Silver nanoparticle protein corona composition compared across engineered particle properties and environmentally relevant reaction conditions†
Richard
Eigenheer‡
a,
Erick R.
Castellanos‡
b,
Meagan Y.
Nakamoto
b,
Kyle T.
Gerner
b,
Alyssa M.
Lampe
b and
Korin E.
Wheeler
*b
aUC-Davis Genome Center, Proteomics Core Facility, University of California, Davis, CA 95616, USA
bDepartment of Chemistry & Biochemistry, Santa Clara University, Santa Clara, CA 95053, USA. E-mail: kwheeler@scu.edu; Tel: +1 (408) 554 4310
First published on 14th March 2014
Abstract
Engineered silver nanoparticles (AgNPs) undergo profound chemical transformations in the environment. Upon exposure to biological systems, a protein corona forms to coat the NPs, altering NP bioavailability, toxicity and environmental fate. Although recent studies have explored the importance of the engineered properties of NPs in the formation of protein coronas, less is known about the impact of environmental conditions. In this work, protein corona populations are compared across AgNPs of varied sizes and surface coatings, as well as in aqueous solutions with additives that mimic environmentally relevant conditions. Bioinformatic analysis of the protein corona populations reveals that, in low buffer concentrations, the formation of the protein corona is dominated by electrostatic interactions between the proteins and NPs. The influence of electrostatics is attenuated by addition of solution additives such as sodium chloride and cysteine. Protein enrichment on the NPs is compared across 6 samples with varied AgNP engineered properties and solution conditions. Clustered results reveal that engineered surface coatings strongly mediate protein corona formation. However, under conditions that mimic varied biological and environmental systems, the protein affinities are similar. Indeed, the AgNP protein coronas characterized under environmentally relevant conditions share 70% of the protein corona population. Results provide insight into the relative importance of engineered NP properties and uncontrollable environmental variables, such as solution reaction conditions, in the formation of protein coronas.
Nano impactThe surface of nanoparticles (NPs) dominates their reactivity in the environment. Yet, proteins at the NP surface are currently poorly characterized under biologically and environmentally relevant conditions. Importantly, the influence of aqueous environments has been largely overlooked thus far in studies of protein coronas. Characterization of NPs in the presence and absence of proteins provides insight into the changing surface chemistry and NP morphology over their lifetime. Using MS proteomics to characterize the protein corona populations of NPs across NPs and reaction conditions, this research not only provides biophysical insights into NP–protein interactions but also enables comparison of the relative importance of controllable engineered NP properties and uncontrollable environmental variables (e.g. aqueous environment) in the formation of protein coronas. |
Introduction
In an effort to aid in the design of sustainable nanoparticles (NPs), a foundational understanding of their environmental fate, transformation, and toxicity is necessary.1–5 The high surface area of NPs makes them attractive to researchers and manufacturers alike, as the surface chemistry often governs chemical reactivity. Similarly, it is the surface chemistry that strongly influences their reactivity in environmental processes. Studies of NP fate are complicated by the fact that NPs in the environment undergo chemical and physical changes, including adsorption interactions with biological systems that leave the particle surface with little resemblance to the original NPs.4,6–9 Of the predominant surface modifiers in the environment, proteins selectively adsorb onto NPs to alter reactivity.6,8–12The adventitious adsorption of proteins to NPs, referred to as the NP protein corona, significantly influences NP uptake, accumulation, and cellular fate. Generally, the formation of a corona lowers NP toxicity in comparison to toxicity of the “bare” particle.13–15 On the other hand, if the protein corona is recognized by the cell, it may trigger activation of specific regulatory pathways.16 In this way, the protein corona influences the ability of NPs to cross biological barriers, alters the biological responses, and determines the environmental and biological fates of NPs. The ability to monitor and predict these phenomena has deep implications for the lifetime of NPs in the environment and NP bioaccumulation. The field, however, is still in its nascent stages. The vast majority of corona studies have been performed with human blood serum,12,17–20 with few characterizations of NP protein coronas within environmentally relevant systems.15,21
Particle size, composition, surface properties,22–25 and even reaction temperatures26 have been shown to influence the composition of the NP protein corona. The corona is best described through the long-lived, equilibrium state, or “hard” protein corona of non-exchangeable proteins adsorbed to the NP surface in a specific environment.9,10,23,27,28 The nature of the NP and its corona is best understood in the context of NP dispersion and aggregation properties, as these factors impact NP biological and environmental reactivity.4
Yeast (Saccharomyces cerevisiae) proteins were chosen as a model system here to investigate the biophysical parameters of NP–protein interactions. Nanotoxicity is not the focus of this study, it is important to note that the nanotoxicity of AgNPs has not been extensively studied in yeast.29 However, some experimental results show that metal oxide fullerenes exhibit little or no yeast toxicity.30–34
Although many studies have detailed the impact of organic small molecules on environmental reactivity of NPs,35–37 the effect of proteins on NPs is a fundamental aspect that requires further investigation. Importantly, the influence of solution conditions, changes in solutes, ionic strength and other natural additives common to changes in water bodies and biological fluids have been largely overlooked thus far in protein corona studies. To fill this gap, we compare the modulation of the protein corona with variants of engineered AgNP properties (size and surface charge) to the protein coronas under several environmentally relevant solution conditions, modeling both fresh water and cellular conditions. To our knowledge, this is the first comprehensive study to examine and compare the NP protein coronas across both NP engineered properties and solution conditions. A comprehensive approach was employed that enables both qualitative and relative quantitative characterization of the protein corona. Bioinformatic and functional classification of proteins within the coronas of various NPs under a spectrum of reaction conditions provides insight into the importance of biophysicochemical parameters in NP corona formation within the environment.
Materials and methods
Nanoparticle characterization
BioPure spherical AgNPs were purchased from Nanocomposix (La Jolla, CA). AgNPs with two different surface coatings were used; anionic, citrate coated particles and cationic branched polyethyleneimine (BPEI) coated NPs are designated with the (−) and (+) prefix, respectively. Two sizes of (−) AgNPs, 10 and 100 nm, were also studied.Monodispersity of the NPs was confirmed by dynamic light scattering size distribution (DLS) and transition electron microscopy (TEM) images. For imaging (Hitachi H-9500 at 300 kV), soluble yeast protein extract (YPE) and NPs were reacted as described below. TEM sample plates were prepared by depositing 4 μL of the homogeneous solution (NPs, or NP–YPE mixture) on a 400 mesh copper grid (SPI Supplies, PA) coated with formvar, and then air-dried at room temperature. For Z-average hydrodynamic diameter and zeta potential measurements (ZetaPlus from Brookhaven Laboratories), YPE and NPs were reacted as described below, and then diluted in the appropriate buffer to obtain optimal DLS measurements. A Smoluchowski model was used to calculate zeta potential from electrophoretic mobility measurements (Fig. S3†).
Isolation of soluble protein extract (YPE) mixture
A 1 L culture of BY4 yeast was inoculated at 30 °C with constant shaking at 150 rpm. The cells were harvested after 24 h via centrifugation (Allegra X-22R) at 2300 × g for 30 min. Cell pellets were then stored at −80 °C.For the extraction of YPE, 5 mL of frozen cells were thawed on ice, and then incubated on a shaker overnight in 5 mM ammonium bicarbonate (AMBIC) buffer, pH 7. Cells were heated at 95 °C for 5 min and pelleted via centrifugation. Cells were washed with ice-cold water, followed by ice-cold 50 mM AMBIC buffer, pH 7. Washed cells were resuspended in 15 μL of protease inhibitors (Thermo Scientific Halt Protease Inhibitor Cocktail) and 10 mL of ice-cold 50 mM AMBIC buffer, pH 7. The resuspended cells were mechanically crushed in liquid nitrogen chilled mortar and pestle. Cell lysate was transferred to an ice-cold beaker, thawed with 20 mL of 50 mM AMBIC buffer, pH 7, and 15 μL of protease inhibitors. Particulates were removed with centrifugation (10 min, 3900 × g). Supernatant (now YPE) was exchanged into 10 mM sodium phosphate buffer, pH 7.4 (Millipore 5K MWCO centrifugal concentrators). Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Scientific).
Protein reaction with nanoparticles
0.25 mg mL−1 NPs and 0.21 mg mL−1 YPE were incubated for 16 h at room temperature. Previous kinetic studies have shown that protein coronas form rapidly and are stable over time.25 Unassociated proteins were separated from tightly associated proteins via centrifugation (3× for 10 min, 3900 × g). Unassociated proteins in the supernatant were transferred and exchanged into 50 mM AMBIC buffer (Millipore 5K MWCO centrifugal concentrators). Tightly associated proteins formed a pellet with NPs; this pellet was washed twice with water and resuspended in 50 mM AMBIC buffer.Mass spectrometry (MS) sample preparation
Sequence grade modified porcine trypsin (Promega, Madison, WI) was added to both the separated non-associated and adsorbed (associated) protein samples for digestion overnight at 38 °C. After digestion, tryptic peptides in NP associated and unassociated samples were separated from any potential remaining NPs via centrifugation (10 min, 3900 × g). The solution was resuspended once in 50 mM AMBIC, which was loaded onto 10K MWCO spin tubes (Millipore). Each spin tube was centrifuged for 15 min at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to remove any remaining NPs or NP fragments (occasionally visible in NP associated samples as a yellow color) and digested peptides spun through. This (clear) flowthrough was dried completely in a vacuum concentrator (Labconco).
000 rpm to remove any remaining NPs or NP fragments (occasionally visible in NP associated samples as a yellow color) and digested peptides spun through. This (clear) flowthrough was dried completely in a vacuum concentrator (Labconco).
Mass spectrometry
Digested peptides were resolubilized in 0.1% trifluoroacetic acid with 2% acetonitrile by 10 min of sonication and centrifuged again at 16000 rpm for 8 min, and then all samples were normalized by weight after quantification on a ND 1000 spectrophotometer (NanoDrop) using the A280 function. Peptides were then analyzed by LC-MS/MS on a Thermo Finnigan LTQ with Michrom Paradigm LC and CTC Pal autosampler. Peptides were separated using a Michrom 200 μm × 150 mm Magic C18 AQ reversed phase column at 2 μl min−1. Peptides were directly loaded onto an Agilent ZORBAX 300SB C18 reversed phase trap cartridge, which, after loading, was switched in-line with a Michrom Magic C18 AQ 200 μm × 150 mm column connected to a Thermo-Finnigan LTQ ion trap mass spectrometer through a Michrom Advance Plug and Play nano-spray source. The nano-LC column was run with a 120 min long gradient using a two buffer system, with Buffer A being 0.1% formic acid and Buffer B 100% acetonitrile, at a flow rate of 2 mL min−1. MS and MS/MS spectra were acquired using a top 10 method, where the top 10 ions in the MS scan were subjected to automated low energy CID. An MS survey scan was obtained for the m/z range 375–1400. An isolation mass window of 2 Da was for the precursor ion selection, and a normalized collision energy of 35% was used for the fragmentation. A 2 min duration was used for the dynamic exclusion.
Three biological replicates were run for each condition for proteins from the NP-associated and non-associated fractions. Samples were normalized for total peptide loading by weight using A280 on a ND1000 Nanodrop spectrometer. Approximately 10 μg of total peptide was loaded on the LTQ for each replicate.
Tandem mass spectra were extracted using XCalibur. All MS/MS samples were analyzed using X! Tandem and a Uniprot 2012 Saccharomyces cerevisiae database. X! Tandem was searched with a fragment ion mass tolerance of 0.40 Da and a parent ion tolerance of 1.8 Da.
Scaffold was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 85.0% probability, with a peptide false discovery fate of less than 0.5%. Protein identifications were accepted if they could be established at greater than 80.0% probability and contained at least 2 identified peptides, which resulted in less than a 5% false discovery rate for proteins.
Data analysis
Normalized spectral abundance factor (NSAF) values were calculated for all proteins identified by MS in both the non-associated protein fractions and the associated fractions. In all analyses, proteins were considered part of the NP protein corona if identified within the NP-associated fractions in all three biological replicates (NSAF > 0 for all three NP associated fractions). The NSAF ratio of NP associated and unassociated fractions was calculated for all proteins. Proteins were considered part of the “enriched protein corona” when the following criteria were both met: 1) the average NSAF ratio (or enrichment factor) was greater than 10, where there was an average of one order of magnitude more protein in the associated fraction than the non-associated protein fraction; and 2) the enrichment factor was greater than one for all three biological replicates. The results of the NSAF ratio values for each protein identified were clustered using Cluster version 3.0 (http://bonsai.ims.u-tokyo.ac.jp/mdehoon/software/cluster/software.htm#ctv) and visualized as heat maps with TreeView software.A full database of proteins, NSAF counts, and designations of affinity can be found in ESI Table S1.†
Results and discussion
Assessment of NP characteristics
The NPs utilized were characterized prior to reaction with YPE to ensure NP stability, size, and shape. In addition, they were extensively purified to remove residual reactants from synthesis possibly deleterious to protein stability. The shape and size of AgNPs were retained across all samples in the reaction buffer and upon addition of YPE, as shown in the TEM images of NPs after YPE reaction, with a small amount of dissolution visible in the 10 nm (−) AgNPs, as indicated by the presence of smaller particles and amorphous features likely formed by precipitation of Ag salts (Fig. 1A).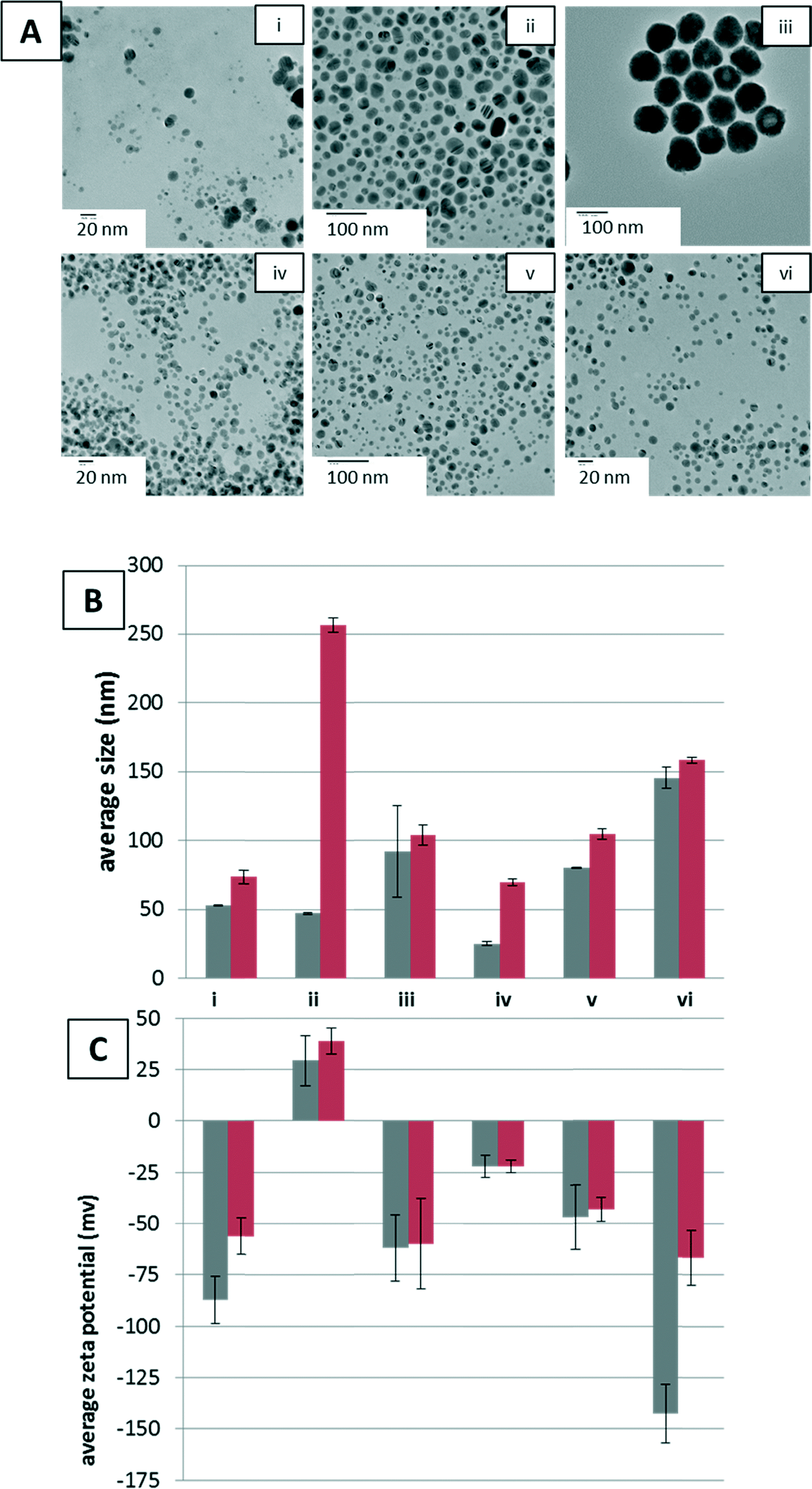 | ||
| Fig. 1 A) TEM images of AgNPs after reaction with YPE. B) Average hydrodynamic radius (Z-average) measured by DLS and C) zeta potential of AgNPs. Measurements for Z-average hydrodynamic diameter and zeta potential are shown with YPE (red bars) and without YPE (grey bars) with error bars indicating standard error. Samples are labeled as follows: [i.] (−) 10 nm AgNPs in 10 mM NaPi at pH 7.4, [ii.] (+) 10 nm AgNPs in 10 mM NaPi at pH 7.4, [iii.] (−) 100 nm AgNPs in 10 mM NaPi at pH 7.4, [iv.] (−) 10 nm AgNPs in 10 mM NaPi at pH 7.4 with 0.1 mM Cys, [v.] (−) 10 nm AgNPs in 10 mM NaPi at pH 7.4 with 0.8 mM NaCl, and [vi.] (−) 10 nm AgNPs in 10 mM NaPi at pH 7.4 with 3.0 mM NaCl. | ||
NP Z-average hydrodynamic diameters were determined by DLS to provide an indication of aggregation of the NPs under reaction conditions, before and after YPE addition (Fig. 1B). Aggregation increased with addition of 0.8 and 3.0 mM NaCl, but, interestingly, aggregation decreased with addition of Cys. Upon addition of YPE, aggregation increased in every sample. Although in most cases YPE induced aggregation was minimal, the average hydrodynamic radius of the (+) AgNPs increased five-fold upon YPE addition. At this point, the cause of aggregation upon YPE addition is unclear. Some studies have, however, reported protein unfolding on the surface of NPs, which causes NP aggregation.38–40 These changes in NP size and stability are likely to influence corona formation as well.
The engineered surface coating of the AgNPs dictated the zeta potential, both before and after addition of YPE. Prior to addition of YPE, anionic (−) AgNPs were found to have more negative zeta potentials than (+) AgNPs in 10 mM sodium phosphate, pH 7.4 (Fig. 1C, i, iii–vi and ii, respectively). After protein adsorption, the NP zeta-potential was altered for all NPs independent of reaction conditions, except when 0.1 mM Cys was added to the reaction buffer (Fig. 1C, iv). For (−) AgNPs in 10 mM sodium phosphate, pH 7.4, and with the addition of 0.8 and 3.0 mM NaCl, zeta-potential decreased upon addition of YPE, regardless of NP size. The (+) NPs showed a slight increase in average zeta-potential upon addition of YPE.
As indicated by changes in Z-average hydrodynamic diameters and zeta potential, the NaCl and Cys additives used here modify both the morphology and the surface chemistry of the AgNPs. Additives such as chloride19,41,42 and cysteine42,43 are known to mediate the dissolution, aggregation, and surface chemistry of AgNPs. For example, AgNP stability is strongly affected by chloride anions, which form a AgCl layer on the AgNP surface that inhibits dissolution at low Cl/Ag ratios.41 Similarly, analysis of AgNPs with Cys suggests the formation of Ag(I)–sulfhydryl bonds.42,43 Thus, changes in the protein corona as a result of solvent conditions are a result of multiple interlinked transformation processes.
Although the origins of differences in zeta-potential upon addition of YPE are not yet known, we believe they are a result of variance in protein populations adsorbed across samples. Along these lines, the zeta potential shows the role of solution additives, e.g. Cys and NaCl, in mediation of protein adsorption to NPs. The addition of YPE to (−) 10 nm AgNPs with Cys in the buffer results in no measurable change in zeta potential. By comparison, the addition of YPE to (−) 10 nm AgNPs with 3.0 M NaCl results in a roughly 70 mV change, decreasing the zeta potential by half.
Identification of NP protein corona and NP unassociated proteins
After incubation with YPE, NPs and adsorbed proteins were pelleted by centrifugation and washed three times to remove NP unassociated and loosely associated proteins. The supernatant from the initial centrifugation step was saved for comparison to the NP adsorbed population (unassociated proteins, vide infra). NP adsorbed proteins were directly trypsin digested off of the NPs to ensure isolation and identification of even strongly adsorbed and low abundance proteins in the corona.Over 500 proteins were identified in each sample, including NP associated and unassociated proteins. The number of NP associated proteins identified here is on the order of that identified in several other studies.11,25 A full list of identified proteins, including their biophysical parameters and relative spectral abundances, can be found in Table S1.† Proteins were considered part of the protein corona if identified in the NP-associated fraction of all three biological replicates (Fig. 2).
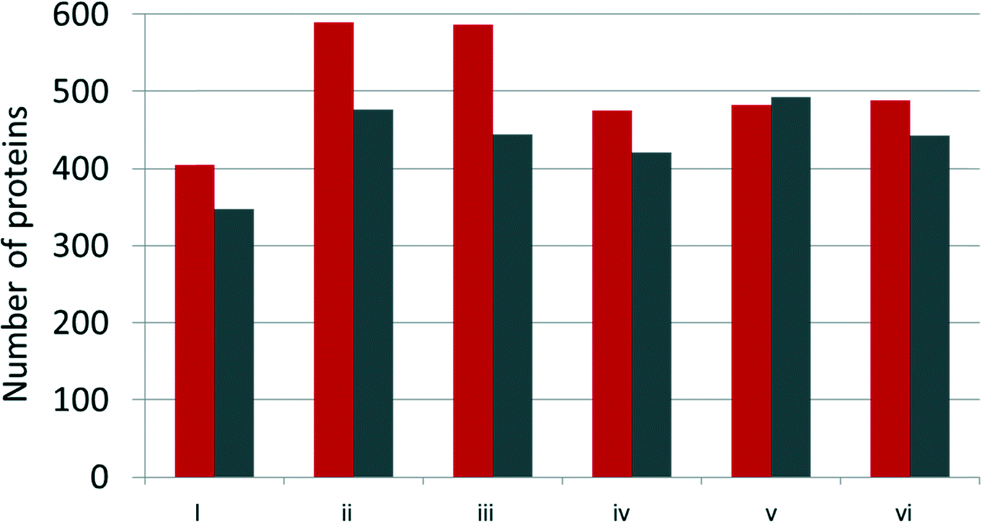 | ||
| Fig. 2 Number of proteins identified by LC-MS/MS within the AgNP protein corona (grey) and AgNP unbound (red) fractions. Samples are labeled as detailed in Fig. 1. | ||
As noted by others characterizing NP protein coronas,9,23–25,44 our analysis showed that binding profiles did not simply correspond to the relative protein concentrations in the YPE. For example, the most abundant protein in yeast, 60 s ribosomal protein L39, is identified within our studies, but is not identified as the most abundant protein within the corona of any NP or condition studied.
Characterization of proteins within the NP corona
The biophysical properties and functional annotations of protein coronas were compared across the NPs and sample conditions. Protein molecular weight, length, pI, and amino acid composition were analyzed for trends in enrichment within the protein corona. No statistically relevant trends were resolved for linking protein adsorption to molecular weight, length, or amino acid composition (Fig. S2–S3†). These results are consistent with previous studies of the protein corona.11,25,44In agreement with Shannahan et al.,44 our data did not reveal an increase in histidines, cysteines, or methionines within proteins in the AgNP protein corona, nor were these factors linked to enrichment of proteins within the corona. These results are surprising, especially given the strength of the silver–thiol bond. AgNP properties other than the NP composition must dominate protein adsorption. To fully explore the theory that NP composition plays a minimal role in the formation of the protein corona, further studies are necessary to evaluate the availability of individual amino acids in the formation of the protein corona, either through assessment of peptide populations interacting with the NP21 or through individual protein–NP interaction studies.45
We also extracted protein function information from each protein's gene ontology (GO) and enzyme commission number. The majority of the detected proteins were classified with a GO molecular function or subcellular location. Protein function was also compared using enzyme commission (EC) numbers for each sample. EC numbers can be compared to provide a straightforward, functional profile of the protein corona (Table 1A). In most samples, the enzymes were largely transferases, oxidoreductases or hydrolases. GO information also provides the subcellular localization of each protein identified (Table 1B). Because YPE population consists of soluble proteins, it is not surprising that the majority of proteins in the protein corona of each sample were cytoplasmic proteins.
| Reaction condition | 10 mM NaPi, pH 7.4 | +0.1 mM Cys | +0.8 mM NaCl | +3.0 mM NaCl | ||
|---|---|---|---|---|---|---|
| AgNP (charge), size | (−) 10 nm | (+) 10 nm | (−) 100 nm | (−) 10 nm | (−) 10 nm | (−) 10 nm |
| EC 1 – oxidoreductases | 10.35% | 11.52% | 8.97% | 9.43% | 7.30% | 8.50% |
| EC 2 – transferases | 10.77% | 10.96% | 9.42% | 8.49% | 7.30% | 8.05% |
| EC 3 – hydrolases | 8.28% | 9.27% | 5.83% | 7.55% | 6.69% | 7.16% |
| EC 4 – lyases | 4.14% | 3.93% | 4.26% | 3.77% | 3.85% | 3.36% |
| EC 5 – isomerases | 3.11% | 3.93% | 2.91% | 3.77% | 2.64% | 3.58% |
| EC 6 – ligases | 7.04% | 6.18% | 7.17% | 4.01% | 3.04% | 3.36% |
| Non-enzymatic | 56.31% | 54.21% | 61.43% | 62.97% | 69.17% | 66.00% |
| Reaction condition | 10 mM NaPi, pH 7.4 | +0.1 mM Cys | +0.8 mM NaCl | +3.0 mM NaCl | ||
|---|---|---|---|---|---|---|
| AgNP (charge), size | (−) 10 nm | (+) 10 nm | (−) 100 nm | (−) 10 nm | (−) 10 nm | (−) 10 nm |
| Nucleus | 21.33% | 21.91% | 19.06% | 18.16% | 20.08% | 19.24% |
| Cytoplasm | 62.94% | 56.46% | 64.57% | 53.77% | 48.07% | 50.34% |
| Mitochondria | 12.01% | 10.39% | 12.33% | 16.27% | 19.27% | 19.02% |
| Endoplasmic reticulum | 1.66% | 1.40% | 1.57% | 1.42% | 1.83% | 1.79% |
| Golgi apparatus | 1.66% | 1.12% | 1.35% | 0.94% | 1.22% | 0.67% |
| Cell membrane | 7.45% | 8.99% | 7.62% | 13.21% | 17.04% | 16.11% |
Although the sample preparations for this study prohibit conclusions bearing on AgNP toxicity in yeast, it is noteworthy that bioinformatic analysis can enable identification of proteins within the corona whose function is vital for survival. As shown in Table 2, there are proteins within the corona of each sample identified within the database of essential genes (DEG)46 as essential for yeast function. Any essential protein identified as part of the NP protein corona in vivo would be pulled out of their normal function in the cell and concentrated on the NP, potentially impacting NP toxicity. Although a DEG is not available for most species, this type of analysis may be useful in future nanotoxicity studies.
| Reaction condition | 10 mM NaPi, pH 7.4 | +0.1 mM Cys | +.8 mM NaCl | +3.0 mM NaCl | ||
|---|---|---|---|---|---|---|
| AgNP (charge), size | (−) 10 nm | (+) 10 nm | (−) 100 nm | (−) 10 nm | (−) 10 nm | (−) 10 nm |
| Corona proteins | 36 | 25 | 27 | 29 | 40 | 36 |
| Enriched proteins | 13 | 15 | 9 | 13 | 18 | 16 |
Proteins identified in the protein corona across all samples are of particular interest, being adsorbed to the NP independent of reaction condition. These ubiquitous proteins are likely to persist and define the biological identity of the particles across the many environmental and biological changes an NP undertakes in its lifetime. Of the proteins found within the protein corona of each sample, 169 proteins were identified in the protein corona of all samples, roughly 30% of the average protein corona. Many of these are amongst the most abundant proteins in the YPE, but most are specifically enriched within the protein coronas of one or more sample(s). The ubiquitous proteins are listed in Table S2.†
Engineered properties of NPs modulate the protein corona profile
In the analysis of the role of engineered properties of AgNPs in the formation of the protein corona, the NP–protein interaction was run at low salt and low buffer concentrations. These conditions were chosen to reduce the influence of the aqueous solution on protein adsorption and increase the ability to isolate the NP properties that mediate interactions with proteins. The protein coronas of (−) 10 nm AgNPs were compared to (+) 10 nm and (−) 100 nm AgNPs to gain insight into the role of surface charge and size, respectively.Evaluation of the protein corona populations makes it clear that AgNP surface charge played a stronger role in the formation of the corona than AgNP size. A Venn diagram comparing protein corona populations across the three types of AgNPs studied provides insight into the similarities and differences in protein corona populations that were a result of changing surface charge and AgNP size (Fig. 3A). The (−) 10 and 100 nm AgNPs were the most similar, with 376 (approximately 83%) proteins shared across their protein corona populations. Although the differences give insight into the relative importance of NP properties in determining the NP protein corona population, it is worth noting that 247 (approximately 50%) proteins were found within the coronas of all three NPs. These data demonstrate that these ubiquitous proteins would be found on the NPs regardless of their surface coating or size, making them an important group of proteins on which to focus future studies.
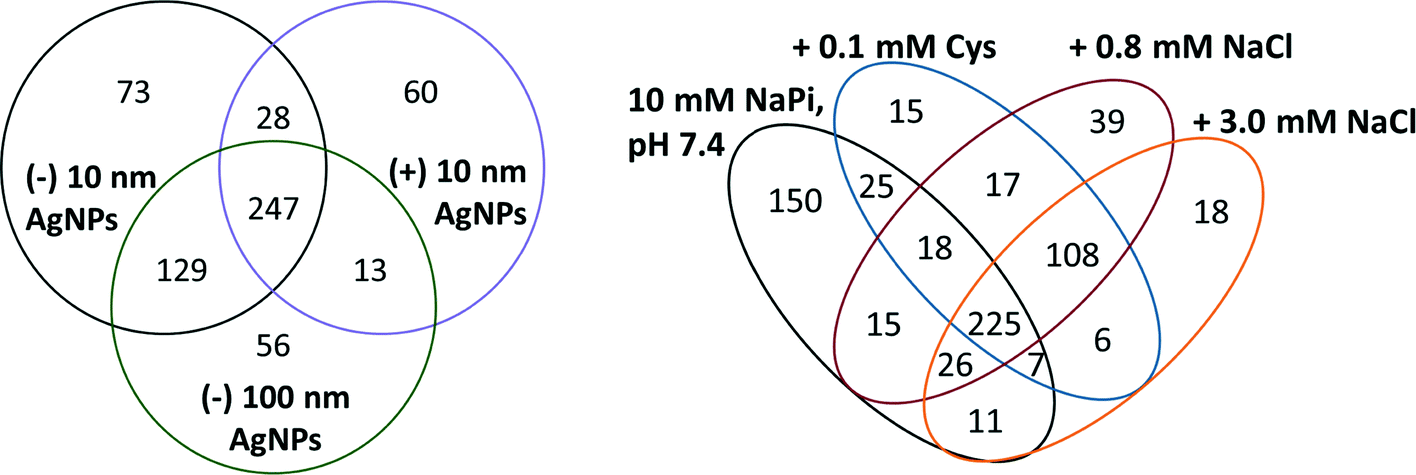 | ||
| Fig. 3 (A, left) Venn diagram of AgNP corona proteins identified across a spectrum of engineered AgNP properties, including surface charge and size. (B, right) Venn diagram of all corona proteins for (−) 10 nm AgNP across reaction conditions. | ||
Review of the biophysical properties of the proteins within the corona of each AgNP emphasizes the importance of AgNP surface charge in the formation of the protein corona. The most readily apparent biophysical property in the proteins studied here that appears to regulate binding to AgNPs is the charge. As shown in Fig. 4, there is an overall increase in the abundance of negatively charged proteins that bind to (+) AgNPs when compared to the 10 and 100 nm (−) AgNPs. Conversely, there is a greater than two-fold decrease in anionic proteins bound to both the 10 nm and 100 nm (−) AgNPs (34% and 20%, respectively), as compared to (+) AgNPs (71%). This effectively demonstrates that electrostatics dominate modulation of protein affinity for AgNPs.
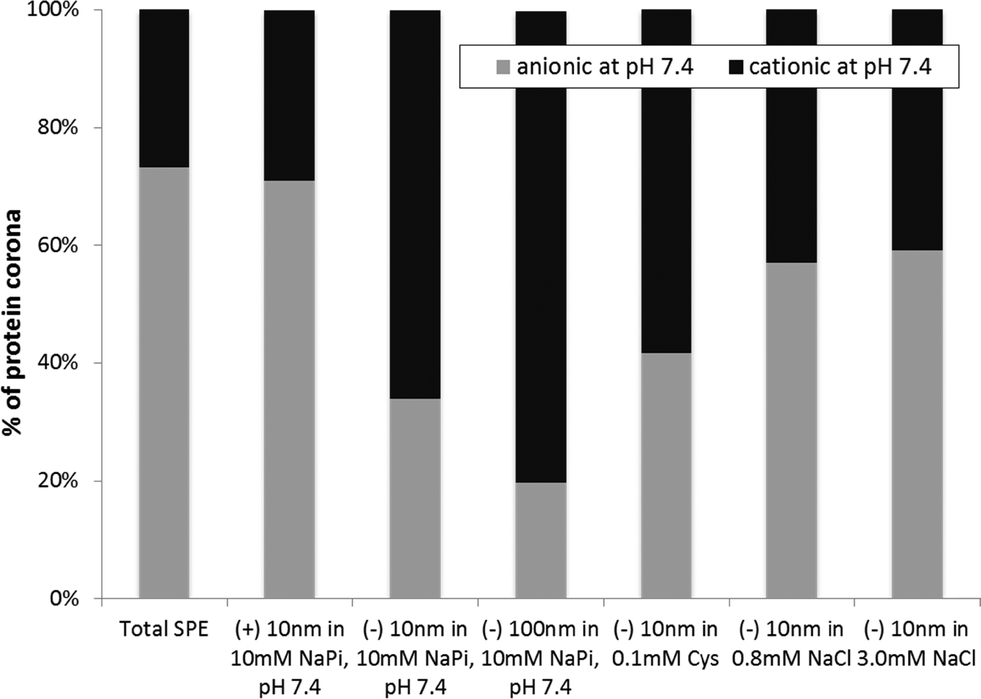 | ||
| Fig. 4 Percent abundance of protein pIs across the six sample conditions tested. | ||
Interestingly, AgNP size also influenced the charges of the proteins within the AgNP protein corona. Although others report changes in the protein corona population as a result of NP size,44,47 this is the first report on the impact of size on the charges of the proteins within the corona population. As seen in Fig. 4, a comparison of the 10 and 100 nm (−) AgNPs reveals a disparity in the abundance of anionic proteins in the corona, with 34% anionic proteins within the corona of the (−) 10 nm and 20% within that of the (−) 100 nm AgNPs. Other factors that influence the changes in the corona population with alterations in NP size are likely related to the varying surface curvature of the NPs and consequent energetics involved in protein adsorption and crowding.
There is no consensus in the literature on the role of engineered NP properties in the formation of the protein corona. Although many have suggested that electrostatic interactions dominate NP protein corona formation,11,47–49 others have reported that NP surface charge did not trend with the charge of bound proteins.25 In addition, we do not report a NP size dependent increase in the protein corona population, as seen by others.44 We propose that, in part, the variable results are due to striking variations in reaction conditions, in addition to differences in protein populations studied. The vast majority of studies were performed in complex sample conditions (e.g. blood serum or cell growth media) that are likely to shield charge and decrease the ability to detect the influence of electrostatics on protein–NP interactions. As detailed below, our own results demonstrate that the addition of just 0.8 mM NaCl changes the charge profile of the protein corona and may be enough to obviate the impact of electrostatics on interaction. These results highlight the importance of solution/reaction conditions, not just particle and protein properties for evaluating forces that drive the formation of the corona.
Environmental conditions mediate NP protein corona formation
Using (−) 10 nm AgNPs, the protein corona was characterized across three different biologically and environmentally relevant conditions, previously evaluated by Liu et al. in the absence of protein:42 0.8 mM NaCl, modeled after freshwater salinity; 3.0 mM NaCl, mimicking mitochondrial salinity; and 0.1 mM Cys, which has been shown to mediate NP toxicity. Comparison of the protein corona populations across these three different conditions provides insight into the impact of the environment in the protein corona formation. Studies outlining the variations in the NP protein corona have previously noted the importance of a diverse number of NP properties (e.g. size, shape, composition, surface roughness, porosity, surface charge) that impact the corona.5,9,11,23,25,44 Reaction conditions, for the most part, have been largely ignored.Solution conditions mediate formation of the corona by altering the number of proteins bound and the population. With 150 unique proteins, the 10 mM sodium phosphate, pH 7.4, conditions resulted in the most diverse protein corona population (Fig. 3B). The addition of NaCl leads to similar protein corona populations, with 108 proteins shared between 0.8 mM and 3.0 mM sodium chloride concentrations. Indeed, the distribution of EC numbers within the protein corona of each sample varies across the studied conditions, with the two sodium chloride conditions more similar than the others (Table 1A). To extrapolate these results from idealized test conditions, the data suggest minimal exchange of proteins within the corona as NPs move from freshwater into cellular conditions.
Although solution conditions influenced the formation of the NP protein corona, it is notable that 225 proteins were found in all four samples containing (−) 10 nm AgNPs (Fig. 3B). These ubiquitous proteins found on (−) 10 nm AgNPs regardless of condition may provide insight into altered reactivity of the NPs, independent of environmental conditions. On average, these ubiquitous proteins had a pI of 7.6, suggesting that overall protein charge was not a determining factor in adsorption. Surprisingly, there is also no significant increase in histidines or cysteines across the ubiquitously bound corona proteins.
Changes in the protein corona upon addition of solutes, NaCl and Cys, can provide insight into the changing biophysicochemical interactions at the NP surface. The solutes impact AgNP aggregation/dissolution processes, as shown in Fig. 1 and discussed earlier. Resulting changes in NP size and surface chemistry will alter protein binding, but the resulting changes in interaction mechanisms are still unclear. The addition of NaCl and Cys does, however, clearly disrupt the otherwise strong trend correlating protein and NP charge (Fig. 4). In other words, upon addition of NaCl and Cys, the number of cationic and anionic proteins within the corona of (−) 10 nm AgNPs becomes roughly equal. This demonstrates the loss of what was an otherwise strong trend suggesting electrostatic interactions dominate protein corona formation. These results can be extrapolated to suggest that as conditions move towards those similar to the natural environment, the role of electrostatics in protein corona formation is minimized.
Comparison of protein corona populations across engineered NP properties and reaction conditions mediated by the environment
The comparison of NSAF values between the NP corona and the NP unassociated protein populations provides insight into the proteins truly enriched within the NP protein corona. This ratio of NSAF values, or enrichment factor, is listed in Table S1.† Those proteins with NSAF values an order of magnitude greater in the bound sample were annotated as enriched within the NP corona. By analysis of protein enrichment within the six protein coronas studied here, we can more readily evaluate protein affinity for NPs across samples, independent of their presence in the corona.Analysis of enrichment factors across the six samples analyzed here enables a comparison of the relevance of engineered NP properties and solution additives. As shown in Fig. 5, the NP protein corona enrichment data for each protein was clustered by similarity of NP protein enrichment across NPs and reaction conditions. The clustered data divides the protein populations from each study into three distinct groups: group 1: (+) 10 nm NPs, group 2: (−) 10 and 100 nm NPs reacted in 10 mM sodium phosphate, and group 3: (−) 10 nm NPs reacted in the presence of cysteine and sodium chloride. These groupings reinforce the more detailed results from bioinformatic analysis of the protein corona populations.
 | ||
| Fig. 5 Heat map representation of protein enrichment across the six NP protein coronas. Protein coronas are clustered from the top: 1. (+) 10 nm NPs, 10 mM NaPi; 2. (−) 10 nm NPs, 10 mM NaPi; 3. 100 nm NPs, 10 mM NaPi; 4. 10 nm NPs, 10 mM NaPi + Cys; 5. 10 nm NPs, 10 mM NaPi + 0.8 mM NaCl; 6. 10 nm NPs, 10 mM NaPi + 3.0 mM NaCl. Proteins identified as enriched within the NP corona are shown in yellow, NP unassociated in blue, and nonenriched in black. Proteins without strong enrichment in one of the six samples were removed from the far left side of the heat map. | ||
Given that the branching distances within the clustering data are indicative of the similarity of the data sets, it is clear that NP charge is the strongest mediator of protein affinity, followed by solution conditions. From this, we can begin to extrapolate the role of engineered NP surface properties in comparison to that of the environment in driving the formation of the NP protein corona. The clear differentiation of the NP coronas by NP surface charge emphasizes the importance of the engineered NP surface properties over their engineered size. The differences in NP protein affinity due to NP surface charges are significant, as detailed by bioinformatics analysis previously discussed. Size changes, on the other hand, are relatively small at low ionic strength, as seen with tightly paired (−) 10 nm and (−) 100 nm NPs. The samples reacted under conditions modeling those within the natural environment show strong similarity in NP protein affinity. It is clear that environmental conditions, whether intra- or extra-cellular, will strongly impact the NP corona. Yet, once the NP protein corona has formed, there is a population of ubiquitous proteins that will influence the NP environmental and biological reactivity independent of the environmental conditions.
Conclusion
NP protein coronas form upon NP interaction with biological systems and mediate chemical reactivity, biological toxicity, and the environmental fate of NPs. It has been established that alterations in the NP corona alter cellular uptake and activity of the nanomaterials.15,46,50 Despite their importance in the role of NPs within the environment, there is little consensus on the biophysicochemical characteristics that mediate formation of the NP protein corona, or the role of environmental reaction conditions that alter NP protein corona formation. A novel approach was implemented here to use MS proteomics to characterize protein corona populations and to evaluate protein enrichment on the NP surface across engineered NP properties under biologically and environmentally relevant reaction conditions.Yeast AgNP protein coronas were sampled across various engineered sizes and surface charges. By analysis of changes in NP aggregation states and surface charges across solution conditions, it is clear that factors impacting biological and environmental reactivity have been altered by the protein coating. For (+) AgNPs, for example, a dramatic increase in particle size upon the formation of the protein corona may lead to increased sedimentation in the environment. Aggregation is similarly exacerbated by addition of Cys and NaCl, which change AgNP morphology and surface chemistry.19,41–43 At the most basic level, these results reiterate the importance of extensive characterization of NPs and reaction conditions when studying the NP protein corona. More broadly, the data suggest that proteins and other biomolecules are likely to play a significant role, beyond toxicity in AgNP environmental availability and reactivity.45,51
We have shown that the AgNP protein corona not only is highly complex but also varies depending upon both engineered NP properties and solution conditions. By evaluation of protein enrichment on the NPs, it is clear that the electrostatics of the NP surface coatings mediate the formation of the protein corona more strongly than the engineered NP size. Despite the importance of engineered NP properties, there is a population of ubiquitous corona proteins that bind independently of engineered properties or solution reaction conditions. It is these proteins that deserve further study, as they will bind throughout the lifetime of the NP and dictate its environmental availability and surface chemistry.
We have shown that conditions under which the NP corona is formed lead to alterations in the protein corona. In the environment, the NP corona is highly dependent upon NP exposure and history.5,9 The interactions and association of proteins with NPs are dominated by electrostatic interactions under low ionic strength conditions. When studied under reaction conditions that model cellular or fresh water conditions protein enrichment on the NPs increased in diversity. Addition of solutes such as Cys and NaCl not only increased NP aggregation states but also impacted the surface charge, complicating biophysical parameters that mediate protein–NP interactions. We cannot control the environmental conditions under which the NP corona is formed, nor can we control the NP corona over the lifetime of the particle in the environment. With an understanding of the biophysicochemical factors that mediate NP protein formation, we lay the foundation for predictive tools to characterize the NP protein corona within the environment.
Acknowledgements
This research was supported by financial assistance from the Keck Foundation and Santa Clara University. E.R.C. was supported by an ALZA fellowship and M.Y.N. was supported by a Clare Boothe Luce fellowship. E.R.C., M.Y.N. and A.M.L also received funding from the Roelandts Grants Program at the Center for Science and Technology at SCU. TEM images were obtained at the UCSC MACS Facility within the Advanced Studies Laboratories (NASA grant NNX09AQ44A to the University of Santa Cruz), with training and support provided by seed funding from the UCSC MACS Facility. We also thank David Hess for supplying our initial yeast stock and Thorsteinn Adalsteinsson (SCU) for insightful discussions about DLS.References
- M. R. Wiesner, G. V. Lowry, P. Alvarez, D. Dionysiou and P. Biswas, Environ. Sci. Technol., 2006, 40, 4336–4345 CrossRef CAS.
- B. Nowack and T. D. Bucheli, Environ. Pollut., 2007, 150, 5–22 CrossRef CAS PubMed.
- V. L. Colvin, Nat. Biotechnol., 2003, 21, 1166–1170 CrossRef CAS PubMed.
- S. Saptarshi, A. Duschl and A. Lopata, J. Nanobiotechnol., 2013, 11, 26 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- I. Lynch, T. Cedervall, M. Lundqvist, C. Cabaleiro-Lago, S. Linse and K. A. Dawson, Adv. Colloid Interface Sci., 2007, 134–135, 167–174 CrossRef CAS PubMed.
- I. Lynch, K. A. Dawson and S. Linse, Sci. Signaling, 2006, 327, pe14 Search PubMed.
- I. Lynch, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2009, 4, 546–547 CrossRef CAS PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- C. Ge, J. Du, L. Zhao, L. Wang, Y. Liu, D. Li, Y. Yang, R. Zhou, Y. Zhao, Z. Chai and C. Chen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16968–16973 CrossRef CAS PubMed.
- W. Hu, C. Peng, M. Lv, X. Li, Y. Zhang, N. Chen, C. Fan and Q. Huang, ACS Nano, 2011, 5, 3693–3700 CrossRef CAS PubMed.
- C. C. Fleischer, U. Kumar and C. K. Payne, Biomater. Sci., 2013, 1, 975–982 RSC.
- Z. J. Deng, M. Liang, M. Monteiro, I. Toth and R. F. Minchin, Nat. Nanotechnol., 2011, 6, 39–44 CrossRef CAS PubMed.
- G. V. Lowry, K. B. Gregory, S. C. Apte and J. R. Lead, Environ. Sci. Technol., 2012, 46, 6893–6899 CrossRef CAS PubMed.
- W.-C. Hou, B. Stuart, R. Howes and R. G. Zepp, Environ. Sci. Technol., 2013, 47, 7713–7721 CrossRef CAS PubMed.
- C. Levard, S. Mitra, T. Yang, A. D. Jew, A. R. Badireddy, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2013, 47, 5738–5745 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- N. S. Wigginton, A. D. Titta, F. Piccapietra, J. Dobias, V. J. Nesatyy, M. J. F. Suter and R. Bernier-Latmani, Environ. Sci. Technol., 2010, 44, 2163–2168 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, M. Foy, T. Berggard, S. C. Donnelly, G. Cagney, S. Linse and K. A. Dawson, Angew. Chem., Int. Ed., 2007, 46, 5754–5756 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed.
- M. Mahmoudi, A. M. Abdelmonem, S. Behzadi, J. H. Clement, S. Dutz, M. R. Ejtehadi, R. Hartmann, K. Kantner, U. Linne, P. Maffre, S. Metzler, M. K. Moghadam, C. Pfeiffer, M. Rezaei, P. Ruiz-Lozano, V. Serpooshan, M. A. Shokrgozar, G. U. Nienhaus and W. J. Parak, ACS Nano, 2013, 7, 6555–6562 CrossRef CAS PubMed.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, ACS Nano, 2010, 4, 3623–3632 CrossRef CAS PubMed.
- S. Milani, F. Baldelli Bombelli, A. S. Pitek, K. A. Dawson and J. Rädler, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS PubMed.
- S. Lee, J. Lee, K. Kim, S.-J. Sim, M. Gu, J. Yi and J. Lee, Biotechnol. Bioprocess Eng., 2009, 14, 490–495 CrossRef CAS PubMed.
- T. Nomura, J. Miyazaki, A. Miyamoto, Y. Kuriyama, H. Tokumoto and Y. Konishi, Environ. Sci. Technol., 2013, 47, 3417–3423 CAS.
- C. García-Saucedo, J. A. Field, L. Otero-Gonzalez and R. Sierra-Álvarez, J. Hazard. Mater., 2011, 192, 1572–1579 CrossRef PubMed.
- K. Kasemets, A. Ivask, H.-C. Dubourguier and A. Kahru, Toxicol. In Vitro, 2009, 23, 1116–1122 CrossRef CAS PubMed.
- H. Schwegmann, A. J. Feitz and F. H. Frimmel, J. Colloid Interface Sci., 2010, 347, 43–48 CrossRef CAS PubMed.
- A. N. Hadduck, V. Hindagolla, A. E. Contreras, Q. Li and A. T. Bakalinsky, Appl. Environ. Microbiol., 2010, 76, 8239–8242 CrossRef CAS PubMed.
- S. M. Louie, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2013, 47, 4245–4254 CrossRef CAS PubMed.
- S. Diegoli, A. L. Manciulea, S. Begum, I. P. Jones, J. R. Lead and J. A. Preece, Sci. Total Environ., 2008, 402, 51–61 CrossRef CAS PubMed.
- J. Fabrega, S. R. Fawcett, J. C. Renshaw and J. R. Lead, Environ. Sci. Technol., 2009, 43, 7285–7290 CrossRef CAS.
- S. Linse, C. Cabaleiro-Lago, W. F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8691–8696 CrossRef CAS PubMed.
- H. Pan, M. Qin, W. Meng, Y. Cao and W. Wang, Langmuir, 2012, 28, 12779–12787 CrossRef CAS PubMed.
- R. Cukalevski, M. Lundqvist, C. Oslakovic, B. Dahlbäck, S. Linse and T. Cedervall, Langmuir, 2011, 27, 14360–14369 CrossRef CAS PubMed.
- X. Li, J. J. Lenhart and H. W. Walker, Langmuir, 2010, 26, 16690–16698 CrossRef CAS PubMed.
- J. Liu, D. A. Sonshine, S. Shervani and R. H. Hurt, ACS Nano, 2010, 4, 6903–6913 CrossRef CAS PubMed.
- A. P. Gondikas, A. Morris, B. C. Reinsch, S. M. Marinakos, G. V. Lowry and H. Hsu-Kim, Environ. Sci. Technol., 2012, 46, 7037–7045 CrossRef CAS PubMed.
- J. H. Shannahan, X. Lai, P. C. Ke, R. Podila, J. M. Brown and F. A. Witzmann, PLoS One, 2013, 8, e74001 CAS.
- A. J. Martinolich, G. Park, M. Y. Nakamoto, R. E. Gate and K. E. Wheeler, Environ. Sci. Technol., 2012, 46, 6355–6362 CrossRef CAS PubMed.
- G. W. Doorley and C. K. Payne, Chem. Commun., 2011, 47, 466–468 RSC.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, P. Aggarwal, J. B. Hall and S. E. McNeil, Mol. Pharmaceutics, 2008, 5, 487–495 CrossRef CAS PubMed.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- F. Wang, L. Yu, M. P. Monopoli, P. Sandin, E. Mahon, A. Salvati and K. A. Dawson, Nanomed.: Nanotechnol., Biol. Med., 2013, 9, 1159–1168 CrossRef CAS PubMed.
- B. Wang, W. Feng, Y. Zhao and Z. Chai, Metallomics, 2013, 5, 793–803 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4en00002a |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |