation of 1.5 μm mean aerodynamic diameter cerium particles the t1/2 of cerium in the liver was estimated to be 11 and 17 days, with 0.5 to 1% of the hepatic burden excreted daily, accounting for approximately 0.09% of the initial cerium body burden.35,36 In contrast, the percent of the dose of a 30 nm ceria in the liver decreased from 7 to 30 days after a single IV administration, but over the next 90 days did not decrease further, while there was little decrease in the bone marrow and an increase in the spleen.18What we know
• After IV infusion, nanoceria rapidly translocates from the blood to the liver, spleen, and bone marrow, from which clearance is slow. Appreciable entry of nanoceria into brain parenchyma was not seen with ≥ 5 nm ceria, but was observed in the cerebellum with a 2.9 nm ceria.Knowledge gaps
• What is the fate of nanoceria in the vascular compartment that has associated with the vascular endothelial cells? Does it dissociate from those cells, perhaps due to protein coating, to re-enter circulating blood and then be cleared by macrophages into MPS organs?• What are the long term fate and effects of nanoceria, for the life of the mammal, given it is quite insoluble and persistent, and its expected low exposure concentration in air and food?
The toxicodynamics of intravenously-administered nanoceria
During the IV infusion of 50, 250, and 750 mg kg−1 of an ~30 nm platelet ceria to rats, clinical toxicity was only observed in some rats receiving the 750 mg kg−1 dose; bruxism and excessive licking were seen in some rats receiving all doses.13 No adverse effects were seen after the infusion and no animals died within 20 h. Kidney and liver weights increased. There was a dose-dependent increase of activated Kupffer cells. This study revealed little acute toxicity from very large doses given intravenously. Intravenous administration of 175 and 250 mg kg−1 of a 5 nm ceria resulted in mortality within 1 h, whereas 85 mg kg−1 was tolerated, with no mortality to 30 days.16,22 An infusion of 100 mg kg−1 30 nm ceria produced mild distress (tachypnea, skittish, not resting well). Doses of 78 to 250 mg kg−1 of 55 nm ceria produced significant toxicity (dyspnea, lethargy), whereas doses of 100 mg kg−1 of the 5, 15, and 30 nm ceria and 50 mg of the 55 nm ceria did not result in mortality, further demonstrating low acute toxicity of IV nanoceria.22Compared to the cerium ion, nanoceria is much less toxic. The LD50 of CeCl3·6H2O in mice was ~13 mg kg−1.33 Intravenous injection into dogs of 50 mg kg−1 cerium chloride resulted in deterioration over 21 days.37 In contrast, doses up to 100 mg kg−1 of 5, 15, 30, and 55 nm ceria were tolerated.22
To assess the potential to produce subchronic toxicity, extensive daily cage side observations were conducted for 90 days after a single 85 mg kg−1 30 nm ceria IV infusion. These revealed no adverse effects. The nanoceria-treated rats gained 5% less weight during the first 7 days after treatment than controls. This 5% differential persisted relatively unchanged throughout the study.18
Histological assessment of toxicity to the liver 1 and 20 h after IV infusion of 50, 250, and 750 mg kg−1 of an ~30 nm ceria showed some hepatocyte degeneration 20 h after the 2 highest doses.13 Intravenous infusion of 85 mg kg−1 of a 30 nm ceria produced nanoceria-laden Kupffer cells. These stimulated CD3+ lymphocyte proliferation in the sinusoids to form granulomata, composed of ceria-laden Kupffer cells and a few non-ceria accumulating mononuclear cells, that were seen 30 days after the single nanoceria dose and persisted without great resolution or progression to 90 days after dosing.18,38 The granuloma were of the non-necrotizing type and did not progress to frank hepatic necrosis. There was a 50% increase in serum ALT levels, but apoptosis was not seen 20 h to 90 days after nanoceria dosing. There was an increase of cell proliferation, particularly 90 days after nanoceria dosing. The hepatic localization and effects of a 5 nm ceria were similarly studied after a single IV administration of 85 mg kg−1.39 Liver cerium increased to 51% of the dose, decreasing to 44% 30 days later (Fig. 1). Similar to the 30 nm ceria, agglomerations of nanoceria (up to 2 um) were seen in activated Kupffer cells. Some of these intermingled with CD3+ cells and mononucleated cells to form granulomas. Although nanoceria agglomerates appeared near the bile canaliculi, translocation into the biliary system was not observed, consistent with the lack of significant decrease of cerium in the liver up to 30 days after its administration.
In contrast to the above findings, no evidence of liver pathology was observed in mice on day 35 that received a total IV dose of 130 mg kg−1 of a citrate/EDTA coated 2.9 nm ceria over 35 days.24
Histological examination of H & E stained thin sections of the brain, lungs, liver, kidneys, spleen, and pancreas 7 days after IV injection of a 3 to 5 nm primary particle sized nanoceria, 0.1 or 0.5 mg kg−1, and 15 days after a second injection, revealed no lesions.20 After IV, IP, or oral administration of 3 to 5 nm primary particle size nanoceria, 0.5 mg kg−1 weekly for 2 or 5 weeks, histology assessment only revealed a reactive Peyer's patch in the small intestine of one IP dosed mouse and a pulmonary hemorrhage in one IV dosed mouse. The authors concluded that this nanoceria dose did not cause overt pathology.19 Intravitreal injection of 344 ng of nanoceria into each eye of rats did not produce adverse effects up to 120 days later, determined as retinal thickness or function.40
One and 20 h after the IV infusion of 50, 250, and 750 mg kg−1 of an ~30 nm ceria, BBB flux markers showed non-significant change, nor was there microscopic evidence of BBB disruption.13 Nanoceria effects on BBB integrity were further assessed in rats up to 20 h after a single IV infusion of the 5 nm ceria described above.16,17 No adverse effects on BBB integrity were seen other than a slight, significant, increase of horseradish peroxidase above control 20 h after the 5 nm ceria.16 Intra-arterial injection of up to 500 μg ml−1 5 nm ceria for 120 seconds at 20 ml min−1 did not alter BBB integrity.14
Twenty h after IV infusion of 250 and 750 mg kg−1 of an ~30 nm ceria, brain protein-bound 4-hydroxy 2-transnonenal (HNE, formed by lipid peroxidation of unsaturated acyl chains of phospholipids) was elevated in the hippocampus but not cortex or cerebellum. In contrast, 3-nitrotyrosine (3NT, produced by nitric oxide reaction with tyrosine) was not changed, and protein carbonyls (a biomarker of oxidative stress) were decreased in the cerebellum.13 To further pursue this nanoceria effect, multiple oxidative stress markers (HNE, 3NT, protein carbonyls, glutathione reductase [GR] level and activity, glutathione peroxidase [GPx] level and activity, superoxide dismutase [SOD] level and activity, and catalase level and activity) were determined 1 and 20 h after the 5 nm ceria. They showed little change, limited to an increase of catalase level 1 h and activity 20 h in the hippocampus and a decrease of catalase activity 1 h in the cerebellum.16 Thirty days after the 5 nm ceria infusion, protein carbonyl and heat shock protein 70 (Hsp70) levels were increased, and GPx and catalase activity and the reduced/oxidized glutathione (GSH/GSSG) ratio were decreased in the hippocampus.17 3NT and inducible nitric oxide synthase (iNOS) levels were increased, and GR level decreased, in the cortex. Protein carbonyl and heat shock protein 70 levels were increased, and GPx levels and activity and catalase levels decreased in the cerebellum. These results suggest greater pro-oxidant effects on the brain 30 days after a 5 nm ceria than at earlier times, in the absence of nanoceria entry into brain parenchyma.
To extend these findings, numerous measures of oxidative stress were measured in rat hippocampus 1 and 20 h and 1, 7, 30, and 90 days after a single IV administration of 30 nm ceria.41 Protein carbonyl, 3NT, heme oxygenase-1 (HO-1), and Hsp70 levels decreased in the first day, then increased up to day 30, whereas GPx and catalase levels and activity showed the opposite trend, increasing in the first day, followed by a decrease up to day 30. IL-1β levels were elevated from days 1 to 30, and pro-caspase-3 levels increased at day 30 and LC-3 AB levels increased from days 7 to 30, indicating apoptosis and cell proliferation. Ninety days after the single nanoceria infusion, nearly all endpoints returned to control levels. These trends can be explained by induction of Tier 1 oxidative stress responses 1 and 20 h after nanoceria, induction of Tier 2 oxidative stress response after 1 to 30 days, and induction of Tier 3 oxidative stress response at 30 days, as previously described.42 Additionally, they suggest activation of a Tier-4 response that restores the cellular redox balance 90 days after the single nanoceria administration. Release of factors such as pro-inflammatory cytokines may lead to these effects distal to the site of nanoceria deposition. These responses were seen in the absence, or at most a very few particles, of nanoceria in the brain.16,17,41
Nanoceria biotransformation in vivo
To address if there are changes in the Ce3+/Ce4+ ratio,43,44 electron energy loss spectrometry (EELS) of the surface of a 5 nm ceria was determined. It showed Ce3+ enrichment, which was not changed after the nanoceria was in the vasculature of the rat's brain for 20 h.16 A 30 nm ceria showed similar Ce3+ enrichment on its surface. Its core had a greater percentage of Ce4+.18,38 These results did not show nanoceria biotransformation over this short time. However, when the liver was examined by HRTEM 90 days after a single IV infusion of a 30 nm ceria, the ceria exhibited rounded edges and corners. Clouds of 1 to 3 nm, (111) and (200) faced, crystalline ceria near the partially biodegraded nanoceria were seen, suggesting a dissolution–precipitation process. EELS showed increased surface Ce3+ consistent with ultrafine nanoceria particles, suggesting greater anti-oxidant potential.45 These observations suggest nanoceria bioprocessing to a more stable, potentially more beneficial, nanoceria form.What we know
• Intravenous nanoceria has low acute toxicity, ~1 order of magnitude less than ionic cerium.• Oxidative stress leading to inflammation is the key toxicological effect, possibly leading to granuloma at high dose levels.
Knowledge gaps
• There is hardly any information whether repeated systemic nanoceria exposure results in a change in response due to prior exposure.• What mediates the changes in brain stress responses in the absence of nanoceria in brain parenchyma?
• How reversible are the observed adverse responses to nanoceria exposures?
Pulmonary deposition and response
Introduction
The most likely route of human unintentional exposure to nanoceria is through inhalation. Following inhalation exposure, nanoceria, being poorly-soluble in body fluids,46,47 deposits within the respiratory tract based on physical properties related to its size distribution and agglomeration/aggregation state.48 Ceria detected in diesel exhaust emissions from a nanoceria-based fuel additive was found to be of nanoscale size (<100 nm).3,49,50 Knowledge of the potential health effects associated with the use of nanoceria, including its use as a diesel fuel additive, is rather limited.51Deposition, translocation, and clearance
Particle deposition following inhalation is dependent on the particle thermodynamic and aerodynamic size, and agglomerate size, generally measured as mass mean (aerodynamic) diameter (MMAD).48 At low to zero air velocity, diffusion dominates nanoparticle deposition rate. However, measurement of MMAD rather than primary particle size distributions and amounts does not predict the delivered dose in the human respiratory tract.In general, nanoscale particles with a primary diameter 1 to 100 nm penetrate deeper into the lung than do larger size particles. Nano-sized particles are deposited by diffusion, primarily in the alveolar and tracheal bronchial regions. Due to their poor solubility they are slowly cleared.3,52 Some of the deposited particles are cleared via the mucociliary escalator to the pharynx and ingested, while others are transported to the tracheobronchial and pulmonary lymph nodes via penetration through the airway epithelium.3,48 Therefore, inhalation of particles leads to secondary exposure to the GI tract and potential uptake from this organ system, although nanoceria uptake from the GI tract is extremely low (see below). Studies in humans on absorption of microscale ceria following inhalation exposure have not been identified. However, case reports and retrospective occupational investigation provide support for the limited absorption of microscale ceria deposited in the lung following inhalation exposure.52 It is possible that nanoceria may dissolve more easily than microscale particles due to their larger surface area.53 However, given the biopersistence of nanoceria in the lung, significant nanoceria dissolution seems unlikely.
Much more attention has been paid to the inhalation of nanoceria than its ingestion, since cerium and nanoceria are poorly absorbed from the intestine,53 described below. For microscale ceria, it is known that the primary targets after inhalation are the lung and the associated lymph nodes. Other organs could also be affected through the blood and intestine following their clearance or translocation from the lung. After absorption into circulation, nanoceria may distribute to the liver, skeleton, spleen, and kidney, as summarized.53 Two hundred μg nanoceria (6.6 nm primary particle size, 12.8 nm hydrodynamic mean diameter; 97% 24 nm and 3% 88 nm in water, ζ = 30 mV) labeled with 141Ce was introduced by intratracheal instillation into male Wistar rats and translocation evaluated 6 h to 28 days later.46 Of the heart, liver, spleen, kidney, brain, testes, and stomach (cerium in the stomach decreased over time), liver had the highest 141Ce at 28 days (~0.1% of the dose) showing low translocation from the lung. Studies in Wistar rats exposed to cerium chloride aerosol (mean diameter 0.1 μm) showed that cerium was deposited in the lysosomes of alveolar macrophages (AMs).54,55
The process of phagocytosis by macrophages is in competition with uptake of particles by alveolar epithelial cells (which can result in increased access of particles to the interstitium, where they may cause damage). Some studies have shown that small particles are more readily taken up by epithelial cells than by macrophages and show greater rates of transfer across the epithelium.3 This route results in interstitialisation with persistence in the submucosal centriacinar region, or transfer to the lymph nodes. The uptake of 20 to 50 nm ceria, 100 ng ml−1 to 100 μg ml−1, by human lung 3T3 fibroblasts was measured in vitro. Nanoceria internalization occurred linearly with exposure time at concentrations as low as 100 ng g−1 cells. Nanoceria was not present outside of the vesicles or flowing freely in the cytoplasm and was present exclusively as agglomerates. The size of the nanoceria (including the agglomeration state) greatly affected the amount of cerium incorporated into the cell, with more efficient uptake of larger particles and agglomerates (at the same mass concentration). Particle size was a more important factor in uptake than particle density and total surface area,56 consistent with lack of saturation of processes moving nanoceria across membranes, noted above.
The tissue distribution of inhaled nano- and microsized ceria particles in rats was assessed in a 28 day exposure study.57 Powder aerosolization resulted in comparable MMAD (1.02, 1.17, and 1.40 μm) for the three types of ceria despite marked differences in primary particle size of 5 to 10, 40, and <5000 nm, respectively. At 24 h after a single exposure, approximately 10% of the inhaled dose was measured in lung tissue, in agreement with estimations by a multiple path particle dosimetry model. Each ceria sample also translocated to liver, kidney, spleen, brain, testis, and epididymis after a single 6 h exposure. No consistent differences in pulmonary or extrapulmonary deposition among the nano- and microparticles (as defined by their primary particle size) were observed, most likely as a consequence of their overlapping aerodynamic sizes. Repeated exposure to ceria resulted in low but statistically significant particle accumulation in extra pulmonary tissues. In addition, tissue clearance was shown to be slow, and, overall, insignificant amounts of cerium were eliminated from the body 48 to 72 h post-exposure. In conclusion, no clear effect of the primary particle size or surface area on pulmonary deposition and extrapulmonary tissue distribution could be demonstrated. This is most likely explained by a similar aerodynamic diameter of the ceria particles in air because of the formation of agglomerates and irrespective of possible differences in surface characteristics. This confirms findings58 that it is highly likely that a substantial fraction of the test atmospheres were micron sized when they entered the deep lung.
In an inhalation study in mice, exposure of nanoceria (15 to 30 nm primary diameter and 30 to 50 m2 g−1 specific surface area [SSA]) for 0, 7, 14, or 28 days at an aerosol concentration of 2 mg m−3 resulted in significant bioaccumulation in pulmonary and extrapulmonary tissues, even 30 days after inhalation exposure.59 Despite the small primary diameter, the test aerosol was characterized by a MMAD of 1.4 μm and a wide distribution, as indicated by a geometric standard deviation of 2.4. Although a substantial fraction of these particles would not have been able to reach the lower airways and lungs due to their large size, it is noteworthy that some nanoceria could be distributed through the body and that a slow clearance was reported.
The implications of size were further investigated as the uptake of 15, 25, 30, and 45 nm ceria into human bronchial epithelial cells (BEAS-2B). It was observed that nanoceria penetrated into the cytoplasm and was found in the perinuclear region as aggregated particles. However, there were no clear signs that primary particle size in this small range affected uptake.4
J774A.1 murine macrophages were studied to determine if 3 to 5 nm ceria could be incorporated intracellularly. Nanoceria particles and agglomerates localized in phagosomes and cytosol, suggesting that one mechanism of uptake is through phagocytosis and that the particles may thereafter diffuse into the cytosol. The presence of nanoceria clusters in the outermost edges of the macrophages suggests that single nanoceria particles may also diffuse directly through the membrane.20
Nanoceria was shown to be internalized through clathrin- and/or caveolae-mediated endocytosis in human keratinocytes and lung epithelial cells (BAES-2B) as well as in mouse macrophages (RAW 264.7 cells).60,61 These and other studies demonstrated that nanoceria co-localizes with mitochondria, lysosomes, endoplasmic reticulum, and the nucleus as well as occurring free in the cytoplasm.62–64
The results described above are not very specific for nanoceria and apply to most, if not all, granular low solubility particles. Uptake is largely driven by their physical properties.
Clearance of inhaled particles depends on their site of pulmonary deposition, their physical and chemical properties, and their impact on biological systems. Phagocytosis of particles is the major mechanism of uptake of insoluble structures such as nanoceria into a cell and the ability will vary among various cell types, macrophages being the most active. This is typically a process that occurs in macrophages, but free particle transport, especially of nanosized particles, cannot be excluded. No information is available concerning pleural transfer of nanoceria. Another pathway to remove particles is by dissolution, followed by absorption in the blood. Through this process, cerium ions might be transported to and may accumulate in various organs. Dissolution might occur either within the AMs or in the extracellular fluids. The dissolution rate is proportional to the particle surface: smaller particles are more likely to be dissolved than larger particles. However, nanoceria is not very soluble in lung lining fluid. In addition, many metallic particles (including cerium compounds) have been shown to dissolve and precipitate inside macrophages as phosphate compounds, thus preventing movement into the bloodstream.3
Nanoceria is removed from the lungs to the GI tract through mucociliary clearance.3 Therefore, a primary route of elimination of inhaled nanoceria is through the feces, with small amounts eliminated in the urine.52 Rapid elimination by mucociliary clearance was shown for nanoscale particles in a study in hamsters exposed to ceria aerosols with an aerodynamic diameter of 60 and 110 nm with decreases in initial body burden of 60 and 95%, respectively, 4 days after exposure.52,65
Some insight into the fate and persistence of worker inhalation of ceria particles can be derived from a case report. Cerium-containing particles accounted for 70% of the particles in AMs in bronchoalveolar lavage (BAL) fluid (BALF). Particles containing cerium were seen in interstitial macrophages and elastic fibers of lung tissue 15 years after termination of occupational exposure to a ceria abrasive powder.66 These results suggest prolonged persistence of ceria in the lung, which is supported by a t1/2 of 235 days after whole body exposure inhalation of 25 mg m−3 of NM-212, a 40 nm uncoated commercial nanoceria.67
The implications of nanoceria particle accumulation for systemic toxicological effects after repeated chronic exposure via ambient air are significant and require further exploration to elucidate the extent and time course of such translocation. In addition these observations are of importance to future risk assessment.
Toxicological data from in vivo inhalation toxicity studies: acute exposure (hours up to one day)
There are very little data published on short-term (up to one day) inhalation exposures. A 4 h inhalation toxicity study of nanoceria (55 nm primary size and 30 to 50 m2 g−1 surface area) in rats following OECD TG403 was reported.68 The test atmosphere was characterized as 55 nm and 641 mg m−3 and a MMAD of 2.28 μm. Neutrophils and AMs overloaded with phagocytosed nanoceria were observed along with non-phagocytosed free nanoceria that was deposited over the epithelial surfaces of the bronchi, bronchiole, and alveolar regions of lungs within 24 h of post-exposure, and were consistent throughout the 14 day observation period. Well distributed, multifocal, pulmonary microgranulomas, due to impairment of the clearance mechanism leading to nanoceria biopersistence, were observed at the end of the 14 day post-exposure period. These results suggest that acute nanoceria exposure via the inhalation route may induce cytotoxicity via oxidative stress and may lead to a chronic inflammatory response, but it must be emphasized that the levels of exposures are orders of magnitude higher than one might expect in a work place environment. Another study exposed rats by inhalation 2 h per day for 4 days to 2.7 mg m−3, that was 1% of the total inhaled dose use in the previously described study.69 The MMAD was 281 nm. It is worth noting that this is the only published study that used a truly nanoscale aerosol exposure atmosphere generated using an industry relevant flame spray pyrolysis system (Harvard VENGES system).70 BAL levels of neutrophils increased 6 fold 24 h post-exposure indicating inflammation, while BAL activity of LDH, a biomarker of cell damage and cytotoxicity, increased 2 fold indicating cytotoxicity. These markers of pulmonary response returned to control levels 84 days post-exposure.A single intratracheal dose of 0.15 to 7 mg kg−1 nanoceria was delivered to male Sprague Dawley rats.71,72 Pulmonary inflammation with AMs containing nanoceria was reported. Fibrogenic responses, evidenced by increased hydroxyproline content, a marker for fibrosis, were induced 84 days after the 3.5 and 7 mg kg−1 doses. The results showed that nanoceria induced fibrotic lung injury in rats, suggesting it may cause potential health effects. It was also observed that at these two dose levels the inflammatory effects induced by nanoceria did not return to control 84 days after exposure, suggesting a relatively low clearance rate. In contrast, aspiration of nanoceria (20 μg per mouse) did not affect BAL neutrophil counts or levels of inflammatory mediators (MCP-1 and IL-6).73 It should be noted that after accounting for the rat lung being approximately 10 times larger than the mouse lung, the nanoceria dose used in this study73 was still about 10-fold lower than reported by others.72 This implies that the LOAEL for a single dose to the lung is in the order of 0.6 g kg−1 bw.
Effects observed in kidneys74 suggest there is risk of development of adverse effects at distal sites. Systemic effects were also noted when rats were intratracheally instilled with nanoceria (up to 7 mg kg−1). However, this study suggested that this single high dose exposure only resulted in liver toxicity, as no effects on heart, spleen, and kidney, based on organ weights and serum biochemistry, were reported.75
Toxicological data from in vivo inhalation toxicity studies: sub-acute exposure (several days to weeks)
Preliminary results of a toxicity study in rats were described.76 Exposure by inhalation for 5 days, 6 h per day, to 27 nm MMAD (Gsd 1.6) ceria caused pronounced effects at a mass concentration of 0.14 mg m−3 compared to the aggregated 0.80 um MMAD (Gsd 1.4) nanoceria or the 0.90 um MMAD (Gsd 1.5) microceria. Increased levels of biochemical parameters and leukocytes in BALF were seen. Although the spark-generated method may not be widely employed to commercially produce nanoceria, it provides useful information on the effects of singlet nanoceria particles. In contrast, aggregated ceria particles of similar primary size induced less and milder changes in BALF at 1.4 mg m−3, a more than 10 times higher mass concentration. Similar mass concentration of the 0.90 um MMAD ceria failed to induce any changes in BALF. These results suggest that true nano-sized ceria aerosols are more toxic than such particles in aggregated form and that the latter are more toxic than micro-sized particles of the same material. To what extent this is due to the differences in (local) pulmonary deposition is not yet fully understood.The effects of coating ceria nanoparticles with silica (silicon dioxide; SiO2) were determined.69 Male Sprague–Dawley rats were exposed to 2.7 mg m−3 of silica-coated ceria (21 nm), uncoated ceria (17 nm), or particle-free environments (controls) (2 h per day, 4 days). Interestingly, while animal exposure to uncoated nanoceria induced considerable PMN infiltration (an indicator of inflammation), compared with the control group, PMN levels for the silica-coated scenario were similar to the particle-free control group levels. These results suggest that freshly generated nanoceria particles possess at least some surface chemistry that results in an inflammatory response which can be prevented by SiO2. This is discussed in more detail below in Engineering safer by design nanoceria.
The comparative hazard of two nanoscale (5 to 10 and 40 nm) and one microscale (<5000 nm) ceria previously studied57 were assessed in 28 day inhalation toxicity studies in rats.77 All three materials gave rise to dose-dependent pulmonary inflammation and lung cell damage but without gross pathological changes on the day following the last exposure. Epithelial cell injury was observed 14 to 21 days following nanoceria exposure. There was no evidence of systemic inflammation or other hematological changes following exposure to any of the three particles. The comparative hazard was quantified by application of the benchmark concentration approach.77 The relative toxicity was explored in terms of three exposure metrics. When exposure levels were expressed as mass concentration, one of the nanoceria was the most potent material, whereas when expression levels were based on surface area concentration, micron-sized ceria induced the greatest extent of pulmonary inflammation/damage. Particles were equipotent based on particle number concentrations. In conclusion, similar pulmonary toxicity profiles, including inflammation, were observed for all three materials with little quantitative differences. Systemic effects were virtually absent in a 28 day OECD TG 412 inhalation study in which the effect of 10 μg ml−1 16 nm ceria added to diesel fuel was investigated.50 Of interest, a recent study reported that use of nanoceria as a fuel additive reduced the number of particles generated by a diesel engine and decreased the size of atherosclerotic plaques resulting from inhalation of diesel engine exhaust in a rodent model.50
An inhalation study in mice showed that nanoceria can induce pulmonary and extrapulmonary toxicity.59 Male CD1 mice were subjected to nose-inhalation exposure of nanoceria (15 to 30 nm primary diameter, MMAD 1.4 μm, and 30–50 m2 g−1 SSA) at an aerosol concentration of 2 mg m−3 for 0, 7, 14, or 28 days with 14 or 28 days of recovery time. Markers of lung injury and pro-inflammatory cytokines (IL-1β, TNFα, IL-6, and macrophage inflammatory protein-2) in BALF, oxidative stress in lungs, bioaccumulation, and histopathology of pulmonary and extrapulmonary tissues were assessed. BALF analysis revealed the induction of pulmonary inflammation, evident by an increase in the influx of neutrophils with a significant secretion of pro-inflammatory cytokines. This led to oxidative stress generation and cytotoxicity, evident by induction of lipid peroxidation, depletion of glutathione, and increased BALF LDH and protein. Histopathological examination revealed that inhaled nanoceria was located throughout the pulmonary parenchyma, inducing a severe, chronic, active inflammatory response characterized by necrosis, proteinosis, fibrosis, and well-formed discrete granulomas in the pulmonary tissue and tubular degeneration leading to coagulative necrosis in kidneys. The study also indicated that significant systemic effects were only detected after 28 days and that these responses remained present during the recovery phase. More subtle effects were also reported for shorter durations, and these became worse with longer exposure durations. Pulmonary responses remained evident during the recovery period. From this, one can conclude that due to an imbalance in deposition and clearance, accumulation can occur leading to a (local) dose that causes toxicity and that clearance is rather slow. In other words, there is a risk that long term exposure to low levels of nanoceria may eventually lead to adverse health effects.
Very few studies have assessed the effects of nanoceria exposure in organs other than the lung. The effects of nanoceria (4 nm primary size, 81 m2 g−1 surface area) on microcirculation were studied as the reactivity of isolated rat coronary and mesenteric arterioles.74 Animals received intratracheal exposure (0 to 400 μg per rat suspended in saline with 5% serum; 197 ± 77 nm). BALF and arterioles were collected 24 h post exposure. The authors calculated that the highest dose is equivalent to the deposition in a person performing light work for 30 years and an exposure of 5 μg m−3. Pulmonary cytotoxicity and inflammation were recorded in the 100 and 400 μg exposure groups, whereas microvascular dysfunction was observed at lower levels where no pulmonary responses were recorded. The underlying mechanism for these observations is not yet known, although it appears to involve depletion of endothelium-derived nitric oxide. The fact that pulmonary responses are less sensitive to lung exposure than systemic vascular responses should be considered in risk assessment.
Toxicological data from subchronic inhalation toxicity studies (several weeks to months)
Toxicological data are available from one subchronic inhalation exposure study using Sprague–Dawley rats exposed to microscale ceria (MMAD = 1.8–2.2 μm), geometric standard deviation = 1.8 to 1.9, and at concentrations of 0, 5, 50.5, or 507.5 mg m−3 for 6 h per day, 5 days per week for 13 weeks.52,78 This study observed an increased incidence of alveolar epithelial hyperplasia. Histological examination revealed dose-related alveolar epithelial and lymphoid hyperplasia and pigment accumulation in the lungs, lymph nodes, and larynx of male and female rats at ≥5 mg m−3.52,78 The metaplasia evident in the larynx was interpreted by the study pathologist as adaptive and reversible. This study suggested that exposure to ceria is associated with antigenic stimulation; however, they did not discuss the possibility of non-antigenic stimulation. This study identified a LOAEL of 5 mg m−3 in rats, based on the increased incidence of lymphoid hyperplasia in the bronchial lymph nodes of male and female rats. A NOAEL was not identified.Toxicological data from chronic (>1 year) inhalation toxicity studies
There are no data on the effects of chronic exposure to nanoceria. However, at the request of the German Government, BASF in Germany started in spring 2013 to perform a 2 year exposure study following OECD TG 453 including four concentrations using a nanoceria (NM-212) with a primary particle diameter of 40 nm and a BET surface area of 27 m2 g−1. It is dosed at 0.1, 0.3, 1, and 3 mg m−3. The highest concentration is expected to result in lung overload. Biodistribution and effects will be determined after 3 month, 1 year, and 2 year exposure, with 3 animals per time point. This material has been utilized in other inhalation studies.77Mechanism of action of respiratory toxicity
Toxic effects of nanoceria may be attributed to two actions: chemical toxicity based on the chemical composition, or due to stress/mechanical irritation or stimuli caused by the surface, size, and/or shape of the particles (physical aspects). However, it is not straightforward to differentiate between these two types of cytotoxicity. Solubility greatly affected cell culture response to nanoceria, which was insoluble.79 An in vitro toxicity assay was used to investigate the involvement of the oxidative stress responding signal transduction pathway and transcription factors in the toxicity of nanoceria in BEAS-2B cells.80 They found that nanoceria may exert toxicity through oxidative stress, as it caused significant increases in cellular reactive oxygen species (ROS) concentrations, subsequently leading to strong induction of HO-1 via the p38-Nrf-2 signaling pathway.80 ROS mediated DNA damage and cell cycle arrest are also suggested to play a major role in nanoceria-induced apoptotic cell death in A549 cells.81 Although it was claimed that nanoceria can act as a free radical scavenger due to reactive sites on its surface, prolonged exposure to nanoceria further induces the production of 8-oxoguanine, a marker for oxidative DNA damage. This may explain the nanoparticulate oxidative stress and resulting altered gene expression found in another study.82 As seen in these studies the biological mechanism of nanoceria's action has been a bit contradictory, which may reflect some as yet unknown unique chemical property of this novel material. One suggestion is the method of nanoceria production. It has been observed that room temperature synthesis of nanoceria results in anti-oxidative activity whereas high temperature synthesis often results in pro-oxidative activity.83 Other nanoceria chemical factors as well as biological status may affect the response to nanoceria.What we know
• In several reports, pulmonary exposure to nanoceria has resulted in pulmonary inflammation, alveolar interstitial fibrosis, and alteration in the ability of the systemic microvasculature to respond to dilators. However, other studies report little or no adverse pulmonary response to exposure of the lung to nanoceria but a systematic comparison of the delivered dose is lacking. The vast majority of the published studies have assessed the toxicity of agglomerates of nanoceria, which would classify nanoceria as a low toxic material. The exception is a single study in which rats were exposed to 27 nm ceria particles which appears to result in marked toxicity compared to larger size aggregates.Knowledge gaps
• What aspects of ceria cause toxicity: surface reactivity and aggregate size?• Nanoceria appears to be quite insoluble and persists in the lungs. What is the rate of clearance from the lung and translocation to, and accumulation in, other organs?
Oral exposure
Oral exposure to nanoceria might occur in an occupational setting, which might employ nanoceria in a pure form, or by environmental exposure, such as nanoceria associated with diesel fuel combustion. No relevant literature using acellular or in vitro models was found addressing nanoceria uptake from, or effects on, the GI tract. Particle absorption from the intestine results from diffusion though the mucus layer, initial contact with enterocytes or M (microfold or membranous specialized phagocytic enterocyte) cells, cellular trafficking, and post-translocation events.84 Absorption of colloidal bismuth subcitrate and colloidal (maltodextran) gold particles appeared to occur through regions of gastric epithelial disruption85 or penetration through gaps created by enterocytes that died and were being extruded from the villus.86 Studies with other nanoscale materials have generally shown very low absorption from the GI tract. Absorption inversely correlated with the size of 50 and 100 nm polystyrene87 and 4 to 58 nm colloidal gold nanoparticles.86Oral bioavailability of nanoceria has been studied in rodents.19,46,88 Thirty nm ceria, 0.1 or 5 g kg−1, was administered orally, suspended in water, to male SD rats, which were evaluated 1, 7, or 14 days later.88 No significant changes in hematology or serum biochemistry were observed 1 day later. No deaths; lesions in the liver, lung, or kidney; or signs of inflammation were seen up to 14 days. Cerium concentrations in the liver, kidney, spleen, lung, testis, and brain were greatest in the lung, and decreased over 14 days, whereas cerium concentrations in the other sampled organs did not. Some lung cerium may have resulted from lung uptake during nanoceria administration rather than translocation from the GI tract. Cerium in the sampled organs accounted for ~0.02% of the 0.1 g kg−1 dose. Nanoceria (6.6 nm, 12.8 nm hydrodynamic size; 97% 24 nm and 3% 88 nm in water, ζ = 30 mV) labeled with 141Ce was introduced into the GI tract of male Wistar rats, which were evaluated 1, 3, or 7 days later.46 Of the heart, liver, spleen, kidney, brain, testes, muscle, bone, blood, and lung, liver had the highest 141Ce. Approximately 0.0003% of the dose was accounted for in these 10 tissues and blood, showing very low oral absorption. Nanoceria produced by methods that result in crystalline 3 to 5 nm primary particles20 were functionalized with carboxyfluorescein that did not alter the individual particle size. CD-1 mice were given 0.5 mg kg−1 nanoceria weekly for 2 or 5 weeks suspended in saline via a tube inserted into the stomach through the mouth.19 Mice were evaluated 1 week after the last dose. Approximately 0.2 and 0.025% of the dose was in the lung (that was most likely due to the gavage procedure or aspiration) and spleen, respectively, with less in the liver, kidney (where clusters were seen in lysosomes), and heart. None was detectible in the brain. Approximately 98% of the ceria appeared in the feces. None was detectible in the urine. Compared to 0.03% cerium ion absorption within 3 days after its oral introduction89 and a report of no intestinal reabsorption,36 the oral absorption of nanoceria does not appear to be different from ionic cerium. Nanoceria that had a hydrodynamic diameter of 53 nm but extensively agglomerated in reconstituted hard water was ingested by C. elegans but could not be detected inside cells.90 It can be concluded that the oral bioavailability of nanoceria is extremely low. It is unlikely that a sufficient amount of intact nanoceria would be absorbed to induce effects at distal sites.
What we know
• Nanoceria absorption from the GI tract is extremely low.Knowledge gaps
• There is no information about repeated, low-dose exposure regarding accumulation of nanoceria and potential effects.• The influence of gastric acidity and GI juices on nanoceria dissolution and absorption has not been reported.
Dermal exposure
In the absence of organic solvents or stretching of the skin, there does not appear to be any good evidence that topically-applied nanomaterial can penetrate through skin into circulatory or lymphatic circulation.91 A commercial 9 nm primary particle size nanoceria intended for use as a diesel fuel additive (Envirox®) was tested using an epidermis model (EpiDerm® System) that contains human-derived epidermal keratinocytes.92 An unreported concentration initially increased cell metabolism (MTT assay) 19% after 960 min exposure, then decreased it 32 and 35% after 1200 and 1440 min. Based on comparison to a reference material (stated to be 20% SLS; whereas the usual positive control is 5% SLS (sodium lauryl sulfate)), that reduced cell viability by 42% in 15 and 94% in 120 min, the authors concluded that their nanoceria did not have potential to be an in vivo skin irritant. However, 0.06 g l−1 of 7 nm ceria decreased viability of human dermal fibroblasts and produced concentration-dependent genotoxicity from 0.00005 to 0.02 g l−1.28What we know
• Based on limited data, there is no evidence for nanoceria uptake through the skin or that it is a skin irritant.Knowledge gaps
• There is very limited published data on the kinetics and effects of nanoceria interaction with skin.Ocular exposure
No information was found on the effects of exposure of the surface of the eye (cornea) to nanoceria. Intravitreal injection of 344 ng of nanoceria into each eye of rats resulted in a much higher concentration in the retina than lens or eyecup tissue and an estimated t1/2, determined from results obtained up to 1 year after the injection, of 525 days in the eye and 414 days in the retina.40What we know
• Nanoceria is retained in the eye for at least a year.Knowledge gaps
• The extent of corneal exposure to nanoceria, and potential adverse outcomes, are unknown.Toxicological data from in vitro toxicity studies using cells
The cytotoxicity of nanoceria to human mesothelioma (MSTO-211H) and rodent fibroblast (3T3) cells was determined by measuring metabolic activity and cell proliferation. Human mesothelioma cells were found to be more sensitive than rodent fibroblast cells when exposed to 6 nm ceria at different concentrations; metabolic activity and DNA content decreased approximately 50% after 3 days of exposure. The mesothelioma and fibroblast cells were, however, not completely killed at 30 μg mL−1. After 6 days, the metabolic activity was not significantly altered and DNA content was increased slightly in the mesothelioma cells. In the rat fibroblasts, the DNA content increased slightly and metabolism was not significantly affected.79 Nanoceria toxicity was also investigated using the human BAL carcinoma-derived alveolar type II cell line A549, which was exposed to 20 ± 3 nm ceria at 3 concentrations and durations.93 Cell viability decreased at all 3 concentrations over 3 days dose- and time-dependently. The elevated ROS levels, increased lipid peroxidation, increased membrane damage, and reduced antioxidant levels are evidence of oxidative stress caused by nanoceria exposure.93The cytotoxicity of several metal oxide nanoparticles was compared using A549 cells.4 The test materials fell into 2 distinct categories based upon surface area: those with a surface area of 500 to 600 m2 g−1, including titanium dioxide and aluminum oxide, and those with a surface area of 50 to 60 m2 g−1, which included copper oxide, ceria, and iron(III) oxide. Unfortunately, no information on the size or shape of these particles was presented. They used a synthetic model of human respiratory-tract lining fluid as well as an ascorbate-only model. The experiment was repeated with soluble chloride salts. The chloride salts were more toxic than the metal oxide forms, but there was no effect on cell viability of nanoceria or soluble cerium chloride at concentrations up to 1000 μM. Nanoceria did not cause any detectable cytotoxicity to epithelial cells based on lactate dehydrogenase (LDH) leakage and did not have a significant stimulating effect on IL-8, which is typically involved in acute inflammation caused by particles.4 Ceria had, like titanium oxide and aluminum oxide, very low effects on antioxidant depletion, whereas copper oxide caused a 50 times higher depletion than the other metal oxides, most likely due to its solubility and, therefore, the availability of free metal ions.
Exposure of A549 cells to nanoceria for 24 h did not affect cell viability.69 A physiological model of the air–liquid interface of A549 cells was reported.82 For the exposure to nanoceria (mean primary particle diameter of 5 to 20 nm), the culture plates were opened for 10, 20, and 30 minutes. LDH measurements and cell morphology did not show a difference between exposed and control cells. The mean total lamellar body (storage sites for alveolar surfactant synthesized by alveolar type II cells) volume per cell was found to be significantly lower in cells exposed for 30 minutes compared to control cells and cells exposed for 10 minutes. Another effect was the reduction of cell–cell contacts after exposure, but the mechanism for this increased epithelial permeability was unknown. Possibly, this indicates a disorganization of the tight junctions rather than dying cells. Prolonged exposure to nanoceria further induced the production of 8-oxoguanine. This may explain the nanoparticulate oxidative stress and resulting altered gene expression found in other studies.82
The cytotoxic effect of nanoceria was assessed using BEAS-2B cells, compared to other cell lines (T98G and H9C2, derived from human brain fibroblasts and rat cardiomyocytes, respectively).94 BEAS-2B cell viability was decreased by treatment with nanoceria (15 to 45 nm) in a time- and dose-dependent manner. Differences in nanoceria size had no effect on cell viability. No cytotoxic effects were found for T98G and H9C2 cells, suggesting that nanoceria has cell-type dependent modes of action. ROS were generated in a dose-dependent manner by 5, 10, 20, or 40 μg ml−1 30 nm ceria particles in BEAS-2B cells. The levels of GSH were decreased in the nanoparticle-treated groups. Caspase-3, which plays a key role in the apoptotic pathway, was increased following treatment with 5, 10, 20, or 40 μg ml−1 30 nm ceria. Increased levels of oxidative stress related genes (catalase, glutathione S transferase, HO-1, and thioredoxin reductase) and chromatin condensation were also observed.94 The effects of nanoceria on BEAS-2B cells were also evaluated in another study.61 Unlike zinc oxide, nanoceria did not generate oxidant stress or affect cell viability.
A different cell type, human aortic endothelial cells (HAECs), was studied to investigate systemic inflammation, since this is critical in the development of cardiovascular pathology.95 HAEC cells were incubated for 4 h with different concentrations of nanoceria (mean size 44 nm). The exposure produced modest mRNA upregulation of the inflammatory markers IL-8 and monocyte chemotactic protein-1 that was to some extent concentration dependent. There was no significant cell loss at any of the concentrations studied.95
No differences were found in cell morphology of J774A.1 murine macrophages between nanoceria-treated cells and controls.20 Likewise, there were no significant differences between apoptosis levels of control and nanoceria-treated cells, and no toxic effects to cells up to 10 μM nanoceria.20 It was demonstrated that higher toxicity was observed with nanoceria localized with lysosomes as compared to the cytoplasm.96
The biological impact of engine emissions using a nanoceria fuel additive compared to that of a reference fuel was determined using an organotypic culture of lung slices from female Wistar rats.97 The impact of 10 nm ceria was tested alone. The authors concluded that there was no impact of nanoceria aerosol on lung tissue viability, glutathione dependent metabolism, superoxide dismutase activity, or pro-inflammatory reactions. Increased catalase activity was observed for the 3 concentrations tested, but this did not induce any loss in cell viability. The authors concluded that the biological effects of the nanoceria fuel additive are very limited. The observed trend on organotypic culture viability and TNF-α was viewed as beneficial, while a limited oxidant activity was observed through catalase induction.97
Eight experiments were conducted using organotypic cultures of rat lung slices by exposing the cells to a continuous flow of nanoceria aerosol (mean particle agglomerate diameter 140 nm) over 3 h.4 No effects were found on the viability of the lung tissue slices, glutathione-dependent metabolism, superoxide dismutase activity, or pro-inflammatory factors.4 It is unclear just how well or to what extent nanoceria reached key pulmonary effector cells in these ex vivo organ studies employing lung slices.
Surface charge is an important factor that has been shown to modulate nanoceria toxicity. Negatively charged nanoceria preferentially entered cancer cells and localized in lysosomes while positively charged and neutral particles were able to enter cancer as well as normal cells and mostly localized in the cytoplasm.96 Moreover negatively charged nanoceria have been shown to induce human lens epithelial cell DNA damage.98,99 In that work, it was demonstrated that, in addition to surface charge, time of exposure had greater impact on DNA damage than particle dose (10 μg ml−1 nanoceria exposure for 72 vs. 48 h exposure at 100 μg ml−1). Other studies also point towards the fact that longer duration exposures may be needed to observe nanoceria-induced toxic responses. Dose (3.5 to 23.3 μg ml−1) as well as exposure duration (24 to 72 h) significantly contributed to the toxic effect of nanoceria towards A549 cells.93 Nanoceria exposure decreased glutathione and alpha-tocopherol levels and increased malondialdehyde and LDH levels.93 In addition to dose dependence (0 to 10 μg ml−1), nanoceria-induced toxicity was time dependent; i.e., 48 h exposure to nanoceria induced significant apoptosis and autophagy in human peripheral blood monocytes whereas 24 h exposure resulted in a very mild toxic response.63 Significantly higher toxicity (decreased mitochondrial activity and DNA content) was reported after 3 to 6 day exposures of MSTO-211H cells or 3T3 fibroblasts to 0 to 30 μg ml−1 nanoceria.79 Nanoceria-induced toxicity was comparable to other non-soluble metal oxide nanomaterials studied (titanium dioxide and zirconium dioxide) but was far less than crocidolite asbestos (fiber toxicity) or zinc oxide nanoparticles (solubility-driven toxicity). Moreover, there was cell type specificity in the persistence of the toxic response, since MTSO cell responses persisted for 6 days while partial recovery of 3T3 cells started after 3 days. Nanoceria was shown to preferentially kill SCL-1 squamous carcinoma cells through ROS production and oxidation of proteins at concentrations (150 μM), which were not toxic to dermal fibroblasts.100 Nanoceria of two sizes (6 and 100 nm) induced size-dependent gene expression changes and apoptosis, inhibition of G1/S transition, as well as growth inhibition in murine HT22 hippocampal nerve cells.101
However, others did not observe any toxic response after nanoceria exposure in BEAS-2B cells, but rather cytoprotective effects and suppression of exogenously-induced oxidative stress.61 Nanoceria also had protective effects when cultured cardiomyocytes were exposed to cigarette smoke extracts.102 Similarly, it was shown that nanoceria protected against hydrogen peroxide-induced apoptosis in a human breast fibrosarcoma cell line (HT-1080 cells) and had no hemolytic potential towards peripheral red blood cells from healthy human volunteers.103 Moreover, it was shown that nanoceria-supported gold nanoparticles exerted higher antioxidant effects than nanoceria particles alone or glutathione in Hep3B and HeLa cells.104 Some other in vitro studies also demonstrated no toxicity of nanoceria.20,73,105
Differences in production methods, particle shape (sharp vs. round edges), and particle size are among many differences in the studies showing oxidative vs. antioxidative effects of nanoceria. We have mentioned above how production method differences lead to differences in physicochemical characteristics, differential uptake and biological responses. Moreover, it has already been shown that the ability of nanoceria to agglomerate in water and cell culture media (RPMI1640) is inversely proportional to their primary particle diameter.106 This could have important repercussions in the nanoceria uptake pathway and handling by cells, resulting in different biological outcomes.
In general, nanoceria can be classified as a substance with very low acute toxicity in realistic exposure levels (albeit in vitro).
Interaction of nanoceria with biological molecules (acellular effects)
Only a few studies have described the interactions of nanoceria with biologically-relevant molecules.107–111 The ability of nanoceria to cleave the phosphate ester bond in p-nitrophenyl phosphate, ATP, and o-phospho-L-tyrosine was demonstrated.109 However, they observed that nanoceria did not dephosphorylate DNA. The authors concluded that the ability to perform such reactions is related to the availability of Ce3+ (not Ce4+) sites on the surface of the particle. They further proposed that DNA may interact with nanoceria in a similar manner to nucleosomes, by wrapping around the nanoparticle.108 However, contrary to these findings, others observed that Ce4+ is able to break phosphorous–oxygen bonds (e.g. in DNA and cAMP).107,112 It was demonstrated that nanoceria exhibits ATPase activity (in acellular conditions as well as inside human vascular endothelial cells), which depends upon its physicochemical properties (including Ce3+/Ce4+ ratio, morphology, and oxygen extraction energy) and surface modifications (hexamethylenetetramine used in its production).113 These authors suggested that ATPase activity should be considered when synthesizing nanoceria for therapeutic applications. Moreover, it was demonstrated that nanoceria catalytic behavior is significantly altered in the presence of phosphate anions.110 As an abundant amount of inorganic phosphate anions are present in biological systems, it was postulated that the catalytic behavior of nanoceria may be significantly altered under physiological conditions. It was demonstrated that nanoceria adsorbs Ca2+ from cell culture medium and is able to transfer it into cells.114 This resulted in the activation of calcium-dependent cysteine proteases (calpains) inside the cells.114 Five commercially-available nanocerias reacted with dopamine.111 Results of spectroscopic and surface characterization methods suggested nanoceria oxidized dopamine followed by chemisorption of the oxidized dopamine. This reduced free dopamine, which could have potential neurological consequences.These findings demonstrate the possibility of not only changes in nanoceria reactivity but also changes in biological molecules after nanoceria contact. As the cellular environment is much more complex inside the organism, the possibility of these, and similar, reactions with other biological molecules (e.g. with lung lining surfactants, enzymes, etc.) could be amplified, resulting in unanticipated outcomes. Such interactions could thus significantly impact biological outcomes and may be the reason for observed discrepancies between in vitro and in vivo findings.69
Mechanisms of nanoceria-induced toxicity
The signaling mechanism behind the oxidative response induced by nanoceria was revealed to be through induction of HO-1 via the p38-Nrf-2 signaling pathway.80 The following chemical reactions for the oxidative stress and lipid peroxidation produced by nanoceria were proposed, where Ared− is a physiologically relevant reductant like thiol or ascorbate and LOOH is lipid peroxide.93| Ce4+ + Ared− → Ce3+ + Aox |
| Ce3+ + O2 → Ce4+ + O2− |
| O2− + O2− → O2 + H2O2 |
| H2O2 + Ce3+ → Ce4+ + OH− + OH˙ |
| LOOH + Ce3+ → Ce4+ + LO˙ + OH− |
Another study with a commercial nanoceria product (7 nm ellipsoidal monocrystallites of cerianite ((HNO3)0.5(H2O)), Rhodia Chemicals, France) and human infant foreskin fibroblasts (transfected human fibroblast mutants with varying levels of NADPH oxidase activity), and murine 3T3 fibroblasts, showed that nanoceria can act as a catalyst in nucleophilic-type reactions, forming the perhydroxyl radical HOO˙, the conjugate acid form of superoxide (O2˙).115
Based on the available literature, the following mechanisms can be proposed for the potential toxic effects of nanoceria:
• Oxidative stress-independent mechanisms of nanoceria toxicity relate to formation of agglomerates in cell culture media, persistence/accumulation of ceria inside cells/organelles, and damage to the organelles. This accumulation in intracellular organelles leads to release of proteases (like cathepsins) and results in the activation of inflammatory and cell death pathways. Indeed, we have recently demonstrated that nanoceria induced apoptosis of human peripheral blood monocytes through oxidative stress-independent mechanisms involving modulation of autophagy and phagolysosomal accumulation (Fig. 2).63 These mechanisms are particularly plausible for high aspect ratio nanoceria. Significant contribution of length (≥200 nm) and aspect ratio (≥22) were demonstrated in the toxicity (progressive pro-inflammatory and cytotoxic response) of ceria nanorods and nanowires towards THP-1 cells.116 The mechanisms behind these responses included lysosomal damage, NLRP3 inflammasome activation, and IL-1β release. Nanoceria affected redox-dependent apoptosis through the presence of Ce3+ ions.117 It was further demonstrated that oxygen vacancies do not contribute to this response. These results further confirm the notion that nanoceria-induced oxidative stress-independent effects would not benefit from its valance configuration (Ce3+/Ce4+ redox reactions) and are one plausible explanation for the toxicity of nanoceria.
 | ||
| Fig. 2 Mechanism of nanoceria-induced toxicity in human peripheral blood monocytes. Nanoceria induces programmed cell death (apoptosis) through a mitochondrial damage pathway (involving activation and mitochondrial localization of Bax, decreased mitochondrial membrane potential (ΔΨm), and overexpression of apoptosis inducing factor (AIF)). Nanoceria-induced autophagy was characterized by increased LC3B-stained autophagic vesicles, monodansylcadaverine-stained autophagosomes/autolysosomes and increased lysosomal production of LC3b+ phagolysosomes. Inhibition of p53 using its pharmacological inhibitor (pifithrin-α) increased the autophagy. Inhibition of autophagy through wortmannin decreased the cytotoxicity, indicating a pro-death role of autophagy. | ||
• Mechanisms of toxicity related to surface valance state configuration change after being in contact with biological molecules. A significant reduction of Ce4+ and appearance of Ce3+ on the surface (i.e. a more redox active state) was proposed to be the basis of genotoxicity towards human fibroblasts.28 These authors observed a significant reduction in surface Ce4+ after interaction with organic molecules in biological media (complemented DMEM). Genotoxicity was observed at 2 to 3 orders of magnitude lower doses than needed to induce cytotoxic effects. Appearance of more Ce3+ on the surface of nanoceria has already been described as a cause of nanoceria-induced contact toxicity of bacteria.118 Indeed, it was reported that increased angiogenesis induction by nanoceria is directly related to the Ce3+/Ce4+ ratio.119
• Short term exposure to nanoceria leads to the induction of phase 2 antioxidant enzymes such as HO-1 and a persistent long-term exposure would result in defective antioxidant defense leading to reactive oxygen species production and toxicity.
• Enzyme mimetic activity of nanoceria, e.g. ATPase-like activity, may contribute to nanoceria toxicity.
• Either one or a combination of these mechanisms can occur in a given situation and may result in toxic effects.
What we know
• Nanoceria induces toxicity through similar mechanisms induced by other low solubility nanomaterials and/or through its unique surface chemical nature. These mechanisms may or may not mediate nanoceria's many beneficial effects, which are described.12 Mechanisms potentially mediating the beneficial and adverse effects are also described in this issue.43 However, identification of the mechanisms contributing to nanoceria toxicity vs. its beneficial effects are not yet clearly delineated.Knowledge gaps
• Existing literature points towards the interaction of nanoceria with biological molecules. Unfortunately, the exact mechanism of these interactions and their impact on the observed outcome under in vitro conditions are not known.• There is a lack of understanding about the so-called “higher sensitivity” of cancer cells to nanoceria as compared to normal cells. One possible explanation could be higher metabolic and growth rates of these cells, making them more susceptible to nanoceria-induced effects.
• Primary cells also demonstrate differential responses to nanoceria. The reasons for this cell type specificity are not clearly understood. It may be that they lie with nanoceria characteristics, the biological environment, cell surface receptors of the cells, or some combination of these.
• It is not known how increased redox activity of nanoceria through increased Ce3+ may lead to toxicity.
• The modifying effects of the protein corona on nanoceria-induced toxicity are not well understood (as is the case for most nanomaterials).
• In vitro studies cannot accurately represent kinetics and biodistribution in a complex organism, and thus little is known about distant or secondary effects of nanoceria exposure.
• It is unknown what mediates distal effects from the site(s) of nanoceria accumulation, where there is little/no nanoceria.
• Responses of environmentally-relevant nanoceria interacting with all the modifications taking place in the “natural” environment (e.g. catalytic converters and diesel exhausts) are not known.
Nanoceria- and cerium-induced human toxicity
No reports were found documenting toxicity to humans from nanoceria or cerium, although cerium has pharmacological properties that could contribute to adverse effects. Cerium has been reported to interfere with calcium function, including an anticoagulant effect, by inhibition of mitochondrial membrane calcium transport via binding to Ca2+/Mg2+-ATPase, inhibition of neuronal low voltage-activated (T-type) calcium channels, binding to calmodulin calcium-binding sites, and substituting for calcium in the regulation of calcium/CaM-dependent enzymes, contributing to reduced cardiac and skeletal muscle contractility. It has the ability to inhibit immune responses.120Engineering safer by design nanoceria
If we have sufficient information to identify the toxicological risk factors, and if they are amenable to incorporation into the design of nanoceria, safer by design becomes possible. The surface properties of nanomaterials are believed to be very important factors influencing response to nanomaterials. Assessing and understanding the interactions of nanoceria with environmental and biological systems, as well as identifying means to reduce potential toxicological outcomes, are pivotal to the sustainability of industries employing nanoceria in their products. However, while significant research has been directed toward understanding nano–bio interactions, research toward devising safer engineered nanomaterial (ENM) formulation concepts that can be readily adopted by the nanotechnology industry is very sparse.121,122A promising approach in this regard is coating potentially toxic ENMs with a nanothin layer of biologically and environmentally inert material. The challenge is to generate “core–shell” ENMs that exhibit the biologically inert surface properties of their shell while preserving certain important intrinsic functional properties (i.e. optical, magnetic, plasmonic, and phosphorescence properties) of their core materials.
Recently, a “safer formulation concept” was developed at Harvard and tailored to a family of ENMs with the highest volume of production,123 namely, flame-generated nanomaterials. ENMs are coated in-flight with a nanothin layer of amorphous silica. Gas phase (flame aerosol) processes are the preferred routes for scalable ENM synthesis as they do not create liquid by-products, offer easier particle collection from gases than liquids, usually include fewer process steps, and result in high purity materials with unique morphologies, including the synthesis of metastable phases.124 However, it is worth pointing out that wet synthesis methods result to more uniform sizes of nanomaterials compared to flame synthesis of nanomaterials. Flame-made nanoparticles typically yield lognormal particle size distributions with a geometric standard deviation of 1.45.125 Amorphous silica has a reduced toxicological footprint and is often used as a negative control material in in vitro ENM screening assays.79 Nanothin silica therefore has potential to shield otherwise potentially toxic core materials from interactions with environmental and biological systems. It was demonstrated in many studies that coating nanoparticles with silica preserves certain important intrinsic functional properties (i.e. optical,126 magnetic,127,128 plasmonic,129 and phosphorescence130) of the core materials.
The applied silica-coating process in flames has major advantages over wet-methods, such as sol–gel,131 and reverse micro-emulsion,132 which are frequently applied to the synthesis of silica-coated nanoparticles by industry. These low yield, multi-step, wet synthesis methods often produce porous silica coatings,133 which cannot sufficiently protect surrounding media from any potential toxic implications of the core materials. A flame-based silica-coating process has recently been explored as a means of high yield scalable nanomanufacturing of silica-coated ENMs, such as titanium oxide, iron oxide, zinc oxide, ceria, and silver.69,134
Silica encapsulation strategy for nanoceria
Fig. 3(a) and (b) illustrate the proposed approach and underlying theory to the one-step nanoparticle synthesis, and in-flight silica-encapsulation, respectively. In brief, core nanoparticles are formed in the gas phase by flame spray pyrolysis of organometallic compounds dissolved in high enthalpy solvents.135 The freshly formed core nanoparticles pass through the silica coating region, where they are encapsulated with a nanothin (1–5 nm) amorphous silica layer by the swirl-injection of hexamethyldisiloxane (HMDSO) vapor-laden N2. | ||
| Fig. 3 ENM synthesis and silica coating fundamentals. (a) A schematic of the coating reactor highlighting the most important elements. Core ENMs are formed through the combustion, by a flame spray pyrolysis (FSP) burner of organometallic compounds dissolved in high enthalpy solvents. The HMDSO vapor is injected into the particulate flow in an in-line silica-coating reactor. (b) The mechanism of the coating and the possible results. The high temperatures obtained in the core particle synthesis provide the necessary energy for the HMDSO conversion to silica vapor. Under optimal silica vapor supersaturation conditions, the silica vapor nucleates heterogeneously onto the core ENM surface, thereby forming a desired silica coating layer of a controlled thickness. Reprinted, with permission, from ref. 69. | ||
Ceria and silica-coated nanoceria synthesis
Uncoated and silica-coated nanoceria particles were synthesized by flame spray pyrolysis of Ce3+ ethylhexanoate (0.05 M) dissolved in xylene and cerium3+ ethylhexanoate (0.04 M) dissolved in xylene:ethylhexanoate (3:1), respectively, as described in detail.69,134
Ex situ characterization of generated ENMs
Fig. 4 shows TEM and SEM images of nanoceria and SiO2-coated nanoceria samples collected in situ (panels c–f). The images illustrate the fractal structure of the agglomerates formed by the flame synthesis method. In the case of silica-coated nanoceria, a smooth and relatively homogenous 2 to 4 nm silica coating layer is visible around the core ceria particles at higher magnification (Fig. 4(g)). The hermetic silica encapsulation of nanoceria was also evaluated using X-ray photoelectron spectroscopy (Fig. 4(b)). As shown in this figure, the Ce 3d electron transition almost entirely disappears in the silica-coated nanoceria survey spectra. Small Ce 3d peaks that remain visible in the silica-coated nanoceria spectra can be attributed to a bulk Ce signal.136 This is in agreement with previous data showing coating thicknesses between 2 to 3 nm resulted in close to hermetic encapsulation of the core nanoceria.134Fig. 4(a) shows X-ray diffraction (XRD) patterns for nanoceria and silica-coated nanoceria. The location of the peaks is identical for both materials, indicating that the coating had minimal effect on core composition and crystal structure. XRD-determined crystal size of uncoated ceria (17.3 nm) was slightly smaller than the silica-coated ceria (21 nm). The SSA of uncoated ceria was 61 m2 g−1 and that of silica-coated ceria was 50 m2 g−1, corresponding to equivalent diameters of 12.8 and 19.2 nm, respectively. Differences between nanoceria and silica-coated BET equivalent diameter are more pronounced than crystal sizes due to silica encapsulation, which is not accounted for when measuring the crystal size. | ||
| Fig. 4 Physicochemical ENM characterization. XRD (a) and XPS (b) spectra of nanoceria (black) and silica-coated nanoceria (red) (dXRD: crystal size measured by X-ray diffraction). In situ SEM (c, d) and TEM (e, f) images of nanoceria and silica-coated nanoceria. Higher resolution TEM image of silica-coated nanoceria (ex situ); 2 to 4 nm silica coating appears as lighter contrast around darker contrast core nanoceria particles (g). Reprinted, with permission, from ref. 69. | ||
In vivo toxicological characterization of ENMs prepared by the safer by design approach
An animal model was used to assess the effect of the silica coating on the toxicological profile of inhaled nanoceria. In brief, male Sprague–Dawley rats were exposed to 2.7 mg m−3 of silica-coated nanoceria, uncoated nanoceria, or a particle-free environment (controls) (2 h per day, 4 days). Twenty-four h post exposure the animals were sacrificed.Alveolar phagocytes and biomarkers of injury and inflammation were obtained by BAL. Complete pathophysiological analysis was performed. Inflammatory and cytotoxicity biomarkers, including cell counts of PMNs and AMs, LDH activity, and albumin content, were measured in the BALF.
Fig. 5 summarizes inflammatory and cytotoxicity biomarkers in the BALF of the animals sacrificed 24 h post exposure. While exposure to uncoated nanoceria induced considerable PMN infiltration compared to the control group, an indicator for inflammation, PMN levels for the silica-coated scenario were similar to the particle-free control group levels (Fig. 5(b)). Similarly, LDH release from the uncoated nanoceria-exposed group was significantly higher than the particle-free exposed control group, as shown on Fig. 5(d). In contrast, LDH release in the silica-coated nanoceria-exposed group was nearly identical to that of the particle-free exposed control group, clear evidence that silica coating results in reduced toxicity (Fig. 5(d)). Furthermore, neither nanoceria nor silica-coated nanoceria exposure appeared to cause air/capillary damage, as shown by similar albumin levels for silica-coated, uncoated nanoceria, and the particle-free control animal group (Fig. 5(c)). This is compelling evidence that the silica encapsulation significantly reduced particle toxicity. Other than a demonstration that titanium doping of nanoceria reduced its superoxide dismutase activity,137 there do not appear to be any reports of the biological effects of nanoceria doping.
 | ||
| Fig. 5 In vivo toxicity. Alveolar macrophages (AM) (a), polymorphonuclear neutrophils (PMNs) (b), albumin (c), and lactate dehydrogenase (LDH) (d) levels in bronchoalveolar lavage (BAL) of rats sacrificed 1 day post-exposure. Error bars represent standard error, * represents p value <0.05, Transmission electron microscopy (TEM) images of AM recovered from the BAL of rats instilled with nanoceria (e) and silica-coated nanoceria particles (f). Reprinted, with permission, from ref. 69. | ||
Knowledge gap
• The stability of the silica coating in vivo and potential risk from chronic silica-coated nanoceria exposure needs to be assessed.Safer by design conclusions
The versatile and scalable process outlined here enables the one step, inflight, hermetic encapsulation of nanoceria with a nanothin silica layer. Preliminary toxicological evidence demonstrates the ability of the proposed concept to reduce the toxicological profile of nanoceria, while maintaining the functional properties of the core materials. The described concept bears great promise for large-scale industrial application as a means of effectively inhibiting nanoparticle toxicity.Research recommendations
1. Address why some studies report toxic effects of nanoceria while others report that nanoceria is of low toxicity and exhibits antioxidant properties. Are these differences due to:a) valance state or synthesis temperature of the nanoceria;
b) the physicochemical properties of the sample tested;
c) the dose, exposure route, or experimental model used?
2. Evaluate the initial site(s) of deposition, clearance, translocation, elimination, and biological effect of nanoceria given by various exposure routes (IV, IP, oral, inhalation). How would various physicochemical properties affect such biokinetics?
3. Determine the effects of interaction of nanoceria with biological fluids (blood, lung lining fluid, etc.) on agglomeration, surface reactivity, and bioactivity.
4. Compare the effects of short-term/high-dose exposure to long-term/low-dose exposure of the same total burden. Determine no effect levels.
5. Characterize the effects of co-exposure (diesel plus cerium fuel additive) on pulmonary and cardiovascular response.
6. Develop predictive in vitro tests that:
a) use doses relevant to in vivo exposure;
b) evaluate mechanisms other than oxidant stress;
c) evaluate genotoxicity and induction of cell proliferation/transformation.
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health.Abbreviations
| ALT | Alanine aminotransferase |
| AM | Alveolar macrophages |
| BAL | Bronchoalveolar lavage |
| BBB | Blood–brain barrier |
| BEAS-2B cells | Human bronchial epithelial cells |
| BET | Brunauer, Emmett and Teller surface area determination |
| EDTA | Ethylenediaminetetraacetic acid |
| EELS | Electron energy loss spectrometry |
| ENM | Engineered nanomaterial |
| FTIR | Fourier transform infrared spectroscopy |
| GI | Gastrointestinal |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GSH/GSSG | Reduced/oxidized glutathione ratio |
| H & E | Hematoxylin and eosin stain |
| HAEC | Human aortic endothelial cells |
| HNE | Protein-bound 4-hydroxy 2-transnonenal |
| HO-1 | Heme oxygenase-1 |
| Hsp70 | Heat shock protein 70 |
| iNOS | Inducible nitric oxide synthase |
| LDH | Lactate dehydrogenase |
| LC-3 AB | Antibody to light chain 3, an autophagy marker |
| MCP-1 | Monocyte chemoattractant protein-1, an inflammatory marker |
| MMAD | Mass mean aerodynamic diameter |
| MPS | Mononuclear phagocyte system |
| 3NT | 3-Nitrotyrosine |
| OECD TG 412 | Organisation for Economic Co-operation and Development, Guideline for the Testing of Chemicals: Subacute Inhalation Toxicity: 28-Day Study |
| OECD TG 453 | Test Guideline 453: Combined Chronic Toxicity\Carcinogenicity Studies |
| ROS | Reactive oxygen species |
| SEM | Scanning electron microscope |
| SOD | Superoxide dismutase |
| SSA | Specific surface area |
| TEM | Transmission electron microscopy |
| XRD | X-ray diffraction |
Acknowledgements
This critical review is a product of a workshop on nanoceria held November 2, 2013 at Fess Parker's Doubletree Resort, Santa Barbara, California which was made possible by financial support from the Sustainable Nanotechnology Organization; NSF grant CBET-1343638 to UCSB; and the Tracy Farmer Institute for Sustainability and the Environment, Department of Pharmaceutical Sciences, Office of the Vice President for Research, and Associate Dean for Research of the College of Pharmacy, University of Kentucky. The authors' research was supported (in part) by US EPA STAR grant RD-833772; the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences; NSF grant 1235806; NIEHS grant P30ES000002; and the Ministry of Infrastructures and Environment, the Netherlands.References
- N. J. Lawrence, J. R. Brewer, L. Wang, T.-S. Wu, J. Wells-Kingsbury, M. M. Ihrig, G. Wang, Y.-L. Soo, W.-N. Mei and C. L. Cheung, Nano Lett., 2011, 11, 2666–2671 CrossRef CAS PubMed.
- B. R. Powell, R. L. Bloink and C. C. Eickel, J. Am. Ceram. Soc., 1988, 71, C104–C106 CrossRef CAS PubMed.
- M. G. Costantini, Evaluation of human health risk from cerium added to diesel fuel, Health Effects Institute, 2001, vol. 9, p. 64 Search PubMed.
- B. Park, K. Donaldson, R. Duffin, L. Tran, F. Kelly, I. Mudway, J. P. Morin, R. Guest, P. Jenkinson, Z. Samaras, M. Giannouli, H. Kouridis and P. Martin, Inhalation Toxicol., 2008, 20, 547–566 CrossRef CAS PubMed.
- V. D. Kosynkin, A. A. Arzgatkina, E. N. Ivanov, M. G. Chtoutsa, A. I. Grabko, A. V. Kardapolov and N. A. Sysina, J. Alloys Compd., 2000, 303–304, 421–425 CrossRef CAS.
- N. N. Dao, M. D. Luu, Q. K. Nguyen and B. S. Kim, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2011, 2, 045013 CrossRef.
- D. Bello, J. Martin, C. Santeufemio, Q. Sun, K. L. Bunker, M. Shafer and P. Demokritou, Nanotoxicology, 2013, 7, 989–1003 CrossRef CAS PubMed.
- K. Reed, A. Cormack, A. Kulkarni, M. Mayton, D. Sayle, F. Klaessig and B. Stadler, Environ. Sci.: Nano, 2014, 1 10.1039/C4EN00079J.
- H. Zhang, X. He, Z. Zhang, P. Zhang, Y. Li, Y. Ma, Y. Kuang, Y. Zhao and Z. Chai, Environ. Sci. Technol., 2011, 45, 3725–3730 CrossRef CAS PubMed.
- Integrated Laboratory Systems Inc., Chemical information profile for ceric oxide [CAS no. 1306-38-3]. Supporting Nomination for Toxicological Evaluation by the National Toxicology Program, Research Triangle Park, NC, 2006, p. 21 Search PubMed.
- Organisation for Economic Co-operation and Development, List of manufactured nanomaterials and list of endpoints for phase one of the sponsorship programme for the testing of manufactured nanomaterials: Revision, in Series on the safety of manufactured nanomaterials, 2010, Number 27, ENV/JM/MONO(2010)46 Search PubMed.
- S. Das, S. Seal, J. Erlichman, K. Heckman, E. Traversa, J. McGinnis and W. T. Self, Catalytic properties and biomedical applications of cerium oxide nanoparticles, presented at the SNO Special Workshop I on Nanoceria, to be submitted to Environ. Sci.: Nano Search PubMed.
- R. A. Yokel, R. L. Florence, J. M. Unrine, M. T. Tseng, U. M. Graham, P. Wu, E. A. Grulke, R. Sultana, S. S. Hardas and D. A. Butterfield, Nanotoxicology, 2009, 3, 234–248 CrossRef CAS.
- M. Dan, M. T. Tseng, P. Wu, J. M. Unrine, E. A. Grulke and R. A. Yokel, Int. J. Nanomed., 2012, 7, 4023–4036 CrossRef CAS PubMed.
- B. D. Chithrani and W. C. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS PubMed.
- S. S. Hardas, D. A. Butterfield, R. Sultana, M. T. Tseng, M. Dan, R. L. Florence, J. M. Unrine, U. M. Graham, P. Wu, E. A. Grulke and R. A. Yokel, Toxicol. Sci., 2010, 116, 562–576 CrossRef CAS PubMed.
- S. S. Hardas, R. Sultana, G. Warrier, M. F. Dan, R. L. P. Wu, E. A. Grulke, M. T. Tseng, J. M. Unrine, U. M. Graham, R. A. Yokel and D. A. Butterfield, Neurotoxicology, 2012, 33, 1147–1155 CrossRef CAS PubMed.
- R. A. Yokel, T. C. Au, R. MacPhail, S. S. Hardas, D. A. Butterfield, R. Sultana, M. T. Tseng, M. Dan, R. L. Florence, J. M. Unrine, U. M. Graham, P. Wu and E. A. Grulke, Toxicol. Sci., 2012, 127, 256–268 CrossRef CAS PubMed.
- S. M. Hirst, A. Karakoti, S. Singh, W. Self, R. Tyler, S. Seal and C. M. Reilly, Environ. Toxicol., 2011, 1–12 Search PubMed.
- S. M. Hirst, A. S. Karakoti, R. D. Tyler, N. Sriranganathan, S. Seal and C. M. Reilly, Small, 2009, 5, 2848–2856 CrossRef CAS PubMed.
- A. S. Karakoti, N. A. Monteiro-Riviere, R. Aggarwal, J. P. Davis, R. J. Narayan, W. T. Self, J. McGinnis and S. Seal, JOM, 2008, 60, 33–37 CrossRef CAS PubMed.
- R. A. Yokel, M. T. Tseng, M. Dan, J. M. Unrine, U. M. Graham, P. Wu and E. A. Grulke, Nanomed.: Nanotechnol., Biol. Med., 2013, 9, 398–407 CrossRef CAS PubMed.
- S. Rojas, J. D. Gispert, S. Abad, M. Buaki-Sogo, V. M. Victor, H. Garcia and J. R. Herance, Mol. Pharmaceutics, 2012, 9, 3543–3550 CrossRef CAS PubMed.
- K. L. Heckman, W. DeCoteau, A. Estevez, K. J. Reed, W. Costanzo, D. Sanford, J. C. Leiter, J. Clauss, K. Knapp, C. Gomez, P. Mullen, E. Rathbun, K. Prime, J. Marini, J. Patchefsky, A. S. Patchefsky, R. K. Hailstone and J. S. Erlichman, ACS Nano, 2013, 7, 10582–10596 CrossRef CAS PubMed.
- R. A. Yokel, J. M. Unrine, P. Wu, B. Wang and E. Grulke, Environ. Sci.: Nano, 2014, 1 10.1039/C4EN00035H , in revision.
- M. Dan, P. Wu, E. A. Grulke, U. M. Graham, J. M. Unrine and R. A. Yokel, Nanomedicine, 2012, 7, 95–110 CrossRef CAS PubMed.
- B. Wang, P. Wu, R. A. Yokel and E. A. Grulke, Appl. Surf. Sci., 2012, 258, 5332–5341 CrossRef CAS PubMed.
- M. Auffan, J. Rose, T. Orsiere, M. de Meo, A. Thill, O. Zeyons, O. Proux, A. Masion, P. Chaurand, O. Spalla, A. Botta, M. R. Wiesner and J.-Y. Bottero, Nanotoxicology, 2009, 3, 161–171 CrossRef CAS.
- J. F. Nyland and E. K. Silbergeld, Hum. Exp. Toxicol., 2009, 28, 393–400 CAS.
- J. E. Riviere, Wiley Interdiscip. Rev.: Nanomed Nanobiotechnol., 2009, 1, 26–34 CrossRef CAS PubMed.
- M. Li, K. T. Al-Jamal, K. Kostarelos and J. Reineke, ACS Nano, 2010, 4, 6303–6317 CrossRef CAS PubMed.
- K. Takada and M. Fujita, J. Radiat. Res., 1973, 14, 187–197 CrossRef CAS.
- K. Bjondahl, Med. Biol., 1976, 54, 454–460 CAS.
- K. Takada, Strahlentherapie, 1977, 153, 195–199 CAS.
- C. S. Lustgarten, R. G. Cuddihy, B. B. Boecker and N. Huber, Biliary excretion of cerium-144 following inhalation of cerium-144 citrate by rats, Annual Report of the Inhalation Toxicology Research Institute (Lovelace Biomedical and Environmental Research Institute), 1974, pp. 57–61 Search PubMed.
- N. Castellino, P. Nizza and A. Aeberhardt, Int. J. Radiat. Biol., 1962, 5, 379–399 CrossRef CAS.
- J. G. Graca, F. C. Davison and J. B. Feavel, Arch. Environ. Health, 1964, 8, 555–564 CrossRef CAS.
- M. T. Tseng, Q. Fu, G. K. Lorc, R. Fernandez-Botran, Z.-B. Deng, U. M. Graham, D. A. Butterfield, E. A. Grulke and R. A. Yokel, Toxicol. Pathol., 2014, 42(6), 984–996 CrossRef PubMed.
- M. T. Tseng, X. Lu, X. Duan, S. S. Hardas, R. Sultana, P. Wu, J. M. Unrine, U. M. Graham, D. A. Butterfield, E. A. Grulke and R. A. Yokel, Toxicol. Appl. Pharmacol., 2012, 260, 173–182 CrossRef CAS PubMed.
- L. L. Wong, S. M. Hirst, Q. N. Pye, C. M. Reilly, S. Seal and J. F. McGinnis, PLoS One, 2013, 8, e58431 CAS.
- S. S. Hardas, R. Sultana, G. Warrier, M. Dan, P. Wu, E. A. Grulke, M. T. Tseng, J. M. Unrine, U. M. Graham, R. A. Yokel and D. A. Butterfield, Nanotoxicology, 2014, 8, 155–166 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Madler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- E. Grulke, K. Reed, M. Beck, X. Huang, A. Cormack and S. Seal, Environ. Sci.: Nano, 2014, 1 10.1039/C4EN00105B.
- A. Kumar, S. Das, P. Munusamy, W. Self, D. R. Baer, D. C. Sayle and S. Seal, Environ. Sci.: Nano, 2014, 1 10.1039/C4EN00052H , in revision.
- U. M. Graham, M. T. Tseng, J. B. Jasinski, R. A. Yokel, J. M. Unrine, B. H. Davis, A. K. Dozier, S. S. Hardas, R. Sultana, E. Grulke and D. A. Butterfield, ChemPlusChem, 2014, 79(8), 1083–1088 CrossRef CAS PubMed.
- X. He, H. Zhang, Y. Ma, W. Bai, Z. Zhang, K. Lu, Y. Ding, Y. Zhao and Z. Chai, Nanotechnology, 2010, 21, 285103 CrossRef PubMed.
- W.-S. Cho, R. Duffin, F. Thielbeer, M. Bradley, I. L. Megson, W. Macnee, C. A. Poland, C. L. Tran and K. Donaldson, Toxicol. Sci., 2012, 126, 469–477 CrossRef CAS PubMed.
- G. Oberdörster, E. Oberdörster and J. Oberdörster, Environ. Health Perspect., 2005, 113, 823–839 CrossRef.
- H. Jung, D. B. Kittelson and M. R. Zachariah, Combust. Flame, 2005, 142, 276–288 CrossRef CAS PubMed.
- F. R. Cassee, A. Campbell, A. J. Boere, S. G. McLean, R. Duffin, P. Krystek, I. Gosens and M. R. Miller, Environ. Res., 2012, 115, 1–10 CrossRef CAS PubMed.
- F. R. Cassee, E. C. van Balen, C. Singh, D. Green, H. Muijser, J. Weinstein and K. Dreher, Crit. Rev. Toxicol., 2011, 41, 213–229 CrossRef PubMed.
- U. EPA, Toxicological Review of cerium oxide and cerium compounds (CAS no. 1306-38-3) In support of summary information on the integrated risk information system (IRIS) U.S. Environmental Protection Agency, 2009 Search PubMed.
- PROSPECT, Global Nanomaterials Safety, Toxicological review of nano cerium oxide, 2010, p. 27 Search PubMed.
- J. P. Berry, M. Meignan, F. Escaig and P. Galle, Toxicology, 1988, 52, 127–139 CrossRef CAS.
- P. Galle, J. P. Berry and C. Galle, Environ. Health Perspect., 1992, 97, 145–147 CrossRef CAS.
- L. K. Limbach, Y. Li, R. N. Grass, T. J. Brunner, M. A. Hintermann, M. Muller, D. Gunther and W. J. Stark, Environ. Sci. Technol., 2005, 39, 9370–9376 CrossRef CAS.
- L. Geraets, A. G. Oomen, J. D. Schroeter, V. A. Coleman and F. R. Cassee, Toxicol. Sci., 2012, 127, 463–473 CrossRef CAS PubMed.
- M. Seipenbusch, A. Binder and G. Kasper, Ann. Occup. Hyg., 2008, 52, 707–716 CrossRef CAS PubMed.
- S. Aalapati, S. Ganapathy, S. Manapuram, G. Anumolu and B. M. Prakya, Nanotoxicology, 2013, 8, 786–798 Search PubMed.
- S. Singh, A. Kumar, A. Karakoti, S. Seal and W. T. Self, Mol. BioSyst., 2010, 6, 1813–1820 RSC.
- T. Xia, M. Kovochich, M. Liong, L. Madler, B. Gilbert, H. Shi, J. I. Yeh, J. I. Zink and A. E. Nel, ACS Nano, 2008, 2, 2121–2134 CrossRef CAS PubMed.
- S. Hussain, F. Al-Nsour, A. B. Rice, J. Marshburn, Z. Ji, J. I. Zink, B. Yingling, N. J. Walker and S. Garantziotis, Int. J. Nanomed., 2012, 7, 1387–1397 CrossRef CAS PubMed.
- S. Hussain, F. Al-Nsour, A. B. Rice, J. Marshburn, B. Yingling, Z. Ji, J. I. Zink, N. J. Walker and S. Garantziotis, ACS Nano, 2012, 6, 5820–5829 CrossRef CAS PubMed.
- S. Hussain and S. Garantziotis, Autophagy, 2013, 9, 101–103 CrossRef CAS PubMed.
- G. Kanapilly and R. Luna, Deposition and retention of inhaled condensation aerosols of 144CeO2 in Syrian hamsters, Annual report of the Inhalation Toxicology Research Institute. Lovelace Foundation for Medical Education and Research, Albuquerque, NM, 1975, pp. 79–83, Report no. LF-52 Search PubMed.
- J. C. Pairon, F. Roos, P. Sebastien, B. Chamak, I. Abd-Alsamad, J. F. Bernaudin, J. Bignon and P. Brochard, Am. J. Ind. Med., 1995, 27, 349–358 CrossRef CAS PubMed.
- J. L. Keller, L. Ma Hock, K. Kuettler, V. Strauss, S. Groeters, K. Wiench, B. van Ravenzwaay and R. Landsiedel, 7th International Nanotoxicology Congress, 2014, Abstract # P223 Search PubMed.
- A. Srinivas, P. J. Rao, G. Selvam, P. B. Murthy and P. N. Reddy, Toxicol. Lett., 2011, 205, 105–115 CrossRef CAS PubMed.
- P. Demokritou, S. Gass, G. Pyrgiotakis, J. M. Cohen, W. Goldsmith, W. McKinney, D. Frazer, J. Ma, D. Schwegler-Berry, J. Brain and V. Castranova, Nanotoxicology, 2013, 7, 1338–1350 CrossRef CAS PubMed.
- P. Demokritou, R. Buechel, R. M. Molina, G. M. Deloid, J. D. Brain and S. E. Pratsinis, Inhalation Toxicol., 2010, 22, 107–116 CrossRef CAS PubMed.
- J. Y. Ma, R. R. Mercer, M. Barger, D. Schwegler-Berry, J. Scabilloni, J. K. Ma and V. Castranova, Toxicol. Appl. Pharmacol., 2012, 262, 255–264 CrossRef CAS PubMed.
- J. Y. Ma, H. Zhao, R. R. Mercer, M. Barger, M. Rao, T. Meighan, D. Schwegler-Berry, V. Castranova and J. K. Ma, Nanotoxicology, 2011, 5, 312–325 CrossRef CAS PubMed.
- H. Zhang, Z. Ji, T. Xia, H. Meng, C. Low-Kam, R. Liu, S. Pokhrel, S. Lin, X. Wang, Y.-P. Liao, M. Wang, L. Li, R. Rallo, R. Damoiseaux, D. Telesca, L. Madler, Y. Cohen, J. I. Zink and A. E. Nel, ACS Nano, 2012, 6, 4349–4368 CrossRef CAS PubMed.
- V. C. Minarchick, P. A. Stapleton, D. W. Porter, M. G. Wolfarth, E. Ciftyurek, M. Barger, E. M. Sabolsky and T. R. Nurkiewicz, Cardiovasc. Toxicol., 2013, 13, 323–337 CrossRef CAS PubMed.
- S. K. Nalabotu, M. B. Kolli, W. E. Triest, J. Y. Ma, N. D. Manne, A. Katta, H. S. Addagarla, K. M. Rice and E. R. Blough, Int. J. Nanomed., 2011, 6, 2327–2335 CrossRef CAS PubMed.
- Y. Staal, L. Ma-Hock, H. Muijser, S. Treumann, V. Strauss and R. Landsiedel, The Toxicologist, 2010, PL 1398, 297 Search PubMed.
- I. Gosens, L. E. A. M. Mathijssen, B. G. H. Bokkers, H. Muijser and F. R. Cassee, Nanotoxicology, 2014, 8, 643–653 CrossRef CAS PubMed.
- Bio-Research Laboratories for Rhone-Poulenc Inc., Final report for a 90-day inhalation neurotoxicity and toxicity study by exposure to a dry powder aerosol of ceric oxide in the albino rat with cover letter dated 013095, 1994 Search PubMed.
- T. J. Brunner, P. Wick, P. Manser, P. Spohn, R. N. Grass, L. K. Limbach, A. Bruinink and W. J. Stark, Environ. Sci. Technol., 2006, 40, 4374–4381 CrossRef CAS.
- H. J. Eom and J. Choi, Toxicol. Lett., 2009, 187, 77–83 CrossRef CAS PubMed.
- S. Mittal and A. K. Pandey, BioMed Res. Int., 2014, 2014, 891934 Search PubMed.
- B. Rothen-Rutishauser, R. N. Grass, F. Blank, L. K. Limbach, C. Muhlfeld, C. Brandenberger, D. O. Raemy, P. Gehr and W. J. Stark, Environ. Sci. Technol., 2009, 43, 2634–2640 CrossRef CAS.
- A. S. Karakoti, P. Munusamy, K. Hostetler, V. Kodali, S. Kuchibhatla, G. Orr, J. G. Pounds, J. G. Teeguarden, B. D. Thrall and D. R. Baer, Surf. Interface Anal., 2012, 44, 882–889 CrossRef CAS PubMed.
- P. H. Hoet, I. Bruske-Hohlfeld and O. V. Salata, J. Nanobiotechnol., 2004, 2, 12 CrossRef PubMed.
- C. U. Nwokolo, J. F. Lewin, M. Hudson and R. E. Pounder, Gastroenterology, 1992, 102, 163–167 CAS.
- J. F. Hillyer and R. M. Albrecht, J. Pharm. Sci., 2001, 90, 1927–1936 CrossRef CAS PubMed.
- P. Jani, G. W. Halbert, J. Langridge and A. T. Florence, J. Pharm. Pharmacol., 1990, 42, 821–826 CrossRef CAS PubMed.
- E.-J. Park, Y.-K. Park and K. Park, Toxicol. Res., 2009, 25, 79–84 CrossRef CAS.
- Y. I. Moskalev, Med. Radiol., 1959, 4, 52–57 CAS.
- M. C. Arnold, A. R. Badireddy, M. R. Wiesner, R. T. Di Giulio and J. N. Meyer, Arch. Environ. Contam. Toxicol., 2013, 65, 224–233 CrossRef CAS PubMed.
- R. A. Yokel and R. C. MacPhail, J. Occup. Med. Toxicol., 2011, 6 Search PubMed.
- B. Park, P. Martin, C. Harris, R. Guest, A. Whittingham, P. Jenkinson and J. Handley, Part. Fibre Toxicol., 2007, 4, 12 CrossRef PubMed.
- W. Lin, Y. W. Huang, X. D. Zhou and Y. Ma, Int. J. Toxicol., 2006, 25, 451–457 CrossRef CAS PubMed.
- E. J. Park, J. Choi, Y. K. Park and K. Park, Toxicology, 2008, 245, 90–100 CrossRef CAS PubMed.
- A. Gojova, H. S. Jung, B. Guo, A. I. Barakat and I. M. Kennedy, Inhalation Toxicol., 2009, 21, 123–130 CrossRef CAS PubMed.
- A. Asati, S. Santra, C. Kaittanis and J. M. Perez, ACS Nano, 2010, 4, 5321–5331 CrossRef CAS PubMed.
- M. Fall, M. Guerbet, B. Park, F. Gouriou, F. Dionnet and J.-P. Morin, Nanotoxicology, 2007, 1, 227–234 CrossRef CAS.
- B. K. Pierscionek, J. Keenan, A. Yasseen, L. M. Colhoun, Y. B. Li, R. A. Schachar and W. Chen, Curr. Anal. Chem., 2010, 6, 172–176 CrossRef CAS.
- B. K. Pierscionek, Y. Li, R. A. Schachar and W. Chen, Nanomedicine, 2012, 8, 383–390 CrossRef CAS PubMed.
- L. Alili, M. Sack, A. S. Karakoti, S. Teuber, K. Puschmann, S. M. Hirst, C. M. Reilly, K. Zanger, W. Stahl, S. Das, S. Seal and P. Brenneisen, Biomaterials, 2011, 32, 2918–2929 CrossRef CAS PubMed.
- T.-L. Lee, J. M. Raitano, O. M. Rennert, S.-W. Chan and W.-Y. Chan, Nanomedicine, 2012, 8, 599–608 CrossRef CAS PubMed.
- J. Niu, K. Wang and P. E. Kolattukudy, J. Pharmacol. Exp. Ther., 2011, 338, 53–61 CrossRef CAS PubMed.
- A. Clark, A. Zhu, K. Sun and H. R. Petty, J. Nanopart. Res., 2011, 13, 5547–5555 CrossRef CAS PubMed.
- C. Menchon, R. Martin, N. Apostolova, V. M. Victor, M. Alvaro, J. R. Herance and H. Garcia, Small, 2012, 8, 1895–1903 CrossRef CAS PubMed.
- A. Kroll, C. Dierker, C. Rommel, D. Hahn, W. Wohlleben, C. Schulze-Isfort, C. Gobbert, M. Voetz, F. Hardinghaus and J. Schnekenburger, Part. Fibre Toxicol., 2011, 8, 9 CrossRef CAS PubMed.
- G. Pyrgiotakis, C. O. Blattmann, S. Pratsinis and P. Demokritou, Langmuir, 2013, 29, 11385–11395 CrossRef CAS PubMed.
- J. Sumaoka, K. Furuki, Y. Kojima, M. Shibata, K. Hirao, N. Takeda and M. Komiyama, Nucleosides, Nucleotides Nucleic Acids, 2006, 25, 523–538 CAS.
- M. A. Berg, R. S. Coleman and C. J. Murphy, Phys. Chem. Chem. Phys., 2008, 10, 1229–1242 RSC.
- M. H. Kuchma, C. B. Komanski, J. Colon, A. Teblum, A. E. Masunov, B. Alvarado, S. Babu, S. Seal, J. Summy and C. H. Baker, Nanomedicine, 2010, 6, 738–744 CrossRef CAS PubMed.
- S. Singh, T. Dosani, A. S. Karakoti, A. Kumar, S. Seal and W. T. Self, Biomaterials, 2011, 32, 6745–6753 CrossRef CAS PubMed.
- A. Hayat, D. Andreescu, G. Bulbul and S. Andreescu, J. Colloid Interface Sci., 2014, 418, 240–245 CrossRef CAS PubMed.
- H. Katada, H. Seino, Y. Mizobe, J. Sumaoka and M. Komiyama, JBIC, J. Biol. Inorg. Chem., 2008, 13, 249–255 CrossRef CAS PubMed.
- J. M. Dowding, S. Das, A. Kumar, T. Dosani, R. McCormack, A. Gupta, T. X. T. Sayle, D. C. Sayle, K. L. von, S. Seal and W. T. Self, ACS Nano, 2013, 7, 4855–4868 CrossRef CAS PubMed.
- M. Horie, K. Nishio, K. Fujita, S. Endoh, A. Miyauchi, Y. Saito, H. Iwahashi, K. Yamamoto, H. Murayama, H. Nakano, N. Nanashima, E. Niki and Y. Yoshida, Chem. Res. Toxicol., 2009, 22, 543–553 CrossRef CAS PubMed.
- M. Culcasi, L. Benameur, A. Mercier, C. Lucchesi, H. Rahmouni, A. Asteian, G. Casano, A. Botta, H. Kovacic and S. Pietri, Chem.-Biol. Interact., 2012, 199, 161–176 CrossRef CAS PubMed.
- Z. Ji, X. Wang, H. Zhang, S. Lin, H. Meng, B. Sun, S. George, T. Xia, A. E. Nel and J. I. Zink, ACS Nano, 2012, 6, 5366–5380 CrossRef CAS PubMed.
- I. Celardo, M. De Nicola, C. Mandoli, J. Z. Pedersen, E. Traversa and L. Ghibelli, ACS Nano, 2011, 5, 4537–4549 CrossRef CAS PubMed.
- A. Thill, O. Zeyons, O. Spalla, F. Chauvat, J. Rose, M. Auffan and A. M. Flank, Environ. Sci. Technol., 2006, 40, 6151–6156 CrossRef CAS.
- S. Das, S. Singh, J. M. Dowding, S. Oommen, A. Kumar, T. X. T. Sayle, S. Saraf, C. R. Patra, N. E. Vlahakis, D. C. Sayle, W. T. Self and S. Seal, Biomaterials, 2012, 33, 7746–7755 CrossRef CAS PubMed.
- M. A. Jakupec, P. Unfried and B. K. Keppler, Rev. Physiol., Biochem. Pharmacol., 2005, 153, 101–111 CAS.
- S. George, S. Pokhrel, T. Xia, B. Gilbert, Z. Ji, M. Schowalter, A. Rosenauer, R. Damoiseaux, K. A. Bradley, L. Madler and A. E. Nel, ACS Nano, 2010, 4, 15–29 CrossRef CAS PubMed.
- T. Xia, Y. Zhao, T. Sager, S. George, S. Pokhrel, N. Li, D. Schoenfeld, H. Meng, S. Lin, X. Wang, M. Wang, Z. Ji, J. I. Zink, L. Madler, V. Castranova, S. Lin and A. E. Nel, ACS Nano, 2011, 5, 1223–1235 CrossRef CAS PubMed.
- K. Wegner and S. E. Pratsinis, Chem. Eng. Sci., 2003, 58, 4581–4589 CrossRef CAS PubMed.
- S. E. Pratsinis, AIChE J., 2010, 56, 3028–3035 CrossRef CAS PubMed.
- G. Beaucage, H. K. Kammler and S. E. Pratsinis, J. Appl. Crystallogr., 2004, 37, 523–535 CrossRef CAS.
- A. Teleki, M. C. Heine, F. Krumeich, M. K. Akhtar and S. E. Pratsinis, Langmuir, 2008, 24, 12553–12558 CrossRef CAS PubMed.
- A. Teleki, M. Suter, P. R. Kidambi, O. Ergeneman, F. Krumeich, B. J. Nelson and S. E. Pratsinis, Chem. Mater., 2009, 21, 2094–2100 CrossRef CAS.
- G. A. Sotiriou, A. M. Hirt, P.-Y. Lozach, A. Teleki, F. Krumeich and S. E. Pratsinis, Chem. Mater., 2011, 23, 1985–1992 CrossRef CAS PubMed.
- G. A. Sotiriou, T. Sannomiya, A. Teleki, F. Krumeich, J. Voeroes and S. E. Pratsinis, Adv. Funct. Mater., 2010, 20, 4250–4257 CrossRef CAS PubMed.
- G. A. Sotiriou, D. Franco, D. Poulikakos and A. Ferrari, ACS Nano, 2012, 6, 3888–3897 CrossRef CAS PubMed.
- Y. Kobayashi, H. Katakami, E. Mine, D. Nagao, M. Konno and L. M. Liz-Marzan, J. Colloid Interface Sci., 2005, 283, 392–396 CrossRef CAS PubMed.
- Y. Han, J. Jiang, S. S. Lee and J. Y. Ying, Langmuir, 2008, 24, 5842–5848 CrossRef CAS PubMed.
- K. Xu, J.-X. Wang, X.-L. Kang and J.-F. Chen, Mater. Lett., 2008, 63, 31–33 CrossRef PubMed.
- S. Gass, J. M. Cohen, G. Pyrgiotakis, G. A. Sotiriou, S. E. Pratsinis and P. Demokritou, ACS Sustainable Chem. Eng., 2013, 1, 843–857 CAS.
- R. Strobel and S. E. Pratsinis, J. Mater. Chem., 2007, 17, 4743–4756 RSC.
- R. Jung, J. C. Lee, G. T. Orosz, A. Sulyok, G. Zsolt and M. Menyhard, Surf. Sci., 2003, 543, 153–161 CrossRef CAS PubMed.
- A. Zhu, K. Sun and H. R. Petty, Inorg. Chem. Commun., 2012, 15, 235–237 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |