 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of available analytical technologies for qualitative and quantitative determination of nitramines
Sofia
Lindahl
*,
Cathrine Brecke
Gundersen
and
Elsa
Lundanes
University of Oslo, Department of Chemistry, P.O. Box 1033, Blindern, NO-0315 Oslo, Norway. E-mail: sofia.lindahl@kjemi.uio.no; Tel: +4722855538
First published on 5th June 2014
Abstract
This review aims to summarize the available analytical methods in the open literature for the determination of some aliphatic and cyclic nitramines. Nitramines covered in this review are the ones that can be formed from the use of amines in post-combustion CO2 capture (PCC) plants and end up in the environment. Since the literature is quite scarce regarding the determination of nitramines in aqueous and soil samples, methods for determination of nitramines in other matrices have also been included. Since the nitramines are found in complex matrices and/or in very low concentration, an extraction step is often necessary before their determination. Liquid–liquid extraction (LLE) using dichloromethane and solid phase extraction (SPE) with an activated carbon based material have been the two most common extraction methods. Gas chromatography (GC) or reversed phase liquid chromatography (RPLC) has been used often combined with mass spectrometry (MS) in the final determination step. Presently there is no comprehensive method available that can be used for determination of all nitramines included in this review. The lowest concentration limit of quantification (cLOQ) is in the ng L−1 range, however, most methods appear to have a cLOQ in the μg L−1 range, if the cLOQ has been given.
Environmental impactNitramines constitute a group of compounds that are classified as emerging environmental pollutants, being potent carcinogens. Nitramines can be formed in the atmosphere due to the release of amines from e.g. post combustion CO2 capture plants. The nitramines may end up in the nature at low levels, and for the determination of these nitramines highly sensitive analytical methods are necessary. We have reviewed the available open literature on methods for the determination of nitramines that might be formed from amines used in post combustion CO2 capture plants. |
1. Introduction
This review compiles the available published analytical methods for qualitative and quantitative determination of some aliphatic (linear and cyclic) nitramines. The interest in nitramines (Fig. 1a) has increased in recent years since they are among the possible degradation products of amines used in post-combustion CO2 capture (PCC) technology.1–3 The human influence is the dominant cause for the observed global warming that is now reported to be more certain than ever.4 CO2 capture, with ultimate long-term storage, has been advanced to be a significant contributor in the global mitigation portfolio.5,6 The amine based CO2 capturing method is currently considered the most feasible and hence the one most likely to be first implemented on a large scale.7 Although there is a promising technology for mitigating excess atmospheric carbon dioxide, concern has been raised since the nitramines are reported as potent carcinogens.8 In comparison with the more well studied compound group of nitrosamines (Fig. 1b), also formed from the use of amines,3,9 the nitramines are believed to be less carcinogenic.8 However, due to the expected longer residence times of the nitramines in the environment, this compound group may pose a higher risk to public health and ecosystems in close vicinity of a PCC plant (<50 km)10 than the short-lived nitrosamines which are subject to rapid photolysis.3 Nitramines are believed to be of concern with regard to chronic exposure, whereas acute toxicity is assumed to be negligible. A number of tests have shown that N-nitrodimethylamine (DMNA) and N-nitromethylamine (MMNA) are carcinogenic.11,12 DMNA and MMNA are listed in the Carcinogenic Potency Database (http://toxnet.nlm.nih.gov/cpdb/), and the TD50 values for rats are 0.55 and 17.4 mg per kg per day, respectively. Due to the lack of toxicology data for nitramines, the Norwegian Institute of Public Health has suggested a combined safety threshold concentration of 4 ng of nitramines and nitrosamine per L in drinking water.10 Other guidelines on safety threshold values are available but mostly for nitrosamines. For example the California Department of Public Health has set the notification level to 10 ng L−1 for N-nitrosodiethylamine, N-nitrosodimethylamine (NDMA) and N-nitrosodipropylamine (NDPA),13 the Drinking Water Inspectorate for England and Wales has set the value to 9 ng L−1 for NDMA14 and the Ministry of Environment of Ontario has set the value to 1 ng L−1 for NDMA.15 Fjellsbø et al.16 studied the eye irritation and skin sensitization/irritation/corrosion of DMNA, MMNA and 2-methyl-2-(nitroamine)-1-propanol (AMP-NO2) and the result was that all three were irritants for the eye.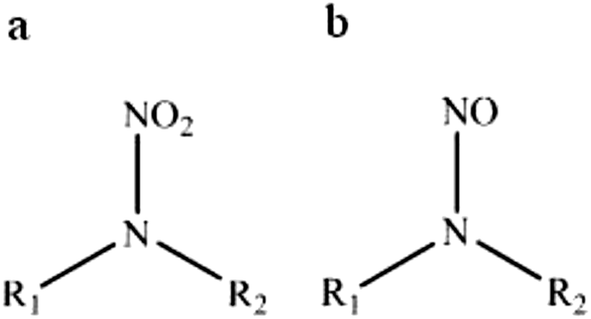 | ||
| Fig. 1 Chemical structure of (a) nitramine and (b) nitrosamine. In primary nitramine/nitrosamine R1 = H and R2 = aliphatic group and in secondary nitramine/nitrosamine R1, R2 = aliphatic group. | ||
From a typical PCC plant, as illustrated in Fig. 2, two different pathways for nitramine formation exist:
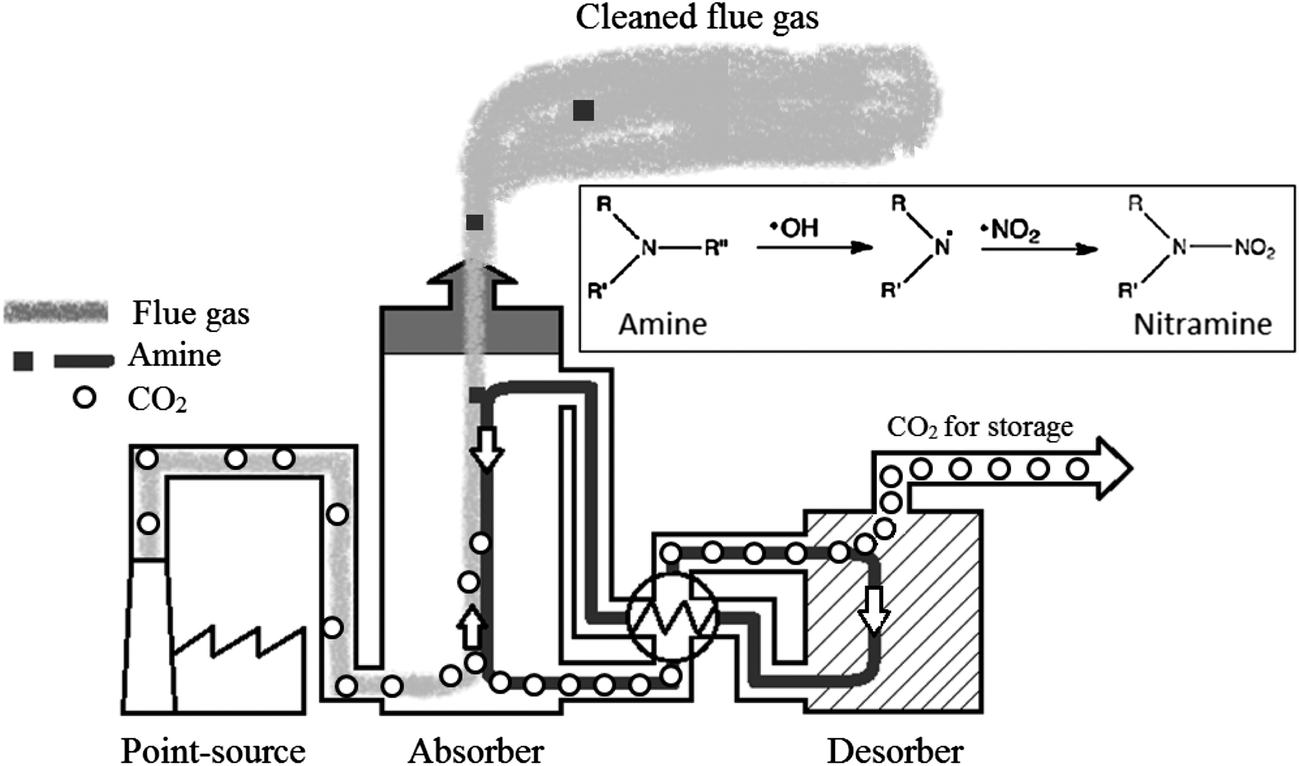 | ||
| Fig. 2 Schematic illustration of the process of amine-based CO2 capture. The flue gas (light grey) from an industrial related point-source is fed into the absorber tower in which it counter-currently contacts a stream of liquid amine solution (dark grey). The CO2 (open circle) is captured from the flue gas by reversibly reacting with amine molecules, forming carbamate complexes. This reaction is subsequently reversed inside the desorber unit by applying heat, resulting in the production of a pure stream of CO2 suitable for long-term storage. The amine solution is reused in the process, but small amounts may be lost along with the cleaned flue gas. In the atmosphere, rapid degradation processes initiated by the photolytically produced hydroxyl radical may result in the formation of nitramines. | ||
1. Atmospheric degradation of amines released with the cleaned flue gas.
2. Oxidative degradation of amines inside the capture unit.
The amines typically used for CO2 capture have low volatility (e.g. alkanolamines),17 but studies have shown that small alkylamines of higher volatility are formed in the capture process18 making concurrent atmospheric emissions with the cleaned flue gas virtually impossible to avoid.
In the atmosphere, amines may be converted to nitramines through reactions with hydroxyl radicals OH* (or NO3/Cl/O3)3 and nitrating agents (NO2) (Fig. 2). The amount of nitramine formed post-emission will therefore depend on the local mixing ratio of NOx.19 The hydrophilic nature of the nitramines indicates deposition with rain and fog droplets,20 and subsequent partitioning between the terrestrial and aquatic phase.
Inside of the capture unit, nitramines may form from oxidative amine degradation in the presence of NOx. The mechanism proposed is through formation of the nitrating agent dinitrogen tetroxide (N2O4).21 The nitramines are, in contrast to the amines, not very volatile and the low volatility largely limits the direct emission from the plant.22 The type of nitramines formed is restricted by the properties of the parent-amines. Primary and secondary amines form stable nitramines, whereas tertiary amines are degraded into secondary amines which can then react to form nitramines.23
2. Determination of nitramines
The number of publications in the open literature on determination of nitramines is quite scarce compared to those on nitrosamines. The analytical methodologies for determination of nitrosamines in water are quite well established and have been reviewed recently.24 There are also a number of published standard methods for the determination of nitrosamines,25–27 and therefore publications on determination of nitrosamines are not included in this review, unless they also cover nitramines. The nitramines included in this review and some of their chemical properties are listed in Table 1. The nitramines included in this review are nitramines that might be formed from amines used in PCC and linear aliphatic nitramines up to n-butyl. The mass solubility of the nitramines at pH 7 and ambient temperature ranges from 1 to 1000 g L−1. But most of the nitramines have high water solubility and a low vapour pressure. Most of the primary nitramines, for example MMNA and N-nitroethanolamine (EtOHNA) are weak acids with a pKa value around 6.2–6.5 (Table 1). Since both the estimation of possible nitramine concentrations in environmental aqueous samples and the threshold value set around the world are in the ng L−1 range, highly sensitive analytical methods are needed for their determination. The challenges for establishing reliable quantification methods for nitramines in environmental aqueous samples are (i) that they are present at very low concentrations (ng L−1), (ii) that they are small polar/hydrophilic highly water soluble compounds and (iii) there is a lack of commercially available standards.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, pKa and chemical structure of nitramines included in the review
P, pKa and chemical structure of nitramines included in the review
| Nitramine (abbreviation) | CAS-no | M (g mol−1) | Solubility in H2O at 25 °C, pH 7a (g L−1) | Vapour pressurea (Torr) | logPa |
pKaa | Structure |
|---|---|---|---|---|---|---|---|
| a Values taken from SciFinder (https://scifinder.cas.org/scifinder/view/scifinder/scifinderExplore.jsf). | |||||||
| N-Nitromethylamine (MMNA) | 598-57-2 | 76.05 | 1000 | 19.5 | −0.598 ± 0.369 | 6.51 ± 0.1, −6.14 ± 0.7 |
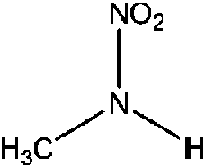
|
| N-Nitrodimethylamine (DMNA) | 4164-28-7 | 90.08 | 176 | 0.887 | −0.447 ± 0.377 | −7.12 ± 0.7 |
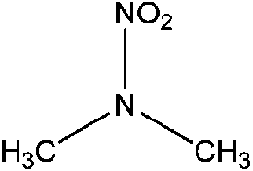
|
| N-Nitroethylmethylamine (EMNA) | 19092-01-4 | 104.11 | 76 | 0.611 | 0.063 ± 0.377 | −6.88 ± 0.7 |
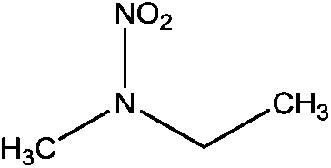
|
| N-Nitroethylamine (MENA) | 19091-98-6 | 90.08 | 652 | 8.45 | −0.089 ± 0.369 | 6.53 ± 0.10, −6.23 ± 0.70 |
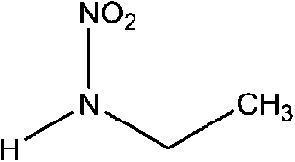
|
| N-Nitrodiethylamine (DENA) | 7119-92-8 | 118.13 | 33 | 0.340 | 0.572 ± 0.377 | −6.63 ± 0.7 |
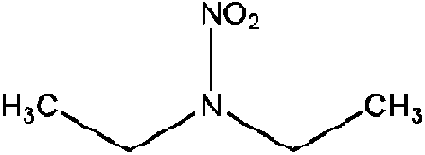
|
| N-Nitrobutylethylamine (BENA) | 52330-08-2 | 146.19 | 5.4 | 0.0481 | 1.591 ± 0.378 | −6.63 ± 0.70 |
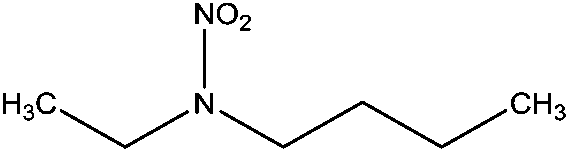
|
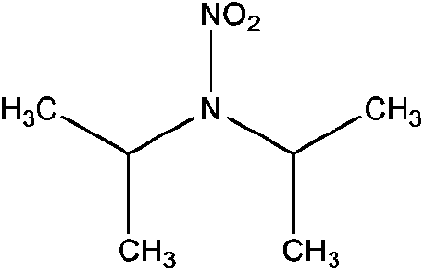
| N-Nitrodiisopropylamine (DIPNA) | 4164-30-1 | 146.19 | 9.4 | 0.108 | 1.280 ± 0.407 | −6.14 ± 0.70 |
|
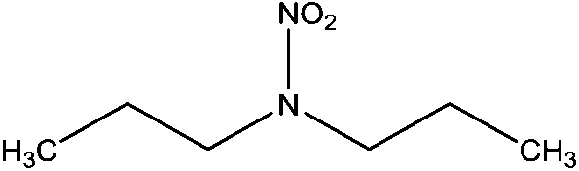
| N-Nitrodipropylamine (DPNA) | 4164-29-8 | 146.19 | 5.4 | 0.0481 | 1.591 ± 0.378 | −6.67 ± 0.7 |
|
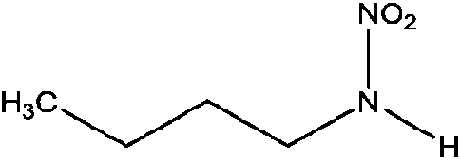
| N-Nitrobutylamine (MBNA) | 3182-75-0 | 118.13 | 100 | 1.2 | 0.930 ± 0.370 | 6.53 ± 0.10, −6.18 ± 0.70 |
|
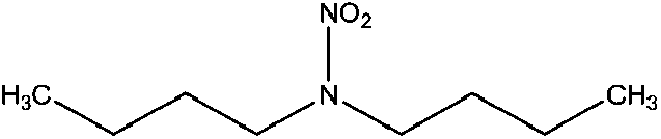
| N-Nitrodibutylamine (DBNA) | 4164-31-2 | 174.24 | 0.98 | 7.34 × 10−3 | 2.610 ± 0.378 | −6.63 ± 0.70 |
|
| N-Nitroethanolamine (EtOHNA) | 74386-82-6 | 106.08 | 1000 | 1.22 × 10−3 | −1.241 ± 0.433 | 6.24 ± 0.1, −8.04 ± 0.7 |
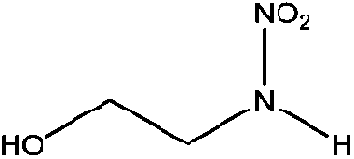
|
| N-Nitrodiethanolamine (diEtOHNA) | 13084-48-5 | 150.13 | 171 | 2.86 × 10−8 | −1.732 ± 0.465 | 13.85 ± 0.10, −9.27 ± 0.70 |
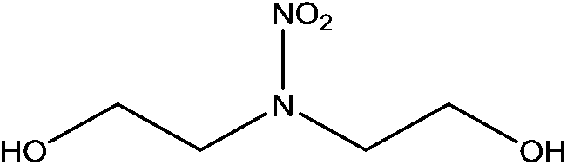
|
| 2-Methyl-2-(nitroamine)-1-propanol (AMP-NO2) | 1239666-60-4 | 134.13 | 1000 | 5.91 × 10−3 | −0.476 ± 0.452 | 6.29 ± 0.1, −7.53 ± 0.7 |
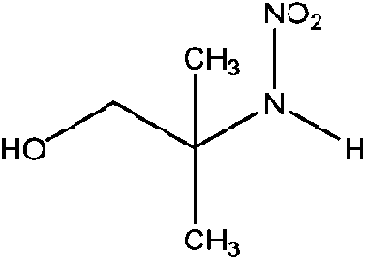
|
| N-Nitropiperazine (NIPZ) | 42499-41-2 | 131.13 | 236 | 6.68 × 10−4 | −0.917 ± 0.495 | 7.58 ± 0.1 |
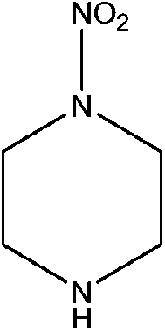
|
| 1-Nitro-4-nitrosopiperazine (1,4-NIPZ/NPZ) | 107938-05-6 | 160.13 | 13 | 7.49 × 10−8 | −1.143 ± 0.458 | −5.61 ± 0.7 |
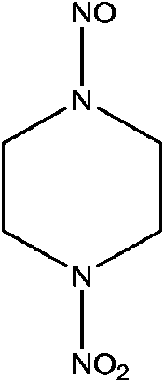
|
| N,N-Dinitropiperazine (1,4-NIPZ) | 4164-37-8 | 176.13 | 1.5 | 2.37 × 10−9 | −1.561 ± 0.442 | −8.80 ± 0.7 |
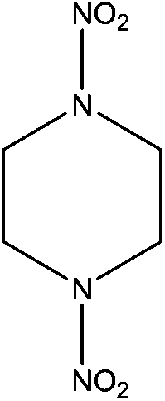
|
| N-Nitromorpholine (NIMOR) | 4164-32-3 | 132.12 | 15 | 1.07 × 10−3 | −1.030 ± 0.456 | −9.21 ± 0.2 |
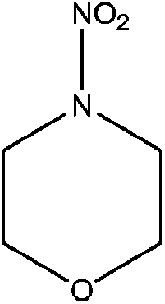
|
| N-Nitropiperidine (NIPIP) | 7119-94-0 | 130.15 | 2.5 | 0.0106 | 0.326 ± 0.369 | −6.67 ± 0.2 |
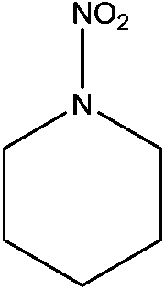
|
| N-Nitropyrrolidine (NIPYR) | 3760-55-2 | 116.12 | 4.1 | 0.0199 | −0.201 ± 0.367 | −6.63 ± 0.2 |
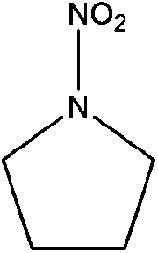
|
| N-Nitrothiazolidine | 104549-75-9 | 134.16 | 3.1 | 1.06 × 10−4 | 0.065 ± 0.596 | −9.63 ± 0.2 |
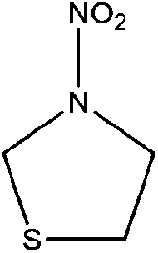
|
Nitramines that are not included in this review are nitramines used in explosives or degradation products of explosives, for example 1,3,5-trinitroperhydro-1,3,5-triazine (RDX).28
Table 2 summarizes the available analytical methods for determination of the nitramines listed in Table 1.
| Nitramines | Sample | Sample preparation method | Method | Column | Mobile phase/Oven temp | Detector | cLOQ in the sample | Ref. |
|---|---|---|---|---|---|---|---|---|
| DMNA | Standard solutions and water samples | SPE: activated coconut charcoal, ES: acetone, Vsample: 500 mL, Cfactor: 500x | GC | Supelcowax 10® 0.53 mm × 15 m × 1 μm | T grad: 40–150 °C | NPD | 10 ng L−1 | 30 |
| DMNA | Standard solutions and synthetic water samples | SPE: activated carbon, ES: DCM, IS: d6-NDMA before SPE and d14-NDPA before analysis, Vsample: 500 mL, Cfactor: 1000x, according to ref. 25 | GC25 | DB-1701, 0.25 mm × 30 m × 1 μm | T grad: 35–280 °C | MS (CI) | N.A. | 42 |
| DMNA | Standards and pool/aquaria samples | SPE: activated carbon, ES: DCM, IS: d6-NDMA before SPE and d14-NDPA before analysis. Vsample: 500 mL, Cfactor: 1000x, according to ref. 25 | GC25 | DB-1701, 0.25 mm × 30 m × 1.00 μm with a deactivated silica column, 0.25 mm × 5 m × 0.25 μm | T grad: 34–250 °C | MS (CI) | 2 ng L−1 | 31 |
| DMNA | Standards and municipal waste water | Reaction quenched. SPE: Ambersorb 572.60 ES: DCM, IS: d6-NDMA before SPE. Vsample: 1000 mL, Cfactor: 1000x. modification of ref. 61 | GC61 | Supelco SPB™ 1701 capillary column 0.25 mm × 30 m × 0.25 μm | T grad: 35–220 °C | MS (CI) | 30 ng L−1 a | 29 |
| DMNA | Standards, water and cell cultures | EPA method 521 (ref. 25) and/or: centrifugation of cell culture. Supernatant resuspended in H2O and extracted using LLE with DCM. Vsample and Cfactor: N.A. (Cfactor of EPA method is 500x (ref. 25)) | GC25 | Not stated | T grad: 34–250 °C | MS (CI) | 120ng L−1 a | 36 |
| DMNA, EMNA, DENA, DPNA, BENA, NIPYR, NIMOR, NIPIP, and DBNA | Standards | LLE of irradiated samples (water) with DCM. IS: NDEA added before LLE and 1,3-dimethyl-2-nitrobenzene added before analysis. Vsample: 5 mL, Cfactor: 0.5x or 0.2x | GC | JW Scientific DB-5 ms | T grad: 45–250 °C | MS (EI) | 3–6 μmol L−1 a, Ex DMNA: 270–540 μg L−1 (not specified detection limit for specific nitramines, only state that detection limit is 1–2 μM) | 44 |
| NIMOR | Standards, amine samples | LLE with DCM. IS: NPYR added before LLE. Similar method as (ref. 62 and 63) Vsample and Cfactor: N.A. | GC | DB-5 capillary column | MS | N.A. | 45 | |
| NIMOR | Standards, amine samples | Addition of Na–ascorbate. LLE with DCM. IS: NPYR added before LLE, Vsample: 5 mL, Cfactor: 5x | GC | 10% Carbowax 20M stationary phase, 2 mm × 1.8 m | T grad: 120 °C and 160 °C | TEA | 2.4 ng a | 37 |
| DMNA | Standard | N.A. | GC | 1% OV-101 on Supelcoport 100–120 mesh, 0.6 cm × 170 cm | T grad: 80 °C, flow: 30 mL min−1 | Nitrogen specific | N.A. | 12 |
| NIMOR | Amine standard bubbled with NO2. | LLE with DCM. IS: NPYR added before LLE. Filter through cotton. Vsample: 5 mL, Cfactor: N.A. | GC | 10% Carbowax 20 stationary phase on chromsorb WHP 80–100 mesh packed in a glass column, 2 mm × 1.8 m | T grad: 120 °C, Tpyr: 475 °C | TEA | N.A. | 46 |
| NIMOR | Amine standard bubbled with NO2. | LLE with DCM. IS: NPYR added before LLE. Filter through cotton, Vsample: 5 mL, Cfactor: 5x | GC | 10% Carbowax 20 stationary phase on chromsorb WHP 80–100 mesh packed in a glass column, 2 mm × 1.8 m | T grad: 120 °C, gas: helium 10 mL min−1, Tpyr: 475 °C | TEA | N.A. | 47 |
| NIMOR | Secondary amines treated with peroxynitrite etc. | Reaction terminated. LLE with DCM or ethyl acetate | GC | 5% FFAP on chromasorb W-HP, 3 mm × 2 m | Gas: argon 30 mL min−1, Tgrad: 120–190 °C, Tpyr: 700 °C | TEA | N.A. | 38 |
| IS: NDBA or N-nitrosopipecolic acid added before LLE. Nitro- and nitroso-proline converted to their methyl derivatives with diazomethane. Vsample: 1 mL, Cfactor: N.A. | GC | HP-1, 0.18 × 25 m × 0.18 μm film | Gas: argon 30 mL min−1, Tgrad: 50–230 °C | MS | 38 | |||
| DENA, DMNA, DBNA | Artificial saliva | Rubber incubated in artificial saliva, HCl and NaOH added. LLE with Extrelut and DCM | GC | 10% Carbowax (20 M) on chromosorb W-AW, 2m × 1/4 inch o.d. | Gas: argon 30 mL min−1, Tgrad: 150 °C, TTEA: 450 °C | TEA | N.A. | 65 |
| IS: N-nitrosodiisopropylamine, added before HCl and NaOH, according to Spiegelhalder et al.64 | GC | Capillary OV-101, 0.2 mm × 12 m × 0.3 μm film | T grad: 30–180 °C | MS | 65 | |||
| DMNA, DIPNA | Standards | No sample prep. | LC | Lichrosorb SI-60, 4.6 × 250 mm | 7:93 EtOH: isooctane |
UV, 250 nm | N.A. | 66 |
| EtOHNA, DMNA, NIPZ | Lab-scale PCC samples | No information about sample pretreatment. Deuterated internal standard used | LC | RP-columns | MeOH, ACN, formic acid, acetate buffer | MS APCI or jet stream ESI | 0.1–1 μg L−1 | 67 |
| EtOHNA, AMP-NO2 | Lab-scale PCC samples | No information | LC | N.A. | N.A. | MS | N.A. | 68 |
| DMNA | Standards, tap water, pool water, waste water etc. | With or without SPE. Some water samples quenched with ascorbic acid, some water samples filtered through cellulose nitrate filters. SPE: HLB and activated carbon. ES: DCM, Vsample: 1000 mL, Cfactor: 1000x. Based on Krauss et al.69 | LC | TSKgel G2500PWxl, 7.8 × 300 mm, 7 μm | Gradient, 25 mM phosphate buffer pH 6.7 and ACN | PCUV, 540 nm | 8.8 μg L−1 without SPE, 12 ng L−1 a with SPE | 32 |
| DENA | Standards | LC | Zorbax-ODS, 4.6 × 150 mm | 9:1 H2O:ACN |
UV, 254 nm | N.A. | 39 | |
| DMNA, MMNA, EtOHNA, AMP-NO2, NIPZ | Standards, air, water, etc. | Adjust the pH of sample to 6–6.5, SPE: activated carbon, ES: MeOH, THF, ethyl acetate/MeOH, Vsample: 30 mL, Cfactor: 20x | LC | HSS T3, 150 × 2.1 mm, 3 μm (Waters), Atlantis T3, 150 × 2.1 mm, 3 μm (Waters) | Gradient H2O:ACN | MS, EtOHNA: ESI− MMNA: ESI− DMNA: APCI + NIPZ: ESI+ AMP-NO2: ESI− | DMNA: 120 μg L−1 a, MMNA: 6.3 μg L−1 a, EtOHNA: 1.5 μg L−1 a, AMP-NO2: 0.9 μg L−1 a, NIPZ: 1.5 μg L−1 a | 33 |
| DMNA | Standard solutions | No sample preparation | LC | RP-18, 250 × 4.6 mm, 5 μm | 3 mM tetrabutylammonium phosphate, pH 7.0. | UV, 232 nm | N.A. | 57 |
| NIPZ, DMNA, NIMOR | Wash water lab scale PCC, standard | Quenching of H2O2 by addition of sodium sulphite | LC | Inertsil ODS-3, 4.6 × 250 mm, 5 μm | MeOH:H2O or MeOH:10 mM phosphate buffer pH 7.7 |
UV, 245 nm | N.A. | 35 |
| EtOHNA, diEtOHNA, 1,4-NIPZ/NPZ, 1,4-NIPZ | Wash water lab scale PCC, standards | V sample/Cfactor: no LLE or SPE | LC | Hi-Plex ligand exchange, 300 × 6.5 mm, 8 μm | H2O | UV, 245 nm | N.A. | 35 |
| 1,4-NIPZ | Standard solutions and thermolysis samples of standards | N.A. | LC | Econosphere C18, 4.6 × 250 mm, 5 μm | MeOH/THF/H2O | UV, 229 nm | N.A. | 56 |
| DMNA, DENA, DIPNA, DPNA | LC | Lichrosorb SI-60, 4.6 × 250 mm | EtOH/isooctane or EtOH/hexane | UV, 254 nm | 56 | |||
| DMNA, DIPNA, NIPIP, NIPYR | GC | DB-05 | T grad: 50/60/80 to 180 °C | FID | 56 | |||
| MMNA | Standard solutions and blood samples | Proteins precipitated with MeOH | LC | RP18, 4.6 × 250 mm, 5 μm | 10 mM tetrabutylammonium phosphate, pH 7.0 | UV, 254 nm | N.A. | 34 |
| DMNA | Standard solutions, blood samples | Addition of NaOH and IS NDPA, SPE: Extrelut (kieselguhr). ES: DCM, Vsample: 20 g, Cfactor: 20x, According to Spiegelhalder et al. (ref. 70) | GC | 15% Carbowax column 2 mm × 1.2 m | Helium 25 mL min−1 (ref. 70), Tgrad : 175 °C (ref. 70), Vinj: 3–5 μL (ref. 70) | TEA | DMNA: 0.42 ng μL−1 a blood | 34 |
| DMNA, MMNA | Standard solutions, cell culture samples | LLE with DCM:acetone, Vsample: 0.5 mL, Cfactor: 0.5x |
LC71 | AnionSep Ice-Ion 310 Fast organic acid column, 6.5 × 150 mm | 1.73 mM sulfuric acid, 35 °C | UV, 225 nm | N.A. | 43 |
| LC71 | Zorbax SB-C18, 0.5 × 150 mm, 5 μm | 20:80 ACN:H2O |
MS, ESI− | 43 | ||||
| GC | 0.33 μm HP-5MS capillary column, 0.2 mm × 50 m | 40–200 °C, helium. | MS, EI | 43 | ||||
| EtOHNA, diEtOHNA | Standard solutions, water samples | pH adjusted to pH 2 and the day after to pH 5.5–6.0. LLE with ethyl acetate. IS: Addition of d8-NDEtOH before LLE. Vsample: 500 mL, Cfactor: 500x | LC | Agilent Hi-Plex ligand exchange, 6.5 × 300 mm, 8 μm | 30 mM formic acid | MS APCI+ | EtOHNA: 75 μg L−1 a, diEtOHNA: 90 μg L−1 a, NDEtOH: 0.6 μg L−1 a | 23 |
| NIPZ | Standard solutions, water samples | No sample prep for analysis of NPZ and NIPZ | LC | Agilent Zorbax 300-SCX 2.1 × 150 mm, 5 μm | 8:2 10 mM Am. Form. pH 3: ACN |
MS APCI+ | NPZ & NIPZ: 390 μg L−1 a,b in deionized water. NIPZ: 1.2 mg L−1 a in matrix-specific | 23 |
| DMNA, NIMOR, 1,4-NIPZ, 1,4-NIPZ/NPZ | Standard solutions, water samples | SPE: activated carbon, ES: DCM,25 IS: d6-NDMA and d8-NMOR added before SPE. Vsample: 500 mL, Cfactor: 500x | GC25 | Agilent DB-1701 | T grad: 35–250 °C, Vinj: 8 μl | MS (CI) | 3 μg L−1 a, except 1,4-NIPZ, 1,4-NIPZ/NPZ 30 μg L−1 a | 23 |
| LLE with DCM. IS: d6-NDMA and d8-NMOR added before LLE. Vsample: 1000 mL, Cfactor: 1000x | ||||||||
| EtOHNA | Standard solutions, soil samples | Nitramine solution mixed with soil. Shaking and sample collection for 192 h. Sample centrifuged and filtered | LC | N.A. | N.A. | MS | N.A. | 72 |
| DMNA | Standard solutions, soil samples | As above and LLE with DCM before GC analysis. Vsample/Cfactor: no information | GC | N.A. | N.A. | MS | N.A. | 72 |
| DMNA, DENA, DBNA | Standard solutions | None | GC | FFAP, 0.25 mm × 2 m | Temp: 120 °C | TEA | N.A. | 54 |
| TLC | Silica gel F254 | Hexane–diethylether–dichloromethane as an eluent | 54 | |||||
| DBNA (GC) | Rat samples (microsomes, hepatocytes and urine) | Microsomes: microsomes obtained.73 Proteins precipitated. Samples analyzed with GC and LC. Supernatant extracted with ethyl acetate before GC analysis | GC | 1.5% OV-17 on chromosorb W | NPD | N.A. | 75 | |
| DENA, MENA, MBNA (LC) | Hepatocytes: nitramines added to isolated hepatocytes. Samples analyzed with GC and LC | LC | LiChrosorb RP-8, 5 μm | 40% ACN | N.A. | 75 | ||
| Urine: urine fractionated by solvent extraction and hydrolysed by β-glucuronidase.74 Samples analysed with GC, LC and TLC | TLC | LiChrosorb SI-100, 5 μm | Hexane:ether and hexane:ether:DCM |
N.A. | 75 | |||
| MMNA, MBNA | Standards | TLC | Silica gel G, 10 × 20 cm, 0.3 mm | Benzene:nitromethane 2:1 or chloroform:nitromethane 10:1 |
UV | N.A. | 53 |
2.1 Sample preparation
The analytical chain for the analysis of a sample is divided into a number of steps: (i) planning, (ii) sample collection, (iii) sample preparation, (iv) separation, (v) detection, (vi) evaluation, (vii) interpretation and (viii) validation. The focus in this review for determination of nitramines is on step ii–v.Sample collection (ii) is an important step regarding the determination of nitramines in environmental samples. But only a few of the articles listed in Table 2 concern nitramine determination in “real” samples, mostly different kinds of water samples. The container material used for sample collection and storage is important and should be evaluated for trace determination of nitramines, to avoid loss of nitramines due to adsorption to the container material. Some of the methods are based on EPA-method 521,25 which is developed for determination of nitrosamines in drinking water. This method recommends to use amber glass containers for collection of water samples, to dechlorinate collected samples at the time of collection and that samples should not be stored above 6–10 °C, but not frozen.25 Schreiber et al.29 collected waste water samples in fluorinated high density polyethylene containers, but why this material was selected is not discussed. Some authors also state for how long the samples are stored before further sample preparation and analysis.30–32 Bjerke et al.33 studied both storage stability in different solvents and possible adsorption of the nitramine standards (DMNA, MMNA, EtOHNA and AMP-NO2) on the filter used in bacterial filtration and on the wall of falcon tubes and 96-well plates. None or only very little adsorption of the nitramines was found.33 Regarding storage, the best solvent was water at −18 °C.33 Possible adsorption of nitramines on glass, which is commonly used for sample collection and storage, has not been published.
The sample preparation might vary depending on the subsequent separation method that will be employed. In the articles listed in Table 2 the sample preparation step is not always described and in the case of determination of nitramines in standard solutions no sample preparation is necessary, except e.g. dilution. For extraction of nitramines from different kinds of liquid samples (e.g. blood and water) either solid-phase extraction (SPE) or liquid–liquid extraction (LLE) has been used (Table 2). However, prior to the extraction sometimes some additional sample preparation steps such as adjusting of pH and/or quenching of reactions have been performed.23,29,32–39
In SPE the nitramines are adsorbed on a solid material and then eluted using a suitable solvent, and by using a smaller elution volume (which may also be further reduced by evaporation) compared to the sample volume applied, the nitramines are enriched. The adsorbent materials that have been used for nitramines are mainly carbon based (Table 2). Bjerke et al.33 tested four different SPE materials for the extraction of DMNA, MMNA, EtOHNA and AMP-NO2, and found an activated carbon based material to be the best adsorbent. However, some of the nitramines were so well adsorbed that only a few percent of the adsorbed amount could be eluted when different elution solvents were evaluated (See Appendix B in Bjerke et al.33). Hence improvement of the method by changing the elution strength of the solvents is needed.33 But if nitrosamines (Fig. 1b) are also included in the analysis method another adsorbent material is recommended to be used, since studies have shown that nitrosamines might be artificially formed if the samples contain amines and nitrites, and active carbon is used as an adsorbent material.23,40,41 Dichloromethane (DCM) has been the most commonly used elution solvent in SPE of nitramines.23,29,31,32,34,36,42 Other solvents that have been used as the eluting solvent in SPE or as the extraction solvent in LLE are for example acetone,30 ethyl acetate,23,38 DCM + acetone43 and methanol (MeOH)/tetrahydrofuran/ethylacetate + MeOH.33
In LLE, the extraction solvent should not be miscible with the sample solvent and the nitramines should preferably have a higher solubility in the extraction solvent than in the sample. For LLE of nitramines DCM has been the most commonly used solvent.23,37–39,44–47 However, Bjerke et al.33 showed that only 5–13% of the nitramines EtOHNA, MMNA and AMP-NO2 were extracted by DCM from water samples, indicating that selection of the extraction solvent is important.
For both samples extracted using SPE and LLE it should also be kept in mind that if the extract is to be analyzed by reversed phase liquid chromatography (RPLC), evaporation of the organic solvent or back extraction23 of the nitramines into water is necessary.
Since most of the samples listed in Table 2 are liquids, SPE and/or LLE have been the only extraction step necessary. But if the sample is a solid, for example soil, a suitable solid–liquid extraction method is necessary. For extraction of nitramines associated with explosives in soil samples organic solvents have been used.48,49
Some of the nitramines listed in Table 1 (DMNA, MMNA, N-nitrodiethylamine (DENA)) have also been determined in air- and aerosol samples and cartridges used for sampling have for example been Thermosorb/N, Tenax and charcoal.9,19,50–52
Unfortunately, only a few studies on stability of nitramines in aqueous environmental samples have been reported. Hence there is a need for investigation of stability and also possible adsorption of nitramines to sample containers. More thorough studies are also needed for evaluating adsorbent materials and elution/extraction solvents, in order to develop methods where several nitramines can be determined simultaneously at low ppt levels.
2.2 Separation and detection
Table 2 summarises the published analytical methodologies used for qualitative and quantitative determination of nitramines. The two most common techniques used are gas chromatography (GC) and liquid chromatography (LC) in combination with different kind of detectors. However, thin layer chromatography (TLC)53,54 has also been used in a few articles. For GC determination the analytes should have a low boiling point (bp)/vapour pressure and be thermally stable at the separation temperature, otherwise a derivatization step might be necessary. A derivatization step may, however, increase the uncertainty of the method since an extra step is added in the sample preparation procedure. From a sustainability point of view it is also advantageous to avoid derivatization since it usually results in the use of extra chemicals and more waste is produced.55 For analytes requiring derivatization for GC, LC is an alternative technique. For determination of nitrosamines (Fig. 1b), GC is commonly used24 since nitrosamines have a lower bp compared to the corresponding nitramine. In the standardised methods for determination of nitrosamines, for example EPA method 521,25 GC is the recommended separation method.The various detectors used for GC determination of nitramines (Table 2) include MS, thermal energy analyzer (TEA), nitrogen–phosphorus detector (NPD) and flame ionization detector (FID). The most frequently used one is MS with either chemical ionization (CI)23,29,31,36,42 or electron ionization (EI).43,44
Other groups have also used various columns for the determination of various nitramines in the same set of samples. Dai et al.23 used several methods for determination of nitrosamines and nitramines associated with PCC. One LC method was used for the determination of nitramines/nitrosamines with alcohol as a functional group, another LC method was used for the determination of NIPZ and N-nitrosopiperazine (NPZ), while a GC-method (EPA method 52125) was used for determination of DMNA, NDMA, N-nitrosomethylethylamine, N-nitrosodiethylamine (NDEA), NDPA, N-nitrosomorpholine (NMOR), N-nitrosopyrrolidine (NPYR), N-nitrosopiperidine, NIMOR, N-nitrosodibutylamine (NDBA), NPZ, NIPZ, 1,4-dinitrosopiperazine, 1,4-NIPZ/NPZ and N,N-dinitropiperazine (1,4-NIPZ).23 For the LC determination of EtOHNA, diEtOHNA and N-nitrosodiethanolamine (NDEtOH) an Agilent Hi-Plex ligand column was used with 30 mM formic acid as the mobile phase.23 The same group published another article half a year later where they used the same type of column for the determination of EtOHNA, diEtOHNA, 1,4-NIPZ and 1,4-NIPZ/NPZ, but this time 100% water was used as the mobile phase.35 The reason for excluding acid in the latter publication was not stated, but better separation was achieved on the Hi-Plex column, which is a strong cation exchange (SCX) column, when acid was excluded. Dai et al.23 used another SCX column for determination of NPZ and NIPZ. The difference between the two SCX columns is the matrix used, the Hi-Plex column is styrene-divinylbenzene based, while the Zorbax SCX-column is silica based. In a later publication, Shah et al.35 used an Intersil ODS-3 column to determine NIPZ, and also NIMOR and DMNA. They do not state why they choose to use different columns and methods in the two publications.
The mobile phases used with the different LC columns are shown in Table 2, and mostly isocratic elution with water and additive (acid/salt) or gradient elution with water and ACN have been used for RP separation. Lee et al.32 used a size exclusion chromatography (SEC) column, but state that the separation of DMNA and the various nitrosamines was not based on the size but on hydrophobic/hydrophilic interactions with the matrix.
The conclusion is that at present there is no column material available for simultaneous LC separation of all the nitramines listed in Table 1.
In all the published LC methods, spectrometric detection, either an ultraviolet (UV)-detector or a MS (see Table 2) has been utilised. An MS is generally much more sensitive than a UV-detector, and thus MS is preferred for analysis of environmental samples where the nitramines might be present at very low concentrations. Another advantage of MS detection is that structural information and hence identification of the analytes can be obtained by analysing the fragmentation pattern of the analyte. UV absorbance spectra of DMNA and EtOHNA are shown in Fig. 3. As seen in Fig. 3 nitramines absorb light in the low UV-range, which might be a problem with gradient elution in LC, since organic solvents and other mobile phase additives often absorb light at low wavelengths. The UV wavelengths that have been used in the published LC-UV analysis (Table 2) range are between 225 nm and 254 nm.
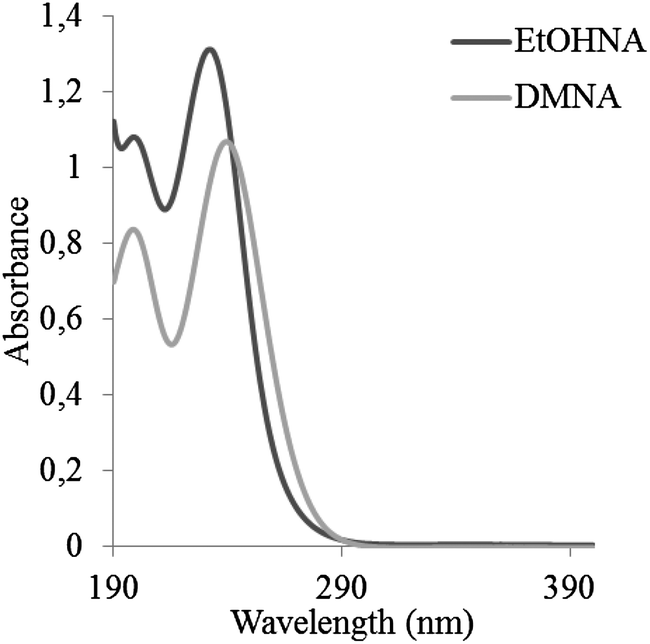 | ||
| Fig. 3 Absorption spectra of EtOHNA (dark grey) and of DMNA (light grey) in aqueous solution at pH 7. | ||
As mentioned above, MS is a more sensitive detection method compared to UV, but it is also more expensive. Detecting nitramines by MS may be hampered by their low molar mass because of many low molar mass interfering ions from for example solvents, LC-MS systems and samples.
Both electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI) have been used for ionization of nitramines (Table 2). Dai et al.23 used an APCI interface with positive ionization for detection of EtOHNA, diEtOHNA and NIPZ. In a report by Bjerke et al.,33 the sensitivity of both APCI and ESI in negative and positive mode was evaluated for DMNA, MMNA, EtOHNA, NIPZ and AMP-NO2. They found that negative ionization was the best for primary nitramines (EtOHNA, AMP-NO2, MMNA), whereas positive ionization was the best for secondary nitramines (DMNA, NIPZ). Highest sensitivity was achieved using ESI compared to APCI, but in the case of DMNA, UV-detection resulted in the highest sensitivity.33 Fournier et al.43 also used ESI for determination of DMNA and MMNA, but negative ionization was employed for both nitramines.
The choice of ionization mode has been more thoroughly studied for nitrosamines. Ripolles et al.58 compared ESI and APCI for determination of eight nitrosamines and the highest sensitivity was achieved using APCI. Zhao et al.59 also compared the use of ESI and APCI for determination of eight aliphatic (linear/cyclic) nitrosamines. Their conclusion was that ESI was a better alternative since the parent ion was not generated for some of the thermolabile nitrosamines in the case of APCI. Evans et al.28 compared the use of ESI and APCI for determination of some explosives, whereof one was a cyclic nitramine (RDX), and the limit of detection (LOD) was better using APCI compared to ESI.
Thus regarding ionization mode and type, no general conclusion can be drawn for nitramines. The best sensitivity obtainable probably depends on a number of factors such as kind of MS and mobile phase composition. The latter has shown to have an effect on the ionization of the analytes in MS (own observations). None of the articles listed in Table 2 discusses the effect of the mobile phase composition on the sensitivity of the LC-MS methods. However, Ripolles et al.58 noticed that for ESI, a H2O/MeOH mobile phase resulted in more abundant peaks compared to a H2O/ACN mobile phase, but this was for determination of nitrosamines. The addition of formic acid to the H2O/MeOH mobile phase improved the signal further. When APCI was used as an ionization source, the sensitivity was better using H2O/ACN compared to H2O/MeOH as the mobile phase.
To date, regarding the choice of the separation method for nitramines associated with PCC, there is no method available in the open literature. A method which can be used for determination of a wide range of nitramines at low concentrations is urgently needed.
2.3 Limit of quantification, enrichment factor and selection of internal standard
The concentration limit of quantification (cLOQ) has only been reported for some of the methods listed in Table 2 and not for all of the nitramines, but in general the cLOQ is in the μg L−1 range. There are however, a few exceptions where the cLOQ is in the ng L−1 range, but only for DMNA.29–32 Greene et al.,30 Schreiber et al.29 and Walse et al.31 used SPE for enrichment and GC in the separation step. With a NPD detector Greene et al.30 found a cLOQ of 10 ng L−1, whereas with MS detection Schreiber et al.29 and Walse et al.31 obtained a cLOQ of 30 ng L−1 and 2 ng L−1, respectively. Lee et al.32 also used SPE, but LC-PCUV (post column UV photolysis) for separation and detection, obtaining a cLOQ of 12 ng L−1. Schreiber et al.29 and Greene et al.30 both obtained an extraction efficiency of 0.6, so by increasing the extraction efficiency the detection limit can be further lowered. Lee et al.32 obtained a higher extraction efficiency of 0.80–0.96. The matrix of the sample can also have an interfering effect on the cLOQ and therefore it is important that the sample preparation not only enriches the nitramines but also removes interfering compounds. An example is that the cLOQ limit for NIPZ was ∼390 μg L−1 in deionized water, but the matrix-specific cLOQ was estimated to ∼1.2 mg L−1.23 NIPZ was determined without any sample preparation and this may explain the higher cLOQ, compared to that of EtOHNA and diEtOHNA.23 The cLOQ of the nitramines is usually higher compared to the corresponding nitrosamines.29,36,37 For example the cLOQ of diEtOHNA was 90 μg L−1, which was approximately 10 times higher than that of NDEtOH.23 The reason for this was not investigated, but the authors speculate that it was due to nitramines being more thermolabile compared to the corresponding nitrosamines.23Another important factor to address in method development is improving the extraction enrichment factor, especially for samples with low concentrations of nitramines. In the four methods where the cLOQ is in the ng L−1 range,29–32 the enrichment factors were 500x or 1000x, using a sample of 500 or 1000 mL, which were reduced to 0.5–1 mL. Dai et al.23 obtained an enrichment factor of 500x or 1000x depending on the analyte and extraction method, but the cLOQ was in the μg L−1 range. The extraction efficiency varied depending on the extraction method used, from 0.25 up to 0.96–1.2.23
The use and selection of the internal standard (IS) is yet another important factor in the quantification. For determination of nitramines it has been most common to use nitrosamines as the IS, either a deuterium labelled nitrosamine of one of those included in the determination or a nitrosamine that is known not to be present in the samples. An IS is added to samples to compensate e.g. for the loss of analytes during sample preparation, control of volume of the sample and to compensate for variation in the MS detection response. Thus it is important to choose an IS that will act in a similar way as the analyte(s) of interest, otherwise there is a risk for under or over estimation of the nitramine concentration. The reason why nitrosamines have been used as an IS is probably due to the fact that in many of the methods listed in Table 2, not only nitramines were determined, but also nitrosamines, and nitrosamine standards are commercially available while that is not the case for nitramine standards. If a nitrosamine is chosen as an IS it is important to evaluate that it “behaves” similar to the nitramines in the different steps of the analysis of the sample. In some cases, e.g. if the extraction efficiency of the analytes of interest in the sample differ, more than one IS can be used. Dai et al.23 for example used two different IS for the determination of DMNA, NIMOR, 1,4-NIPZ, 1,4-NIPZ/NPZ and 11 nitrosamines. A d6-labelled NDMA was used for early eluting analytes in the GC-method and a d8-labelled NMOR for the late eluting analytes.23 More than one IS can also be use to compensate for different parts of the method, for example as in EPA method 521 where one IS is added before SPE and one before GC analysis.25 Since the commercial availability of the deuterium labelled nitramine standard is very limited, another alternative is to use the so-called standard addition method as Bjerke et al.33 have published. Even though the standard addition method is time consuming it compensates for matrix effects. If external calibration is used, and the calibration standards are prepared in solvents only, which appears to be the case in several of the publications, there is no compensation for possible matrix effects.
3. Concluding remarks
After reviewing the open literature regarding determination of nitramines a number of conclusions can be drawn. To date, there is no universal method published where all nitramines listed in Table 1 can be determined. The lowest reported cLOQs are obtained with GC-MS determination, but GC might not be a viable method for all nitramines due to their low volatility. Therefore, LC-MS seems to be a more promising method if you want to determine several nitramines within one analysis. Because most of the interest today is on nitramine determination in aqueous environmental samples (e.g. lake), LC is better suited since exchanging the water to a more volatile solvent is not necessary. For the determination of very low concentrations of nitramines, an extraction step before analysis is necessary. Either SPE or LLE can be used, but so far there is no adsorbent material or extraction solvent, respectively, available where the extraction efficiency of several nitramines is high. Regarding SPE, an alternative can be to use a mixture of adsorbents or to couple SPE columns with different adsorbents in series. Future research has to focus on the evaluation of the SPE-material and the LLE solvent, respectively, and to find an LC column material which separates a broad range of nitramines. Regarding detection, MS seems to be the alternative to use, but the ionization method and selection of the mobile phase will have an effect on the sensitivity of the method, and need to be addressed.Since the threshold values for nitramines in environmental samples is in the ng L−1 range there is a need for new more sensitive analytical determination methods. The main challenge in the development of new methods is that the nitramines are small and polar, and present in very polar matrices (water).
Abbreviation list
| 1,4-NIPZ | N,N-Dinitropiperazine |
| 1,4-NIPZ/NPZ | 1-Nitro-4-nitrosopiperazine |
| ACN | Acetonitrile |
| AMP-NO2 | 2-Methyl-2-(nitroamine)-1-propanol |
| APCI | Atmospheric chemical ionization |
| BENA | N-Nitrobutylethylamine |
| bp | Boiling point |
| C factor | Concentration factor |
| CI | Chemical ionization |
| cLOQ | Concentration limit of quantification |
| d14-NDPA | N-d14-Nitrosodi-n-propylamine |
| d6-NDMA | N-d6-Nitrosodimethylamine |
| DBNA | N-nitrodibutylamine |
| DCM | Dichloromethane |
| DENA | N-Nitrodiethylamine |
| diEtOHNA | N-Nitrodiethanolamine |
| DIPNA | N-Nitrodiisopropylamine |
| DMNA | N-Nitrodimethylamine |
| DPNA | N-Nitrodipropylamine |
| EI | Electron ionization |
| EMNA | N-Nitroethylmethylamine |
| ES | Elution solvent |
| ESI | Electrospray ionization |
| EtOH | Ethanol |
| EtOHNA | N-Nitroethanolamine |
| FID | Flame ionization detector |
| GC | Gas chromatography |
| IS | Internal standard |
| LC | Liquid chromatography |
| LLE | Liquid–liquid extraction |
| LOD | Limit of detection |
| M | Molar mass |
| MBNA | N-Nitrobutylamine |
| MENA | N-Nitroethylamine |
| MeOH | Methanol |
| MMNA | N-Nitromethylamine |
| MS | Mass spectrometry |
| N.A. | Not available |
| NDBA | N-Nitrosodibutylamine |
| NDEA | N-Nitrosodiethylamine |
| NDEtOH | N-Nitrosodiethanolamine |
| NDPA | N-Nitrosodipropylamine |
| NIMOR | N-Nitromorpholine |
| NIPIP | N-Nitropiperidine |
| NIPYR | N-Nitropyrrolidine |
| NIPZ | N-Nitropiperazine |
| NMOR | N-Nitrosomorpholine |
| NPD | Nitrogen–Phosphours detector |
| NPYR | N-Nitrospyrrolidine |
| NPZ | N-Nitrosopiperazine |
| PCC | Post-combustion CO2 capture |
| PCUV | Post column ultra violet |
| RDX | 1,3,5-Trinitroperhydro-1,3,5-triazine |
| RPLC | Reversed phase liquid chromatography |
| SCX | Strong cation exchange |
| SEC | Size exclusion chromatography |
| SPE | Solid phase extraction |
| TEA | Thermal energy analyzer |
| T grad | Temperature gradient |
| T pyr | Temperature pyrolyzer |
| T TEA | Temperature thermal energy analyzer |
| THF | Tetrahydrofuran |
| TLC | Thin layer chromatography |
| UV | Ultraviolet |
| V inj | Injection volume |
| V sample | Volume of the sample |
Acknowledgements
Project/Research funded by VISTA – a basic research programme funded by Statoil, conducted in close collaboration with The Norwegian Academy of Science and Letters. The scientific discussions with Professor Claus J. Nielsen at University of Oslo are highly appreciated.References
- J. N. Pitts, D. Grosjean, K. Van Cauwenberghe, J. P. Schmid and D. R. Fitz, Environ. Sci. Technol., 1978, 12, 946–953 CrossRef CAS.
- S. Knudsen, M. Karl and S. Randall, Summary Report: Amine Emissions to Air During Carbon Capture. Phase I: CO2 and Amines Screening Study for Effects to the Environment OR 8/2009, NILU, 2009 Search PubMed.
- C. J. Nielsen, H. Herrmann and C. Weller, Chem. Soc. Rev., 2012, 41, 6684–6704 RSC.
- IPCC, Summary for Policymakers, IPCC, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013 Search PubMed.
- IPCC, IPCC Special Report on Carbon Dioxide Capture and Storage. Prepared by Working Group III of the Intergovernmental Panel on Climate Change, IPCC, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2005 Search PubMed.
- International Energy Agency, CO2 Capture and Storage: A Key Carbon Abatement Option, OECD Publishing, 2008 Search PubMed.
- M. Wang, A. Lawal, P. Stephenson, J. Sidders and C. Ramshaw, Chem. Eng. Res. Des., 2011, 89, 1609–1624 CrossRef CAS PubMed.
- M. Låg, B. Lindeman, C. Instanes, G. Brunborg and P. Schwarze, Health Effects of Amines and Derivatives Associated with CO2 Capture, ISBN 978-82-8082-462-2, The Norwegian Institute of Public Health, 2011 Search PubMed.
- J. N. Pitts, D. Grosjean, K. Van Cauwenberghe, J. P. Schmid and D. R. Fitz, Environ. Sci. Technol., 1978, 12, 946–953 CrossRef CAS.
- Y. Maree, S. Nepstad and G. de Koeijer, Energy Procedia, 2013, 37, 6265–6272 CrossRef CAS PubMed.
- H. R. Scherf, E. Frei and M. Wiessler, Carcinogenesis, 1989, 10, 1977–1981 CrossRef CAS PubMed.
- S. S. Mirvish, O. Bulay, R. G. Runge and K. Patil, Natl. Cancer Inst., J., 1980, 64, 1435–1442 CAS.
- Drinking Water Notification Levels and Response Levels: An Overview, California Department of Public Health Search PubMed.
- Guidance on the Water Supply (Water Quality) Regulations 2000 a specific to N-nitrosodimethylamine (NDMA) concentrations in drinking water, Drinking Water Inspectorate, 2008 Search PubMed.
- Technical Support Document for Ontario Drinking Water Standards, Objectives and Guidelines, Ontario's Ministry of the Environment, 2003 Search PubMed.
- L. M. Fjellsbø, A. R. Van Rompay, J. Hooyberghs, I. Nelissen and M. Dusinska, Toxicol. in Vitro, 2013, 27, 1205–1210 CrossRef PubMed.
- A. L. Kohl and R. B. Nielsen, Gas purification, Gulf Publ. Co., Houston, Tex., 1997 Search PubMed.
- B. Fostås, A. Gangstad, B. Nenseter, S. Pedersen, M. Sjøvoll and A. L. Sørensen, Energy Procedia, 2011, 4, 1566–1573 CrossRef PubMed.
- C. J. Nielsen, B. D'Anna, C. Dye, M. Graus, M. Karl, S. King, M. M. Maguto, M. Müller, N. Schmidbauer, Y. Stenstrøm, A. Wisthaler and S. Pedersen, Energy Procedia, 2011, 4, 2245–2252 CrossRef CAS PubMed.
- E. Gjernes, L. I. Helgesen and Y. Maree, Energy Procedia, 2013, 37, 735–742 CrossRef CAS PubMed.
- B. C. Challis and S. A. Kyrtopoulos, J. Chem. Soc., Perkin Trans. 1, 1979, 299–304 RSC.
- E. F. d. Silva, K. A. Hoff and A. Booth, Energy Procedia, 2013, 37, 784–790 CrossRef PubMed.
- N. Dai, A. D. Shah, L. Hu, M. J. Plewa, B. McKague and W. A. Mitch, Environ. Sci. Technol., 2012, 46, 9793–9801 CrossRef CAS PubMed.
- J. Nawrocki and P. Andrzejewski, J. Hazard. Mater., 2011, 189, 1–18 CrossRef CAS PubMed.
- J. W. Munch and M. V. Bassett, Method 521 determination of nitrosamines in drinking water by solid phase extraction and capillary column gas chromatography with large volume injection and chemical ionization tandem mass spectrometry (MS/MS), U. S. E. P. Agency, 2004 Search PubMed.
- Method 8070A Nitrosamines by gas chromatography, U.S. Environmental Protection Agency, 1996 Search PubMed.
- G. D. Foley, NISOH Manual of Analytical Methods (NMAM): Nitrosamines Method: 2522, Issue 2, 1994 Search PubMed.
- C. S. Evans, R. Sleeman, J. Luke and B. J. Keely, Rapid Commun. Mass Spectrom., 2002, 16, 1883–1891 CrossRef CAS PubMed.
- I. M. Schreiber and W. A. Mitch, Environ. Sci. Technol., 2007, 41, 7039–7046 CrossRef CAS.
- B. Greene, D. Mast and D. L. Baker, in JANNAF 33rd PEDCS, Sandestin, FL, United States, 2006, pp. 6–10 Search PubMed.
- S. S. Walse and W. A. Mitch, Environ. Sci. Technol., 2008, 42, 1032–1037 CrossRef CAS.
- M. Lee, Y. Lee, F. Soltermann and U. von Gunten, Water Res., 2013, 47, 4893–4903 CrossRef CAS PubMed.
- A. Bjerke, B. Kristin, O.-A. Braathen, O. G. Brakstad, M. Dušinská, C. Dye, L. M. B. Fjellsbø, G. Hammerseth, B. H. Hansen, S.-H. Hansen, P. Kaur, H. Leknes, S. Manø, Z. Magdolenova, M.-B. Pedersen, E. R. Pran, S. Ravnum, M. Schlabach, Y. Stenstrøm, T. Syversen, M. Vadset and K. Zahlsen, Nitramine analysis procedures development and screening toxicity study, Scientific report OR 15/2011, NILU Norwegian Institute for Air Research, 2011 Search PubMed.
- M. Hassel, E. Frei, A. J. Streeter and M. Wiessler, Carcinogenesis, 1990, 11, 357–360 CrossRef CAS PubMed.
- A. Shah, Environ. Sci. Technol., 2013, 47, 2799 CrossRef CAS PubMed.
- J. L. Weidhaas, M. J. Zigmond and R. R. Dupont, Biorem. J., 2012, 16, 74–85 CrossRef CAS.
- R. V. Cooney, P. D. Ross, G. L. Bartolini and J. Ramseyer, Environ. Sci. Technol., 1987, 21, 77–83 CrossRef CAS.
- M. Masuda, H. F. Mower, B. Pignatelli, I. Celan, M. D. Friesen, H. Nishino and H. Ohshima, Chem. Res. Toxicol., 2000, 13, 301–308 CrossRef CAS PubMed.
- H. Takanashi, H. Homma and M. Matsui, Chem.-Biol. Interact., 1988, 66, 49–59 CrossRef CAS.
- L. Padhye, P. Wang, T. Karanfil and C.-H. Huang, Environ. Sci. Technol., 2010, 44, 4161–4168 CrossRef CAS PubMed.
- L. P. Padhye, B. Hertzberg, G. Yushin and C.-H. Huang, Environ. Sci. Technol., 2011, 45, 8368–8376 CrossRef CAS PubMed.
- J. M. Kemper, P. Westerhoff, A. Dotson and W. A. Mitch, Environ. Sci. Technol., 2009, 43, 466–472 CrossRef CAS.
- D. Fournier, J. Hawari, S. H. Streger, K. McClay and P. B. Hatzinger, Appl. Environ. Microbiol., 2006, 72, 6693–6698 CrossRef CAS PubMed.
- S. P. Mezyk, B. Razavi, K. L. Swancutt, C. R. Cox and J. J. Kiddle, J. Phys. Chem. A, 2012, 116, 8185–8190 CrossRef CAS PubMed.
- P. A. Chandan, J. E. Remias, J. K. Neathery and K. Liu, Environ. Sci. Technol., 2013, 47, 5481–5487 CrossRef CAS PubMed.
- R. V. Cooney, P. D. Ross and G. L. Bartolini, Cancer Lett., 1986, 32, 83–90 CrossRef CAS.
- R. V. Cooney and P. D. Ross, J. Agric. Food Chem., 1987, 35, 789–793 CrossRef CAS.
- E. Holmgren, S. Ek and A. Colmsjo, J. Chromatogr. A, 2012, 1222, 109–115 CrossRef CAS PubMed.
- A. Hilmi, J. H. T. Luong and A. L. Nguyen, J. Chromatogr. A, 1999, 844, 97–110 CrossRef CAS.
- C. J. Nielsen, B. D'Anna, R. Bossi, A. J. C. Bunkan, L. Dithmer, M. Glasius, M. Hallquist, A.-M. K. Hansen, A. Lutz, K. Salo, M. M. Maguta, Q. Nguyen, T. Mikonivy, M. Müller, H. Skov, E. Sarrasin, Y. Stenstrøm, Y. Tang, J. Westerlund and A. Wisthaler, Atmospheric Degradation of Amines (ADA) Summary report from atmospheric chemistry studies of amines, nitrosamines, nitramines and amides, CLIMIT project no. 208122, University of Oslo, Norway, 2012 Search PubMed.
- C. J. Nielsen, B. D'Anna, C. Dye, C. Gorge, M. Graus, A. Hansel, M. Karl, S. King, M. Musabila, M. Müller, N. Schmidbauer, Y. Stenstrøm and A. Wisthaler, Atmospheric Degradation of Amines (ADA) Summary report: gas phase photo-oxidation of 2-aminoethanol (mea) climit project no. 193438 OR 8/2010, NILU Norweigan Institute for Air Research, 2010 Search PubMed.
- C. J. Nielsen, B. D'Anna, M. Karl, M. Aursnes, A. Boreave, R. Bossi, A. J. C. Bunkan, M. Glasius, M. Hallquist, A.-M. K. Hansen, K. Kristiansen, T. Mikoviny, M. M. Maguta, M. Müller, Q. Nguyen, J. Westerlund, K. Salo, H. Skov, Y. Stenstrøm and A. Wisthaler, Atmospheric Degradation of Amines (ADA) Summary report: photo-oxidation of methylamine, dimethylamine and trimethylamine, climit project no. 201604 OR 2/2011, 2011 Search PubMed.
- J. A. Bell and I. Dunstan, J. Chromatogr., 1966, 24, 253–257 CrossRef CAS.
- G. B. Pliss, M. A. Zabezhinski, A. S. Petrov, V. V. Khudoley and G. B. Pliss, Archiv für Geschwulstforschung, 1982, 52, 629–634 CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- J. C. Oxley, A. B. Kooh, R. Szekeres and W. Zheng, J. Phys. Chem., 1994, 98, 7004–7008 CrossRef CAS.
- E. Frei, F. Gilberg, M. Schroder, A. Breuer, L. Edler and M. Wiessler, Carcinogenesis, 1999, 20, 459–464 CrossRef CAS PubMed.
- C. Ripolles, E. Pitarch, J. V. Sancho, F. J. Lopez and F. Hernandez, Anal. Chim. Acta, 2011, 702, 62–71 CrossRef CAS PubMed.
- Y.-Y. Zhao, J. Boyd, S. E. Hrudey and X.-F. Li, Environ. Sci. Technol., 2006, 40, 7636–7641 CrossRef CAS.
- I. M. Schreiber and W. A. Mitch, Environ. Sci. Technol., 2005, 39, 3811–3818 CrossRef CAS.
- W. A. Mitch, A. C. Gerecke and D. L. Sedlak, Water Res., 2003, 37, 3733–3741 CrossRef CAS.
- J. W. Dallinga, C. A. M. Krul, A. Tenfelde, E. J. C. Moonen, I. T. M. Vermeer, D. van Doorn and R. C. Schothorst, Eur. J. Cancer Prev., 2001, 10, 265–268 CrossRef CAS PubMed.
- Method 8270D semivolatile organic compounds by gas chromatography/mass spectrometry (GC/MS), 2007 Search PubMed.
- B. Spiegelhalder, II.6.c Isolation and determination of N-nitrosamines and nitrosatable compounds in rubber and plastic commodities after migration into a saliva test solution, IARC scientific publications, 1983, pp. 265–273 Search PubMed.
- J. B. Westin, M. J. J. Castegnaro and M. D. Friesen, Environ. Res., 1987, 43, 126–134 CrossRef CAS.
- J. C. Oxley, M. Hiskey, D. Naud and R. Szekeres, J. Phys. Chem., 1992, 96, 2505–2509 CrossRef CAS.
- L. Sørensen, E. F. d. Silva, O. G. Brakstad, K. Zahlsen and A. Booth, Energy Procedia, 2013, 37, 683–690 CrossRef PubMed.
- A. Einbu, E. DaSilva, G. Haugen, A. Grimstvedt, K. G. Lauritsen, K. Zahlsen and T. Vassbotn, Energy Procedia, 2013, 37, 717–726 CrossRef CAS PubMed.
- M. Krauss and J. Hollender, Anal. Chem., 2008, 80, 834–842 CrossRef CAS PubMed.
- B. Spiegelhalder, G. Eisenbrand and R. Preussmann, II.3.d Volatile N-nitrosamines in beer and other beverages by direct extraction using a kieselguhr column, IARC scientific publications, 1983, pp. 135–142 Search PubMed.
- D. Fournier, A. Halasz, J. Spain, R. J. Spanggord, J. C. Bottaro and J. Hawari, Appl. Environ. Microbiol., 2004, 70, 1123–1128 CrossRef CAS.
- C. W. Mohr and R. D. Vogt, Sorption of Nitramines to Soil – Final report of a prelimianry study, Environmental Chemistry Group, Department of Chemistry, University of Oslo, Oslo, Norway, 2012 Search PubMed.
- E. Suzuki, M. Mochizuki, Y. Wakabayashi and M. Okada, Gann, 1983, 74, 51–59 CAS.
- E. Suzuki and M. Okada, Gann, 1980, 71, 863–870 CAS.
- E. Suzuki, M. Mochizuki, N. Sekiguchi and M. Okada, Metabolism of N-nitrodialkylamines, IARC scientific publications, 1984, pp. 485–490 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |