Effect of natural organic matter on the disagglomeration of manufactured TiO2 nanoparticles†
Received
22nd October 2013
, Accepted 9th January 2014
First published on 28th January 2014
Abstract
One major concern in the fate of nanomaterials in aquatic systems is the lack of data on nanomaterial transformations under relevant environmental conditions. The disagglomeration of aggregates composed of manufactured anatase titanium dioxide nanoparticles is investigated here in the presence of alginate and Suwannee River humic acids at varying concentrations using dynamic light scattering and electrophoretic measurements. Stability of TiO2 nanoparticle agglomerates at typical environmental concentrations of natural organic matter is discussed at a pH value corresponding to the point of zero charge of TiO2 nanoparticles. In this scenario, the surface charge of TiO2 is neutralized, allowing the nanoparticles to form large agglomerates. Alginate and Suwannee River humic acids exhibit a negative structural charge under this pH condition and adsorption of both natural polyelectrolytes on the surface of nanoparticle agglomerates leads to disagglomeration and significant redispersion of TiO2 nanoparticles into fragments. Results indicate that both electrostatic forces and steric interactions play key roles during the disagglomeration process and that the physicochemical properties of natural organic matter are found to influence the kinetics and importance of fragmentation in the disagglomeration process. Most importantly, our data indicate that the presence of natural organic matter at typical environmental concentrations induces significant disagglomeration of large submicron nanoparticle agglomerates. Such a result constitutes an important outcome with regards to the risk associated with manufactured nanoparticles by including the possible transformations of the micron size range structures they can form.
Nano impact
One of the main problems in the ecological risk assessment of nanomaterials is the lack of important information on their environmental (bio)physicochemical transformations. The disagglomeration of manufactured titanium dioxide nanoparticles is investigated here in the presence of alginate and Suwannee River humic acids at realistic environmental concentrations. Under such concentration conditions, the adsorption of these compounds is found to induce disagglomeration and significant redispersion of TiO2 nanoparticles into fragments. Such a result constitutes an important outcome with regards to the risk associated with manufactured nanoparticles by considering one important life-cycle transformation of micron size range structures composed of nanoparticles in aquatic systems.
|
1. Introduction
Nowadays, manufactured nanoparticles (NPs) are part of our everyday life. Due to their specific surface properties, high efficiency and low cost, they are employed in countless consumer goods.1,2 Unfortunately, the mass production of NPs3 and the lack of regulation concerning their production, use and recovery have resulted in the rapid release of these potentially harmful substances into the environment.4 Once present in ecosystems, NPs are subject to interaction with other entities such as natural colloids and (micro)organisms1 and undergo significant transformations during their life cycle as they move into different environmental or biological compartments. Despite the fact that these transformations have received little attention to date, interactions between NPs and natural colloids in aquatic environments are expected to strongly influence NP stability.5 Fate, environmental impact, bioavailability, and stability, i.e. dispersion vs. agglomeration, of manufactured nanoparticles and the agglomerates they can form will depend on the physicochemical properties of the medium. Indeed, pH, ionic strength, temperature and the presence of natural organic matter (NOM)6–9 will control transport and toxicity towards living organisms.10,11
As a result, to achieve a better understanding of the fate, transport and impact of manufactured NPs once released into an aquatic system, their interaction and potential transformation in the presence of NOM have to be investigated for risk evaluation.12–14 Natural organic matter is mainly composed of humic substances (humic acids (HA) and fulvic acids (FA) as soluble entities) and non-humic substances such as polysaccharides. Humic substances play a very important role in aquatic systems. They can act as pH regulators and can also influence a contaminant's mobility when adsorbed on their surfaces.15 Composition, conformation and structure of HA are still ambiguous. HA are sometimes described not only as macromolecules adopting random coil conformation in solution16 but also as supramolecular structures composed of small entities linked to each other by weak interaction forces.17 These forces are thought to result from ion–dipole and hydrophobic interactions which confer a micelle like structure when hydrated.18–20 HA are also described as semi-rigid heterogenous amphiphile macromolecules composed of diverse functional groups such as phenol and carboxylic acid.21,22 Among non-humic substances, exopolysaccharides are an important and abundant class of organic compounds found in aquatic environments.23 Alginate, a model polysaccharide, is a linear block copolymer extracted from the cell walls of brown seaweed. Alginate comprises 1,4-linked β-D-mannuronic acid and α-L-guluronic acid residues. Alginate is a semi-flexible linear polysaccharide with a homogenous charge distribution.22 Alginate is widely used in the food industry as a thickening agent and stabilizer and in the pharmaceutical industry as drug carriers.24–26
NOM was found to strongly interact with NPs in suspension through electrostatic forces and steric interactions, thus modifying significantly their transport and bioavailability. Once the surface is coated with natural polymers or humic substances, the NPs are stabilized against aggregation except in the presence of divalent salts which are found to promote aggregation via bridging mechanisms.5,27–29 NOM characterization is important and higher molecular weight fraction of NOM was found to greatly enhance stabilization of NPs by steric effects in comparison with lower molecular weight NOM fraction.30 NOM adsorption was also found to be mainly promoted by ligand exchange.9
TiO2 NPs are the most produced NPs to date.4 TiO2 NPs are used in many consumer goods including cosmetics and paints and as a UV protection agent31 and are thus inexorably entering aquatic environments.4 Previous studies have demonstrated that TiO2 NP stability is governed by the physicochemical medium parameters, i.e. pH, ionic strength and the presence of multivalent salt as well as the NP intrinsic properties, i.e. size, shape and crystalline form.8,32 The destabilization of TiO2 NPs, which results in NP agglomerate formation, usually happens at a pH close to the point of zero charge (PZC) of the metal oxide. This is due to the predominance of van der Waals attractive interactions over the electrostatic repulsive forces as described by the classic Derjaguin–Landau–Verwey–Overbeek (DLVO) colloid stability theory.33,34 Another way to induce TiO2 NP agglomeration is to increase the ionic strength of the solution8,35 to reduce the Debye length and potential at the shear plan by charge screening effects. Studies indicate that divalent cations enhance agglomeration of TiO2 NPs due to specific adsorption, which results in NP surface charge neutralization.8,28
It is generally assumed that agglomerated NPs are less toxic to aquatic organisms than single NPs. It should be noted here that most of the studies related to NP stability in aquatic systems have focused on aggregation conditions representative of the physicochemical properties prevailing in freshwater.36 NPs that have been incorporated in products and then released in aquatic systems will enter either in their original form or in an altered form due to industrial processes. For several reasons (autoaggregation, difficulties to disperse powders, etc.), it is expected that a given amount of NPs will enter aquatic environments in an agglomerated and potentially less toxic form. However, natural processes may considerably alter the stability of such agglomerates with the possibility to disperse them, thus increasing diffusion and potential toxicity of the NPs.11,37 Consequently, investigating and understanding the role of NOM on already agglomerated NPs are of primary importance to better evaluate the fate, impact and potential transformations of NPs in aquatic systems.
The present study focuses on the disagglomeration process of manufactured TiO2 NPs in the presence of alginate and Suwannee River humic acids. The effect of NOM concentration and physicochemical properties is studied by considering the fragmentation process of TiO2 NP agglomerates at pH = pHPZC,TiO2. Alginate and Suwannee River humic acids belong to two distinct models and classes of natural organic matter. The disagglomeration process and its importance in determining TiO2 NP stability are examined by measuring in a systematic way the evolution of the electrophoretic mobility and hydrodynamic diameters of the TiO2 fragments with respect to time and NOM concentration.
2. Materials and methods
2.1. Materials
Anatase TiO2 NPs (Nanostructured & Amorphous Materials Inc., USA) were obtained as a 170 g L−1 suspension of 15 nm (nominal diameter) NPs in water. Stock suspensions of 1 g L−1, pH 6.2 of alginate (A2158, Sigma Aldrich, Switzerland) and 500 mg L−1, pH 9.8 of Suwannee River humic acids (Standard II, International Humic Substances Society, USA) were stirred for 24 hours and filtered through a 0.45 μm cellulose acetate filter (VWR, Switzerland). Low viscosity alginate and SRHA average molecular weight are equal to 50 kDa and 1066 Da, respectively.38,39 Sodium chloride (NaCl, 99.5%, Acros Organics, Switzerland) was employed to adjust the final ionic strength to 0.001 M. Sodium hydroxide (1 M NaOH, Titrisol®, Merck, Switzerland) and hydrochloric acid (1 M HCl, Titrisol®, Merck, Switzerland) were used after dilution to adjust the pH. All suspensions were prepared with deionized (R > 18 MΩ cm) Milli-Q water (Millipore, Switzerland). Experiments were performed in 50 mL polypropylene tubes, 29 × 115 mm (VWR, Switzerland), with a crosshead single 8 × 10 mm magnetic stirrer (VWR, Switzerland).
2.2. Zeta potential and size distribution measurements
Zeta (ζ) potential values and hydrodynamic z-average diameters were determined with a Zetasizer Nano ZS (Malvern Instruments, UK). Triplicate measurements were performed on each sample. Determination of the ζ potential values and size distribution were achieved with 15 and 12 runs, respectively. The Smoluchowski approximation model was used to calculate the ζ potential values40–42 from the electrophoretic mobility measurements. The characterization of the TiO2 NPs as well as the ζ potential values and z-average diameters as a function of NOM concentration was made at 50 mg L−1 TiO2 mass concentration at a constant 0.001 M salt concentration, whereas NOM characterization was performed at 100 mg L−1 alginate and SRHA mass concentration. Before starting, but also during the experiments if necessary, the pH of all suspensions was adjusted to the value of interest (HQ 40D, Hach Lange, Switzerland).
2.3. TEM image analysis
A Hitachi A7650 transmission electron microscope (TEM) working at 120 kV was used for image analysis. The samples were prepared by dropping 20 μL of 100 mg L−1 TiO2 suspensions onto a 200 square mesh copper grid coated with a thin film of carbon (CF200-Cu Electron Microscopy Sciences, USA). The drops were then left to dry prior to TEM analysis. When analyzing samples containing alginate and SRHA, a 1 minute treatment was performed with osmium tetroxide (4% in water, Electron Microscopy Sciences, USA) as a staining agent in order to enhance the NOM contrast.43 The acquisition time and the image resolution were set to 5 s and 3284 × 2600 pixels, respectively.
3. Results and discussion
3.1. TiO2, alginate and Suwannee River humic acid characterization
TiO2 NPs.
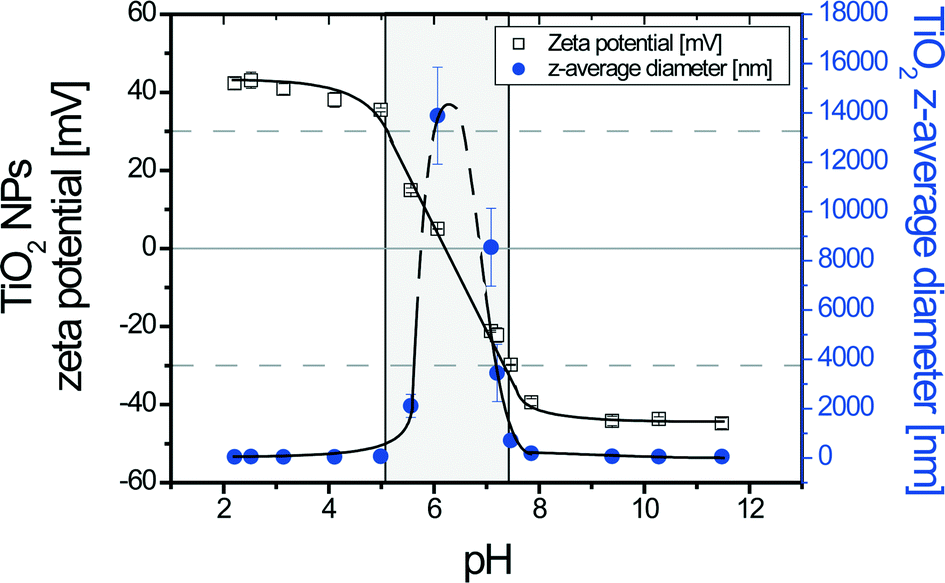
In order to get an insight into TiO2 stability as a function of pH, titration curves for pH values in the range 2–11 were realized. In Fig. 1, it is shown that for a pH value lower than 5, TiO2 NPs exhibit strong and stable positive zeta potential values (black squares) of +40.0 ± 3.1 mV (mean ± standard deviation). As the pH increases, the zeta potential rapidly decreases to the point of zero charge at pH = pHPZC,TiO2 = 6.2 ± 0.1, which is in good agreement with the literature.44 Further pH increase leads to charge inversion and surface charge stabilization is observed for a pH range of 9–11 with a ζ potential value equal to −44.2 ± 1.2 mV. The hydrodynamic z-average diameter variation as a function of pH is also shown in Fig. 1 (blue dots). For pH values between 2 and 5, the TiO2 NPs are stable and the z-average diameter is 52 ± 9 nm. Then, the z-average diameter increases rapidly to a maximum value at pH = pHPZC,TiO2, corresponding to the formation of large agglomerates in the micron size range. A further pH increase rapidly leads to the decrease of TiO2z-average diameter. A plateau corresponding to a z-average of 57 ± 7 nm is obtained at pH > 9. A domain of TiO2 destabilization is found here, as indicated by the gray shaded area in Fig. 1, for zeta potential values between +30 mV and −30 mV. TEM images of stable (pH < pHPZC,TiO2) and agglomerated TiO2 (pH = pHPZC,TiO2) NPs are shown in the ESI† in Fig. S1 and S2, respectively. Out of the destabilization domain, individual NPs as well as small NP agglomerates composed of a few NPs are present, which is in good agreement with our dynamic light scattering measurements (52 ± 9 nm).
 |
| | Fig. 1 Zeta potential and z-average diameters of TiO2 NPs as a function of pH. The point of zero charge (pHPZC,TiO2) was found at pH 6.2 ± 0.1. An important TiO2 NP agglomeration domain (gray shaded area) was found between −30 mV and +30 mV; [TiO2] = 50 mg L−1; I = 0.001 M. | |
Alginate.
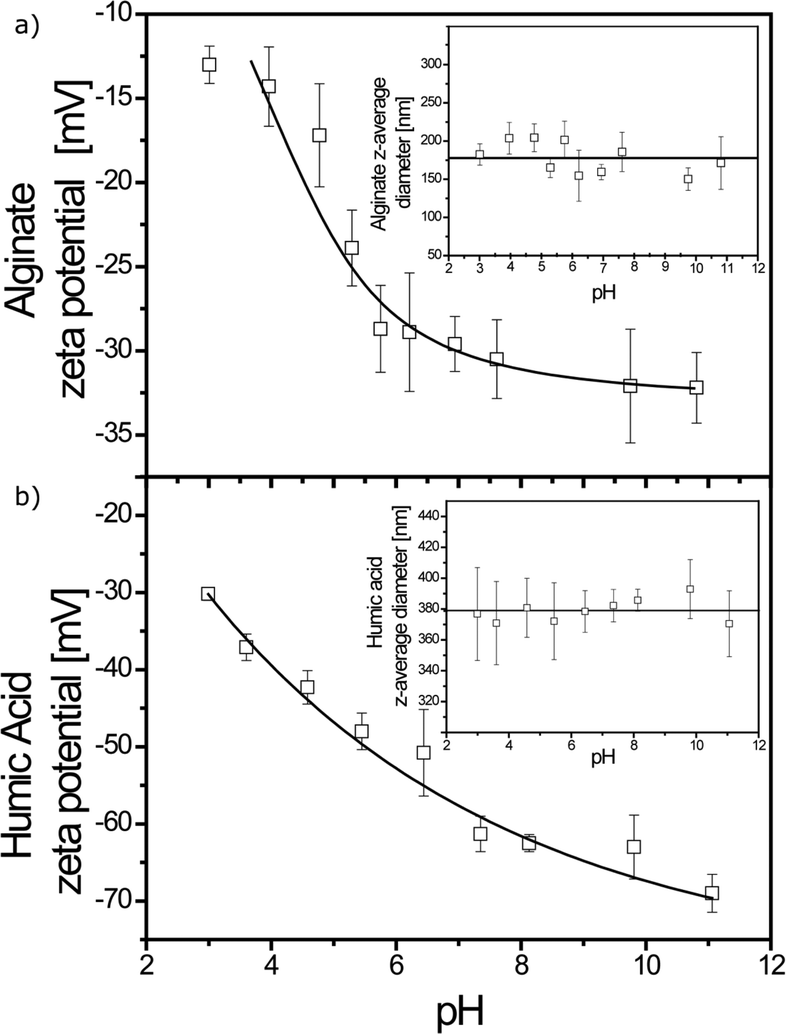
The pH was adjusted from 11 to 3 with diluted HCl at variable concentrations. As shown in Fig. 2a, alginate exhibits a negative surface charge in the full domain. For a pH range of 11–5.75, ζ potential values are found to be stable (−30.3 ± 1.5 mV), whereas at a lower pH, values increase continuously as a result of protonation of the carboxyl functional groups present in the α-L-guluronate and β-D-mannuronate monomers with pKa values of 3.65 and 3.38, respectively.45 The z-average diameter is found to be constant with a mean z-average diameter equal to 178 ± 21 nm (Fig. 2a, inset).
 |
| | Fig. 2 a) Zeta potential of alginate as a function of pH. No PZC was found here. z-Average diameters of alginate as a function of pH (inset). The z-average diameter had a constant value of 178 ± 21 nm. b) Zeta potential of SRHA as a function of pH. No PZC was found here. z-Average diameters of SRHA as a function of pH (inset). The z-average diameter had a constant value of 379 ± 19 nm. [Alginate] = [SRHA] = 100 mg L−1; I = 0.001 M. | |
Suwannee River humic acids.
In the 11 to 3 pH range, SRHA have a significant negative charge and a constant z-average diameter as can be seen in Fig. 2b. The ζ potential increases from −69.0 ± 2.4 mV at pH 11 to −30.2 ± 0.8 mV at pH 3. No ζ potential plateau is obtained due to the heterogeneity of SRHA functional groups. The z-average diameter is 379 ± 19 nm (Fig. 2b, inset). Both ζ potential and z-average diameter values are in agreement with previous studies.46,47
3.2. TiO2 NP disagglomeration in the presence of alginate and SRHA
To investigate the disagglomeration process, the pH of the TiO2 suspension was adjusted to pH = pHPZC,TiO2 in order to form large agglomerates via surface charge neutralization (Fig. 1). The effect of NOM on TiO2 agglomerate stability was then studied by recording ζ potential and size distribution variations as a function of time for different NOM concentrations. The agitation speed was set to obtain a low velocity of mixing only to gently homogenize the suspensions.
TiO2 NP disagglomeration in the presence of alginate.
As shown in Fig. 3, the presence of alginate induces the disagglomeration of the TiO2 aggregates and two distinct regimes are found. In the first regime, an important decrease of z-average diameters is observed, which is due to the fragmentation of TiO2 agglomerates during the first 45 min after alginate addition. Then, a plateau is reached, corresponding to a second regime. In the second regime, the system is at a long-time state, according to particle sizes and ζ potential values, and no further disagglomeration is observed as indicated by the stabilization of z-average diameters and ζ potential values. Increasing the alginate concentration also enhances the importance of the disagglomeration process with maximum efficiency obtained for alginate concentrations ≥3 mg L−1. In such conditions, the corresponding final z-average diameters are equal to 500 nm. Thus, only a partial, but significant, TiO2 NP disagglomeration occurs with z-average diameters for high alginate concentrations an order of magnitude greater than the individual size of TiO2 NPs (500 nm vs. 50 nm). Another interesting point to mention, at high concentrations in the first regime (Fig. 3b), is the continuous decrease of ζ potential values resulting from the continuous adsorption of alginate. The effect of alginate concentration on TiO2 disagglomeration at t = 60 min (in the long-time state plateau) is represented in Fig. 4. It is shown that maximum disagglomeration is achieved with an alginate concentration ≥3 mg L−1. A TEM image of TiO2 agglomerate fragments in the presence of alginate is given in Fig. S3.†
 |
| | Fig. 3 a) z-Average diameters of TiO2 agglomerates as a function of time at pHPZC,TiO2 (6.2 ± 0.1) for different alginate mass concentrations. b) Zeta potential of TiO2 agglomerates in the presence of alginate as a function of time at pHPZC,TiO2. Equilibrium time for alginate induced disagglomeration was 45 min. [TiO2] = 50 mg L−1; I = 0.001 M. | |
 |
| | Fig. 4 Final z-average diameters of TiO2 NPs as a function of alginate mass concentration at pHPZC,TiO2 (6.2 ± 0.1). Measurements were made 60 min after the addition of alginate, i.e. after equilibrium time for NP disagglomeration. Maximum disagglomeration occurs at [alginate] ≥3 mg L−1, which corresponds to a z-average diameter of 500 nm. [TiO2] = 50 mg L−1; I = 0.001 M. | |
TiO2 NP disagglomeration in the presence of SRHA.
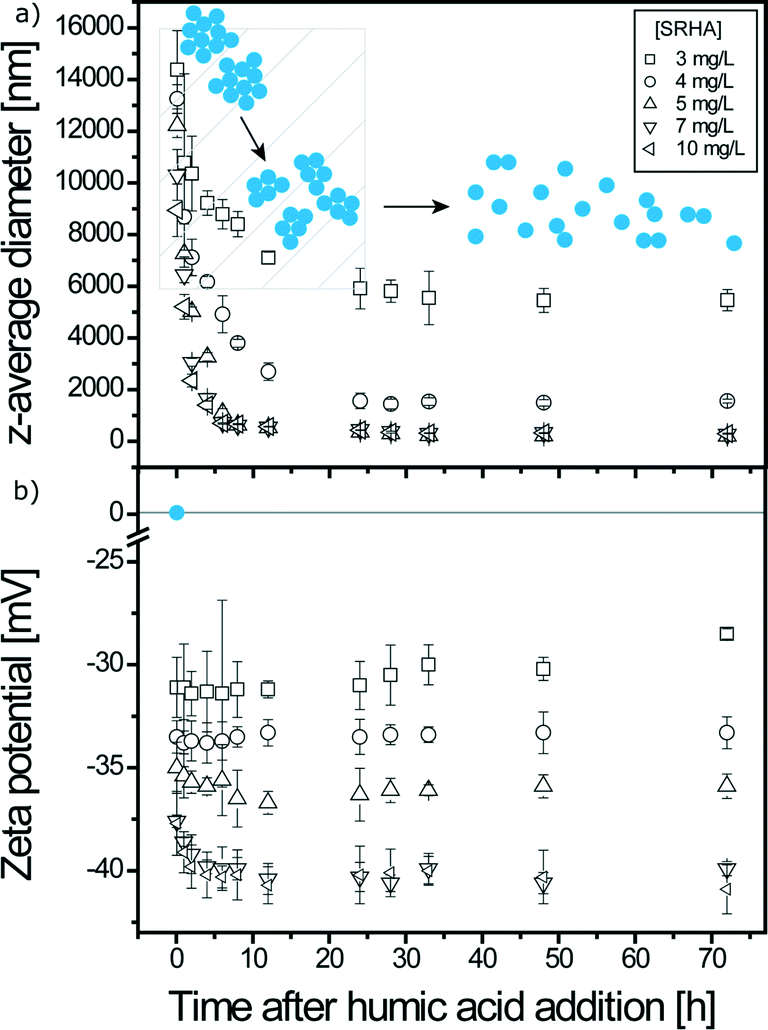
As shown in Fig. 5, two regimes are also present and the time needed to reach system long-time state after SRHA addition is now equal to 24 h. Maximum TiO2 disagglomeration is obtained for SRHA concentrations ≥5 mg L−1. Once the system is at the long-time state, the z-average diameters are equal to 250 nm, i.e. smaller than with alginate. Disagglomeration is therefore much important with SRHA than with alginate. SRHA is also continuously adsorbed onto the surface of TiO2 NPs as denoted by the decrease of the ζ potential values in the first regime for high SRHA concentrations. The TiO2z-average diameter at the long-time state depends on the amount of SRHA added as observed in Fig. 6 where the variation of TiO2z-average diameters with respect to SRHA concentration 48 h after SRHA addition is presented. A TEM image of TiO2 agglomerate fragments in the presence of SRHA is given in Fig. S4.†
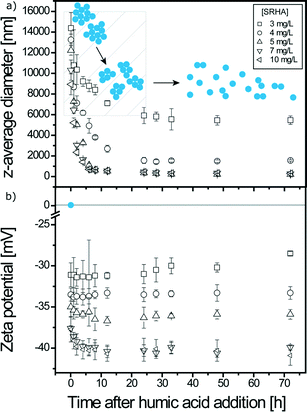 |
| | Fig. 5 a) z-Average diameters of TiO2 agglomerates as a function of time at pHPZC,TiO2 (6.2 ± 0.1) for different SRHA mass concentrations. b) Zeta potential of TiO2 agglomerates in the presence of SRHA as a function of time at pHPZC,TiO2. Equilibrium time for SRHA induced disagglomeration is 24 h. [TiO2] = 50 mg L−1; I = 0.001 M. | |
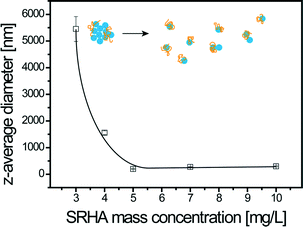 |
| | Fig. 6 Final z-average diameters of TiO2 NPs as a function of SRHA concentration at pHPZC,TiO2 (6.2 ± 0.1). Measurements were made 48 hours after the addition of SRHA, i.e. after equilibrium time for NP disagglomeration. SRHA induced disagglomeration and maximum disagglomeration occurred at [SRHA] ≥5 mg L−1, which corresponds to a z-average diameter of 250 nm. [TiO2] = 50 mg L−1; I = 0.001 M. | |
Influence of NOM properties on TiO2 disagglomeration.
Two important differences in TiO2 NP disagglomeration are observable when comparing the effects of alginate (Fig. 3 and 4) and SRHA (Fig. 5 and 6). Firstly, the time needed to reach system long-time state is 45 min in the case of alginate, whereas it is 24 h for SRHA. The disagglomeration process is expected to be controlled by both adsorption of NOM onto the TiO2 agglomerates and its ability to reach the inner structure of the agglomerates to produce fragments. As shown by Pefferkorn, the interpenetration of polyelectrolyte chains within agglomerates plays a key role in the kinetic process of disagglomeration.48 Alginate is a semi-rigid linear biopolymer with a homogenous charge distribution, whereas SRHA is a semi-rigid globular macromolecule.22 Thus, the ability to adsorb alginate on the TiO2 agglomerate surface and penetrate inside the aggregates through conformational changes (reptation) is greater for alginate in comparison to SRHA which is subject to more important steric hindrance. Secondly, alginate is found to be less efficient than SRHA for disagglomeration when comparing the final NOM–TiO2 fragment sizes at high NOM concentrations (Fig. 7). Indeed, SRHA exhibits a higher ζ potential value and thus, once adsorbed onto the TiO2 agglomerate, restabilizes the NPs more efficiently owing to greater electrostatic repulsion forces. Here, NP surface charge is found to be an essential ingredient in the disagglomeration processes.
 |
| | Fig. 7 Disagglomeration of TiO2 NPs in the presence of alginate and SRHA at pH = 6.2 = pHPZC,TiO2. Influence of adsorption and interpenetration on disagglomeration kinetic and NOM physicochemical properties on final fragment size (TiO2/NOM ratio numbers are only illustrative). | |
In the present study, TiO2 disagglomeration is expected to mainly result from random disagglomeration leading to a rapid decrease in z-average diameters. Nonetheless, even if attrition, i.e. the production of small fragments, has not been identified using DLS analysis, such a fragmentation mechanism cannot be excluded.
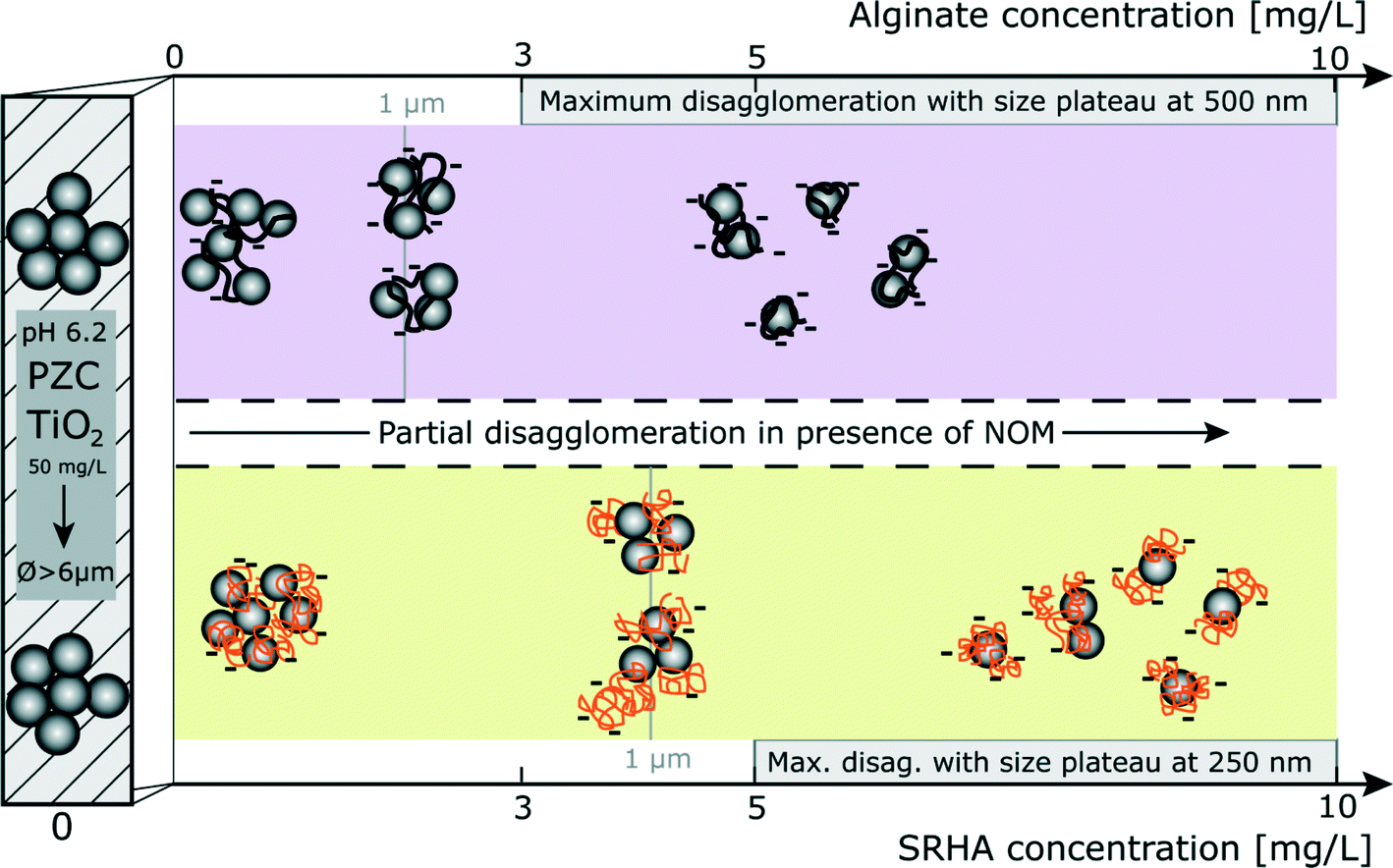
The stability diagram which is presented in Fig. 8 gives an overview of the disagglomeration processes and highlights the main differences between alginate (purple area) and SRHA (orange area). Agglomeration occurs at the PZC before addition of NOM with agglomerates in the micron range. NOM addition induces partial disagglomeration, as indicated by the black arrow, at relatively low concentrations which are representative of NOM concentrations found in aquatic systems. Indeed, to obtain NP fragments of 1 μm, concentrations of alginate and SRHA should be equal to 2 mg L−1 and 4.25 mg L−1, respectively (grey vertical lines). The increase of the NOM concentration results in smaller fragment sizes and maximum disagglomeration is obtained for concentrations ≥3 mg L−1 for alginate and ≥5 mg L−1 for SRHA.
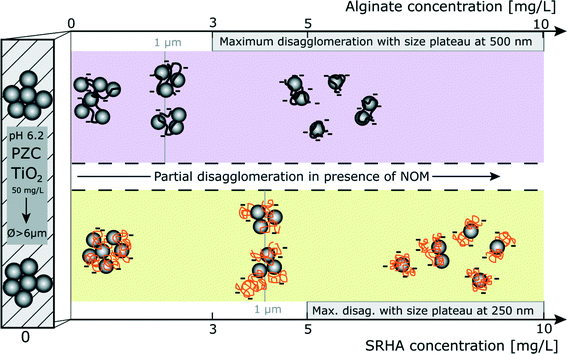 |
| | Fig. 8 Stability of TiO2 agglomerates in the presence of alginate (purple area) and SRHA (orange area) at pH = pHPZC,TiO2. Agglomeration is represented by hatched areas. In both cases, the presence of NOM promotes TiO2 disagglomeration at NOM environmental concentrations. [TiO2] = 50 mg L−1; I = 0.001 M (TiO2/NOM ratio numbers are only illustrative). | |
3. Conclusions
The stability of TiO2 agglomerates was investigated by determining the evolution of the ζ potential and z-average diameter values as a function of alginate and Suwannee River humic acid concentrations. Typical environmental NOM concentrations were used here, and it was clearly shown that NOM significantly modifies the stability of TiO2 NP agglomerates (50 mg L−1 TiO2 suspensions) by promoting their disagglomeration. The physicochemical properties of NOM strongly influenced the kinetics and rates of TiO2 disagglomeration which was found to be mainly governed by electrostatic repulsive forces and steric interactions. Disagglomeration was found to occur on a relatively short time scale and different mechanisms were discussed when comparing the two NOM models. Since disagglomeration processes are expected to be influenced by the fractal dimensions of agglomerates and bonding energies between the NPs, one important aspect to be developed in future work concerns the agglomerate fractal dimension importance and detailed mechanisms of disagglomeration in the presence of NOM. Overall, our results clearly indicate the importance of NOM on the transformation of agglomerates composed of NPs and underline the need to reevaluate, in some circumstances, the fate, transport and impact of nanomaterial assemblies.
Acknowledgements
The authors are grateful to Daniel Azmoon and Nadia Von Moos for stimulating discussions and to Sabrina Lacomme and Etienne Gontier for technical assistance and the contribution of BioImaging Center at the University of Bordeaux 2. We also acknowledge the financial support received from the Swiss National Foundation (project 200021_135240) and nanoMILE FP7 Project.
References
- Y. Ju-Nam and J. R. Lead, Sci. Total Environ., 2008, 400, 396–414 CrossRef CAS PubMed.
- M. Auffan, J. Rose, J.-Y. Bottero, G. V. Lowry, J.-P. Jolivet and M. R. Wiesner, Nat. Nanotechnol., 2009, 4, 634–641 CrossRef CAS PubMed.
- M. C. Roco, J. Nanopart. Res., 2011, 13, 427–445 CrossRef.
- F. Gottschalk, T. Sonderer, R. W. Scholz and B. Nowack, Environ. Sci. Technol., 2009, 43, 9216–9222 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, J. Colloid Interface Sci., 2007, 309, 126–134 CrossRef CAS PubMed.
- Y. Liu, M. Tourbin, S. Lachaize and P. Guiraud, Ind. Eng. Chem. Res., 2012, 51, 1853–1863 CrossRef CAS.
- I. Chowdhury, Y. Hong, R. J. Honda and S. L. Walker, J. Colloid Interface Sci., 2011, 360, 548–555 CrossRef CAS PubMed.
- R. A. French, A. R. Jacobson, B. Kim, S. L. Isley, R. L. Penn and P. C. Baveye, Environ. Sci. Technol., 2009, 43, 1354–1359 CrossRef CAS.
- K. Yang, D. Lin and B. Xing, Langmuir, 2009, 25, 3571–3576 CrossRef CAS PubMed.
- S.-R. Chae, Y. Xiao, S. Lin, T. Noeiaghaei, J.-O. Kim and M. R. Wiesner, Water Res., 2012, 46, 4053–4062 CrossRef CAS PubMed.
- N. R. von Moos and V. Slaveykova, Nanotoxicology, 2014, 8, 605–630 CrossRef CAS PubMed.
- B. Nowack and T. D. Bucheli, Environ. Pollut., 2007, 150, 5–22 CrossRef CAS PubMed.
- V. L. Colvin, Nat. Biotechnol., 2003, 21, 1166–1170 CrossRef CAS PubMed.
- M. R. Wiesner, G. V. Lowry, P. Alvarez, D. Dionysiou and P. Biswas, Environ. Sci. Technol., 2006, 40, 4336–4345 CrossRef CAS.
- J. Pertusatti and A. G. S. Prado, J. Colloid Interface Sci., 2007, 314, 484–489 CrossRef CAS PubMed.
- R. S. Cameron, B. K. Thornton, R. S. Swift and A. M. Posner, J. Soil Sci., 1972, 23, 394–408 CrossRef CAS.
- A. Piccolo, Soil Sci., 2001, 166, 810–832 CrossRef CAS PubMed.
- J. Peuravuori, Environ. Sci. Technol., 2005, 39, 5541–5549 CrossRef CAS.
- R. Sutton and G. Sposito, Environ. Sci. Technol., 2005, 39, 9009–9015 CrossRef CAS.
- D. Smejkalova and A. Piccolo, Environ. Sci. Technol., 2008, 42, 699–706 CrossRef CAS.
- A. Piccolo, Adv. Agron., 2002, 75, 57–134 CAS.
- J. Buffle, K. J. Wilkinson, S. Stoll, M. Filella and J. W. Zhang, Environ. Sci. Technol., 1998, 32, 2887–2899 CrossRef CAS.
- C. Lamelas, M. Benedetti, K. J. Wilkinson and V. I. Slaveykova, Chemosphere, 2006, 65, 1362–1370 CrossRef CAS PubMed.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS PubMed.
-
T. Helgerud, O. Gåserød, T. Fjæreide, P. O. Andersen and C. K. Larsen, in Food Stabilisers, Thickeners and Gelling Agents, ed. A. Imeson, Wiley-Blackwell, Oxford, UK, 2009, pp. 50–71 Search PubMed.
- M. Rajaonarivony, C. Vauthier, G. Couarraze, F. Puisieux and P. Couvreur, J. Pharm. Sci., 1993, 82, 912–917 CrossRef CAS.
- R. F. Domingos, C. Peyrot and K. J. Wilkinson, Environ. Chem., 2010, 7, 61–66 CrossRef CAS.
- K. L. Chen, S. E. Mylon and M. Elimelech, Environ. Sci. Technol., 2006, 40, 1516–1523 CrossRef CAS.
- F. Mohd Omar, H. Abdul Aziz and S. Stoll, Sci. Total Environ., 2014, 468–469, 195–201 CrossRef CAS PubMed.
- S. M. Louie, R. D. Tilton and G. V. Lowry, Sci. Total Environ., 2013, 47, 4245–4254 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- I. Chowdhury, S. L. Walker and S. E. Mylon, Environ. Sci. Technol., 2013, 15, 275–282 CAS.
- B. V. D. A. L. Landau, Acta Physicochim. URSS, 1941, 14, 633–662 Search PubMed.
- E. J. W. Verwey, J. Phys. Colloid Chem., 1947, 51, 631–636 CrossRef CAS.
- K. A. D. Guzman, M. P. Finnegan and J. F. Banfield, Environ. Sci. Technol., 2006, 40, 7688–7693 CrossRef.
- J. Hammes, J. A. Gallego-Urrea and M. Hassellöv, Water Res., 2013, 47, 5350–5361 CrossRef CAS PubMed.
- I. Roemer, T. White, M. Baalousha, K. Chipman, M. R. Viant and J. R. Lead, J. Chromatogr. A, 2011, 1218, 4226–4233 CrossRef CAS PubMed.
- M. A. LeRoux, F. Guilak and L. A. Setton, J. Biomed. Mater. Res., 1999, 47, 46–53 CrossRef CAS.
-
J. Aiken, P. A. Brown, T. I Noyes and D. J. Pickney, in Molecular size and weight of fulvic and humic acids from Suwannee River, ed. R. C. Averett, Leenheer, J. A., McKnight, D. M. and Thorn, K. A., Georgia, 1989, pp. 163–180 Search PubMed.
- M. Baalousha, Sci. Total Environ., 2009, 407, 2093–2101 CrossRef CAS PubMed.
- H. Ohshima, Adv. Colloid Interface Sci., 1995, 62, 189–235 CrossRef CAS.
- H. Ohshima, Colloids Surf., A, 1995, 103, 249–255 CrossRef CAS.
- M. Baalousha, M. Motelica-Heino, S. Galaup and P. Le Coustumer, Microsc. Res. Tech., 2005, 66, 299–306 CrossRef CAS PubMed.
- G. A. Parks, Chem. Rev., 1965, 65, 177–198 CrossRef CAS.
-
A. Haug, PhD Thesis, Norwegian Institute of Technology, 1964 Search PubMed.
- M. Hosse and K. J. Wilkinson, Environ. Sci. Technol., 2001, 35, 4301–4306 CrossRef CAS.
- N. E. Palmer and R. von Wandruszka, Fresenius' J. Anal. Chem., 2001, 371, 951–954 CrossRef CAS.
- E. Pefferkorn, Adv. Colloid Interface Sci., 1995, 56, 33–104 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3en00061c |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.