
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion-exchange membranes in electrochemical energy systems†
John R.
Varcoe
*a,
Plamen
Atanassov
b,
Dario R.
Dekel
c,
Andrew M.
Herring
d,
Michael A.
Hickner
e,
Paul. A.
Kohl
f,
Anthony R.
Kucernak
g,
William E.
Mustain
h,
Kitty
Nijmeijer
i,
Keith
Scott
j,
Tongwen
Xu
k and
Lin
Zhuang
l
aDepartment of Chemistry, University of Surrey, Guildford, GU2 7XM, UK. E-mail: j.varcoe@surrey.ac.uk; Tel: +44 (0)1483 686838
bDepartment of Chemical and Nuclear Engineering, University of New Mexico, Albuquerque, NM 87131-0001, USA
cCellEra, Caesarea Business and Industrial Park, North Park, Bldg. 28, POBox 3173, Caesarea, 30889, Israel
dDepartment of Chemical and Biological Engineering, Colorado School of Mines, Golden, CO 80401, USA
eDepartment of Materials Science and Engineering, The Pennsylvania State University, University Park, PA 16802, USA
fSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332-0100, USA
gDepartment of Chemistry, Imperial College London, South Kensington, London, SW7 2AZ, UK
hDepartment of Chemical & Biomolecular Engineering, University of Connecticut, Storrs, CT 06269-3222, USA
iMembrane Science and Technology, University of Twente, MESA+ Institute for Nanotechnology, P.O. Box 217, 7500 AE, Enschede, The Netherlands
jSchool of Chemical Engineering and Advanced Materials, Faculty of Science, Agriculture and Engineering, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
kSchool of Chemistry and Material Science, USTC-Yongjia Membrane Center, University of Science and Technology of China, Hefei, 230026 P. R. China
lDepartment of Chemistry, Wuhan University, Wuhan, 430072, P. R. China
First published on 4th August 2014
Abstract
This article provides an up-to-date perspective on the use of anion-exchange membranes in fuel cells, electrolysers, redox flow batteries, reverse electrodialysis cells, and bioelectrochemical systems (e.g. microbial fuel cells). The aim is to highlight key concepts, misconceptions, the current state-of-the-art, technological and scientific limitations, and the future challenges (research priorities) related to the use of anion-exchange membranes in these energy technologies. All the references that the authors deemed relevant, and were available on the web by the manuscript submission date (30th April 2014), are included.
 John R. Varcoe | Professor John Varcoe (Department of Chemistry, University of Surrey, UK) obtained both his 1st class BSc Chemistry degree (1995) and his Materials Chemistry PhD (1999) at the University of Exeter (UK). He was a postdoctoral researcher at the University of Surrey (1999–2006) before appointment as Lecturer (2006), Reader (2011) and Professor (2013). He is recipient of an UK EPSRC Leadership Fellowship (2010). His research interests are focused on polymer electrolytes for clean energy and water systems: more specifically, the development of chemically stable, conductive anion-exchange polymer electrolytes. He is also involved in the University's efforts on biological fuel cells. |
 Dario R. Dekel | Dr Dario Dekel (Co-Founder and VP for R&D and Engineering, CellEra, Israel) received his MBA, MSc and PhD in Chemical Engineering from the Technion, Israel Institute of Technology. He was the chief scientist and top manager at Rafael Advanced Defense Systems, Israel, where he led the world's second largest Thermal Battery Plant. He left Rafael in 2007 to co-found CellEra, leading today a selected group of 14 scientists and engineers, developing the novel Alkaline Membrane Fuel Cell technology. He currently holds $3M government research grants from Israel, USA and Europe. Dr Dekel holds 14 battery and fuel cell patents. |
 Michael A. Hickner | Professor Michael Hickner (Associate Professor, Department of Materials Science and Engineering, Pennsylvania State University, USA) focuses his research on the relationships between chemical composition and materials performance in functional polymers to address needs in new energy and water purification applications. His research group has ongoing projects in polymer synthesis, fuel cells, batteries, water treatment membranes, and organic electronic materials. His work has been recognized by a Presidential Early Career Award for Scientists and Engineers from President Obama (2009). He has co-authored seven US and international patents and over 100 peer-reviewed publications with >5400 citations. |
 Paul. A. Kohl | Professor Paul Kohl (Hercules Inc./Thomas L. Gossage Chair, Regents' Professor, Georgia Institute of Technology, USA) received a Chemistry PhD (University of Texas, 1978). He was then involved in new chemical processes for silicon and compound semiconductor devices at AT&T Bell Laboratories (1978–89). In 1989, he joined Georgia Tech.'s School of Chemical and Biomolecular Engineering. His research includes ionic conducting polymers, high energy density batteries, and new materials and processes for advanced interconnects for integrated circuits. He has 250 papers, is past Editor of JES and ESSL, past Director MARCO Interconnect Focus Center, and President of the Electrochemical Society (2014–15). |
 Tongwen Xu | Professor Tongwen Xu (University of Science and Technology of China) received his BSc (1989) and MSc (1992) from Hefei University of Technology and his Chemical Engineering PhD (1995) from Tianjin University. He then studied polymer science at Nankai University (1997). He was visiting scientist at University of Tokyo (2000), Tokyo Institute of Technology (2001) and Gwangju Institute of Science and Technology (Brain Pool Program Korea award recipient). He has received a “New Century Excellent Talent” (2004) and an “Outstanding Youth Foundation” (2010) Chinese awards. His research interests cover membranes and related processes, particularly ion exchange membranes and controlled release. |
 Lin Zhuang | Professor Lin Zhuang (Department of Chemistry, Wuhan University, China) earned his electrochemistry PhD (1998) at Wuhan University. He was then promoted to lecturer, associate professor (2001) and full professor (2003). He was a visiting scientist at Cornell (2004–05) and is an adjunct professor at Xiamen University. He is an editorial board member of Science China: Chemistry, Acta Chimica Sinica, and Journal of Electrochemistry. He was recipient of a National Science Fund for Distinguished Young Scholars. He was vice-chair of the physical electrochemical division of the International Society of Electrochemistry (2011–12) and China section chair of the Electrochemical Society (2010–11). |
Broader contextMany electrochemical devices utilise ion-exchange membranes. Many systems such as fuel cells, electrolysers and redox flow batteries have traditionally used proton-/cation-exchange membranes (that conduct positive charged ions such as H+ or Na+). Prior wisdom has led to the general perception that anion-exchange membranes (that conduct negatively charged ions) have too low conductivities and chemical stabilities (especially in high pH systems) for application in such technologies. However, over the last decade or so, developments have highlighted that these are not always significant problems and that anion-exchange membranes can have OH− conductivities that are approaching the levels of H+ conductivity observed in low pH proton-exchange membrane equivalents. This article reviews the key literature and thinking related to the use of anion-exchange membranes in a wide range of electrochemical and bioelectrochemical systems that utilise the full range of low to high pH environments. |
Preamble
There is an increasing worldwide interest in the use of anion-exchange membranes (including in the alkaline anion forms), in electrochemical energy conversion and storage systems. This perspective stems from the “Anion-exchange membranes for energy generation technologies” workshop (University of Surrey, Guildford, UK, July 2013), involving leading researchers in the field,1 that focussed on the use of AEMs in alkaline polymer electrolyte fuel cells (APEFCs),2 alkaline polymer electrolyte electrolysers (APEE),3 redox flow batteries (RFB),4 reverse electrodialysis (RED) cells,5 and bioelectrochemical systems including microbial fuel cells (MFCs)6 and enzymatic fuel cells.7Conventions used in this perspective article
In this article the following terminology is defined:• AEM is used to designate anion-exchange membranes in non-alkaline anion forms (e.g. containing Cl− anions);
• AAEM used to designate anion-exchange membranes containing alkaline anions (i.e. OH−, CO32− and HCO3−);
• HEM is used to designate hydroxide-exchange membranes and should only be used where the AAEMs are totally separated from air (CO2) and are exclusively in the OH− form (with no traces of other alkaline anions such as CO32−); this is not the case in most of the technologies discussed in the article (a possible exception being APEEs);
• AEI is used to designate an anion-exchange ionomer which are anion-exchange polymer electrolytes in either solution or dispersion form: i.e. anion-exchange analogues to the proton-exchange ionomers (e.g. Nafion® D-52x series) used in proton-exchange membrane fuel cells (PEMFCs). AEIs are used as polymer binders to introduce anion conductivity in the electrodes (catalyst layers).
• CEM is used to designate cation-exchange membranes in non-acidic form (e.g. containing Na+ cations);
• PEM is used to designate proton-exchange membranes (i.e. CEMs specifically in the acidic H+ cation form);
• IEM is used to designate a generic ion-exchange membrane (can be either CEM or AEM).
Note that in this review, all electrode potentials (E) are given as reduction potentials even if a reaction is written as an oxidation.
AEMs and AEIs for electrochemical systems
Summary of AEM chemistries used in such systems
AEMs and AEIs are polymer electrolytes that conduct anions, such as OH− and Cl−, as they contain positively charged [cationic] groups (typically) bound covalently to a polymer backbone. These cationic functional groups can be bound either via extended side chains (alkyl or aromatic types of varying lengths) or directly onto the backbone (often via CH2 bridges); they can even be an integral part of the backbone.The most common, technologically relevant backbones are: poly(arylene ethers) of various chemistries8 such as polysulfones [including cardo, phthalazinone, fluorenyl, and organic–inorganic hybrid types],9 poly(ether ketones),10,11 poly(ether imides),12 poly(ether oxadiazoles),13 and poly(phenylene oxides) [PPO];14 polyphenylenes,15 perfluorinated types,16,17 polybenzimidazole (PBI) types including where the cationic groups are an intrinsic part of the polymer backbones,18 poly(epichlorohydrins) [PECH],19 unsaturated polypropylene20 and polyethylene21 types [including those formed using ring opening metathesis polymerisation (ROMP)],22 those based on polystyrene and poly(vinylbenzyl chloride),23 polyphosphazenes,24 radiation-grafted types,25 those synthesised using plasma techniques,26 pore-filled types,27 electrospun fibre types,28 PTFE-reinforced types,26g,29 and those based on poly(vinyl alcohol) [PVA].19a,30
The cationic head-group chemistries that have been studied (Scheme 1), most of which involve N-based groups, include:
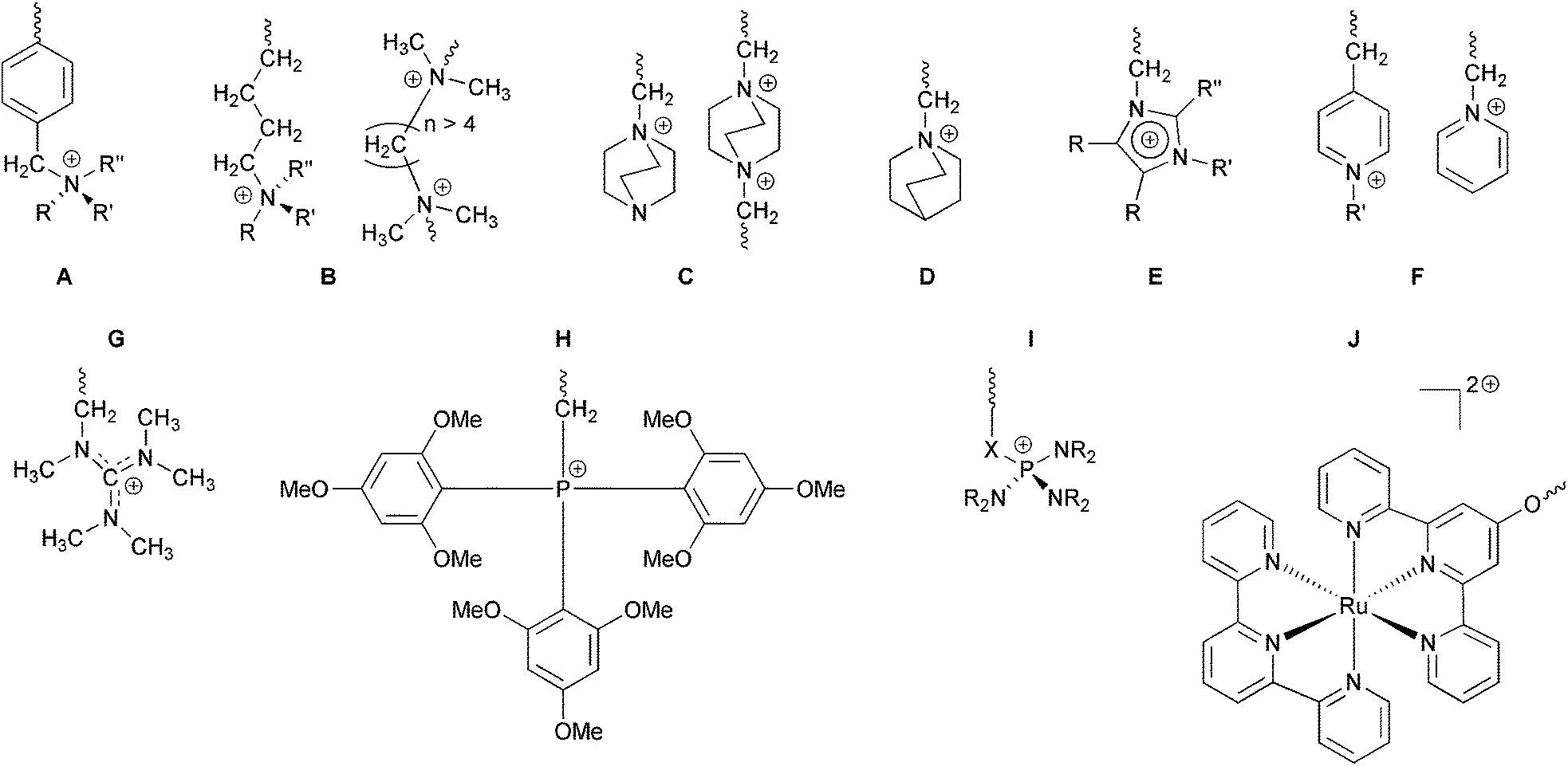 | ||
| Scheme 1 Commonly encountered AAEM/AEI cationic head-groups (those containing N–H and P–H bonds are omitted): A = benzyltrialkylammonium (the benchmark benzyltrimethylammonium is where R, R′, and R′′ are methyl groups); B = alkyl-side-chain (benzene-free) quaternary ammonium (QA) and crosslinking diammonium groups (where the link chain is >C4 in length [preferably >C6 in length]); C = DABCO-based QA groups (more stable when only 1 N atom is quaternised [crosslinked systems where both Ns are quaternised are also of interest but are less stable in alkali]); D = quinuclidinium-based QA groups; E = imidazolium groups (where R = Me or H and R′, R′′ = alkyl or aryl groups [not H]); F = pyridinium groups; G = pentamethylguanidinium groups; H = alkali stabilised quaternary phosphonium groups; I = P–N systems (where X = –SO2– or –NR′– groups and where R = alkyl, aryl, or unsaturated cyclic systems); and J is an exemplar metal containing cationic group. | ||
(a) Quaternary ammoniums (QA) such as benzyltrialkylammoniums [benzyltrimethylammonium will be treated as the benchmark chemistry throughout this report],2,31 alkyl-bound (benzene-ring-free) QAs,21a,b and QAs based on bicyclic ammonium systems synthesised using 1,4-diazabicyclo[2.2.2]octane (DABCO) and 1-azabicyclo[2.2.2]octane (quinuclidine, ABCO) (to yield 4-aza-1-azoniabicyclo[2.2.2]octane,14b,25f,h,27g,32 and 1-azoniabicyclo[2.2.2]octane {quinuclidinium}19c,33 functional groups, respectively);
(b) Heterocyclic systems including imidazolium,10a,13,23a,25g,31,34 benzimidazoliums,35 PBI systems where the positive charges are on the backbone (with or without positive charges on the side-chains),18b,d,f,h,36 and pyridinium types (can only be used in electrochemical systems that do not involve high pH environments);26h,i,30i,37
(c) Guanidinium systems;16c,38
(d) P-based systems types including stabilised phosphoniums [e.g. tris(2,4,6-trimethoxyphenyl)phosphonium]11,14d,32a,39 and P–N systems such as phosphatranium16d and tetrakis(dialkylamino)phosphonium40 systems;
(e) Sulfonium types;41
(f) Metal-based systems where an attraction is the ability to have multiple positive charges per cationic group.42
General comments on the characterisation of AEMs
Given that OH− forms of AAEMs quickly convert to the less conductive CO32− and even less conductive HCO3− forms when exposed to air (containing CO2 – see eqn (1) and (2)), even for very short periods of time,25d,43 it is essential that CO2 is totally excluded from experiments that are investigating the properties of AAEMs in the OH− forms. This includes the determination of water uptakes, dimensional swelling on hydration, long-term stabilities, and conductivities [see specific comments in the below sections].| OH− + CO2 ⇌ HCO3− | (1) |
| OH− + HCO3− ⇌ CO32− + H2O | (2) |
Additionally, when converting an AEM or AEI into a single anion form, it is vital to ensure complete ion-exchange. An IEM cannot be fully exchanged to the desired single ion form after only 1× immersion in a solution containing the target ion, even if a concentrated solution containing excess target ion is used: the use of only 1× immersion will leave a small amount of the original ion(s) in the material (ion-exchange involves partition equilibria). IEMs must be ion-exchange by immersion in multiple (at least 3) consecutive fresh replacements of the solution containing an excess of the desired ion. Traces of the original (or other contaminant) ions can have implications regarding the properties being measured.44
One of the main properties that must be reported for each AEM/AEI produced is the ion-exchange capacity (IEC), which is the number of functional groups (molar equivalents, eq.) per unit mass of polymer. In the first instance, it is highly recommended that the IEC of the Cl−form of the AEM being studied is measured (the form typically produced on initial synthesis).19e,31,45 This is so that the AEMs have not been exposed to either acids or bases that may cause high and low pH-derived degradations (even if such degradations are only slight) and to avoid significant CO2-derived interferences: both acid and bases are required for the use of the classical back-titration method of determining IECs of AAEMs.46 Additionally when using Cl− based titrations, methods are available to measure the total exchange capacities, quaternary-only-IEC and non-quaternary (e.g. tertiary) exchange capacities.47 These techniques will be useful for AAEM degradation studies where QA groups may degrade into polymer-bound non-QA groups (such as tertiary amine groups). However, there can be discrepancies between IECs derived from titration experiments and other techniques such as those that use ion-selective electrodes or spectrophotometers.48 NMR data can also be used to determine IECs with soluble AEMs and AEIs.49,50
Perceived problems with the use of AAEMs
The two main perceived disadvantages of AAEMs are low stabilities in OH− form (especially when the AAEMs are not fully hydrated)51 and low OH− conductivities (compared to the H+ conductivity of PEMs, especially [again] when the AAEMs are not fully hydrated).19d,52,53 The former is going to be challenging problem to solve if the electrochemical system in question requires the conduction of OH− anions (i.e. a strong nucleophile) as the polymer electrolytes contain positively charged cationic groups (i.e. good leaving groups!). Conductivity issues are not insurmountable with improved material and cell designs. While conductivities of ca. 10−1 S cm−1 are needed for high current density cell outputs, operating electrochemical devices with membranes that have intrinsic conductivities of the order of 5 × 10−2 S cm−1 is not out of the question. Conductivities of 10−2 S cm−1 may, however, be too low for many applications.It is apparent in Scheme 2 that even simple benzyltrimethylammonium cationic functional groups (the most commonly encountered, Scheme 1A) can undergo a number of degradation processes in the presence of OH− nucleophiles. The main degradation mechanism for benzyltrimethylammonium groups is via direct nucleophilic substitution (displacement). The formation of intermediate ylides (>C−–N+R3) have been detected via deuterium scrambling experiments and these can potentially lead to Sommelet–Hauser and Stevens rearrangements;51c however, such ylide-derived mechanisms rarely end in a degradation event.51a Hofmann elimination reactions cannot occur with benzyltrimethylammonium as there are no β-Hs present; this is not the case with benzyltriethylammonium groups,51b,56 which contain β-Hs [even though the R–N+(CH2CH3)3 OH− groups may be more dissociated than R–N+(CH3)3 OH− groups]. As an aside, neopentyltrimethylammonium groups (on model small compounds, i.e. not polymer bound) contain a long alkyl chain but with no β-Hs: Hofmann elimination cannot occur, but the degradation of this cationic group appears to be even more complex with unidentified reaction products detected.51b
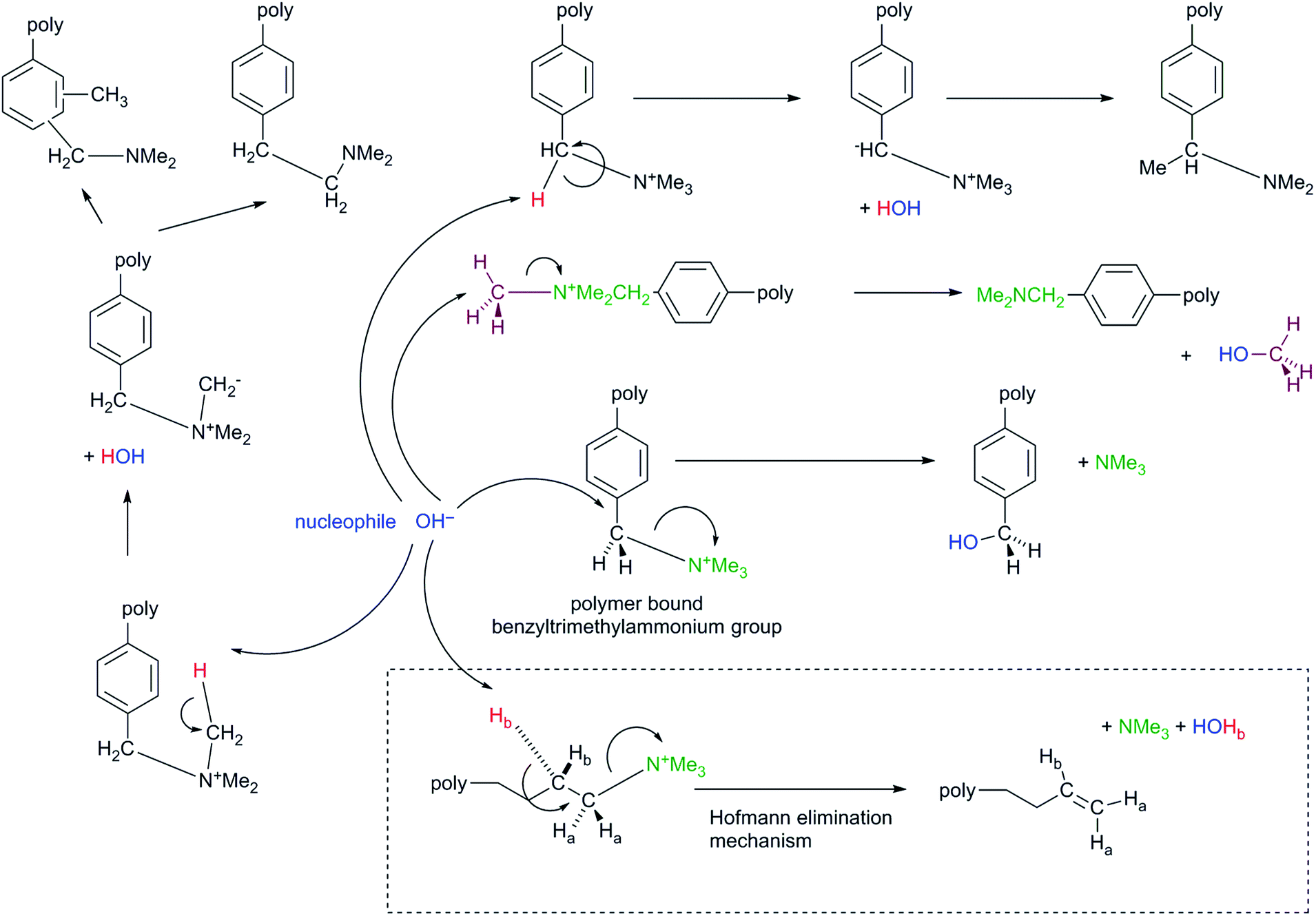 | ||
| Scheme 2 Degradation pathways for the reaction of OH− nucleophiles with benzyltrimethylammonium cationic (anion-exchange) groups.51 The inset [dashed box] shows the additional Hofmann Elimination degradation mechanism that can occur with alkyl-bound QA groups (that possess β-H atoms). | ||
Historically, due to concerns about facile Hofmann elimination reactions, QAs bound to longer alkyl chains were considered to be less stable than those bound to aromatic groups via –CH2– bridges.56 However, more recent evidence suggests that this may not be the case and that QA groups that are tethered (or crosslinked) with N-bound alkyl chains that are >4 carbon atoms long (C4, see Scheme 1B) can have surprisingly good stabilities in alkali.13a,47a,57,58 A hypothesis is that the high electron density around the β-Hs in longer alkyl chains can inhibit Hofmann elimination reactions47a and that steric shielding in the β-positions may also play a role in the surprising stability imparted by longer alkyl chains.57g
The search for alkali stable AAEMs/AEIs is the primary driver for the study of alternative cationic head-group chemistries. An alternative QA system is where DABCO is used as the quaternisation agent (Scheme 1C). This system contains β-Hs but due to the rigid cage structure, the β-Hs and the N atoms do not form the anti-periplanar confirmation required for facile Hofmann Elimination to occur (Scheme 3).19e,59,60 It is suspected that AAEMs/AEIs containing 4-aza-1-azoniabicyclo[2.2.2]octane groups, where only 1 N of the DABCO reactant is quaternised, are more stable than R–N+Me3 analogues.32e,60 However, it is not easy to produce AAEMs/AEIs where only 1 × N of the DABCO reagent reacts (although low temperatures may be helpful in this respect).59 The tendency is for DABCO to react via both N atoms forming crosslinks, which will produce materials with low alkali stabilities.32e This has led to the recent interest in quinuclidinium-(1-azoniabicyclo[2.2.2]octane) systems (Scheme 1D), which is a DABCO analogue containing only 1 N atom.19c,33 However, quinuclidine is much harder to synthesis than DABCO (harder to “close the cage”) and this is reflected in the price: quinuclidine = US$775 for 10 g vs. DABCO = US$34 for 25 g (laboratory scale prices, not bulk commodity prices).61 Also, quinuclidine is highly toxic (e.g. Hazard statement H310 – Fatal in contact with skin).62 This must be taken into account if quinuclidinium-containing AAEMs/AEIs degrade and release any traces of quinuclidine.
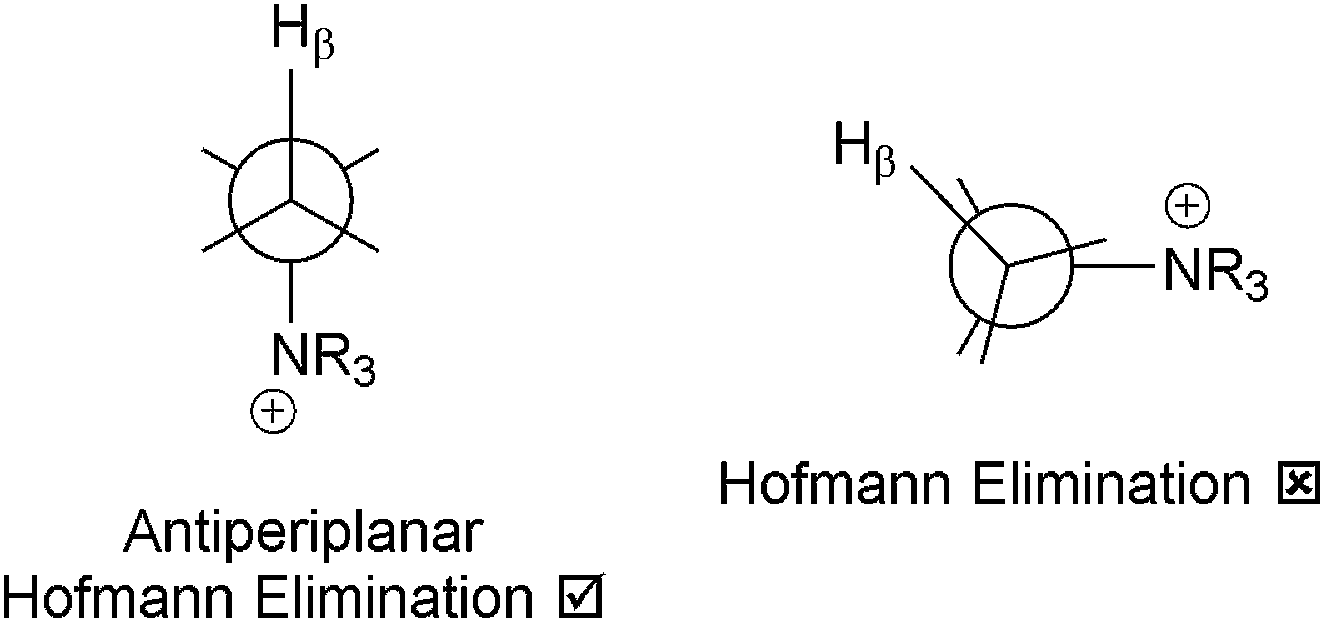 | ||
| Scheme 3 The antiperiplanar confirmation required for facile Hofmann elimination reactions. | ||
Systems involving >1 N atoms and “resonance stabilisation” have been evaluated with the desire of developing alkali stable and conductive AAEMs/AEIs. Firstly, non-heterocyclic pentamethylguanidinium systems (made using 1,1,2,3,3-pentamethylguanidine) have been reported,38 including perfluorinated AAEM examples.16c However, more recent studies suggest that this system may not be as alkali stable as originally reported.38e Polymer bound benzyltetramethylguanidinium (i.e. addition of a benzyl substituent) is reported to be more alkali stable than polymer bound pentamethylguanidinium groups.63 However, other reports indicate that guanidiniums bound to the polymer backbone via phenyl groups may be more stable than those bound via benzyl groups and perfluorosulfone groups.16c,64 These prior reports indicate that new degradation pathways (cf. QA benchmarks) are available with this cationic head-group.
The other multiple N atom system that has been extensively reported is the heterocyclic imidazolium system (Scheme 4). This includes where imidazolium groups have been used to introduce covalent crosslinking into the system.65 Imidazolium systems where R2, R4, and R5 are all H atoms are unstable to alkali.31,34c,34f,66 Polymer bound imidazolium groups with R2 = H can degrade via imidazolium ring-opening in the presence of OH− ions.34l,67 Replacement of the protons at the C2 position (e.g. R2 = Me or butyl group) increases the stability of the imidazolium group.34c,d,68 Different substituents at the N3 position (R3 = butyl, isopropyl, amongst others) can also affect the alkali stability of the imidazolium group:68a,69 systems where R3 = isopropyl or R2 = R3 = butyl groups are reported to be more stable options.
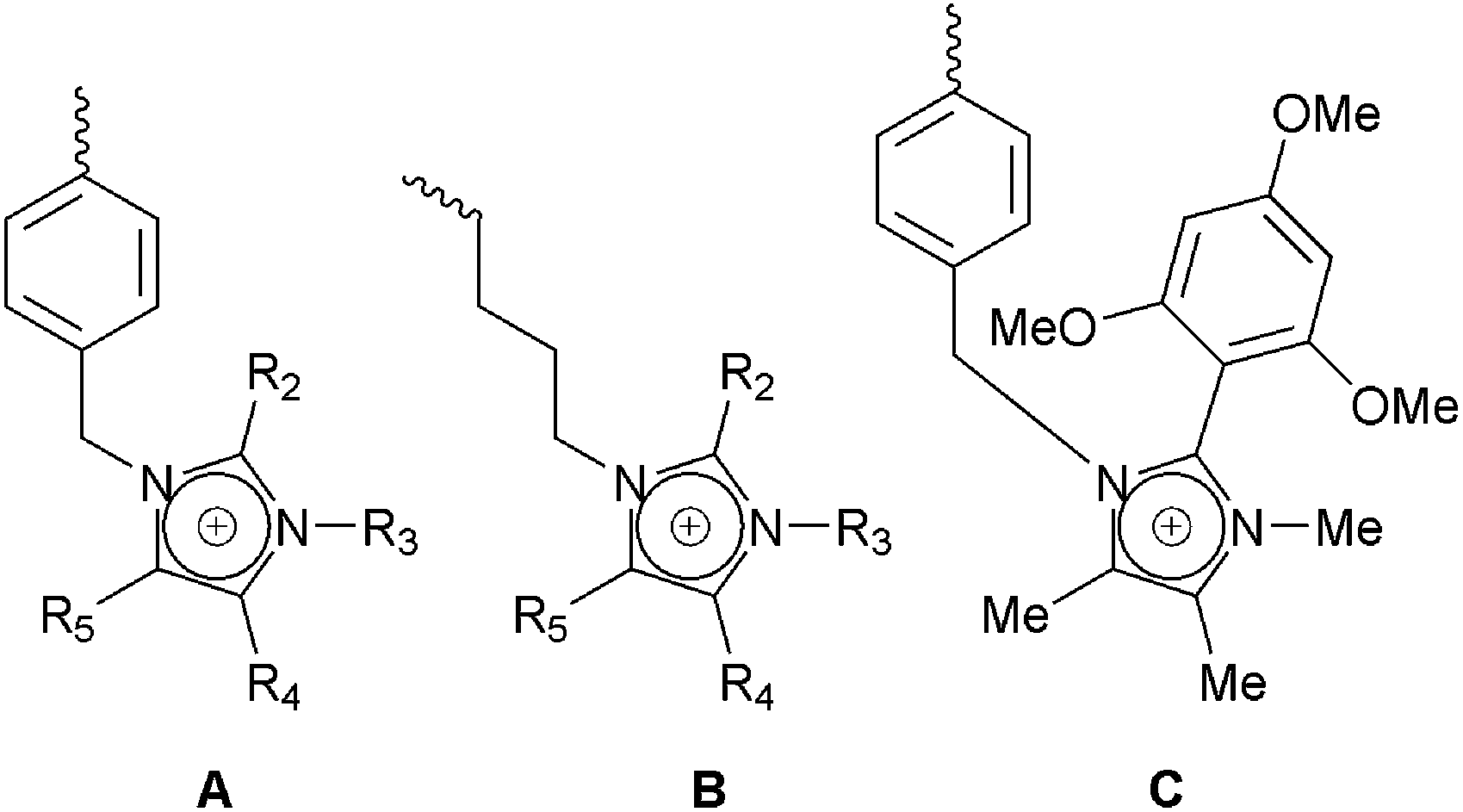 | ||
| Scheme 4 Imidazolium-based cationic head-group chemistry: A = benzyl-bound imidazolium groups; B = alkyl-bound imidazolium groups; and C the alkali stabilised imidazolium group reported by Yan et al.34b | ||
Yan et al. has recently reported an alkali stable PPO-bound imidazolium group [made using 1,4,5-trimethyl-2-(2,4,6-trimethoxyphenyl)imidazole] that contains no C–H bonds on the imidazolium ring and no C2 methyl group (Scheme 4C).34b This sterically bulky functional group was at least as stable as QA benchmarks. This claimed alkali stability is also backed up by DFT measurements in another recent study by Long and Pivovar, which suggests that similar C2-substituted imidazoliums will have superior alkali stabilities.70 Alkyl-2,3-dimethylimidazolium groups (R2 and R3 = Me) that are bound via long alkyl chains34k may also be more alkali stable than benzyl-bound analogues: the latter undergo facile removal of the imidazolium rings via nucleophilic displacement reactions34c (as well as degradation via imidazolium ring-opening).
However, contrary to the above, a study of small molecule imidazolium species by Price et al. suggests that adding steric hindrance at the C2 position is the least effective strategy;71 this study reports that 1,2,3-trimethylimidazolium cations appear to be particularly stable and this matches our experience in that the 1,2,3,4,5-pentamethylimidazolium cation appears to be reasonably stable in alkali. Furthermore, it has been reported that as you add more bulky cations, such as the 1,4,5-trimethyl-2-(2,4,6-trimethoxyphenyl)imidazole highlighted above, the anion transport switches from Arrenhius-type to Vogel–Tamman–Fulcher-type behaviour (i.e. the anions become less dissociated).72 Other studies that have looked into the alkali stability of various imidazolium-based ionic liquids, however, report that all 1,3-dialkylimidazolium protons (R2,R4, and R5 = H) can undergo deuterium exchange (i.e. represent alkali stability weak spots) and that even C2 methyl groups (R2 = –CH3) can undergo deprotonation in base.73 Further fundamental research into these and related systems is clearly still warranted.
The stabilised PBI system poly[2,2′-(m-mesitylene)-5,5′-bis(N,N′-dimethylbenzimidazolium)], where the cationic group is part of the polymer backbone, has recently been reported with promising alkali stabilities.18d This research has led to the development of other PBI-type ionenes that contain sterically protected C2 groups and are soluble in aqueous alcohols but insoluble in pure water;74 they are reported to have “unprecedented” hydroxide stabilities.
Regarding P-based systems, phosphonium AAEMs/AEIs are also common in the recent literature. Yan et al. first reported an alkali-stabilised polymer-bound phosphonium system made using tris(2,4,6-trimethoxyphenyl)phosphine as the quaternising agent (Scheme 1H) where the additional methoxy groups are electron donating and provide additional steric hinderance.11,14d,32a,39,66 This stabilisation is important as simple trialkylphosphonium and triphenylphosphonium analogues (e.g. small molecule benzyltriphenylphosphonium cations) will degrade in aqueous OH− solutions at room temperature in only a few hours; the thermodyanamic driving force being the formation of phosphine oxide via the Cahours–Hofmann reaction (especially in the presence of organics).73,75 However, recent spectroscopic studies suggest this bulky (high molecular weight) head-group still degrades in alkali.66 Initial results with the P–N tetrakis(dialkylamino)phosphonium system [poly-(Me)N–P+(–N(Me)Cy)3 where Cy = cyclohexane] first reported by Coates et al. suggests that this type of cationic head-group chemistry may be stable to alkali40 as indicated by early reports on small molecule studies.76
Prior thinking was that the alkali stability of the cationic head-groups could be treated separately to the chemical stability of the polymeric backbone (e.g. once an alkali stable head-group is found it can be attached to whatever polymer backbone is required and the polymer backbone and head-group will remain alkali stable). However, recent results suggest a much more complex situation with a symbiosis between the stability of the head-groups and the polymer backbone. For example, polysulfone itself is stable when exposed to aqueous alkali but is destabilised and degrades in high pH environments when QA groups are attached to the polymer backbone (via –CH2– linkages): the polymer backbone becomes partially hydrophilic, allowing close approach of the OH− anions.77 The electron withdrawing sulfone linkage has a profound negative influence on the stabilities of the resulting AAEMs.78 The hydrophobicity of unfunctionalised plastics lends significant resistance to alkali and it therefore stands to reason that more OH− uptake into the polymer structure will induce greater degradation. The degradation of AAEM backbones in alkali have been observed for other systems.15b,33b,34f The alkali stabilities of the following backbones containing pendent trimethylammonium cationic groups appear to decrease in the following order (Scheme 5): polystyrene > PPO > polysulfone (and all were less stable than the model small molecule p-methylbenzyltrimethylammonium).77 Note that with polysulfone AAEMs, strategies are now being developed to move the QA group away from the polysulfone backbone, where an additional benzyl group is located between the QA group and the backbone.79 Backbone stability may also be enhanced if phase segregated systems are developed (see later). The development of cationic side-chains containing multiple positive charges may help due to the ability to widely disperse the cationic side chains along the polymer backbone (charged groups placed further apart from each other without changing the IEC).80Therefore, when evaluating the alkali stabilities of new AAEMs/AEIs, the head-group and backbone must be evaluated together in combination.
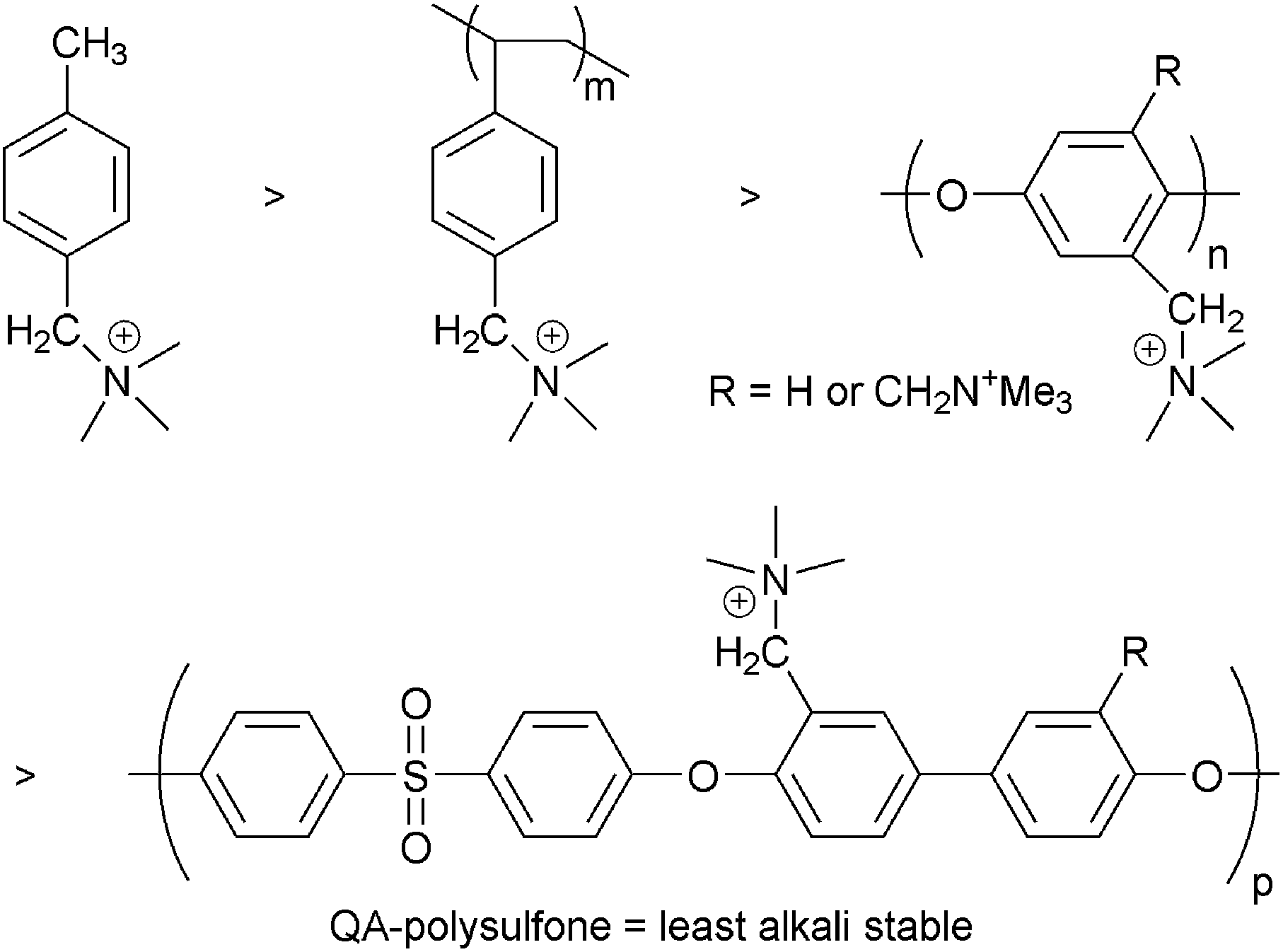 | ||
| Scheme 5 The relative alkali stability of various polymer backbones when containing pendent trimethylammonium groups.77 | ||
Another problem with evaluating the ex situ alkali stabilities of different AAEMs/AEIs with different chemistries is the broad range of different methodologies used throughout the literature. A common approach is to evaluate the change of ionic conductivity of the materials with increasing immersion times in aqueous alkali. This can be a useful measure of alkali stability but there is a risk of false positives: if the degraded membranes exhibit ionic conductivity, then the original AAEM/AEI may appear more alkali stable than it really is. A more useful measure of alkali stability is the measurement of IEC with increasing immersion times in aqueous alkali. This will be even more useful if changes in both quaternary IEC and non-quaternary exchange capacities are studied (see earlier discussion on IECs by titration). However, the authors recommend that such secondary measurements of alkali stability (changes in ionic conductivity and IEC) are always supplemented with multiple spectroscopic measurements (e.g. NMR,30f,33b,34c,66,69,77,81 IR,34k,82 and Raman31,34c).
Clearly as more alkali stable AAEMs/AEIs are developed, ex situ accelerated test protocols must be developed, i.e. immersion in concentrated alkali at high temperatures [e.g. aqueous KOH (6 mol dm−3) at 80–90 °C] with/without addition of peroxy/radical-based degradation agents. However, it must be kept in mind that if the aqueous alkali is too concentrated, viscosity effects may come into play and interfere with the stability measurements (e.g. diffusion of OH− nucleophiles towards the cationic groups is retarded). Data from accelerated degradation studies conducted inside NMR spectrometers (with soluble AAEMs/AEIs) will allow the simple and quick production of useful stability data (including an idea of the degradation mechanism that is operating).77 All of these ex situ stability measurements must be validated/benchmarked against in situ real-world and accelerated durability tests (in the spirit of DOE protocols for PEMFCs).55
It should also be kept in mind that the AAEMs/AEIs inside APEFCs are in an environment in the absence of excess metal (e.g. Na+ or K+) hydroxide species. Therefore, ex situ stability data at high temperatures with the AAEMs/AEIs in OH− forms in the absence of additional/excess NaOH or KOH species is also useful (e.g. an OH− form AAEM submerged in deionised water at 90 °C). The challenge here will be to ensure the AAEMs/AEIs remain in the OH− form (i.e. CO2 is totally excluded from all stages of the experiments [not easy to achieve]) as the AAEMs/AEIs will be more stable in the HCO3− and CO32− anion forms. Warder titration methods43b will be useful as these measure the relative contents of OH−, CO32− and HCO3− anions in the polymer electrolyte materials with time (example data given in Fig. 1 for an AAEM [originally in the OH− form] that is exposed to air); control experiments can be run alongside the degradation experiments where additional AAEMs/AEIs samples, originally in the OH− forms and kept in the same environment as the primary degradation samples, are monitored for a reduction in OH− content and an increase in CO32−/HCO3− content.
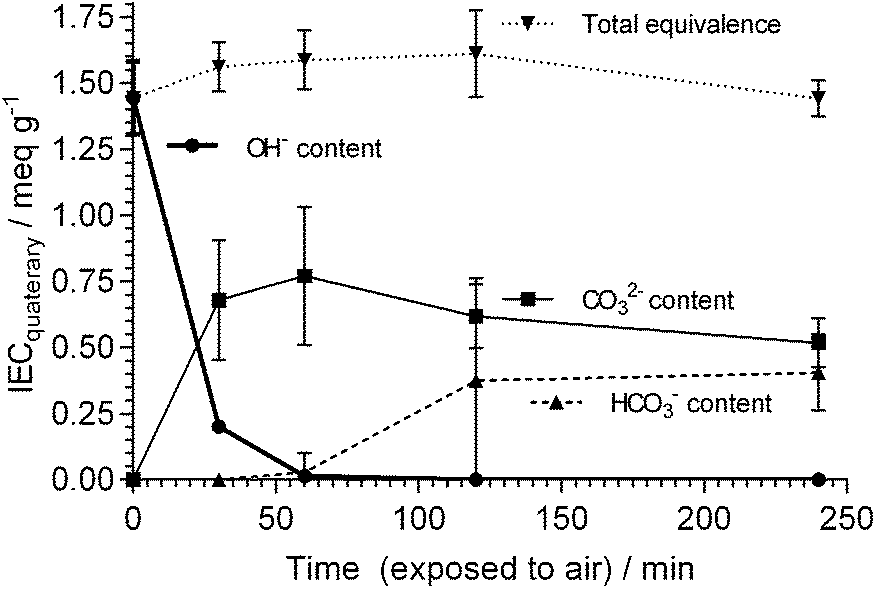 | ||
| Fig. 1 IECs (quaternary) of different alkali anions for a benzyltrimethylammonium-type ETFE-radiation-grafted AAEM (80 μm thick) that was initially exchanged to the OH− form and then directly exposed to air. IECs determined using Warder titration methods.43b,83 Error bars are sample standard deviations (n = 3 repeats). | ||
However, despite all of the studies into the different chemistries, the benzyltrimethylammonium hydroxide group may be stable enough for some applications (even those that contain alkali environments) as long as the benzyltrimethylammonium head-groups are kept fully hydrated (the OH− anion is less nucleophilic when it possesses a full hydration shell).51d This is more true for the use of this cationic group in the AAEMs but less true for use in the AEIs that are exposed to gas flows (much more difficult to maintain the AEIs in the fully hydrated state). Tailoring the hydrophobicity of the cationic group's environment may well have an impact.56 The challenge for applications such as APEFCs, where it is difficult to keep the polymer electrolyte components fully hydrated (unlike in APEEs), is to develop AAEMs and (especially) AEIs that are stable (and conductive) in the presence of OH− when less than fully hydrated.
(a) The lower mobility of OH− (and HCO3−/CO32−) vs. H+ (see Table 1);84
| Ion | Mobility (μ)/10−8 m2 s−1 V−1 | Relative mobilitya (relative to K+) | Ref. |
|---|---|---|---|
| a Calculated from the mobility data to the left and in general agreement with the relative mobility data presented in ref. 89. | |||
| H+ | 36.23 | 4.75 | 87 and 88 |
| OH− | 20.64 | 2.71 | 87 and 88 |
| CO32− | 7.46 | 0.98 | 87 and 88 |
| HCO3− | 4.61 | 0.60 | 87 |
| Na+ | 5.19 | 0.68 | 88 |
| Cl− | 7.91 | 1.04 | 88 |
| K+ | 7.62 | 1.00 | 88 |
(b) The lower levels of dissociation of the ammonium hydroxide groups (cf. the highly acidic R–SO3H groups in PEMs).
With regards to (a) above, the intrinsically lower mobilities are traditionally offset by using AEMs with higher IECs compared to PEMs because ionic conductivity ∝ ion mobility × ion concentration. AEMs typically possess IECs much higher than 1.1 meq. g−1 (cf. Nafion®-11x series of PEMs = 0.91–0.98 meq. g−1)85 apart from of the state-of-the-art phase segregated systems discussed in detail later. This can lead to problems with high water uptakes and dimensional swelling (hydrated vs. dehydrated) and this leads to AEMs with lower mechanical strengths and a difficulty in maintaining the in situ integrity of membrane electrode assemblies (MEAs) containing AEMs (especially for APEFCs that use gas feeds [the MEAs are not in continuous contact with aqueous solutions/water]).86
Regarding (b) above and “lower levels of dissociation”, it is often stated that “trimethylamine is a weak base” as it has a pKb value (in aqueous solutions) of only ca. 4.2 (pKa [≈ 14 − pKb] = 9.8 for the conjugate trimethylammonium [Me3N+H] cation):88
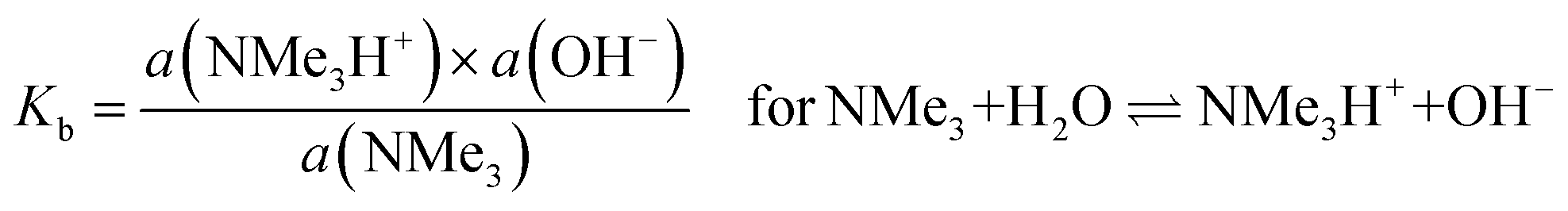 | (3) |
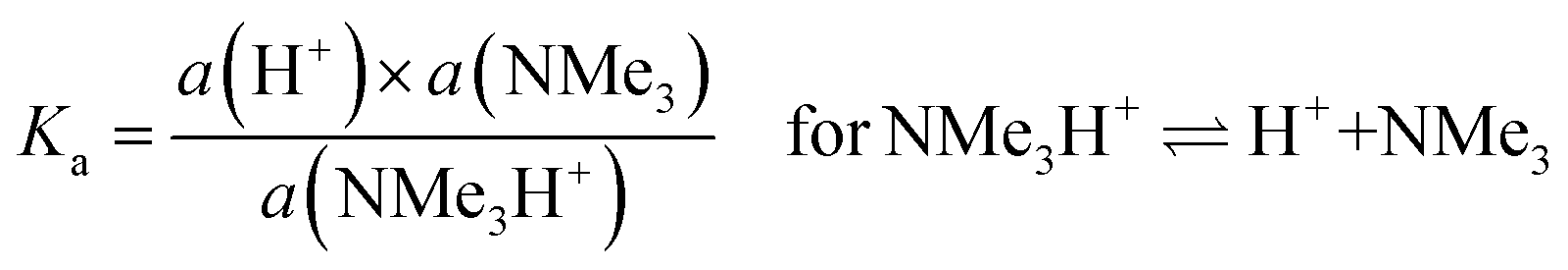 | (4) |
| RNMe3OH ⇌ RNMe3+ + OH− | (5) |
Indeed, AEMs in the OH− (and F−) forms appear to be completely dissociated at high hydration levels (in CO2-free conditions) unlike AEMs in the I−, Cl−, Br−, and HCO3− forms and the OH− ions conduct mainly via structure diffusion (approaching half the conductivities of H+ in PEMs).92 Therefore, concerns over the low levels of dissociation of OH− for N–H bond free QA hydroxide groups (in AAEMs) are generally overstated.
As the AAEMs quickly convert to less conductive CO32−/HCO3− forms when exposed to CO2 (i.e. air, recall Fig. 1),25d,43,93 it is essential that CO2 (air) is totally excluded from conductivity determinations of OH− form AAEMs. It is clear from the literature that this is rarely the case and that different laboratories use different set-ups probably with different levels of CO2 exclusion. This creates problems with regard to inter-laboratory comparisons of OH− conductivities. Most groups are likely underestimating the OH− conductivities of their AAEMs/AEIs due to [difficult to obtain] incomplete CO2 exclusion (i.e. they are measuring the conductivities of mixed alkali [OH−/HCO3−/CO32−] anion forms). Therefore to aid inter-laboratory comparisons, we make the recommendation that HCO3− conductivities are always reported for AAEMs/AEIs alongside conductivity data in other anion forms, such as Cl− and OH− (the water uptake of the material in each of the anion forms must also be measured to understand the conductivity changes in the material). The rationale is the AAEMs remain predominantly in the HCO3− form in the presence of air (CO2) and that the OH− conductivities can be estimated from the measured HCO3− conductivities.31,53 However, caution is required with such estimates as OH− conductivities for AAEMs containing benzyltrimethylammonium cations have been measured to be higher than the size of the cation would normally indicate.93 There is also the added complication with materials of a hydrophobic nature as ion-exchange is often incomplete and small amounts of residual anions can have profound effects on the mobility of the ion that you think you are studying.44
It should also be kept in mind that the conductivities of most relevance to electrochemical devices are the through plane conductivities as the ions move from one electrode to the other through the thickness of the AAEM. The measurement of in-plane conductivities (typically using 4-probe techniques) can sometimes lead to an overestimation of the ionic conductivities (i.e. conductivities are often anisotropic) with a bias towards the conductivities across the surface layers of the membranes (sometimes the most functionalised parts).94 However, we acknowledge that the measurement of through-plane conductivities can be tricky (difficult to isolate the membrane resistance from the electrode interfacial resistance when the membrane thicknesses are smaller than the dimensions of the electrodes) and that in-plane measurements have their place as they are often much more repeatable (and yield results that are less likely to be misinterpreted).
In devices where the AEMs/AEIs are not in continuous contact with liquid water (e.g. APEFCs) it is essential that they can conduct at lower relative humidities (RH). This will be a big challenge as the conductivities (and chemical stabilities) of AAEMs drop off much more rapidly with RH than with PEMFCs.19d,52 Hence, measurements in liquid water are not always relevant because fuel cell developers want ionic conductivities reported with the membranes in water vapour (reviewers often push that conductivity measurements where the membranes are immersed in liquid water should be reported). These are much harder to conduct especially when the measurement of the OH− conductivities of AAEMs/AEIs in RH ≤ 100% atmospheres is desired (the use of glove boxes with CO2-free atmospheres are essential).15a
Fundamental studies (including modelling studies) related to anion conductivity, the effect of water contents and transport, and the effect of CO2 on the properties of AAEMs have been undertaken.19d,46a,95 These should continue in order to understand what is required to maintain high conductivities under lower humidity environments (low water content per exchange group) and the effect of the presence of CO2 on AAEM conductivity (see APEFC section later). For fundamental studies, it is often useful to normalise conductivities to other factors such as, water contents,50 IECs39b and mobilities.46a Additional experiments such as the measurement of NMR T1 and T2 relaxation times for water in AAEMs can also be useful.49 Short water relaxation times can lead to improved AAEM conductivity (even with lower IECs) as they indicate more close interaction between the water molecules and the solid polymer. Too high water uptake (often via excessively high IEC) can mean that much of the H2O is inactive (not interacting with polymer) and is actually diluting the conductive species (leading to a lowering of the conductivity).
Various strategies have been proposed to enhance the conductivities of AAEMs without employing excessively high IECs and water uptakes (dimensional swelling). The development of phase-segregated AAEMs, containing hydrophobic phases interspersed with hydrophilic ionic channels and clusters (à la Nafion®), is rapidly becoming the de facto strategy for developing the high conductivity AAEMs with low IECs and water uptakes (see next section). It should be noted that phase separation is not always essential for high AAEM/AEI conductivities.20 Covalent crosslinking is an alternative strategy, which can additionally reduce gas crossover but may also lead to less desirable attributes such as insolubility, reduced flexibility and embrittlement (leading to poor membrane processability), and even a loss of conductivity (if it interferes with phase segregation).10d,13a,14b,d,25a,96 Other strategies include ionic crosslinking,10b maximising the van der Waals interactions (to minimise swelling without the use of crosslinks),14d using 1,2,3-triazoles to link the QA groups to the polymer backbone,97 and enhancing the number of positive charges on the side-chains.42d,80
Phase segregated AEMs98
A realistic strategy to enhance the ionic conductivity of AAEMs is to improve the effective mobility of OH− rather than increasing the IEC (to avoid excessive water uptakes and dimensional swelling on hydration).2e As shown in Table 1, the mobility of OH− in dilute KOH solution is actually rather high and is only inferior to that of H+ but much superior to that of other ions. However, in AAEMs, the motion of OH− can be retarded by the polymer framework where the effective mobility of OH− is often much lower than that in dilute solutions. This is a common drawback of polymer electrolytes including Nafion® (where the effective mobility of H+ is only about 20% of that in dilute acids).The conduction of ions, such as H+ or OH−, relies on the presence of water so the structure of hydrophilic domains in a polymer electrolyte is the predominant factor for ion conduction. It is believed that the outstanding ionic conductivity of Nafion® is attributed to its phase segregation morphological structure.99 Specifically, the presence of both a highly hydrophobic fluorocarbon polymer backbone and flexible side chains (that contain the ionic groups) drives the formation of a hydrophilic/hydrophobic phase separation structure, where ion-containing hydrophilic domains overlap and form interconnected ionic channels. Although the nominal IEC of Nafion® is only ca. 0.92 meq. g−1, the localised H+ concentration in the ionic channels is much greater, which significantly increases the efficiency of H+ hopping conduction.
Since the OH− conduction operates via a similar mechanism to H+ conduction,100 a phase segregated (self-assembled) structure is expected to improve ion conduction in AAEMs.98 However, the formation of phase segregated structures in AEMs is more challenging as most AEMs are based on hydrocarbon backbones with lower hydrophobicities compared to fluorocarbon-based AEMs. The hydrophobicities are often even lower again as the cationic groups are commonly connected to the hydrocarbon backbones via short links (often –CH2–), e.g. the quaternisation used to form QA polysulfone AAEMs markedly changes the alkali stability and hydrophobicity of the polysulfone backbone (relatively hydrophobic when unmodified). Elongating the length of the link between the polymer backbone and the cationic functional groups should, in principle, assist in the formation of phase segregation structures. However, this requires an entire change in material synthesis methods as a significant number of AEMs reported in the literature are prepared using a polymer modification protocol; for example, a commercially available polymer (such as polysulfone) is functionalised with a reactive group (commonly –CH2Cl formed using some form of chloromethylation reaction [often using highly carcinogenic reagents such as chloromethylether]) and then further reacted (with reagents such as trimethylamine) to yield the final AEM containing polymer bound cations (Scheme 6a).101 This typical process is not easily adapted to yield Nafion®-like pendent cations (i.e. a QA attached to the polymer backbone through a long side chain).
 | ||
| Scheme 6 General strategies for the development of phase segregated AEMs (b–g) compared to a benchmark homopolymer system (a). The rectangles represent a polymer block. | ||
Phase segregated morphologies generally exist in block (Scheme 6b/d/e/g) and graft copolymers (Scheme 6f),46a,102 which results from the enthalpy associated with the demixing of incompatible segments.103 Regarding the development of copolymers, it is clear from the literature that the formation of phase segregated morphologies is much more successful for block copolymers compared to random copolymers (if comparable systems are compared).14a,49,104 For example, the phase separated morphology of a polysulfone block copolymer (IEC = 1.9 meq. g−1, high λ = 32 value [λ values give the number of water molecules per cationic head-group] has been reported to give a very high hydroxide conductivity of 144 mS cm−2 at 80 °C (over 3 times higher than an IEC = 1.9 meq. g−1 random copolymer benchmark).104e Coughlin et al. have shown that block copolymers can yield well defined lamella phase separation mophologies.23b Such high level organisation is, however, not mandatory given that perfluoro QA AEMs can also phase separate (just like perfluorosulfonic acid [PFSA] PEMs like Nafion®).16a QA-functionalised poly(hexyl methacrylate)-block-poly(styrene)-block-poly(hexyl methacrylate) systems have also been shown to possess a highly developed phase separation (using SAXS and TEM techniques) and this yielded relatively high OH− ion diffusion coefficients (comparable to PEM benchmarks).105 However, due to the insufficient mechanical strengths, such olefin types can only serve as models to assess the effects of molecular architecture on performances.
Beyer et al. reported strongly self-segregating covalently crosslinked triblock copolymers with high conductivities (120 mS cm−1 at 60 °C for the fully hydrated sample with IEC = 1.7 meq. g−1 and λ = 72).106 Similarly, Bai et al. have reported conductivities >100 mS cm−1 at 60 °C and >120 mS cm−1 at 80 °C for QA-PPO/polysulfone/QA-PPO triblock AAEM (IEC > 1.83 meq. g−1, fully hydrated but with a much lower λ = 16).107 Coates et al. have also developed block copolymers but with additional crosslinking (via the cationic groups) and this yielded AAEMs with equally exceptional OH− conductivities (up to 110 mS cm−1 at 50 °C).21c Guiver et al. produced polysulfone block copolymer AAEMs with higher OH− conductivities at lower λ values, water uptakes, and dimensional swelling compared to a non-block copolymer QA polysulfone benchmark AAEM.108 Li et al. developed another class of block copolymer where the QA group was separated from the polymer chain by a triazole group (Scheme 6g).97 The triazole formation stemmed from the use of Cu(I) catalysed “click chemistry”. This produced AAEMs with excellent conductivities at room temperature when fully hydrated: an IEC = 1.8 meq. g−1 AAEM gave a OH− conductivity of 62 mS cm−1 (and an interestingly high CO32− conductivity of 31 mS cm−1). However, the addition of triazole links increased the water uptakes (cf. triazole-free examples).
Binder et al. have also developed “comb shaped” block copolymers where the QA groups contained a long hydrocarbon tails (Scheme 6d).109 AAEMs with OH− conductivities up to 35 mS cm−1 (room temperature, fully hydrated, IEC = 1.9 meq. g−1) were reported. Even more interestingly, the water contents, λ, appeared to be independent to IEC (λ = 5.2–5.9 over the IEC range 1.1–1.9 meq. g−1); these were much lower than a benchmark block copolymer AAEM where the QA was a polymer bound trimethylammonium (IEC = 1.4 meq. g−1, λ = 10.4, OH− conductivity = 5 mS cm−1). Hickner et al. also investigated “comb shaped” AEIs for APEFCs where an increasing number of long alkyl chains (C6, C10 and C16 in length) were attached to the QA groups.50 Higher performances were obtained with the 1 × C16 AEI (IEC = 1.65–1.71 meq. g−1, 21 mS cm−1 at room temperature in water) but this AEI had less in situ durability compared to a different 1 × C6 example (IEC = 2.75–2.82 meq. g−1, 43 mS cm−1). The AEIs with multiple long alkyl chains on the QA groups exhibited lower conductivities and water uptakes compared to AEIs of similar IECs that contain only a single long QA alkyl chain. Hickner et al. also showed that introducing crosslinking into comb shaped AEMs can enhance their stability towards alkali.110
Recent studies by Xu et al. investigated graft copolymers (Scheme 6f) for AAEMs, which displayed superior fuel cell related properties.102c Dimethyl-PPO-based copolymers with poly(vinylbenzyl trimethylammonim) grafts were synthesized via atom transfer radical polymerization (ATRP).111 AAEMs with OH− conductivities up to 100 mS cm−1 at 80 °C were produced with high graft densities and optimised graft lengths (IEC = 2.0 meq. g−1). Knauss et al. have also produced a PPO block copolymer AAEM but where the hydrophobic blocks contained additional phenyl side-groups (not aliphatic hydrocarbon side chains). A high OH− conductivity of 84 mS cm−1 was obtained with an IEC = 1.3 meq. g−1 AAEM.14a Importantly, this conductivity was produced with the AAEM in a RH = 95% environment (rather than the normally encountered fully hydrated condition where the AAEM is fully immersed in water).
Recently, Zhuang et al. reported a new and simple method for achieving highly efficient phase segregation in a polysulfone AEM.112 Instead of elongating the cation-polymer links or adding hydrophobic chains to the QA groups, long hydrophobic side chains were directly attached to polymer backbone at positions that are separated from the cationic functional group (Scheme 6c). This polysulfone phase segregated AEM was designated aQAPS. This structure is not categorised as a block copolymer, but rather a “polysurfactant” where the hydrophilic cationic head-groups (e.g. QA) are linked through the polymer backbone but the long hydrophobic tails are freely dispersed. This concept was inspired by the structure of Gemini-type surfactants, where enhanced ensemble effects are seen when properly tying up two single surfactant molecules.113 The effect of phase segregation of the aQAPS design was identified using TEM and SAXS data (Fig. 2). The TEM image of the QAPS polysulfone copolymer benchmark, where there was no hydrocarbon side chain on the hydrophobic blocks (analogous to Scheme 6b), was uniform (Fig. 2a), which indicates the lack of clear phase segregation. However, the hydrophilic domains in the aQAPS system (dark zones in Fig. 2b, dyed using I− before TEM measurement) were clustered and separated from the hydrophobic polymer framework (light background in Fig. 2b). This strong phase segregation resulted in a long-distance structural ordering, as indicated by the SAXS pattern (Fig. 2c).
 | ||
| Fig. 2 TEM images of polysulfone-based AEMs: (a) QAPS [Scheme 6b] and (b) aQAPS [Scheme 6c]. (c) The resulting SAXS patterns.112 | ||
As a consequence of the phase segregation, the ionic conductivity of aQAPS was 35 mS cm−1 at 20 °C and >100 mS cm−1 at 80 °C (Fig. 3) in comparison to non-phase-segregated QAPS (15 mS cm−1 at 20 °C and 35 mS cm−1 at 80 °C). These are very high conductivities for such a low IEC AAEM (1 meq. g−1). The ionic conductivity of aQAPS(OH−) at room temperature was ca. 57% of that of Nafion® (very close to the mobility ratio between OH− and H+ in diluted solutions). This indicates that the ionic channels in aQAPS are as efficient as that in Nafion® (i.e. the difference in ionic conductivity being just the mobility difference between OH− and H+). However, at temperatures that are more relevant to fuel cell operation (60–80 °C), the IEC normalised ionic conductivity of aQAPS(OH−) was as high as that of Nafion®(H+). This shows that the OH− conduction can be as fast as that of H+ at elevated temperatures, provided the ionic channels in the AAEM are optimised. This significant finding demonstrates that OH− conductivities in AAEMs are not intrinsically inferior those of H+ in PEMs.
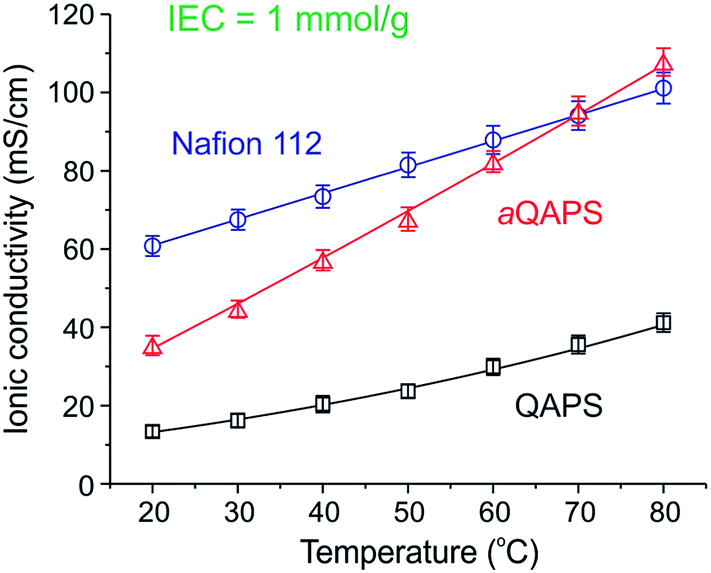 | ||
| Fig. 3 IEC normalised conductivities of Nafion®, aQAPS, and QAPS.112 | ||
The need for AEIs in electrochemical systems
Before the discussions move onto application specific items, the subject of the need for solubilised/dispersible AEIs needs to be introduced. To maximise the catalyst utilisation (optimal tri-phase interface [gas diffusion + ionic conduction + electronic conduction pathways available to a maximum amount of catalyst surface]) and introduce the required level of ionic conduction and hydrophobicity into the electrodes of low pH electrochemical devices such as PEMFCs, Nafion® dispersions (e.g. D-521/D520) are commercially available, scientifically well known, and widely used as acidic ionomers.99,114 For AEM/AAEM containing systems, the availability of commercial-grade AEIs is more restricted and less optimised for application in electrochemical applications. The usage of Nafion® CEM ionomers with AAEMs in APEFCs is a non-ideal situation.115 Both Tokuyama116 and Fumatech have developed AEIs.117 Other researchers have developed their own concepts or solubilised the materials used to make the AAEMs themselves (where possible).15c,28b,118 However, it is important to keep in mind that if production of an AEI technology is to be scaled up (for commercialisation) then it is vital that the AEI is supplied most desirably in an aqueous-based form (dispersion or solution). This is for safety considerations: the presence of both organic solvents and large quantities of finely divided (nano) catalysts in the scaled up manufacture of MEAs present will present a significant hazard.119AAEMs in (chemical) fuel cells2
H2/air(O2) alkaline polymer electrolyte fuel cells (APEFCs)
AAEMs and AEIs are used in APEFC technology.2 In the literature this class of fuel cell is also called Alkaline Membrane Fuel Cell (AMFC), Anion Exchange Membrane Fuel Cell (AEMFC), or Solid Alkaline Fuel Cells (SAFC). In principle APEFCs are similar to PEMFCs, with the main difference that the solid membrane is an AAEM instead of a PEM. With an AAEM in an APEFC, the OH− is being transported from the cathode to the anode, opposite to the H+ conduction direction in a PEMFC. The schematic diagram in Fig. 4 illustrates the main differences between the PEMFC and the APEFC. In the case of a PEMFC, the H+ cations conduct through a solid PEM from the anode to the cathode, while in the case of an APEFC the OH− anions (or other alkali anions – see later) are transported through a solid AAEM from the cathode to the anode. The use of solid electrolytes also prevents electrolyte seepage, which is a risk with traditional alkaline fuel cells (AFCs) that use aqueous Na/KOH electrolytes.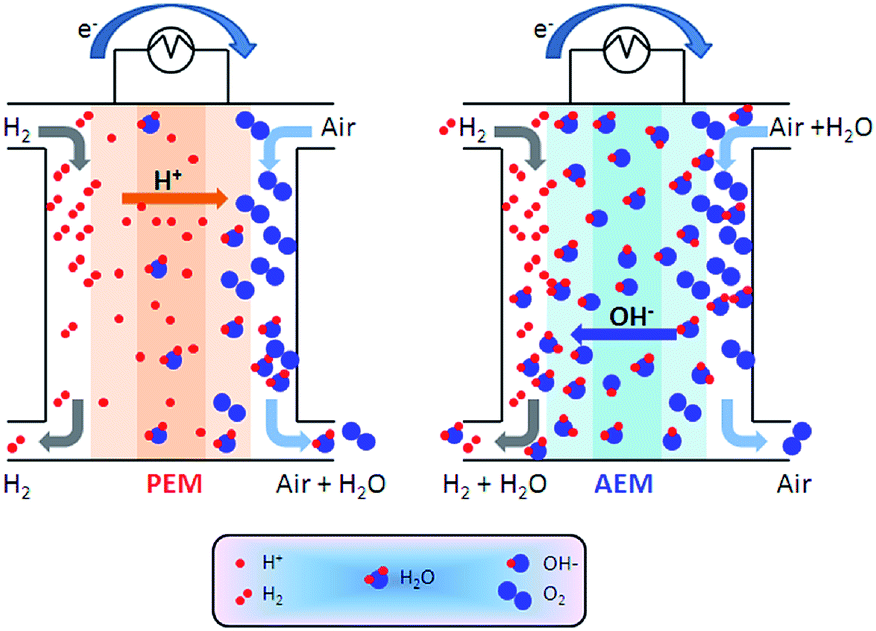 | ||
| Fig. 4 Schematic comparison of a proton exchange membrane fuel cell (PEMFC, left) and an alkaline polymer electrolyte fuel cell (APEFC, right) that are supplied with H2 and air.120 | ||
The ORR121 and hydrogen oxidation reaction (HOR) for a PEMFC (HOR = eqn (6) and ORR = eqn (7)) are compared to an APEFC (HOR = eqn (8) and ORR = eqn (9)) below [recall, all E values are given as reduction potentials even if a reaction is written as an oxidation]:
| 2H2 → 4H+ + 4e−E = 0.00 V vs. SHE | (6) |
| O2 + 4H+ + 4e− → 2H2O E = 1.23 V vs. SHE | (7) |
| 2H2 + 4OH− → 4H2O + 4e−E = −0.828 V vs. SHE | (8) |
| O2 + 2H2O + 4e− → 4OH−E = 0.401 V vs. SHE | (9) |
| 2H2 + O2 → 2H2O Ecell = 1.23 V (both acid and alkali) | (10) |
Although the overall reaction (eqn (10)) is the same for both types of fuel cells, the following differences in both technologies are very important:
(a) Water is generated at cathode side of PEMFCs but is generated at the anode in APEFCs;
(b) While there is no need for water as a direct reactant in PEMFCs, water is a reactant in APEFCs as it is consumed in the cathode reaction.122
In principle, the advantages of APEFCs over PEMFCs are related to the alkaline pH cell environment of the APEFCs:
(a) Enhanced ORR catalysis, allowing for the use of less expensive, Pt-free catalysts such as those based on inorganic oxides including perovskites, spinels and MnOx, as well as those based on Fe, Co, Ag, and doped graphene (among others);123
(b) Extended range of (available) cell and stack materials such as cheap, easily stamped metal (e.g. Ni and uncoated stainless steel bipolar plates);
(c) A wider choice of fuels in addition to pure H2 (e.g. hydrazine hydrate and “dirty” H2 including H2 containing traces of ammonia – see later sections).
The most critical concerns for APEFC technology are the low conductivities and the relatively poor stabilities of the AAEMs that were developed in the first years of the APEFC development.2,124 However, as discussed above, significant advances have been made in recent years that have promoted APEFC development.
| O2 + 2e− + H2O → HO2− + OH− | (11) |
Alternatively, catalysts that are active in reduction of peroxide at low overpotentials (bi-functional catalyst) are also deemed advantageous for alkaline systems.123
In one of the very few studies investigating HOR activities of platinum in both acidic and alkaline media, Sheng et al. found that in alkaline electrolyte the HOR kinetics are several orders of magnitude slower than in acid electrolyte.130 More recently, this finding has been confirmed and quantified by Rheinländer et al. who reported that ultra-low loadings Pt in aqueous KOH (1 mol dm−3) might exhibit prohibitively large losses of 140 mV at 40 mA cm−2.137 Moreover, when looking for non-Pt HOR catalysts, it was found that Pd-based catalysts exhibit 5 to 10× lower activity than Pt in alkaline medium.137 However, a recently reported PdIr/C catalyst showed a HOR activity comparable to Pt/C.138
Rare studies investigating non-PGM catalysts for HOR in H2/O2 APEFCs have been carried out by Zhuang et al.127a,c The authors reported that by decorating Ni nanoparticles with Cr, they succeeded to tune the electronic surface of Ni, making it possible to operate in the anodes of APEFCs. They reported a single preliminary test in real APEFCs, showing a maximum peak power of 50 mW cm−2 at a cell temperature of 60 °C (Ni-based anode and Ag-based cathode). Although the power densities were still low, these are the first published reports on APEFCs that used non-Pt catalysts for both the HOR (anode) and the ORR (cathode) in a single cell, hence they demonstrate the potential of the APEFCs. A more recent ex situ experimental and theoretical study by Yan et al. indicates that CoNiMo catalysts hold promise for use as a HOR catalyst in APEFCs with the potential to outperform Pt-catalysts (when at high loadings).139 All of these fundamental HOR studies strongly emphasise the need for alternative, inexpensive HOR-catalysts for the successful development of the technology. This is a major research priority.
In all cases reported, however, the stability (and durability) of the non-Pt HOR catalyst has been a major limiting factor. All authors of published works suggest morphological changes in the process of catalyst operation as a major source of instability of the interface. The challenge in practical non-Pt HOR design is that no catalyst has been shown to be active at comparable rates in both the HOR and hydrogen evolution reaction (HER). The search for a true breakthrough continues!
As the AEIs will be in intimate contact with the catalysts, another consideration is the influence of the cationic functional groups on the electrochemical activities of the catalysts; this will vary for each catalyst/AEI-cationic-group combination. Recent ex situ electrochemical studies have investigated the effect of fully dissolved cationic small molecules (not bound to polymers) on bulk polycrystalline141 and Pt/C ORR catalysts [1 mmol dm−3 cationic molecules dissolved in aqueous KOH (1 mol dm−3)].142 Even though these studies are not directly comparable to the in situ situation in APEFCs (i.e. Nafion® [cation-exchange] ionomer dispersion were used in the formulation of the catalyst inks [rather than using AEIs as a binder] and cationic molecules were fully dissolved in aqueous KOH electrolytes with excess spectator ions), the studies have provided some useful indicators of issues that need to be considered:
(a) Unlike with acid electrolytes,143 Cl− anions (the anion present in all of the experiments) did not have a major effect on the ORR on Pt (1 mmol dm−3 Cl− concentrations tested);
(b) Benzene-ring-free QA cations (e.g. tetramethylammonium) have a low impact on the ORR on Pt, whereas benzene-ring-analogues (e.g. benzyltrimethylammonium) lead to impeded ORR performances;
(c) Imidazolium cations (e.g. benzyl-3-methylimidazolium) lead to severe reductions in the ORR performance of Pt; these cations also change the mechanism so the level of undesirable (peroxide generating) n = 2e− ORR increases (eqn (11));
(d) Pt catalysts oxidise the organic cations at high potentials and the degradation products may also have an impact of the ORR performances (degradation of organic components at the anode may also have an effect on the HOR). This also suggests that research needs to be conducted into the electrochemical stability of the cationic head-groups bounds to the AEIs that are in contact with the catalysts (especially at higher cathode potentials). In this respect, Pt (that tends to catalyse a broad range of things very efficiently) may well be the worst choice of catalyst;
(e) As expected,144 polycrystalline (bulk) Pt gave higher specific activities (electrochemically active surface area normalised current densities) and exchange current densities than Pt/C nanocatalysts (comparisons with each cation additive).
Similarly, a study by Shao et al. investigated the effect of 1-methylimidazole and triethylamine (but not charged imidazolium and quaternary ammonium species) on the ORR and HOR on Pt/C in aqueous KOH.145 Similarly, Konopka et al. looked at the effect on the ORR of polycrystalline Pt of 1,1,3,3-tetramethylguanidinium cations.146
Other studies have looked at the effect of AEIs themselves on the performances of various catalysts towards various reactions at high pH (where Nafion® ionomer is not present). These experiments are closer to the conditions in APEFCs. These studies involved AEIs such a commercial QA types (Tokuyama AS-4)145–147 in-house synthesised polysulfone-imidazolium types,145 and a phosphonium type.123d A more recent study investigated the effect of polymer backbone of QA-AEIs on the performance of an iron-cyanamide-derived catalyst.148 The poly(phenylene) and Nafion®-based AEIs led to higher performances compared to the polysulfone AEI. For another example, the use of AS-4 and the imidazolium AEI both reduced the ORR mass activity on Pt/C compared to the use of Nafion® ionomer.145 However, the HOR mass activities were increased with the use of AEIs compared to Nafion® (with the imidazolium AEI yielding the best HOR performance). Lemke et al. looked at the use of AS-4 as the AEI with Ag-nanowire ORR catalysts.147 The presence and loading of the AEI was observed to have an effect on both the catalyst activity and the number of e− per O2 reduction (n = 2 [eqn (11)] vs. n = 4 [eqn (9)]). Yan et al. concluded that phosphonium cationic groups poison Ag ORR catalysts much less when polymer bound (as part of the AEI) compared to when part of dissolved small molecules.123d
Note: these prior studies did not compare AEIs containing different head-groups but the same polymer backbone (and IEC). The next stage of this series of investigations should consider the ORR and HOR kinetics on fuel cell catalysts when bound using AEIs containing different cationic head-groups (QA vs. imidazolium etc.) in Nafion®-ionomer-free systems (and without addition of fully solubilised cationic molecules). An AEI concept is now available that would facilitate such a study that uses a selection of bulk producible AEIs containing different cationic head-groups (but with the same IEC and polymer backbone chemistry).149
The trace CO32−/HCO3− anions, generated at the cathode from the reaction of the CO2 in the air supply and the OH− anions present in the electrolytes, diffuse away and accumulate at the anode side of the MEA. This sets-ups an undesirable pH gradient where the anode side of the MEA has a lower effective pH (higher concentration of CO32−/HCO3− species) than the cathode side (retains a higher OH− content than the anode side);151 thermodynamically (cell voltage wise) it is better to have a lower pH at the cathode and a higher pH at the anode. However, experimental and modelling studies43,152 show that the CO32−/HCO3− contents in the AAEMs and AEI can be purged from the anode (“self-purging” and CO2 release) with the rapid and continuous generation of the OH− anions at the cathode at high current densities. This self-purging can actually be exploited if AEMs with suspected low stabilities to high concentrations of OH− [as in the commonly encountered aqueous KOH (0.5–1 mol dm−3) ion-exchange solutions] are to be tested in APEFCs: the AAEMs and AEIs can be initially converted to the CO32− forms, installed in the fuel cell, and then activated at high currents (in situ conversion of the AAEM and AEI into the OH− forms).152
In fact, the situation may be even more complex: while APEFC performances drop when the cathode supply is switched from O2 to air, there is evidence that APEFC performances can actually increase when CO2 is deliberately added to an O2 cathode supply (at higher CO2 concentrations than those found in air).153 This intriguing effect certainly warrants more detailed study. There is also a feeling in the APEFC community along with some anecdotal evidence that being able to raise the APEFC operating temperatures to > 80 °C may well increase the tolerance of these systems to CO2 in the air supplies (reduce the performance gap between APEFC operation with air compared to O2). Again, this needs to be rigorously investigated, especially when development of AAEMs/AEIs that are stable in the OH− forms at temperatures of >80 °C for long periods of time has been achieved.
| Peak power density achieved/mW cmgeo−2 | Current density/mA cmgeo−2 (voltage/V) at peak power | Gas supplies anode/cathode (back pressure) | AAEM | Cell temperature/°C | Catalyst type and loading/mg cmgeo−2 | References | |||
|---|---|---|---|---|---|---|---|---|---|
| Anode | Cathode | ||||||||
| 77 | 158 (0.49) | H2/air | ”Perfluorinated piperazinium” | 70 | Pt | 0.5 | Pt | 0.5 | 156 |
| 70 | 170 (0.41) | H2/O2 | QA polysulfone | 70 | Pt/C | 0.4 | Pt/C | 0.4 | 2n |
| 180 | 400 (0.45) | H2/O2 (1 barg) | QA polystyrene copolymer | 70 | Pt/C | 0.4 | Pt/C | 0.5 | 157 |
| ≈90 | ≈170 (0.53) | H2/O2 (2 barg) | QA polyphenylene | 80 | Pt | 2 | Pt | 2 | 15c |
| ≈40 | ≈60 (0.67) | H2/air (2 barg) | |||||||
| 138 | ≈280 (0.49) | H2/O2 (2.5 barg) | Stabilised phosphonium polysulfone | 50 | Pt | 0.2 | Pt | 0.2 | 39f |
| 196 | ≈380 (0.52) | H2/O2 (2.5 barg) | 80 | 0.5 | 0.5 | ||||
| 28 | 60 (0.47) | H2/air | Crosslinked QA polysulfone | 60 | Pt/C | 0.5 | Pt/C | 0.5 | 57f |
| 30 | ≈65 (0.46) | H2/air | Crosslinked QA polysulfone | 60 | Pt/C | 0.5 | Ag/C | 2 | 57e |
| 230 | 600 (0.38) | H2/O2 | QA radiation-grafted ETFE | 50 | Pt/C | 0.5 | Pt/C | 0.5 | 158 |
| 50 | 100 (0.50) | H2/O2 (1.3 barg) | QA polysulfone | 60 | Ni–Cr | 5 | Ag | 1 | 127c |
| 823 | ≈1800 (0.46) | H2/O2 | QA radiation-grafted low density polyethylene | 60 | Pt/C | 0.4 | Pt/C | 0.4 | 25e |
| 506 | ≈1000 (0.51) | H2/air (1 barg) | 60 | Pt/C | 0.4 | Pt/C | 0.4 | ||
One of the best indicators of the potential of this technology comes from the industrial sector. After all, there are commercial (fuel cell relevant) AAEMs available including those by Tokuyama, Solvay and Fumatech.154 Tokuyama showed a maximum peak power of 450 mW cm−2 and 340 mW cm−2 for H2/O2 and H2/air (CO2 free) respectively at 50 °C.43b Even though those results were obtained with Pt/C catalysts (0.5 mgPt cm−2), they show a power high enough for practical applications (such as backup power for stationary applications including in the telecoms industry). At same time, Kim reported interesting results with his polyphenylene based membranes.155 With 3 mg cm−2 Pt black catalyst in both the anode and cathode, a maximum power density of 577 mW cm−2 (at 1 A cm−2) and 450 mW cm−2 (at ≈0.8 A cm−2) at 80 °C with H2/O2 and H2/air conditions respectively. This power density is very similar to that achieved and reported by Yanagi and Fukuta.43b
Although these Pt-based APEFCs already exhibit performances that are good enough for practical fuel cell application, the need for these performances (or better) with alternative and inexpensive catalysts is paramount. One of the very few results presented on H2-based APEFCs with non-Pt catalysts was recently obtained at CellEra (an Israeli company developing AMFCs).127b Using dry H2 and CO2 filtered ambient air, Dekel reported a peak power density of 700 mW cm−2 (at 1.5 A cm−2) with 3 barg/1 barg H2/air at 80 °C with an APEFC containing a Pt anode catalyst, and a peak power density of 500 mW cm−2 (at 1.6 A cm−2) with 3 barg/1 barg H2/air at 80 °C with an entirely non-Pt (confidential catalysts) APEFC.127b This data also demonstrates that AAEMs can withstand considerable pressure differentials between the anode and cathode (CellEra have data that shows this to be true with 3 bar differentials). Tokuyama also report that their A201 AEM has a burst strength of 0.4 MPa.159
Performances of APEFCs can be increased with the use of active water management,160i.e. control of anode RHs with different anode RHs used at different current densities (lower anode RH at high current densities to prevent anode flooding – recall the anode is where the H2O is electro-generated). Water management and APEFC performances should increase with the use of thinner AAEMs.161 However, our experience to date is that AAEMs that are thinner than ca. 30 μm (e.g. Tokuyama A901 is ca. 10 μm in thickness)116b does not always lead to the expected increases in performance (even with well optimised APEFC systems). This mystery needs to be investigated further. A major in situ problem with the use of AAEMs and AEIs is when they are exposed to low RH environments in APEFCs: the conductivities significantly drop even with only small drops in RH (i.e. AAEMs and AEIs are much more sensitive to drops in humidity than PEMs and H+-conducting ionomers).52 The alkali stabilities of the AAEM and AEIs also decrease dramatically with lower hydration levels (OH− is stronger nucleophile when not fully hydrated). AEI degradation occurs mostly in the cathode catalyst layer as this is where most dehydration occurs; it appears to be hard to keep the cathode AEI hydrated even when more than enough H2O is transported back from anode and the cathode gas supply is fully hydrated.
In summary, extremely rapid and promising achievements in H2 APEFC technology have been shown. Based on the above described (recent) results that have been obtained with first prototypes, it seems that APEFC technology is not just a future promise, but a present reality. APEFCs development has been rapid in the past five or so years where experimental work is now being followed up with detailed theoretical and modelling studies.43a,161,162 This technology promises to solve the cost barriers (of PEMFCs), which is one of the main “pain-points” of fuel cell technology. While H2/air APEFC cells and stacks have achieved practical performances (for a few applications such as back-up power), there are still development challenges, which must be addressed to enable the large-scale introduction of APEFCs. These mainly include:
(a) AAEMs and AEIs with improved stabilities and conductivities at higher temperatures and especially when exposed to lower RH environments;
(b) Pt-free highly efficient catalysts, especially for the HOR.
Advances related the above two issues will assure rapid entrance of APEFCs into existing market opportunities.
AAEM fuel cells fuelled with C-based fuel alcohols2d,l,o,163
There are several drivers for the use of alcohol fuels in AAEM-based fuel cells (primarily for portable power applications):(a) Alcohols are liquid fuels with high volumetric energy densities (Table 3), even when compared to cryogenic H2(l);
| Fuel option | Specific energy density/kW h kg−1 | Density at 20 °C/g cm−3 | Volumetric energy density/kW h dm−3 | E cell /V | n |
|---|---|---|---|---|---|
| a Maximum thermodynamic cell potential at 25 °C on oxidation in a cell with O2 as the oxidant. b The number of e− per fuel molecule obtained on full oxidation. c Gaseous H2 at 200 bar. d Depending on the conditions. e For a solution of 500 g NaBH4 in 1 dm3 solution at 25 °C. (l) = liquid and (s) = solid. | |||||
| H2 (l) | 33 | Gas | 2.37 (0.53c) | 1.23 | 2e− |
| Methanol CH3OH (l) | 6.1 | 0.79 | 4.8 | 1.21 | 6e− |
| Ethanol CH3CH2OH (l) | 8.0 | 0.79 | 6.3 | 1.15 | 12e− |
| Propan-1-ol CH3CH2CH2OH (l) | 9.1 | 0.81 | 7.4 | 1.13 | 18e− |
| Propan-2-ol CH3CH(OH)CH3 (l) | 9.0 | 0.79 | 7.1 | 1.12 | 18e− |
| Ethylene glycol HOCH2CH2OH (l) | 5.2 | 1.11 | 5.8 | 1.22 | 10e− |
| Glycerol HOCH2CH(OH)CH2OH (l) | 5.0 | 1.26 | 6.4 | 1.09 | 14e− |
| Formate HCOO− (s) | 0.9 | Solid | — | 1.45 | 2e− |
| Hydrazine N2H4 (l) | 5.4 | 1.00 | 5.4 | 1.62 | 4e− |
| Ammonia (l) | 3.3 | Gas | 3–5d | 1.17 | 3e− |
| Ammonia borane H3N:BH3 (s) | 8.4 | Solid | — | 1.62 | 6e− |
| Sodium borohydride NaBH4 (s) | 9.3 | Solid (1.07e) | (10e) | 1.64 | 8e− |
| Typical gasoline (l) | 12.8 | 0.71–0.77 | 9.5 | — | — |
(b) The maximum theoretical efficiencies are widely stated to be high when based on the higher heating value (>97% for alcohols compared to 83% for H2 and 91% for NaBH4). However, please consider that these numbers can be misleading and not practical as only free energy in and free energy out (i.e. exergy) matters;
(c) Because alcohol oxidation is generally more facile in high pH environments164 and can be structure insensitive,165 there is a large repertoire of potentially cheaper and more abundant anode catalysts that can be used.
(d) A larger range of ORR catalysts increases the odds of finding alcohol tolerant options (including MxOy types);123f,132,166
(e) The conductive ions (OH−) move through the AAEM in a direction (cathode → anode) that is contrary to alcohol crossover (anode → cathode). This may mitigate against alcohol (and alcohol electro-oxidation product) crossover, especially at higher currents. Alcohol oxidation at the cathode can lower the cathode potential and/or poison the cathode catalyst. This is different to the situation in PEM-based Direct Alcohol Fuel Cells (DAFC) where the H+ ions move from anode → cathode thereby enhancing alcohol crossover (due to electro-osmotic drag). This is serious when materials such as Nafion are used (with high methanol permeabilities of >10−6 cm2 s−1 and methanol uptakes).167 Well designed and engineered systems, where the alcohol concentration in the anode catalyst layers is low (due to efficient and rapid oxidation), suffer less from alcohol crossover effects;
(f) In AAEM-DAFCs, H2O is consumed in the cathode catalyst layers (recall eqn (9)) and electro-generated at the anode where there is already a liquid reactant supply present (the reverse situation to PEM-DAFCs). This change in water balance may be beneficial in reducing flooding at the cathode: there is electro-osmotic drag of a large number of H2O molecules (>19) per H+ when aqueous alcohol solutions are supplied to the anode in PEM-DAFCs (a significant problem).168 However, modelling suggests that insufficient H2O transports through the AAEM (to the cathode) to sustain high currents in AAEM-DMFCs (Direct Methanol Fuel Cell).169 Related to this, Smart Fuel Cells (Germany) has a patent for a “fuel cell combination”, where the cathode of a PEM-DMFC is located next to the cathode of an AAEM-DMFC. This is to efficiently utilise, in the AAEM-DMFC cell, the large quantities of H2O transported through the membrane (anode → cathode) of a PEM-DMFC cell.170
(g) A small amount of alcohol penetrating into the membranes may protect the membranes (particularly hydrocarbon types) against peroxide attack.171 The presence of alcohols may also assist with the “cold start-up” of DAFCs (i.e. starting up the fuel cell from sub-zero temperatures).172
Methanol, ethanol, ethylene glycol, and glycerol are the more common alcohols to have been tested in AAEM-based fuel cells; however, higher alcohols such as the propanols have also been considered for electro-oxidation in alkali.173 The oxidation reactions (in alkali) for methanol, ethanol, ethylene glycol, and glycerol are given in eqn (12)–(16):
| CH3OH + 6OH− → CO2 + 5H2O + 6e−E = −0.81 V vs. SHE | (12) |
| CH3OH + 8OH− → CO32− + 6H2O + 6e− | (13) |
| CH3CH2OH + 16OH− → 2CO32− + 11H2O + 12e−E = −0.75 V vs. SHE | (14) |
| HOCH2CH2OH + 14OH− → 2CO32− + 10H2O + 10e−E = −0.82 V vs. SHE | (15) |
| HOCH2CH(OH)CH2OH + 20OH− → 3CO32− + 14H2O + 14e−E = −0.69 V vs. SHE | (16) |
Alcohol oxidation (assuming full electro-oxidation) produces CO2 leading to significant concentrations of CO32−/HCO3− in the anodes of the AAEM-based DAFCs. The resulting pH gradient (high pH cathode → low pH anode) will produce thermodynamically derived voltage losses.151 It has been calculated that a pH difference of 6.1 would exist across the AAEM at 20 °C corresponding to a thermodynamic voltage loss of ca. 360 mV, which drops to a pH difference of 4.1 (voltage loss of ca. 290 mV) at 80 °C. Such voltage losses can be offset with the improved kinetics at temperatures ≥80 °C (especially if alcohol crossover is suppressed).
As will be evident from the below discussions, acceptable power performances are only obtained when large amounts of Na/KOH are added to the aqueous alcohol fuel supplies. Assuming full oxidation of the alcohols, the presence of such additional quantities of additional OH− will lead to CO32− being the predominant product (rather than CO2):58 hence eqn (13)–(16) are written as such. As the point of using AAEMs/AEIs in fuel cells was to eliminate the use of aqueous Na/KOH, it must be questioned if the use of AAEMs is needed when the addition of Na/KOH to the fuel supplies cannot be avoided.174 After all, methanol has been used as a fuel in AFCs in the past. The review by Koscher and Kordesch details historic work involving alkaline methanol–air systems with liquid caustic electrolytes.175
Alongside the many studies with Pt-based catalysts,182 Pd183,184 and Au185 catalysts have also been considered for MOR in alkali. However, due to cost, the big push has been towards the development of non-PGM catalysts. Ni-based catalysts are a commonly encountered option.186 Spinner et al. have studied NiO-based catalysts for MOR in alkali. They found that activities were higher in aqueous CO32− electrolyte compared to aqueous OH− electrolytes.180 Minteer et al. have reported that the inclusion of magnetic composites into Ni-based electrocatalysts can boost MOR in alkali.187 Perovskite-based MOR catalysts have also been considered.188
As well as the conductive species opposing methanol crossover in AAEM-DMFCs, there have been many studies into finding AAEMs with low methanol (and other alcohol) permeabilities.10f,65,189 High methanol diffusion through the anode AEI is, however, desirable.190 Many of these studies mention an ex situ “selectivity” or “DMFC performance” parameter, which is the ratio of 2 intrinsic properties:10f,65,102b,191 ionic conductivity to methanol permeability (= σ/PMeOH). An ideal AAEM will have a high ionic conductivity and low methanol permeability yielding a high performance parameter. However, caution is required with this parameter as an AAEM with a high performance parameter value but with a very low ionic conductivity will not be suitable for application (i.e. a low conductivity material with an extremely low methanol permeability). Obviously there will be analogous ex situ performance parameters for the other fuel options detailed below.
Without the addition of metal OH− (MOH) salts to the methanol anode supply, the performances of AAEM-DMFCs are generally poor with typical peak power densities of <20 mW cm−2 (even with reactant pressurisation).115,174,192 The chances of 6/8 OH− anions diffusing/migrating through the hydrated components of the anode AEI at the same time (to a localised site on the catalyst surface) to allow rapid oxidation of a single methanol molecule is deemed very low in the absence of an additional source of OH− ions. This is different to PEM-DMFCs where the 6 × H+ (generated on oxidation of a methanol molecule) have to transport away from the anode catalyst surface sites. Despite this, Benziger et al. report a reasonable methanol (2 mol dm−3 and OH−-free)/O2 AAEM-DMFC performance with a peak power density of 31 mW cm−2 (OCV = 0.84 V) at room temperature using an imidazolium-PEEK AAEM (95 μm thickness and IEC = 2.0 meq. g−1) and AEI and Pt catalysts with loadings of 0.5 mgPt cm−2 (the catalyst coated membrane [CCM] method was used).10a Analytical modelling of AAEM-DMFCs suggests that MOH-free performances can be improved when the anode side faces upwards due to more facile removal of the CO2 bubbles (higher temperatures also facilitate CO2 bubble removal).193
The performances are generally higher when MOH is added to the aqueous methanol fuel supplies. The main contributor to this improved performance is an improved anode potential (>300 mV reduction in anode overpotential is possible). Katzfuß et al. obtained a peak power density of 132 mW cm−2 (OCV ca. 0.9 V) in AAEM-DMFCs at 80 °C containing both an in-house synthesised DABCO-crosslinked PPO AAEM (10 μm thick, IEC = 1 meq. g−1, 80% degree of crosslinking) or a Tokuyama A201 AAEM (28 μm thick);14b the fuel cells contained an Acta 4010 (6% Pd/CeO2/C) anode catalyst, and Acta 3020 (4% FeCo/C) cathode catalyst and were supplied with aqueous methanol (4 mol dm−3) containing KOH (5 mol dm−3) at the anode and dry O2 at the cathode. The same group obtained a similar performance (120 mW cm−2) under the same test conditions using a 4 component DABCO-crosslinked PBI-polysulfone membrane that was more stable to alkali than the prior DABCO-crosslinked PPO AAEM.194 Prakash et al. reported a methanol/O2 AAEM-DMFC containing Tokuyama's A201 AAEM and AS-4 AEI that yielded a higher peak power density of 168 mW cm−2 (OCV = 0.9 V) at 90 °C when supplied with an aqueous methanol (1 mol dm−3) anode feed containing KOH (2 mol dm−3).116a However, these performances were obtained using PtRu (HiSpec 6000) anode and Pt (HiSpec 1000) cathode catalysts (cell optimised with a teflonised cathode and non-teflonized anode).
As such, there has been a lot of research into more active EOR catalysts for use in alkali media.199 As can be expected, Pt-based catalysts have been considered.182c,200 Pd and Pd alloy catalysts are of increasing prevalence in the literature as non-Pt options where C–C bond breakage has been reported (under conditions such as lower NaOH concentrations).164b,183,201 It is believed that the sites of the Pd where there are adsorbed OH (OHads) species are the catalytic active sites rather than the Pd atoms themselves.202 However, the in situ durability testing (500 h) of Pd/C anode catalysts in AAEM-DEFCs show that the performance losses observed are due to the growth in Pd particle size (the performance of the Fe–Co cathode catalyst used remained constant).203 Oxide supported Pd catalysts have also been considered where Pd–NiO was reported to be a good option for EOR.204,205 It is believed that the formation of OHads on the metal oxide can transform carbonaceous species to CO2 at lower potentials (releasing Pd sites for further reaction).206 PdAu (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) catalyst yielded a higher AAEM-DEFC performance compared to PdAu catalysts with other ratios and Pd- and Au-only benchmark catalysts. The presence of gold oxide/hydroxyl species is thought to be important.207 Datta et al. reported that PdAuNi catalyst produced improved AAEM-DEFC performances compared to Pd-, PdNi, and Pd–Au benchmarks;208 this catalyst also produced higher yields of acetate and CO32− (cf. acetaldehyde) compared to the other catalysts. Zhao et al. have reported a PdIrNi high performance EOR catalyst for AAEM-DEFCs.209 This catalyst performed much better compared to Pd, PdNi and PdIr benchmarks. PdRu has also been studied (see later).210 Li and He report that in situ reduction of Pd-layers on Ni foam gave higher AAEM-DEFC performances compared to conventional brushed Pd/Ni-foam anodes (164 mW cm−2 compared to 81 mW cm−2 at 60 °C when tested in comparable ethanol–O2 fuel cells containing a Tokuyama A201 AAEM).211 Au- and Ni-base catalysts (Pt- and Pd-free) have also been considered as catalysts for EOR in alkali.129,212 As an example, RuNi catalysts have been reported to produce 8–9e− per ethanol molecule on average (mixture of H3COO− and CO32− as products).213
:10) catalyst yielded a higher AAEM-DEFC performance compared to PdAu catalysts with other ratios and Pd- and Au-only benchmark catalysts. The presence of gold oxide/hydroxyl species is thought to be important.207 Datta et al. reported that PdAuNi catalyst produced improved AAEM-DEFC performances compared to Pd-, PdNi, and Pd–Au benchmarks;208 this catalyst also produced higher yields of acetate and CO32− (cf. acetaldehyde) compared to the other catalysts. Zhao et al. have reported a PdIrNi high performance EOR catalyst for AAEM-DEFCs.209 This catalyst performed much better compared to Pd, PdNi and PdIr benchmarks. PdRu has also been studied (see later).210 Li and He report that in situ reduction of Pd-layers on Ni foam gave higher AAEM-DEFC performances compared to conventional brushed Pd/Ni-foam anodes (164 mW cm−2 compared to 81 mW cm−2 at 60 °C when tested in comparable ethanol–O2 fuel cells containing a Tokuyama A201 AAEM).211 Au- and Ni-base catalysts (Pt- and Pd-free) have also been considered as catalysts for EOR in alkali.129,212 As an example, RuNi catalysts have been reported to produce 8–9e− per ethanol molecule on average (mixture of H3COO− and CO32− as products).213
Sun et al. considered the use of an alkali-doped PBI membrane (APM) for use in AAEM-DEFCs.214 An et al. compared a CEM (Nafion®-211), an AEM (Tokuyama A201), and an APM in DEFCs.215 They concluded that AAEM had the best conductivity and mechanical properties and the lowest Na+ permeability, the CEM has the lowest ethanol permeability and the APM had the best thermal stability but the poorest species permeability. Overall the AAEM case was considered to have the best balance of characteristics for application in DEFCs but the thermal stability of the AAEMs need improvement.
In 2009, Bianchini et al. reported a AAEM-DEFC that gave a peak power density of 170 mW cm−2 at 80 °C with the selective production of acetate when fuelled with ethanol (10% mass) in aqueous KOH (2 mol dm−3) and with an active supply of O2 to the cathode;216 the DEFC utilised an anode containing a PdNiZn/C catalysts on Ni foam, an Acta Hypermec™ K-14 cathode, and a Tokuyama A201 AAEM. The performance dropped to a still respectable 58 mW cm−2 in a passive (air-breathing) AAEM-DEFC at 20 °C. More recently Chen et al. have reported a peak power density of 176 mW cm−2 (OCV > 0.8 V) in a AAEM-DEFC at 80 °C when fuelled with ethanol (3 mol dm−3) in aqueous KOH (3 mol dm−3) and supplied with dry O2 at the cathode;210 the fuel cell utilised a CCM containing a Tokuyama A201 AAEM, a Pd3Ru/C anode catalyst on Ni foam (with a Nafion® ionomer binder), and a MnO2 nanotube cathode catalyst (with Tokuyama AS-4 AEI). The enhancement of performance over the use of a benchmark Pd/C anode catalyst was attributed to the formation of RuOxHy at low potentials and weak CO adsorption.
Zhao et al. reported an AAEM-DEFC peak power performance of 185 mW cm−2 at a lower 60 °C temperature using a Acta “non-Pt anode catalyst” supplied with ethanol (3 mol dm−3) in aqueous KOH (5 mol dm−3) and a Pd3Au/CNT catalyst supplied with O2 supply at the cathode;217 the DEFC contained a Tokuyama A201 AAEM and Tokuyama A3 was used as the AEI. The performance was higher than those obtained when using a Pd/CNT or a Au/CNT cathode catalysts. The Pd3Au catalyst where the Pd and Au were physically mixed gave better performances than the alloyed and core–shell analogues. This performance was still lower than Zhao et al.'s alkaline-acid DEFC concept where a peak power performance of 360 mW cm−2 at 60 °C was obtained with a Nafion-212 CEM, a PdNi/C–Ni-foam anode, and a Pt/C (60% mass) cathode catalyst (Nafion ionomers used at both electrodes);218 the fuel supply was ethanol (3 mol dm−3) in aqueous NaOH (5 mol dm−3) and cathode supply was H2O2 (4 mol dm−3) in aqueous sulfuric acid (1 mol dm−3).
Zhao et al. have recently reported a AAEM-based DEGFC that yielded a peak power density of 112 mW cm−2 at 90 °C when supplied with O2 at the cathode and EG (1 mol dm−3) dissolved in aqueous KOH (7 mol dm−3) at the anode.18c The fuel cell contained an APM, PdNi/C/Ni-foam anode catalyst, an Acta Hypermec™ non-PGM cathode catalyst. The peak power density dropped to 90 mW cm−2 when air was used instead of O2 at the cathode.
Glycerol can have higher ex situ activities on Au electrodes in alkali compared to other alcohols (methanol, ethanol, and ethylene glycol) and higher than on Pt.229 There is spectroscopic evidence that full oxidation to CO2/CO32− may be achieved on polycrystalline Au in alkali.230 A recent study into PdxBi catalysts (synthesised using the sacrificial support method) reports that Pd4Bi displays the highest activity towards glycerol oxidation.231 This report shows that the catalyst is selective towards production of aldehydes and ketones at low potentials, hydroxypyruvate at medium potentials, and CO2 at high potentials in the forward voltammetric sweeps. However, the catalyst is history dependent and carboxylates are selectively produced on the reverse voltammetric sweep. Glycerol oxidation mass activities are also high on PdIn catalysts.223
The power densities of glycerol fuelled fuel cells are generally <200 mW cm−2 and with low Faradic efficiencies (due to incomplete oxidation to CO2/CO3−). However, there are reports of power densities of 265 mW cm−2 being obtained.232 There are even reports that glycerol can self-polymerise inside the fuel cell. Li et al. obtained 184 mW cm−2 at 80 °C with an AAEM-based glycerol/O2 fuel cell containing a Tokuyama AAEM and AEI, a Pt/C anode, and an Acta Hypermec™ non-PGM cathode;224c the anode was supplied with an aqueous solution containing crude glycerol (1 mol dm−3) and KOH (6 mol dm−3) at 30 psi back pressure. The use of crude glycerol (88% mass glycerol, a by-product of soy-bean biodiesel production) did not produce a drop in performance compared to much more expensive high purity glycerol (99.8%). The Pt/C anode catalyst clearly had stability in the presence of the impurities of the crude glycerol (e.g. methanol, fatty acids [e.g. soaps], transesterification catalyst residues, and element such as K, Ca, Mg, Hg, P, S, As etc.).
Non-alcohol C-based fuels
Formate [HC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O−],233 glucose (C6H12O6),234 and urea [H2NC(O)NH2] and urine235 have also been studied as alternative C-based fuels in AAEM-containing fuel cells. Dimethylether has also been studied in fuel cells containing Nafion® CEMs but where performances were highest when the dimethylether was dissolved in aqueous alkaline anolytes (cf. acid anolytes).236 The power densities achieved with glucose-fuelled AAEM-based fuel cells are currently below 50 mW cm−2. For example, Zhao et al. achieved a peak power density of 38 mW cm−2 at 60 °C when supplying a fuel cell containing a Tokuyama A201 AAEM (PdNi anode and Acta Hypermec™ K14 cathode catalysts) with aqueous glucose (0.5 mol dm−3) containing added KOH (7 mol dm−3). Obviously, there is a wide range of reaction pathways that are possible that will produce a variety of electrochemical intermediates and products. 24e− would be needed for the full oxidation of glucose, which is highly unlikely to occur. Glutonate (n = 2e− reaction) is the most common product in alkali.234d The urea (urine) fuel cells containing alkaline polymer electrolyte materials produced peak power densities of <15 mW cm−2:235 however, as with many of the fuels mentioned above, this fuel cell concept is in a very early stage of development.
O)O−],233 glucose (C6H12O6),234 and urea [H2NC(O)NH2] and urine235 have also been studied as alternative C-based fuels in AAEM-containing fuel cells. Dimethylether has also been studied in fuel cells containing Nafion® CEMs but where performances were highest when the dimethylether was dissolved in aqueous alkaline anolytes (cf. acid anolytes).236 The power densities achieved with glucose-fuelled AAEM-based fuel cells are currently below 50 mW cm−2. For example, Zhao et al. achieved a peak power density of 38 mW cm−2 at 60 °C when supplying a fuel cell containing a Tokuyama A201 AAEM (PdNi anode and Acta Hypermec™ K14 cathode catalysts) with aqueous glucose (0.5 mol dm−3) containing added KOH (7 mol dm−3). Obviously, there is a wide range of reaction pathways that are possible that will produce a variety of electrochemical intermediates and products. 24e− would be needed for the full oxidation of glucose, which is highly unlikely to occur. Glutonate (n = 2e− reaction) is the most common product in alkali.234d The urea (urine) fuel cells containing alkaline polymer electrolyte materials produced peak power densities of <15 mW cm−2:235 however, as with many of the fuels mentioned above, this fuel cell concept is in a very early stage of development.
| HCOO− + 3OH− → CO32− + 2H2O + 2e−E = −1.05 V vs. SHE | (17) |
| 2HCOO− + O2 + 2OH− → 2CO32− + 2H2O Ecell = 1.45 V | (18) |
Zhao et al. achieved a peak power density in an alkaline direct formate fuel cell (ADFFC) at 80 °C of 130 mW cm−2 with aqueous potassium formate (5 mol dm−3) as the fuel and when using a Pd/C anode catalyst, commercial Acta Hypermec™ K14 cathode catalyst, QA polysulfone AAEM and AEI, and dry O2 supply to the cathode;233a this raised to >250 mW cm−2 with the addition of KOH (1 mol dm−3) to the fuel supply. Haan et al. achieved a peak power density of 267 mW cm−2 at 60 °C in a ADFFC with a fuel supply consisting of HCOO−K+ (1 mol dm−3) in aqueous KOH (2 mol dm−3), a catalyst coated Tokuyama A201 AAEM (Pd black anode catalyst, Pt black cathode catalyst, and Tokuyama AS-4 AEI), and a humidified O2 cathode supply;233b the performance again decreased when the KOH was removed from the fuel supply (157 mW cm−2).
AAEM fuel cells supplied with N-based fuels237
N–NH2) and hydrazide (–C(O)–NH–NH2) functional groups]. The N2H4 can released on addition of solvents when required.241
Hydrazine hydrate (N2H4·H2O) is, however, considered stable enough to be viable as a fuel for AAEM-DHFCs.242 N2H4·H2O is an industrially used chemical reagent (20 ktons per year distributed in Japan) and it is less volatile than alcohols (so air emissions will be potentially lower). It has a freezing temperature of −50 °C (so it can be easily used in cold climates) and it has a of flame point of 74 °C (at 1 atm). Hence, at aqueous concentrations of <60%, N2H4·H2O is not flammable. The carcinogenic risk of N2H4·H2O (class 2B by International Agency for Research on Cancer) is equivalent to petroleum so the careful handling of the fuel is no more than currently accepted guidelines for petroleum.
The full (4e−) electro-oxidation of N2H4/N2H4·H2O results in the generation of harmless N2 and H2O products (eqn (19) – the complete cell reaction is given by eqn (20) [along with the ORR given in eqn (9)]), while H2 and NH3 are produced on partial oxidation (eqn (21)–(24)).243 It has also been proposed that the use of high pH conditions suppresses undesirable hydrolysis reactions (eqn (25)).244 Unlike with the B-containing fuel vectors (see below), no product (BO2−) accumulation occurs at the anode that requires spent fuel treatment, as all products are gaseous. The potential of the N2H4/O2 cell reaction is larger than the width of potential window of stability of water (on Pt) and so there is a risk of the HER occurring (eqn (26)). Despite this, OCV values in the range 0.8–1.0 V are typically observed.
| N2H4 + 4OH− → N2 + 4H2O + 4e−E = −1.21 V vs. SHE | (19) |
| N2H4 + O2 → 2H2O + N2Ecell = 1.61 V | (20) |
| N2H4 + OH− → NH3 + ½N2 + H2O + e− | (21) |
N2H4 + OH− → N2 + ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) H2 + H2O + e− H2 + H2O + e− | (22) |
| N2H4 + 2OH− → N2 + H2 + 2H2O + 2e− | (23) |
| N2H4 + 2OH− → N2 + ½H2 + 3H2O + 3e− | (24) |
| N2H4 + H2O ⇌ N2H5+ + OH− | (25) |
| 4H2O + 4e− → 2H2 + 4OH−E = −0.83 V vs. SHE | (26) |
A variety of metal catalysts have been studied for use as anode catalysts for N2H4/N2H4·H2O oxidation in DHFCs (Ni, Co, Fe, Cu, Ag, Au, and Pt).245 Ni-based242,246 and Co-based29e,118c catalysts appear most promising (Co catalysts have also been examined in the cathodes). For example, Sakamoto et al. used a combinatorial electrochemistry approach with 79 catalyst candidates and report that Ni0.87Zn0.13, Ni0.9La0.1, Ni0.8Zn0.1La0.1, and Ni0.6Fe0.2Mn0.2 catalysts have N2H4·H2O oxidation activities that are more active than a Ni/C reference catalyst.242a Cu-based catalysts have also been proposed for use at the anode of DHFCs.247 For example, a totally irreversible and diffusion-controlled oxidation of N2H4 is reported on Cu-nanocubes on graphene paper with N2 and H2O as the reaction products.248 The in situ formed copper hydroxide/oxide layers surface layers on the Cu catalysts are thought to enhance the activity and durability of the electrocatalyst. Selectivity to N2 and H2O as sole products of hydrazine oxidation, as opposed to forming NH3 (eqn (21)), is of critical importance in practical DHFC development.242,246The promise of such fuel cells as a “clean energy technology” will easily be compromised by a single digit ppm of NH3in the exhaust of a DHFC vehicle.
DHFC are liquid fuel fed fuel cells with a highly reactive fuel. As a result, the fuel crossover is a natural concern of all designs. The requirement for no hydrazine oxidation on the cathode is a very strong one. As with other types of AAEM-based fuel cells, non-PGM catalysts, such as Ag, can be used in the cathodes and can have low N2H4 oxidation activities (mitigating against fuel crossover effects).241
Peak power densities of over 600 mW cm−2 have been achieved by Daihatsu Motor Co. Ltd. (Japan) in an AAEM-DHFC at 80 °C containing a QA-polyolefin AAEM with Ni anodes and Co-polypyrrole/C cathodes (supplied with humidified O2) and a fuel supply of an aqueous solution of N2H4 (5% vol) containing KOH (1 mol dm−3).243 As a comparison involving the use of H2O2 at the cathode and a Nafion®-117 PEM, a PEM-DHFC performance of >1000 mW cm−2 has been achieved at 80 °C (PtNi/C anode and Au/C cathode).249 Performances can be increased on increasing NaOH concentration in the anode fuel supply up to concentrations of 4 mol dm−3 (reduced anode overpotentials).244 However, concentrations higher than these reduce performances as viscosity increases and this causes mass transport voltage losses at the cathode.
| 2NH3 + 6OH− → N2 + 6H2O + 6e−E = −0.77 V vs. SHE | (27) |
| 2NH3 + O2 → N2 + 3H2O Ecell = 1.17 V | (28) |
Power densities with NH3 have been low to date (<15 mW cm−2) with OCVs < 1.0 V; these values are lower compared to the historic use of KOH as an aqueous electrolyte (ca. 50 mW cm−2).237 (NH4)2CO3 has also been considered for use in AAEM-based fuel cells but power densities were <1 mW cm−2.251
A major problem is the very sluggish ammonia oxidation reaction in alkali.252 Only PtIr, PtRu, and PtRh catalysts show a reasonable ability to oxidise NH3 with the least serious affinity towards the surface poison Nads. There can be recovery from such Nads surface poisoning of Pt with H2 treatment of the anode.250c The presence of Pt(100) surfaces appears to be important.252a,f,g Reactive azides species may also be generated as intermediates.252h Oxygenated (Oads and OHads) can form on the catalyst surface with the presence of water and this (along with Nads) also inhibits the catalytic performance.253 Interestingly, NH3 oxidation reaction is remarkably different on Pt when studied in non-aqueous media (the formation of surface oxygenated species is prevented) yielding N2 as the dominant product and allowing the Pt to remain continuously active.253 This study also shows that Pd becomes highly active towards NH3 oxidation in non-aqueous media (a low activity is seen with Pd in aqueous KOH due to severe surface passivation).
APEFCs fuelled with H2 that is produced from NH3 reformation may have technological promise. AFCs can tolerate NH3 (several %) in the H2 fuel.254 However, PEMFCs are poisoned by traces (1–2 ppm) of NH3 and performances decay (reversible but only after several days of operation with pure H2):255 the PEMs themselves will slowly convert to the NH4+ forms lowering conductivity and water contents.256 Hence, using H2 derived from NH3 in PEMFCs would require prohibitively expensive scrubbing of the reformed H2 supplies.
AAEM fuel cells fuelled with B-based fuels
| BH4− + 2H2O → BO2− + 4H2 | (29) |
| 4H2O2 + 8e− + 8H+ → 8H2O E = 1.77 V vs. SHE (acid cathode) | (30) |
Traditionally, the absence of AAEMs with adequate properties has meant that research efforts have traditionally focused on using CEMs in the DBHFCs (in Na+ form when exposed to aqueous solutions containing NaBH4 and NaOH); this includes radiation-grafted CEMs.260 This configuration leads to the build-up of NaOH at the cathode over extended operational times with a concomitant and undesirable reduction of pH at the anode (in the absence of an engineering solution that resupplies NaOH to the anode from the cathode). However, CEM-DBHFCs can yield reasonable fuel efficiencies due to minimised BH4− crossover (CEMs are persmelective towards positive charged ions).
Replacement of the CEM with an AAEM allows a balance:
| BH4− + 8OH− → BO2− + 6H2O + 8e−E = −1.24 V vs. SHE | (31) |
| BH4− + 2O2 → BO2− + 2H2O Ecell = 1.64 V | (32) |
| 4HO2− + 4H2O + 8e− → 12OH−E = 0.87 V vs. SHE | (33) |
The hydrolysis reaction is a serious problem in DBHFCs and is catalysed on many metals. This inevitably limits the selection of an anode catalyst. The BH4− oxidation and hydrolysis side reactions will happen in parallel to varying extents (as a function of temperature, concentration, catalyst and anode potential etc.):
| BH4− + H2O → BH3OH− + H2 | (34) |
| BH4− + 2OH− → BH3OH− + H2O + 2e− | (35) |
| BH3OH− + H2O → BO2− + 3H2 | (36) |
| BH3OH− + 3OH− → BO2− + H2 + 2H2O + 3e− | (37) |
| BH3OH− + 6OH− → BO2− + 5H2O + 6e− | (38) |
| BH4− + xOH− → BO2− + (x − 2)H2O + (4–½x)H2 + xe− | (39) |
The formal reduction potential for the 8e− BH4−/BO2− redox couple is >300 mV negative to the formal potential of the HER (eqn (26)). It should therefore not be a surprise that the reduction of H2O is thermodynamically favourable (a 1.64 V cell potential is wider than the potential window of stability for H2O on Pt). Operating DBHFCs at high current densities (lower cell potential and higher anode potentials) will help to minimise parasitic HER. There are reports that the addition of small quantities of thiourea acts as a hydrolysis inhibitor.262
All of these factors reduce the number of e− that are extracted from each BH4− molecule (reduces fuel efficiency). The HER is especially problematic in fuel cell stacks263 as H2 evolution in the early cells of the stack can affect the operation of cells farthest away from the inlet (i.e. losses occur due to uneven fuel distributions). The evolution of H2 gas must also be controlled unless it is desired to produce H2 for oxidation at the anode of a PEMFC (an indirect DBHFC).264 Therefore, n < 8e− oxidation of BH4− is generally unavoidable and OCVs will not approach the theoretical maximum (due to a mixed potentials from the presence of both BH4−/BO2− and H2O/H2 redox couples).
Pt, Pt-alloys, Ag, Au, Zn, Ni, Pd, Os, Cu, and hydrogen storage alloy based catalysts have all been studied for BH4− oxidation.257 The common claim that Au catalysts oxidise BH4−via 8e− without competing hydrolysis reactions may not be universally true, while Pt can actually achieve near full BH4− oxidation (e.g. at low anode potentials);265 however, studies with Au and Pt may be complicated by catalyst poisoning by BH4− oxidation intermediates (possibly BH3OH−).266 Ag-based catalysts are less active and the presence of oxidised surface oxide species appears to be a prerequisite for borohydride oxidation.267 Pd-based catalysts can achieve higher peak power densities and reduce the rate of H2 evolution.268 The need for a rational design of binary anode catalysts, such as Au2Cu(111), has been identified.269
In general for AAEM-based DBHFCs, peak power densities up to 250 mW cm−2 are reported (OCVs values tend to be in the range 0.8–1.0 V).262b,270 Huang et al. argue that the use of KBH4 can give improved performances over NaBH4 in AAEM-DBHFCs (lower KBH4 permeability and higher KOH-conductivity in the PVA AAEMs).270a The highest AAEM-based DBHFC performance reported to date is by Zhang et al. who achieved 321 mW cm−2 (at 700 mA cm−2) at 40 °C in a BH4−/O2 fuel cell [anode supply = NaBH4 (1 mol dm−3) in aqueous NaOH (3 mol dm−3) and Pt/C catalysts used in both electrodes, AAEM = a guanidinium-poly(silsesquioxane)-PTFE composite (IEC = 1.14 meq. g−1 and 65 mS cm−1 at 60 °C)].271
For comparison, Miley et al. achieved 680 mW cm−2 (OCV of 1.95 V) in a Nafion®-based NaBH4/H2O2 DBHFC stack at 60 °C with a Pd anode catalyst and an Au cathode catalyst (anode = 18% mass NaBH4 in alkali and cathode = 17% mass H2O2 in acid).272 The concept of using an alkali anode and an acid cathode is a recurring theme273 but may not be relevant to AAEM-containing DBHFCs. Liu et al. have achieved 663 mW cm−2 at 65 °C in a BH4−/O2 (i.e. non-peroxide) DBHFC containing a porous polymer fibre membrane (PFM) separator and using aqueous KBH4 (0.8 mol dm−3)/KOH (6 mol dm−3) as the fuel feed [CoO cathode catalyst and LiNiO3 anode catalyst];274 the performance dropped to (a still respectable) 390 mW cm−2 when Nafion® NRE-212 was used instead of the PFM.
Alkaline direct ammonia borane (AB = H3B←NH3) fuel cells have also been proposed.250b,276 AB (also known as borazane) is a water soluble white crystalline solid containing 19.5% mass hydrogen (AB is a proposed hydrogen storage material), which is stable at high pHs.277 Au and Ag catalysts again look promising for AB oxidation in alkali (MnO2 catalysts appear unaffected by AB so again has promise as a cathode catalyst).276e,278 The reactions (and side-reactions) involved in the oxidation of AB in alkali (and for an overall AB-AFC) are given below:
| BH3NH3 + 7OH− → BO2− + NH3 + 5H2O + 6e−E = −1.22 V vs. SHE | (40) |
| BH3NH3 + OH− + O2 → BO2− + NH3 + 2H2O Ecell = −1.62 V | (41) |
| BH3NH3 + 4OH− → BO2− + NH3 + H2 + 2H2O + 3e− | (42) |
| BH3NH3 + 2H2O → BO2− + NH4+ + 3H2 | (43) |
The latter non-electrochemical reaction (eqn (43)) is thermodynamically spontaneous and so it is important that catalysts that are active for AB oxidation but not AB hydrolysis. An AB-AFC is less efficient than a BH4−-AFC due to the lower number of e− extracted per fuel molecule (6 [or 3 if eqn (42) is operating] vs. 8), despite similar Ecell values. Lu et al. first reported an AAEM-based fuel cell fuelled with AB,276e which concluded that the 3e− process (eqn (42)) was predominant (along with some capacity loss via hydrolysis). More recently Xu et al.276c reported that a direct AB fuel cell containing a Tokuyama AEI (IEC = 2 meq. g−1) and a Tokuyama 27 μm thick AAEM (IEC = 1.8 meq. g−1) achieved an OCV of 0.9 V and a peak power density of >110 mW cm−2 at 45 °C [Pt-based anode and cathodes, anode feed containing AB (0.5 mol dm−3) in aqueous NaOH (2 mol dm−3), humidified cathode feed]; however, H2 evolution was still a problem. In reality, and as expected from the discussion above, the BH3 component of AB is more easily oxidised than the NH3 component. Considering AB is expensive to produce (or regenerate from BO2−), this fuel may only be suitable for niche applications.
Finally, other B-based fuel options exist but have not been considered as a fuel option in AAEM-based fuel cells. This includes the volatile liquid borazine [(BH)3(NH)3, a cyclic molecule that is isoelectronic and isostructural with benzene]. Borazine is normally a trace PEMFC poison when AB is being used as a H2 storage material and it can also polymerise into polyborazylene “gums”.
The operation of carbonate-cycle APEFCs
Even though there is growing awareness that CO32− does not generally have such a serious deleterious effect on APEFC performance,9e,152,279 HCO3−, however, has a strong negative effect.43a,116a,280 HCO3− has a much larger hydration radius than OH− (ca. 4 vs. 3 Å respectively) with the same valence,281 which significantly reduces conductivity and device performance in the presence of CO2.93 On the other hand, CO32− anions have double the valence of OH−, which means that despite its larger hydration radius,281b there will be lesser effect on AAEM conductivity.152 In addition, even commercial AEMs (that were not developed specifically for highly alkaline environments, such as that found in APEFCs), and those that are not stable in the presence of aqueous OH− (such as a number of phosphonium exemplars), are far more stable in HCO3−/CO32− compared to OH− environments/forms.9e,75,279b
All of this suggests the possibly of an alternative high impact (disruptive) approach: to abandon OH− anions altogether and transition to low temperature devices that use CO32− conduction (cycles). The deliberate utilisation of CO32− in low temperature electrochemical systems has only been investigated since 2006, with the lion's share of the effort concentrated on the development of a room temperature carbonate fuel cells (RTCFC).282 However, there has been a recent but slow increase in the amount of work being conducted to investigate alkaline electrochemical devices that purposefully utilise CO32− anions for energy generation (fuel cells), to facilitate new chemical synthesis pathways, and for CO2 separation.279,283
| [RTCFC Cathode] O2 + 2CO2 + 4e− → 2CO32− | (44) |
| [RTCFC Anode] 2CO32− + H2 → H2O + CO2 + 4e− | (45) |
Such catalysts, including when doped with Bi, exhibit intriguing behaviour in the presence of CO32− in aqueous alkali.285 However, Ca2Ru2O7 catalyst still has many issues. Most notably, it is hard to synthesise and it has a CO2 adsorption strength that is too large (leads to an optimum CO2 concentration at the cathode of 10% mol, which yields a very low CO2 activity in the cell).
The direct formation of CO32− in these devices is important since the competing indirect pathway (i.e. involving the chemical reaction between CO2 and electrochemically generated OH−) still involves OH− desorption (implications for the durability of the AAEM and AEI). The indirect route also risks the production of HCO3−, which will lower the ionic conductivity of the AAEM and AEI. However, researchers have also failed to make AAEMs with the appropriate functionality to maintain the anions as CO32−. The equilibrium balance between OH−, CO32− and HCO3− in the membrane is dictated by the effective pKa values of the cationic functional groups. All existing AEMs have effective pKa values that are either too high (leading to mostly OH−) or too low (leading to mostly HCO3−). Thus, even if CO32− were produced with 100% selectivity at the catalyst, the lack of CO32−-specific membranes would cause the concentration of CO32− to shift towards HCO3− and OH− (+CO2) anyway. This suggests that a concerted materials design effort is needed in this area.
Hybrid AAEM/PEM fuel cells
As described in the previous sections, fuel cells and other APEFCs operating at high pH using AAEM materials have attracted attention due to their favourable operating parameters with major advantages including the use non-noble metals at the cathode and a wider range of fuel options at the anode.2,288 However, the lower ionic conductivity of AAEMs compared to PEMs (such as Nafion®) at lower RHs is a concern because it may lower the performance.52 PEMFCs and APEFCs require careful water management because water is consumed at the cathode in APEFCs and water is needed for ion hydration (in both). Eqn (8) and (9) show the reactions at an APEFC anode and cathode, respectively, while eqn (6) and (7) give the acidic PEMFC analogues. In both cases, the overall reaction is given in eqn (10). The analogous anode reactions for methanol fuelled systems are given by eqn (12)/(13) and (46):| CH3OH + H2O → CO2 + 6H+ + 6e− | (46) |
Water management is challenging in the PEMFCs because water is produced at the cathode and needed at the anode (for hydration of the proton and production of carbon dioxide in the case of methanol). APEFCs are also challenged by water management because water is consumed at the cathode (to make the OH− anions) and needed for hydration of the ions. Water is produced at the anode for both H2 and methanol fuelled AAEM-based fuel cells. Thus, significant resources or careful system design are required to recycle the water from one electrode to the other for both PEM-based and AAEM-based fuel cells. In both cases, more water is produced than consumed due to the net production of water (eqn (10)).
| H+ + OH− → H2O | (47) |
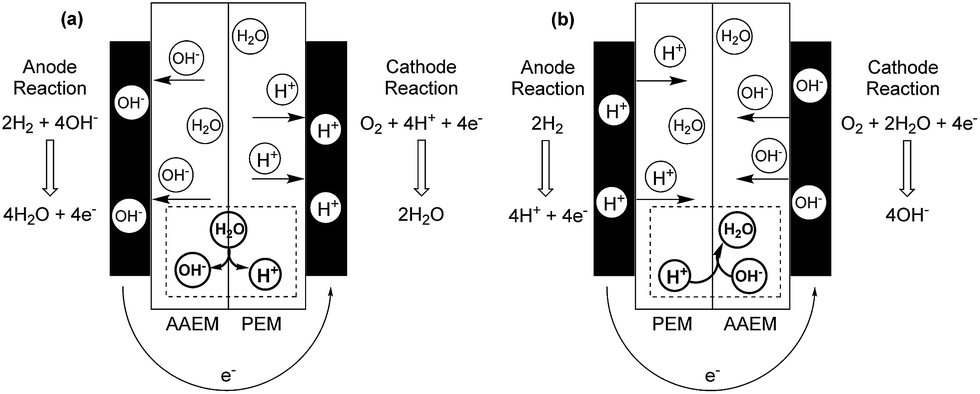 | ||
| Fig. 5 Details of hybrid membrane fuel cells comprising of: (a) high pH AAEM anode + low pH PEM cathode; and (b) low pH PEM anode + high pH AAEM cathode. | ||
This contribution allows the use of non-Pt catalysts at the cathode and an opportunity to generate the water at a junction that can be located close to where the H2O is consumed at the alkaline cathode.291 In principle, the PEM|AAEM junction can be placed anywhere within the structure. The water created at the PEM|AAEM junction is dual use and contributes to both the self-hydration of the membrane and the ORR at the cathode. Excess water can leave the system through either side of the structure. Thus, this structure uses a PEM anode (eqn (6)) and AAEM cathode (eqn (9)). The overall full cell reaction (sum of eqn (6), (9) and (47)) is the same as for PEMFCs and APEFCs (eqn (10)). In the case of a methanol fuelled system, the acid anode is given by eqn (46) and the overall full cell reaction at steady state is the same as for AAEM- or PEM-based DMFCs.
The equilibrium cell potential (Ecell) for the hybrid (bipolar) configuration needs to account for the reactions at each electrode and the junction potential (Ej) developed at the PEM|AAEM interface:
| Ecell = ENernst + Ej = EC − EA + Ej | (48) |
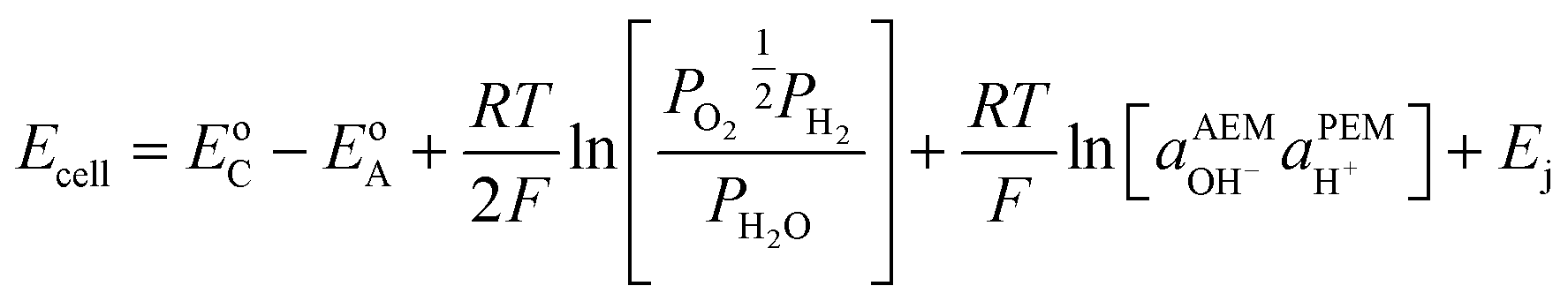 | (49) |
 | (50) |
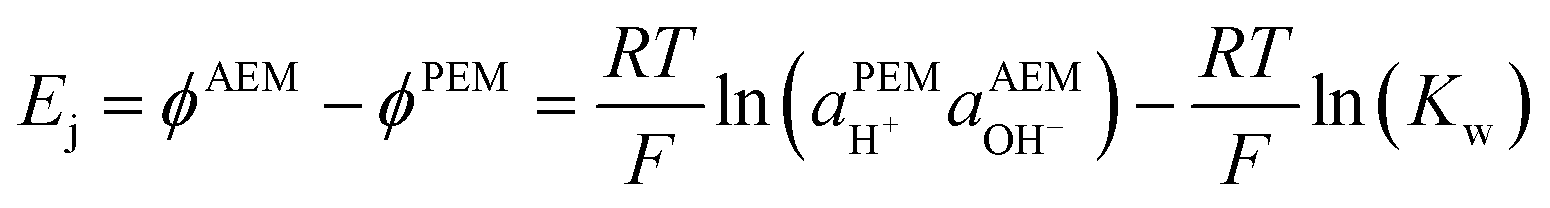 | (51) |
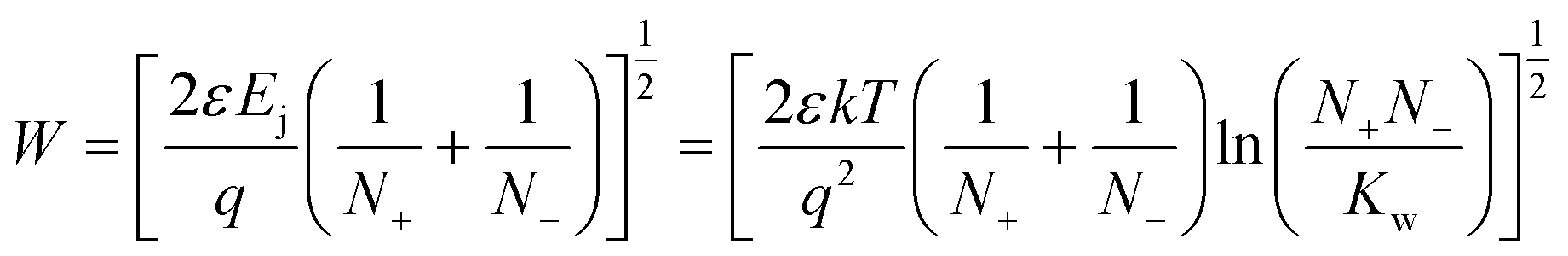 | (52) |
| N+/− = NA × IEC+/− × ρm | (53) |
 | ||
| Fig. 6 Top: Cell voltage vs. current density curves at 60 °C for a hybrid PEManode/AAEMcathode fuel cell with RH levels ranging from 0% to 100% (symmetrical for both H2 and O2 gas supplies).292 Bottom: Corresponding in situ a.c. impendance spectra at Vcell = 600 mV. | ||
In situ a.c. electrochemical impedance spectroscopy was used to help understand the change in cell performance with relative humidity. For all humidity conditions, the impedance spectra (600 mV cell voltage at 60 °C) exhibited semi-circular loops, as seen in Fig. 6(top). The high frequency x-axis intercepts (predominantly the MEA ohmic resistance derived mainly from the ionic resistance of the membrane)293 showed that the ionic resistance of the membrane was nearly constant over the gas RH range RH 0–100%. However, the radius of the semi-circular loop increased with RH. The difference between the x-intercept values of the semi-circular responses at high and low frequency is mainly governed by interfacial ORR kinetics, ionic conductivity and diffusion limitations within the depletion layer.293b Since the decrease in ionic conductivity of the PEM at high humidity is not expected, diffusion limitations within the catalyst layers is a likely reason of the increased low frequency resistances at higher RHs.
These results demonstrate that the water generated at the PEM|AAEM interface maintains adequate hydration in the hybrid membrane when the inlet gases are dry. Hydration of the gas streams results in excess water within the membrane, flooding of the electrodes, and limited O2 diffusion in the cathode catalyst layer. This is a significant result because the performances of conventional PEMFCs (and APEFCs) often have to rely on fully humidified gas feeds. Hydration or wicking of water (from one electrode to the other) can increase complexity and lead to a loss in efficiency.
Beyond fuel cells, the hybrid (bipolar) structures could contribute to more efficient water electrolysers, salt-splitting technologies, electrochemical separators (such as CO2 pumps), and solar-to-fuel applications. This is because the pH of each electrode can be taken to extreme values and optimised for the highest efficiency with the materials present. In addition, the bipolar structures may improve permselectivities compared to single ion conducting membranes because the two kinds of ions migrate in opposite directions. One example of the potential optimisation of the electrolyte based on the materials present is the solar-to-fuels systems proposed by Spurgeon et al. (Fig. 7).294 Sunlight is absorbed at each of the two photoelectrodes. p-type Si in a PEM environment was proposed for the photocathode because it is cathodically stable in acidic media.295 The photoanode may be a metal oxide semiconductor, which could be unstable in an acidic environment but stable at high pH.296 Irradiation with sunlight produces H2 gas at the PEM cathode and O2 at the AAEM anode. The direction of ion flow is opposite to that shown in Fig. 5b and water is split into H+ and OH− ions at the junction (Fig. 5a). A hygroscopic junction between the PEM and AAEM materials is desired due to water consumption there.
 | ||
| Fig. 7 A solar-to-fuel system that could incorporate bipolar membranes.294 | ||
The design and analysis of an efficient, low-loss, bipolar structure is important. Charge neutralisation occurs at the PEM|AAEM junction resulting in a drop in conductivity. The width of the junction region (eqn (52)), is important because the high resistance in this region will result in ohmic losses. The placement of the junction within the electrochemical device is important because water will either be generated or consumed there (depending on the type of device).289 The location of the junction within the membrane should consider the transport of water within the system. In the case of the fuel cell described above, this includes the consumption of water at one electrode and the efficient removal of excess water. The water balance is a function of the RH of the gas feed streams at the two electrodes. The analysis of the width of the junction, location of ions, and impact of conductivity across the junction is challenging. There are few analytical techniques with adequate spatial resolution, chemical specificity, and sensitivity to address the problem. High surface area, non-planar junctions may be of value to minimise the ohmic losses at the junction (i.e. where the real surface area is greater than the superficial surface area) and may provide additional or alternative locations for water generation/consumption (depending on changes in relative humidity of the feed streams).
The need for carbon free catalyst supports at high pHs
Carbon is the favoured material for many fuel cell components for PEMFC and APEFCs.297 However, carbon is thermodynamically unstable across virtually the entire potential range in which fuel cells operate. Under standard conditions (pH = 0, 298 K), carbon is thermodynamically stable only over the potential range −0.05–0.15 V (vs. SHE) as depicted in the relevant Pourbaix diagram (Fig. 8). The fact that carbon has become such an important material for catalyst supports, reactant transport layers, and bipolar plates is related to its relatively low cost, moderate electrical conductivity, and significant kinetic barrier to corrosion. The barrier to corrosion comes about because carbon, once oxidised, has a preferred oxidation state of +4 (i.e. carbon oxidation requires 4e−) and the ultimate product is an oxide, the formation of which is hindered by large activation energy barriers in acidic environments. Furthermore, there are no intermediate states of oxidised carbon that are soluble or produced in significant quantities along the reaction pathways leading to CO2 (e.g. methanol, formaldehyde, and formic acid are not produced in appreciable quantities):| C + 2H2O ⇌ CO2 + 4H+ + 4e−E = 0.118 V vs. SHE | (54) |
| C + 6OH− ⇌ CO32− + 3H2O + 4e−E = −0.854 V vs. SHE | (55) |
 | ||
| Fig. 8 Pourbaix diagram for carbon at 298 K showing the hatched domain of stability. Predominate gaseous (italicised) and solution phase species (non-italicised) are shown in the domains where the activity (or partial pressure) is 10−6. (a) Corresponds to the lower stability window of water (HER) and (b) corresponds to the upper stability limit of water (oxygen evolution reaction, OER). | ||
The corrosion properties of carbon are relatively well appreciated in PEMFCs with the issue being viewed as a concern for the longevity (durability) of these systems. The most extreme events that challenge carbon stability are faced during start-up and shutdown procedures. Fuel starvation events can lead to the cathode potentials rising up to potentials of 1.2–1.5 V (vs. RHE).298
However, carbon shows even less thermodynamic stability under alkaline conditions (as can be seen from Fig. 8); this is in addition to the fact that carbons are active towards the undesirable n = 2e− ORR at high pHs. This is not the end of the matter either as the kinetics for carbon oxidation actually accelerates in alkaline environments due to OH− anions being excellent nucleophiles. Indeed, carbon has been suggested (as far back as 1896 by Jacques) as anode fuel in molten NaOH carbon air batteries;299 such systems were constructed that provided up to 1.5 kW of power.300 More recently, the prospect of using carbon as an anode fuel in a molten AFC has received further attention.301
As might be expected from the Pourbaix diagram, the degradation of carbon is liable to be more extreme at high pHs due to the greater thermodynamic stability of CO32− compared to CO2. Decomposition of fuel cell components in aqueous electrolyte AFCs was moderately well studied in the 1970s and 80s. Long-term corrosion rate studies of Black Pearls 2000 and Vulcan XC 72R in aqueous KOH (12 mol dm−3) at 80 °C for >1500 h showed that the carbons are more severely attacked (cf. in aqueous H3PO4 under the same conditions): after 2300 h, the BET surface area of the Vulcan XC 72R reduced from 225 to 125 m2 g−1.302 In a more recent study, a carbon composite bipolar plate material (30% polymer filler, 5% XC-72R C black, 5% Toray carbon fibres, and 60% graphite powder) was tested under simulated APEFC conditions [aqueous NaOH (1 mol dm−3)] and compared to PEMFC conditions [aqueous H2SO4 (1 mol dm−3)]. An ∼18 fold increase of corrosion current was observed with the alkaline conditions.303
Despite the above, the current preferred material for reactant transport layers in APEFCs is carbon as the issues with carbon corrosion in the reactant transport layers are normally minor (as the electrolyte is not mobile and hence does not tend to wet the transport layer). As the transport layer is not exposed to overly harsh alkaline conditions, carbon is suitable for such application. However, other alternatives are possible and may be borrowed from the field of liquid (aqueous) AFCs: e.g. see the review by Bidault et al.2q For example Nickel coated PTFE shows an electronic conductivity of 300 S m−1;304 Ni foam may also be a suitable alternative.305
One particularly interesting area is the production of combined catalyst/reactant transport layers utilising a porous Ag membrane.306 As Ag has the highest electrical and thermal conductivity of any metal, its use in the reactant transport layers may alleviate some of the issues found in high performance fuel cells caused by local heating effects. A 50 μm thick Ag reactant transport layer with a porosity of 60% yielded a sheet resistance (0.8 mΩ □−1 [where □ indicates square, a commonly used unit for sheet resistances measured using 4-point methods]) that was 400× lower than that of standard carbon-based reactant transport material (Toray TGP-060, 294 mΩ □−1). Hence, it is not necessary to keep the gas supply channels so narrow. Indeed, when using the Ag gas transport layer mentioned above, channels could be 20 mm wide without incurring larger ohmic loses compared to the use of a carbon based gas transport layer with 1 mm wide channels. The cost of the Ag reactant transport layer is approximately 3× the cost of the Toray carbon-paper and so this is not a critical issue. A major benefit of using a Ag reactant transport medium is that it also functions as the catalyst, producing impressive electrochemical performances both in the absence and in the presence of additional catalyst.307 Yan et al. has also advocated support-less Ag nanowire ORR catalysts.308
AAEMs in alkaline electrolysers3
H2 electrolysers containing AAEMs and/or AEIs
For alkaline electrolysers, the individual electrode reaction that produces H2 (HER)313 at the cathode is given in eqn (26), while the O2 producing reaction (oxygen evolution reaction, OER)121 at the anode is given by:
| Anode (OER): 4OH− → O2 + 2H2O + 4e−E = 0.40 vs. SHE | (56) |
In the cell, a separator is used to keep the H2 gas isolated from the O2 gas (to avoid formation of a potentially explosive mixture). As with APEFCs (Fig. 9a), an alkaline electrolyte enables the use of low cost non-PGM catalysts, such as Ni, which helps to keep capital costs of the cells relatively low.
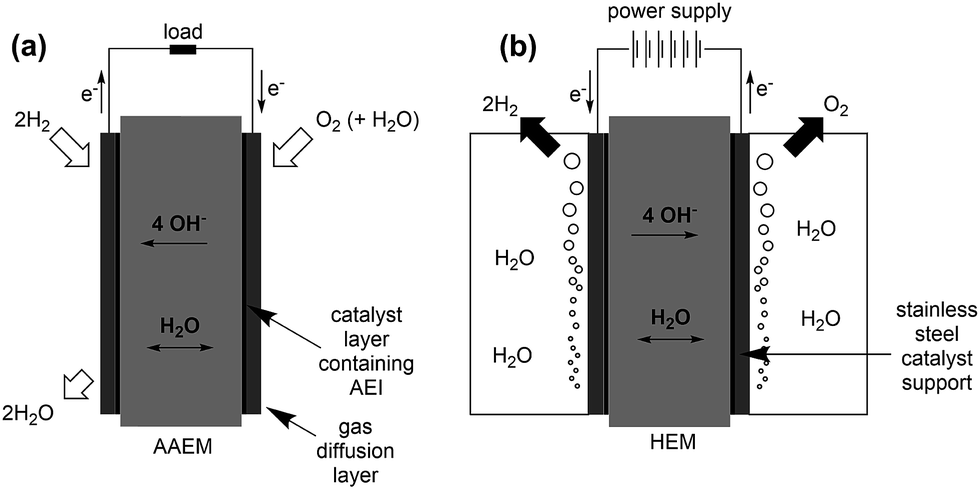 | ||
| Fig. 9 Comparison of: (a) a H2/O2 fuel cell containing an AAEM [APEFC] with (b) an alkaline electrolyser containing an AAEM/MEM [APEE]. | ||
With the use of acid electrolytes, electrolysers typically use solid polymer electrolyte (SPE, e.g. a PEM) and not an aqueous electrolyte (e.g. sulphuric acid).314 In acid electrolysis precious metal catalysts are used to achieve efficient electrolysis, which generally means that high rates of H2 production (per unit area of electrode) are required to minimise the capital costs of the cells. There are clear similarities between the cells used for water electrolysis and those used in PEMFCs because the central component, the polymer electrolyte membrane, is essentially the same for both. Consequently, technological developments in polymer electrolyte based fuel cells can often be transferred to SPE electrolysers.
Replacing the separator/diaphragms mentioned above with an IEM can offer advantages such as reduced gas crossover and area resistances (especially when using thin membranes). Applying AAEMs to electrolysers (Fig. 9b) provides the opportunity to consolidate the advantages of both types of traditional electrolysers. The costs will be reduced with the use of the lower cost electrode materials and catalysts that are used in alkaline electrolysers, while the AAEM electrolyser (APEE) would not be affected by the presence of cationic species (present in the feed-water). The latter is a major issue with PEM-based electrolysers: a major reason for gradual deterioration in their performances relates to the cations binding to the proton conducting (exchange) sites of the PEM (which reduces its conductivity).318 In operation, PEM-based electrolysers are fed with pure water at the anode which decomposes to O2 and H+ cations; the latter pass through the PEM and are subsequently converted to H2 gas. The absence of a corrosive (aqueous/liquid) electrolyte is one of the major features of SPE-type water electrolysers, which adds to the simplicity of operation (and reduced costs of components such as bipolar plates).
The central component of the electrolyser is an MEA (similar to PEMFCs and APEFCs). The IEM serves as the ion (but not electronically) conducting electrolyte and also as the separator for H2 and O2 gases. Ideal electrodes require the following attributes:
(a) Good electronic conductors;
(b) High structural integrity;
(c) Corrosion resistance with the electrolyte being used and at operating potentials that are appropriate for the cathode (reducing) and the anode (oxidising);
(d) Contain high performance electrocatalysts for both the HER and OER;
(e) High (reaction assessable) surface areas to facilitate high current densities and/or H2 production rates;
(f) Stable performance over extended periods of operation (both on and off load).
SPE electrolyser cells require an intimate contact between the phases of the MEA to achieve: optimal ionic and electronic conductivities, high active surface areas of the catalysts, and rapid gas release. High electrocatalyst specific areas are required to decrease overpotentials and to avoid the appearance of hot spots (that drastically shorten the life span of the MEA).319 For SPE water electrolysers, both the anode and cathode electrocatalysts are deposited as thin (several μm thick) coherent layers that are bonded to either side of the IEM.
The main challenge regarding the widespread use of H2 in small to medium size applications is cost reduction (needed to increase the commercial appeal of such H2 generators). Low-price domestic electrolysers can be realised through high production/sales volumes, but this will only occur after economical, efficient, and durable prototypes have been demonstrated. Adopting AAEM-based technology can open up opportunities for low cost electrolysers systems with: low membrane, catalyst (Pt-free), and bipolar plate manufacturing costs; higher energy efficiencies (towards “zero gap”);320 durable, long life operation (unlike with APEFCs [fed with gas supplies of various RHs], the AAEMs are in an environment where they remain fully hydrated in APEEs); flexibility to respond to dynamic load operation; and compact system integration and control. The use of AAEMs may also facilitate the simultaneous production of H2 and useful chemicals (e.g. potassium acetate) when electrolysing aqueous alcohol solutions using only ⅓ of the energy required by traditional H2/O2 electrolysers.321
A solid state water electrolyser has recently been reported that achieved 399 mA cm−2 at a cell voltage of 1.8 V and nearly 1000 mA cm−2 at 2.0 V when using Tokuyama A-201 AAEM, Tokuyama AS-4 AEI, and aqueous KOH (1 mol dm−3) solutions at 50 °C.323 This system, however, used high loadings of precious metal catalysts (RuO2 at the anode and Pt at the cathode). An alkaline electrolyser that used nickel iron oxide coated anodes, Pt cathodes, and a developmental AAEM (ITM Power, 160 μm thick) with aqueous KOH (4 mol dm−3) solutions at 60 °C has been reported to achieve a cell voltage of 2.12 V at 1000 mA cm−2.324 The use of KOH electrolyte is seen as important for achieving performances that approach those of Nafion® PEM-base systems where cell voltages between 1.6–1. 7 V are possible at 1000 mA cm−2. The use of NiMo and RuO2 coatings on nickel or stainless steel micro-meshes have been examined as electrocatalysts for HER in conditions similar to those in “zero gap alkaline water electrolysers”.325 The NiMo and RuO2 coatings gave performances that were comparable to Pt and also stable over 10 d of electrolysis; the performance of an electrolyser containing an AAEM (ITM Power, 160 μm thick) and a NiFe (OH)2 coated anode (OER) with aqueous KOH (4 mol dm−3) was 2.1 V at a current density of 1 A cm−2. Jang et al. report ca. 150 mA cm−2 at 1.9 V with aqueous KOH (1 mol dm−3) feeds with MEAs containing a Tokuyama A201 AAEM and electrodes consisting of Ni that is electrodeposited on carbon papers with very low Ni loadings (8.5 μgNi cm−2).322
Recent work at Newcastle University has used a methylated melamine quaternised grafted poly(vinylbenzyl chloride) AAEM in an APEE containing a Cu0.7Co2.3O4 OER catalyst (Fig. 10);326 1 A cm−2 was achieved at a voltage of 1.8 V in aqueous KOH (1 mol dm−3) at 25 °C. In order to develop APEEs, a polymethacrylate-based OH− conducting AEI ionomer binder (conductivity = 59 mS cm−1 at 50 °C) was synthesised.327,328
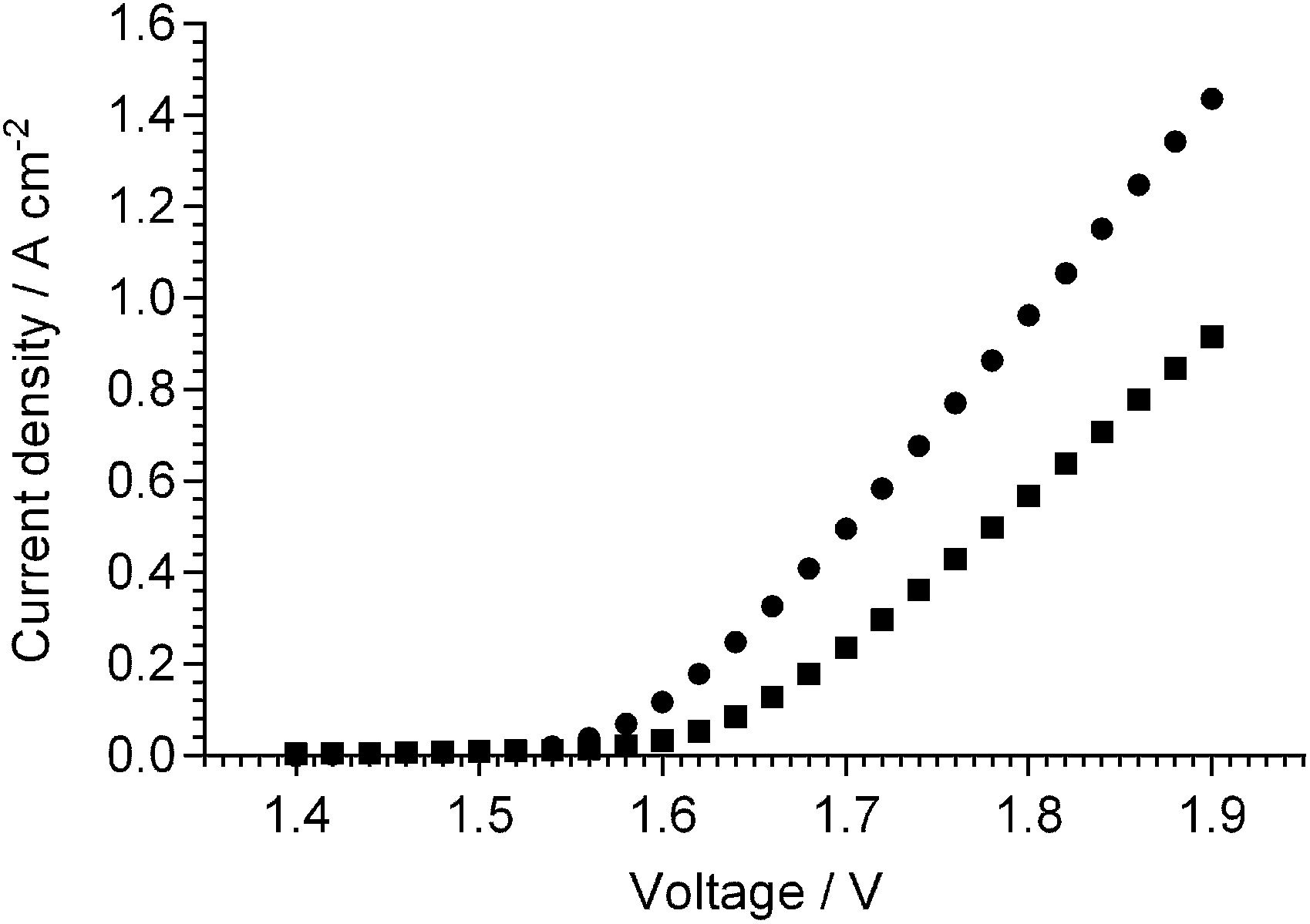 | ||
| Fig. 10 Electrolyser performance with a OH− ion conducting AAEM and a Cu0.7Co2.3O4 cobalt oxide OER catalyst (circles) [cf. squares = Co3O4 catalyst data]. T = 25 °C and aqueous KOH (1 mol dm−3) electrolyte at both the anode and cathode.326 | ||
A very recent study has reported the use of a dilute aqueous K2CO3 solution in conjunction with an AAEM (A-201, from Tokuyama Corporation).329 The MEA was based around low-cost transition-metal catalysts. The HER and OER catalysts were commercially available materials (manufactured by Acta SpA) and based on Ni/(CeO2–La2O3)/C and CuCoOx mixed oxides respectively. This system exhibited a similar performance to a PGM-catalyst benchmark. The best performance reported at 43 °C was a cell voltage of <1.95 at 500 mA cm−2 (using 7.4 mg cm−2 of cathode catalyst). This approach uses a CO2 tolerant and less aggressive alkaline electrolyte (i.e. no OH− anions). This again highlights that CO32− electrolyte systems warrant further detailed studies.
It is of increasing interest to employ AAEMs into RFCs where non-precious-metal catalysts can be used in the electrodes. A recent study used a pore-filling AAEM made using porous PTFE filled with a QA polymethacrylate AEI.335 The composite membrane exhibited a lower swelling ratio (thickness and area variation), stronger tensile strength, but lower ionic conductivity compared to a polymethacrylate-only AAEM. However, the composite membrane was ultra-thin and therefore exhibited a lower in situ MEA ionic (area) resistance and improved current densities. In fuel cell mode, the peak power densities were 0.11 and 0.16 W cm−2 at 20 and 45 °C respectively. In water electrolyser mode, cell voltages at a current density of 100 mA cm−2 were 1.61 V and 1.52 V at 22 and 50 °C respectively, while the degradation rate was only 40 μV h−1 at a current density of 100 mA cm−2 at 22 °C.
(a) Efficient OER and HER catalysts with low activation overpotentials and containing new catalyst structures or metal alloys (resulting in lower noble metal loadings);
(b) AAEM with improved conductivities and alkali stabilities, low gas crossover, and high mechanical stabilities;
(c) High performance AEIs for use in the catalyst layers;
These basic components are needed to fabricate the MEAs. A key challenge is to bond high surface area catalysts, using an AEI, to either the AAEM or a suitable metal supporting electrode substrate. The MEAs must enable efficient gas release to prevent gas bubble adhesion blocking the catalyst surface (and avoid undesirable increases in polarisations in the cell). Thus the surface of the MEA and AAEM should be hydrophilic. The development of AAEM-electrolyser technology can benefit from existing research and know-how in the field of electrocatalysts for electrolysers containing liquid electrolytes.
000 per tonne),342 the anodes are not, as a rule, manufactured from solid Ni but are based on an electrolytically deposited non-porous nickel coatings. Increasing the electrode surface area (i.e. roughness factor – defined as the ratio of its real surface area to its apparent, or geometric, area) of Ni anodes have been achieved, for example, by sintering fine Ni powders prepared from Ni(CO)4 decomposition.343 Coated electrodes (steel and Ni) can be made by applying particulate metal coatings to the metal substrates. Applying paint-like suspensions of metal particles can yield uniform coatings of controlled thickness. Use of mild steel as an anode substrate requires the steel surface to be protected from corrosion (during OER) using fabrication methods such as alloying.344 Raney Ni has been used to produce high surface area anode coatings. Raney Ni is made by alloying Ni with metals such as Al or Zn, which are subsequently leached out in alkaline electrolyte yielding the desired high surface area structures with high electrochemical activities.345
Several material types have been studied for alkaline electrolysers with decreased overpotentials, many of which are metal oxide electrocatalysts for the OER in alkali. Catalytically active materials such as NiCo2O4 have been applied.346 Catalysts such as NiCo2O4, Li-doped Co3O4,347 and perovskite La1−xSrxCoO3 types (and substituted variants) have been tested extensively.348 Mixed Cu–Fe–Mo oxide OER catalysts have also been studied.349 Further research of this type may lead to important catalyst modifications or the development of entirely new catalyst systems. Excellent OER activities have been reported with oxides having the pyrochlore structure (described by the general formula A2[B2−xAx]O7−y, where A = Pb or Bi, B = Ru or Ir, O < x < 1, and O < y < 0.5). A typical Pb2[Ru2−xPbx]O6.5 catalyst evolved oxygen at an overpotential of only 120 mV at a current density of 100 mA cm−2 (and exhibited lower overpotentials than Pt black, RuO2, or NiCo2O4).350 These Pb/Ru electrocatalysts are a reasonable precious-metal-based OER catalyst option for alkaline electrolysis as ruthenium metal is relatively inexpensive (ca. 10% of that of Pt).
Non-H2 electrolysers containing AAEMs/AEIs and involving CO2 reduction
As discussed above, low temperature CO32− electrochemical systems are in their technological infancy but there are increasing reports of electrochemical cells that involve CO32− reactions such as a CEM system for electrolysis of Na2CO3 and NaHCO3 for the production of NaOH.351 The immature state of development of room temperature AEM-based CO32− systems makes them a potentially high impact area that is ripe for rapid growth. Perhaps the best evidence for this is preliminary work with low temperature carbonate electrochemical devices related to methane activation to various high value products including CO or methanol (Fig. 11):283a,352| 2CH4 + 2CO32− → 2CH3OH + 2CO2 + 4e− | (57) |
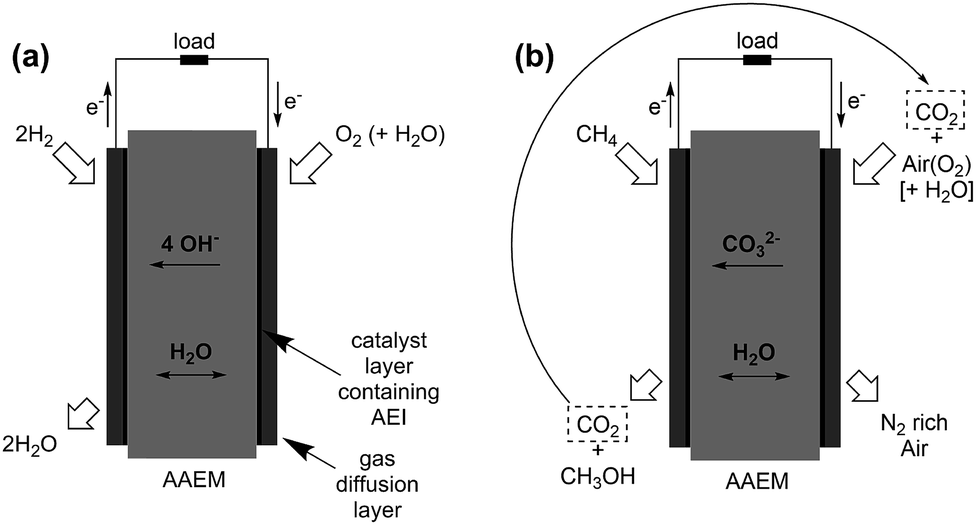 | ||
| Fig. 11 Schematic comparison of (a) an APEFC and (b) a CO32− cycle reactor for the partial oxidation of methane into methanol (refer to eqn (44) and (57) for desired cell reactions). | ||
This synthesis process involves a CO2 cycle when in conjunction with the CO32− generating reaction in eqn (44) (rather than conversion of all of the CO2 into products) and is theoretically galvanic; in reality, with the overpotentials involved, it will proceed via a low overpotential [driven] electrolytic process.
CO2 emissions make the greatest contribution to greenhouse gases. Processes which convert CO2 to useful products are thus desirable from a perspective of sustainability, and environmental protection. This type of system falls into the category of Carbon Dioxide Utilisation (CDU)353 as opposed to Carbon Capture and Storage (although OH− containing polymer electrolytes have also been proposed for reversible CO2 capture).354 Overall in the production of chemicals from CO2, energy is required to break the CO bond to produce various chemicals (the conversion of CO2 into CO32− is a rare example where the conversion of CO2 into something else is thermodynamically “downhill”). The electrochemical reduction of CO2 is the conversion of CO2 to a more reduced chemical species using electrical energy. Electrochemical reduction of CO2 is considered a possible means to produce chemicals or fuels from CO2, making it a feedstock for the chemical industry.355 In prior studies, direct electrochemical reduction of CO2 in low temperature electrolysis has led to production of formic acid, methanol, hydrocarbons and oxalic acid. Such transformations make the electroreduction of CO2 of interest in a carbon energy cycle.356 However, such low temperature electrochemical reduction requires a relatively large amount of electrical energy.
No direct electrochemical CO2 reduction process has been successfully commercialized, although academic and commercial efforts continue using a variety of homogenous and heterogeneous catalysts. Generally speaking, the processes developed to date either have poor thermodynamic efficiencies high overpotentials, low current efficiencies, low selectivity, slow kinetics, and/or poor stability. An exception to this is the production of formate on carbon electrodes at high pH. Det Norske Veritas and the Mantra Venture Group are both developing systems based on Sn cathodes that allow for the conversion of CO2 to formate:357
| CO2 + H2O + 2e− → HCOO− + OH− −1.05 V vs. SHE | (58) |
The process is believed to use two phase electrochemical reactors with Sn-based cathodes. The equilibrium potentials for CO2 reduction are not too different to that for H2 evolution. This however hides the fact that its reduction does not occur easily and at much more negative potentials than the equilibrium values.
Previous research on electrochemical CO2 reduction in aqueous solutions has evaluated a wide range of metals.358 “1st group” metals (such as In, Sn, Hg, and Pb) selectively form formic acid/formate. “2nd group” metals (such as Zn, Au, and Ag) form CO as a major product, while Ag, Au, In, Zn, and Sn can produce CO and carbonate. Ni, Pd and Pt form CO selectively. For example, a set-up involving an AEM was used to investigate the electrochemical reduction of CO2 to C2+ products on Cu surfaces.359,360 All of this suggests that the application of AAEMs and AEIs into low temperature electrolyser systems involving the electrochemical reduction of CO2 (rather than the more well-known high temperature solid oxide and solid proton conductor CO2 electrolyser systems)361 may have a major impact.
AAEMs in alkaline batteries
Metal–air batteries (e.g. Al-, Zn-, Mg-, and Fe–air),362 and other battery types such as Ni-MH (MH = metal hydride) and Ni–Zn cells, often contain alkaline electrolytes and can have higher energy densities and capacities compared to other batteries such as Li-ion batteries. Catalysts such as Ag-MnOx/C can be used for the O2 reactions when air electrodes are employed along with alkaline electrolytes.363 In the last decade, there have been a small number of reports of the use of AAEMs in such batteries. One of the motivations for using an alkaline solid polymer electrolyte is to prevent undesirable changes in the electrodes (such as dendritic growth at the anode [growing towards the positive electrode] in Ni–Zn cells).364 AEMs have even been reported in Li–O2 batteries (to suppress direct LiOH deposition in the air electrodes).365 As with APEFCs/APEEs, the membranes need to have high ionic conduction and high alkali stabilities.Early examples of the use of solid polymers electrolyte in alkaline primary and secondary batteries were reported by Fauvarque et al. who used KOH-doped poly(ethylene oxide) polymers and copolymers.366 Other examples include the work by Yang et al. who used a KOH-doped PVA-based copolymers in a primary solid-state Zn–air battery and a Ni–MH battery,367 while Arof et al. similarly used a KOH-doped PVA electrolyte in a Ni–Zn cell.364 A recent study investigated the use of KOH-doped poly(acrylic acid) polymer electrolyte for all-solid-state Al–air batteries.368 However, these are still metal-cation-containing (KOH-based) systems.
Yasuda et al. investigated a reversible air electrode concept, containing an AAEM with covalently bound cationic groups, for use in secondary air batteries;369 the AAEM was a hydrocarbon-based QA-type (IEC = 1.4 meq. g−1, thickness = 27 μm). The rational for the use of such an AAEM-based air electrode was to reduce ORR/OER performance losses by: blocking the cations from penetrating into the air electrode (reduces carbonate precipitation), reducing the penetration of the alkaline solution into the air electrode, and preventing neutralisation of the alkaline solution via the CO2 in the air electrode supply. A Pt–Ir catalyst provided reduced overpotentials in the air–electrode and the concept did exhibit a reduced (negative) influence from the presence of CO2.
AEMs in redox flow batteries (RFB)2a,4
IEMs play an important role as separators in some types of RFBs4c,d,370,371 where a high degree of reactant isolation is required between the anolyte and catholyte compartments. Microporous separators can be used in RFBs to provide a barrier between the two liquid streams (since the anolyte and catholyte usually contain high concentrations of acids, bases, or other electrolytes to facilitate ionic conductivity). However, while being generally inexpensive and having low ionic resistances when immersed in concentrated electrolyte solutions, microporous separators are prone to high crossover of electroactive species that result in cell performance loses and severe battery capacity fade.372Thus, there has been a concerted effort to apply high-conductivity, low crossover IEMs in a number of different RFB systems. IEMs have been investigated for use in iron/chromium, hydrogen/bromine, vanadium/bromine, non-aqueous,373 and other types of RFB chemistries.370 Nafion® and other perfluorinated IEM variants have been the most heavily-studied IEM in RFBs and most new membrane research (beyond Nafion®) has centred on all-vanadium redox flow batteries (VRFB). In VRFBs, vanadium cations (in various oxidation states) are used to reversibly store and release electrical charge according to eqn (59) and (60) (Fig. 12):
| V2+ ⇌ V3+ + e−E = −0.26 V vs. RHE | (59) |
| VO2+ + H2O ⇌ VO2+ + 2H+ + e−E = 1.00 V vs. RHE | (60) |
 | ||
| Fig. 12 Schematic of a VRFB. Typical concentrations for the vanadium electroactive species (usually sulfate salts) are 1.7 mol dm−3 in supporting aqueous H2SO4 (3.3 mol dm−3) electrolyte. Dashed arrows indicate the valance change of the vanadium species during cell discharge (the positive and negative electrolytes are named after the discharge reactions), while solid lines represent cell charge processes. Undesirable vanadium cation crossover can occur, while SO42− anions can additionally transport through the IEM when an AEM is used instead of a PEM. | ||
Flow batteries are inherently flexible energy storage systems because the system power can be scaled by the size of the electrochemical stack, while the energy storage capacity of the battery system can be varied with the size of the electrolyte holding tanks. Hence such flow batteries have many similarities to fuel cells. The principal degradation mechanisms for VRFBs are from the mixing of the vanadium redox couples by vanadium cation crossover between the flow compartments or the oxidative degradation of the cell components and membrane. The fact that VRFBs have vanadium based couples at each electrode [V(II)/V(III) couple at the negative electrode and V(IV)/V(V) redox couple at the positive electrode] give the ability to regenerate the electrolytes should crossover occur: the electrolyte volumes in each compartment can simply be mixed and electrochemically re-activated to regain the capacity of the battery.374 A primary goal in membrane research for VRFBs is to limit the vanadium cation diffusion through the membrane, while maintaining high oxidative stability in the presence of VO2+ (where vanadium is in the +5 oxidation state) and related species.375
The VRFB system is being heavily considered for grid-scale energy storage due to the fast kinetics of the redox reactions and simple mitigation of electrolyte contamination (due to the all-vanadium chemistry). However, there are hurdles to overcome such as identifying a low-cost source of vanadium (perhaps as a by-product from metal refining) and sourcing an inexpensive but high performing membrane for MW-scale installations (that will require 1000s or even 10000s of m2 of membrane material).
The problem with Nafion® and aromatic CEMs in RFBs
Nafion® and other PFSA IEMs have been deemed currently too expensive for large-scale grid battery systems where capital cost is a primary consideration for large-scale stationary electrical energy storage applications.2a,4d To potentially lower the cost of these systems compared to PFSA benchmarks, aromatic CEMs have been explored to good effect in a range of studies.4cFig. 13 shows a VRFB performance comparison of a sulfonated aromatic CEM versus a Nafion® NRE-212 membrane.376 In this work, the aromatic backbone was selectively fluorinated to prevent oxidative degradation of the aromatic CEM structure that has been reported for aromatic CEMs exposed to high oxidation state vanadium.375,377 Because of the distinctly different ionic domain morphology in the sulfonated aromatic CEM,378 there is much less vanadium ion crossover in aromatic IEMs compared to the Nafion® benchmark. The lower vanadium crossover mitigates the capacity loss with cycling therefore helping to maintain reasonable ionic conductivity. Similar to the membrane proton conductivity/methanol permeability selectivity parameter used in DMFCs,379 the proton conductivity/vanadium permeability electrochemical selectivity of the membrane is an important figure of merit for these systems.380 Despite their excellent in situ RFB performance compared to PFSA membranes, the drawback of aromatic CEMs is that their oxidative lifetime stability below that of Nafion® or that needed for long-term grid storage applications. | ||
| Fig. 13 Low capacity fade observed in a VRFB using a low-crossover sulfonated and selectively fluorinated aromatic CEM (filled symbols) compared to Nafion® NRE-212 (open symbols). | ||
Optimisation of AEMs for low crossover and high conductivity
Because the electroactive vanadium species in VRFBs are positively charged, AEMs with fixed cationic groups are attractive alternatives to CEMs; they have been investigated as ultra-low vanadium crossover IEMs.381 Additionally, if the vanadium cations cannot easily penetrate into the membrane, the degradation of aromatic AEMs (on exposure to the vanadium species) may be mitigated, resulting in a high cell cycle life.382 Typically, the electroactive vanadium species are dissolved in high concentrations of aqueous H2SO4 or HCl as the supporting electrolyte. The charge/discharge reactions of the VRFB in eqn (59) and (60) can be balanced by H+ diffusion between the electrode compartments, or correspondingly, the charge can be balanced by the shuttling of anions across the AEM.AEMs can display some proton-mediated ionic conductivity because their transport numbers are not unity for anions. However, in most AEM-based VRFBs, the majority of the current will be carried by anions, such as sulphate, traversing the cell.383 Because the anions present will have lower intrinsic mobilities than protons, high conductivity AEMs and the optimisation of the conductivity/crossover selectivity ratio is critical in these systems. A series of benzyltrimethylammonium-containing AEMs based on chloromethylated poly(sulfone) were synthesised with different IECs.384 Varying the IEC yielded materials with different conductivity/permeability ratios (Table 4). It is apparent that an AEM with too low conductivity (e.g. QA-Radel 1.7 from Table 4) will induce high ohmic losses and decrease the power density of the cell (Fig. 14). On the other hand, too high a crossover can also negatively impact the power output of the cell (as is observed with NRE-212 or AEMs with too high an IEC). The QA-Radel 2.0 AEM had intermediate crossover and conductivity compared to the other samples and produced the most power across the set of membranes examined. Similar observations can be made for thickness optimization of IEMs in VRFBs where the resistance and crossover must be balanced for a given set of VRFB conditions.385 Because cell designs and desired operating points vary across different RFB technologies, there is no one ideal membrane for all applications. The operating envelope of the cell must be considered in order to arrive at an optimized membrane configuration.
| IECa/meq. g−1 | Gravimetric water uptakeb (%) | Ionic conductivityc/mS cm−1 | VO2+ permeability/m2 s−1 | |
|---|---|---|---|---|
| a Measured using 1H NMR. b Liquid water, room temperature. c Equilibrated in aqueous VOSO4 (1.4 mol dm−3) + H2SO4 (2.0 mol dm−3) solution. nm = not measureable. | ||||
| Nafion® NRE-212 | 0.9 | 28 | 44 | 3.2 × 10−12 |
| QA-Radel 2.0 | 2.0 | 29 | 41 | 3.7 × 10−14 |
| QA-Radel 1.7 | 1.7 | 16 | 24 | nm |
 | ||
| Fig. 14 Performance optimisation (trade-off between conductivity and vanadium cation crossover) in a VRFB system. IEMs: circles = QA-Radel polysulfone AEM (IEC = 1.7 meq. g−1), squares = QA-Radel polysulfone AEM (IEC = 2.0 meq. g−1), and triangles = Nafion® NRE-212. | ||
The study described above demonstrates that AEMs have a large role to play in VRFB technology. In fact, there are a number of other VRFB studies showing the utility of AEMs in these types of energy storage.9f,386 In membrane development work for RFBs, to date studies have focused on basic descriptions of AEM performance in RFBs in order to codify the structure–property relationships for these materials in the unique environment of a redox flow cell. More advanced studies into the optimisation of the polymer-tethered cationic groups and membranes with engineered physical structures (to decrease thickness and increase mechanical strength) are ongoing to continue to boost the performance of the membrane in these systems.32a,373b,387
In most AEM RFB studies, high current densities above 200 mA cm−2 are still to be demonstrated.388 High current densities in cells with CEMs are becoming more common with close attention to the membrane thickness and cell design.389 In many regards the challenges of direct methanol and other liquid-fed fuel cell membranes mirror those of VRFBs. Strategies for high conductivity, low crossover membranes are needed. However, while these issues can be managed, the largest challenge for new generation VRFB membranes, lifetime, will be harder to overcome. Stationary energy storage systems will be cycled for tens of thousands of cycles over thousands of hours of operation. Because most experimental VRFB membranes are based on aromatic structures, long lifetimes have not been proven to date. For example, post-mortem spectroscopic analysis of a cardo-poly(ether ketone) AEM used in a VRFB (100 h of testing) showed a 15% decrease in the QA content.390 The technical and practical challenges for developing stable membranes with proven stability over long periods of time remain and necessitate further research. As new experimental AEMs become more available for evaluation in electrochemical applications (with materials developments towards thinner membranes with higher conductivities), further systematic studies on various types of RFBs will continue to push the application of AEMs in energy storage devices forward. This will require the clarification and management of degradation issues.
AEMs in reverse electrodialysis (RED) cells
Power generation using RED5
Salinity gradient energy (SGE) uses the Gibbs free energy of mixing of two salt solutions with different salinity to generate energy (i.e. energy can be extracted where river water flows into the sea). SGE can also be extracted from industrial processes where more concentrated salt solutions are generated. SGE is a non-polluting (no emissions of CO2, SO2 or NOx), sustainable technology that is available worldwide. The estimated global energy potential from estuaries only is estimated to be 2.6 TW,391 which is approximately 20% of the worldwide energy demand.392Two technologies are available to harvest the energy from the mixing of two solutions with different salinity:5b,392
(a) Pressure Retarded Osmosis (PRO)393 uses a semi permeable membrane allowing the transport of water only, while the solute (salt) is retained;
(b) Reverse electrodialysis (RED – Fig. 15)394 uses IEMs (both CEMs and AEMs) that selectively transport cations and anions only. In this electrochemical technology review, the focus is on RED cell technology.
 | ||
| Fig. 15 Schematic of a reverse electrodialysis (RED) cell. SW = sea water and FW = fresh water. | ||
In RED cells (Fig. 15), a number of CEMs and AEMs are stacked together in an alternating pattern between an anode and a cathode with salt water and fresh water flowing between the membranes. Due to the chemical potential difference between the two solutions, anions are transported through the AEM and cations diffuse through the CEM (from the seawater to the river [fresh] water channels). In the electrode compartment, the ionic charge transport is converted into an electrical charge transport (electrons) using a reversible redox reaction (rinse solution), often based on the redox couple Fe2+/Fe3+; the use of capacitive electrodes has also been explored.395
Pattle394 was the first to demonstrate the principle of RED and Weinstein and Leitz (in the 1970s)396 investigated the effect of the composition of the salt solutions on the power output. They stated that large-scale application of RED could become feasible, but only after improvements in IEM manufacturing and optimization of the operating conditions. In the early 1980s, Lacey397 concluded that membranes for RED should be low cost and have a low electrical [ionic] resistance and a high selectivity combined with a long service lifetime, acceptable strength, and dimensional stability. Audinos398 compared the RED performance of two different types of electrodialysis membranes (i.e. homogeneous and heterogeneous membranes) for different salt solutions (NaCl vs. ZnSO4) and obtained a maximum power output of 400 mW m−2. Since the early 2000s, RED has regained attention as a technology option for sustainable energy production.399−405 Veerman et al.406 determined the power outputs and thermodynamic efficiencies for a series of six commercially available IEM pairs and used a response parameter (the product of these two parameters) to rank the different membranes; however, this could not related directly to the properties of the individual membranes.
Most research focuses on generating power from mixing aqueous NaCl solutions but the effect of using thermolytic solutions (e.g. ammonium bicarbonate) instead of NaCl has also been investigated.403 Additionally, the combination of RED with other technologies (e.g. RED with seawater desalination and solar ponds,404 RED with reverse osmosis (RO),405 closed-loop ammonium carbonate RED cells for energy efficient H2 recovery when combined with an OER anode,407 or RED combined with microbial fuel cell technology [see later]) have also been reported. Most reports focus on the technological aspects of RED rather than the membranes. Experiments are usually performed using commercially available IEMs that have not been specifically designed for RED (usually they have a background in traditional electrodialysis). Such membranes are usually robust, mechanically strong and consequently rather thick. Moreover, membrane chemistry, structure, and properties are tailored for different applications. These can, however, play an essential role in determining the power output obtainable in a RED cell (not only in terms of ionic flux but also in relation to membrane fouling, mixing characteristics, and water composition). As such, they are key factors determining the net power output obtainable in a RED cell (discussed below).
Membrane chemistries for RED
Długołęcki etal.408 were first to systematically investigate different commercially available membranes for RED applications. They experimentally determined the ionic resistance, permselectivity, and charge density of a wide range of CEMs and AEMs, and used these experimental values as input for model calculations to predict power densities obtainable in RED. Their main conclusion was that in order to obtain high power densities, the IEM resistance is critical and should be as low as possible (preferably area resistances r < 1–2 Ω cm2), while the permselectivity is of minor importance (this parameter is already high at >95%). This was later confirmed experimentally by a study of Güler et al.409 who measured the power densities obtainable for a large series of different AEMs and CEMs (Fig. 16). | ||
| Fig. 16 Experimental power densities obtainable in RED cells for different membrane couples as a function of the flow rate (number of cells = 5; spacer thickness = 200 μm).409 Highlighted examples: (a) University of Twente in-house developed CEM = sulfonated PEEK and AEM = quaternary ammonium poly(epichlorohydrin); (b) commercially available Tokuyama CEM = CMX and AEM = AMX; (c) commercially available Ralex CEM = CMH and AEM = AMH. | ||
RED cells provide an important contrast to the use of AEMs and CEMs in chemical fuel cells (and other electrochemical systems containing high and low pH environments) where the AEMs are typically in alkaline anion form (AAEM) and the CEMs are in the acidic PEM form: the IEMs in RED cells are commonly in the Na+ and Cl− forms. H+ and OH− ions have an additional conduction mechanism available (the Grotthuss mechanism) so the conductivities of aqueous solutions containing H+ and OH− ions are significantly higher than those containing other ions such as Na+ and Cl− (recall Table 1 gives the ion mobilities, μ, at 25 °C in aqueous solutions). Hence, the IEMs will yield higher area resistances in RED cells compared to when the same IEMs are used in chemical fuel cells. This contrast also exists for microbial electrochemical cells vs. abiotic, chemical, fuel cells (see later). The situation is even worse for RED cells utilising thermolytic solutions as HCO3− ions are less mobile than Cl− ions (this can be mitigated by designing polymers that swell more in ammonium bicarbonate compared to NaCl solutions).403
As well as using commercially available membranes, Güler et al. also, for the first time, tested tailored made membranes with chemistries targeted for RED.410 The AEMs and CEMs synthesised for use in RED cells were systematically investigated to study the effect of charge density, resistance, and permselectivity in relation to the power output.409,410 Although the authors observed a reasonable statistical correlation between the thickness of all (tailor made and commercial) membranes and the area resistance, no significant correlations between both resistance and permselectivity to power output could be extracted. The results, however, clearly showed that IEM resistance is more important than permselectivity.
The CEMs synthesized by Güler et al. were based on sulfonated poly(ether ether ketone) (SPEEK), which is a common cation exchange polymer frequently used in electro-membrane processes. With a degree of sulfonation of 65%, the SPEEK membranes had an area resistance of 1.22 Ω cm2 and a permselectivity of 89%.409 For AEMs, a synthesis using a halogenated polyether [polyepichlorohydrin (PECH)] and DABCO was employed to simultaneously introduce anion exchange functionality and crosslinks into the polymer membrane (during amination);410 poly(acrylonitrile) was also used (as an inert polymer matrix) to further improve the strength and stability of the materials. Area resistances of this series of AEM ranged from 0.82 to 2.05 Ω cm2 (with permselectivities of 87–90%). As shown in Fig. 16, this first attempt to use tailor made membranes resulted in the highest power output so far, for the different membranes studied, with a value of 1.27 W m−2.409 Previous research showed that the thickness of the membranes is a critical parameter as well (as it directly influences the area resistance). In a non-optimized RED stack (inflow limitations dominate the power output), a decrease in membrane thickness from 130 to 33 μm resulted in an increase in power output of about 20%; with an optimized stack design, a more pronounced effect is predicted.410
Geise et al.411 investigated the ionic resistance and permselectivity of a series of synthesized quaternary ammonium PPO- and poly(phenylsulfone)-based AEMs. They aimed to develop structure–property relationships between transport properties and water content and fixed charge concentration that can be used to assess the membranes with respect to their applicability in a wide range of electro-membrane processes. It was reported that the water content of the membranes turned out to be essential and that the polymers with higher water content tended to have lower ionic resistances and lower permselectivities. This relationship was not, however, straightforward as it was highly dependent on the membrane chemistry.
Kwon et al.412 used a nanoporous polycarbonate track-etch membrane in a parallel structure with nanofluidic channels and a membrane diameter of 10 mm. These membranes are characterized by their low thickness, straight pores, high flexibility, and mechanical stability. The pore sizes investigated were 15, 50, and 100 nm. The authors report that the mechanism for selective ion transport is based on the formation of a charged electrical double layer (EDL) on the inner surface of the negatively charged pores. When the EDLs of both pore surfaces overlap, counter-ions (anions in the case of a positively charged surface) are preferentially transported while co-ions (cations) are mostly retained due to electrostatic repulsion. The authors showed this principle for CEMs, but indicate it should also work with AEMs. Although the exact type of membrane, material and concept is not very well addressed in the paper, the mechanism seems to work best for the smallest pore size (15 nm) and the power output significantly decreased on application of membranes with larger pore sizes. Although not measured in a real RED stack, the concept was evaluated in a simple two compartment cell (where only a CEM was used with salt solutions with different salinity on either side); the authors reported a maximum power of ca. 5 μW (5.8 μW cm−2).
Microstructured (profiled) membranes
Usually, the IEMs in a RED stack are separated by non-ionically-conductive “spacers”, which block part of the membrane area available for ion transport (the so-called spacer shadow effect).399,413 To overcome this approach, Długołęcki et al.413 proposed the use of profiled IEMs (that contain intrinsic “spacer” functionality) and recently Vermaas et al.414 (Fig. 17) and Güler et al.415 demonstrated this concept in practice. These microstructured IEMs (with integrated spacer functionality) were developed either by the hot pressing of commercially available membranes or by solution casting of the previously described PECH-based AEMs on a structured mould. Although the boundary layer resistances were higher and the mixing was poorer inside a stack containing profiled IEMs with non-optimised microstructures (compared to a stack using conventional spacers), higher stack power densities were nonetheless observed.414 Despite the small improvements observed, profiled IEMs have a strong future development potential as they lead to lower pressure drops (loss of power) in the stack and more optimised structure designs (in terms of mixing and improved boundary layer resistances) can be envisaged. A very recent study that has looked into optimising the microstructure design is the start of efforts to address this.416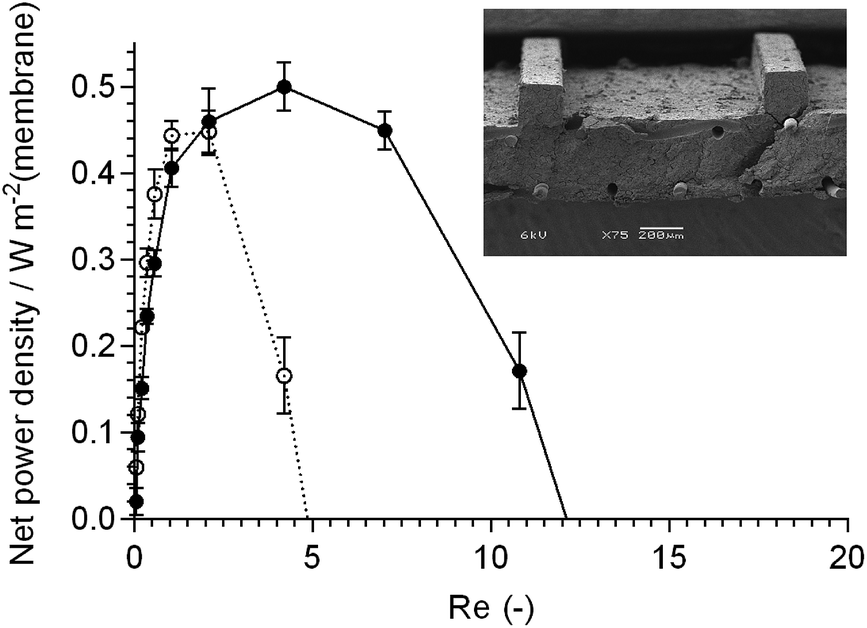 | ||
| Fig. 17 The improved net power output in a RED cell system using profiled IEMs (solid circles, i.e. an IEM with integrated spacer functionality [see inset SEM micrograph]) in comparison to a standard system containing non-conductive spacers (open circles).414 SEM: scale bar = 200 μm, 75× magnification. | ||
Multivalent ions and fouling in RED cells
Although most experiments in laboratory conditions are performed using artificial sea and river water (e.g. containing only NaCl), real world application is different and natural waters contain significant amounts (ca. 10%) of multivalent ions (predominantly MgSO4) and potential foulants such as humic acids, clay, colloids, and scale inducing minerals. Post et al.417 performed laboratory experiments with feeds containing not just NaCl but also MgSO4 or MgCl2. A major effect was observed when the multivalent ions were present where the resistance increased and the stack voltage decreased. This was attributed to the transport of these multivalent ions from the dilute solution side to the concentrated solution side (against the concentration gradient). In a follow-up study, Vermaas et al.418 characterized this transport as “uphill transport”, in accordance with other membrane processes. They experimentally and theoretically investigated the effect of increasing the fraction of MgSO4 (0, 5, 10, 25, 50, and 100%), alongside the NaCl, on the open circuit voltage and power density with three different membrane pairs. The presence of MgSO4 in the river water compartment was shown to have an especially significant negative (performance deteriorating) effect. For example, the presence of only 10% MgSO4 yielded a decrease in stationary state power output by 29–50% compared to the benchmark (NaCl only) case. In addition, switching from a NaCl only solution to a mixture of NaCl and MgSO4, led to voltage response times in the range of tens of minutes and several hours (due to ion exchange processes between the membranes and the feed water). Both researchers concluded that the presence of MgSO4 in natural feed waters is a serious factor that needs to be taken into account and that the development of monovalent selective membranes (that preferentially allow the transport of monovalent ions over multivalent ions) are required to minimise this undesirable characteristic.Vermaas et al.419 also performed long term (25 days) experiments with realistic natural feed waters. Before entering the RED stack, these were filtered through a 20 μm filter (to remove particulates only). Both a system with IEMs and spacers and a system with profiled IEMs were investigated. The power output showed a major (40–60%) decrease in only the first few hours (Fig. 18). In all cases involving realistic water feeds, deposition of remnants of diatoms (algae) and clay minerals, organic fouling, and scaling were observed, depending on the type of IEM being used. The AEMs were mainly covered with diatoms and clay minerals, whereas scaling dominated with the CEM. Although fouling was observed for both profiled IEM and spacer-containing systems, it was less severe with the use of profiled membranes. In addition, recent work has shown that anti-fouling strategies (e.g. periodic feed water switching or air sparging) can be very effective in maintaining higher RED cell power outputs.420
 | ||
| Fig. 18 Peak power densities as function of the number of days after the start of the experiment for a stack with spacers (open circles) and a stack with profiled membranes (filled circles).419 | ||
Future perspective
The importance of IEMs that are specifically designed for use in RED cells is evident. Specifically tailored IEMs are mandatory if power outputs (for an economically and commercially viable process) are to be achieved at the target values of 2–3 W m−2 (normalised to membrane geometric area). Modification of membrane chemistry is a strong tool to allow this goal to be reached. Focus should be on decreasing the membrane resistance rather than increasing permselectivity. New chemistries would not only allow the design of membranes with improved ionic conduction properties, but would also make it possible to combine this with the additionally desirable development of monovalent selective (to mitigate against the negative effect of multivalent species such as MgSO4) and “anti-fouling” (chemical and biological) IEMs.421 Efforts on IEM, including AEM, development should be focused at these research directions.AEMs in biological energy systems
All of the above are chemical, abiotic systems. However, in the last decade there has been resurgence in the development of biological fuel cells and related systems. Such systems normally operate at more neutral pHs compared to most of the abiotic electrochemical energy systems discussed above, apart from RED. As with RED, high pH and low pH degradation processes are less of a concern. Most of the biotic systems that report the use of AEMs contain a microbial component (Fig. 19).422,423 There are, however, non-microbial biotic systems that have used AEMs. For example, there has been a notable report of the use of a polysulfone-AEM in a methanol-fuelled enzymatic fuel cell.7 This system contained a fuel tolerant enzyme-based cathode (laccase from Rhus vernificera with an enzyme loading of 0.22 mg cm−2) along with Pt/Ru-based anodes. This enzymatic fuel cell demonstrated a very promising power performance (for a biological fuel cell): 8.5 mW cm−2 with a 290 h lifetime.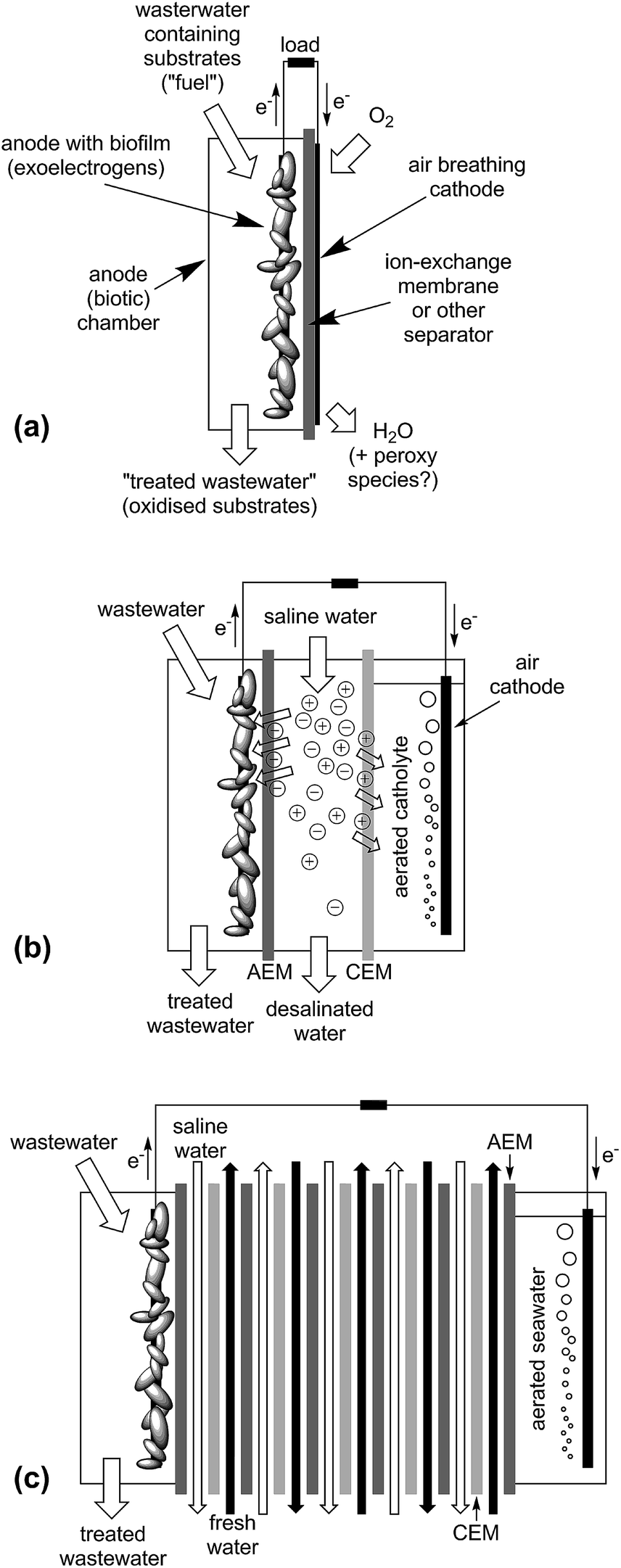 | ||
| Fig. 19 Simple schematics of: (a) a Microbial Fuel Cells [MFC] in air-breathing single-chamber mode [alternative configuration {not shown}: a 2-chamber MFC would contain a cathode chamber containing an aerated catholyte and a submerged cathode]; (b) the simplest configuration of a 3-chamber Microbial Desalination Cell [MDC]; (c) the simplest configuration of a microbial reverse electrodialysis cell [MRC]. Engineering components, such as gaskets and end plates, are not shown. | ||
Microbial fuel cells
Microbial fuel cells424 (MFCs, see Fig. 19a), are generally being developed for energy efficient treatment of various wastewaters (containing a variety of substrates [fuels], such as acetate or sucrose) rather than energy generation per se. They can also be operated with additional added value functions (e.g. water softening, NH3 remediation422a,g or electrosynthesis – see later). MFCs typically contain carbon-based anodes that have been inoculated with either: (a) a microbial consortia that contain electroactive microbial species (commonly designated as exoelectrogens and more historically as electricigens) for real world applications involving a supply of target wastewater (include human wastewater) that requires treatment; or (b) single species [monoculture] exoelectrogens (such as Escherichia coli [E. coli], Shewanella oneidensis, or Geobacter Sulfurreducens) for more fundamental studies. The exoelectrogens oxidise the substrates and use the anode as the terminal electron acceptor when they are located in the target anaerobic (or anoxic) environment of the anode chamber. This avoids the microbes using dissolved O2 as the terminal electron acceptor (as with normal microbial respiration), which would be the case if the anode chamber contains an aerobic environment.The electrons then pass through an external circuit to the cathode, which is most commonly an ORR type. The cathode can be either “air breathing” (supplied directly with air [passively or actively]) or solution-based (uses O2 [or other electron accepting species] that is dissolved in a catholyte). MFCs, therefore operate because the O2 is separated away from the microbes. The cathodes can contain a variety of catalysts.425 They can be abiotic: e.g. contain Pt-, MnxOy-,422b,426 or CoTMPP-422p based catalysts or they can even be “non-catalysed” (i.e. metal-free and carbon-based).427 They can also be biotic: i.e. either microbial (e.g. containing autotrophic bacteria)422c or enzymatic (e.g. containing laccase including that excreted from the white-rot fungus Coriolus versicolor).428 Cathodes can also be photocatalytic (e.g. contain algae or cyanobacteria).429
MFCs can be constructed with a variety configurations including packed-bed,422d single chamber, or 2 chamber; 3 or more chambers are often found with related systems that involve added-value functions (see later). A large proportion of these configurations contain an IEM (or other non-ionic and/or porous separator). One of the earliest reports of a comparison of 2-chambered MFCs containing AEMs vs. CEMs vs. non-ionic ultrafiltration membranes was by Logan et al. in 2007.422q This study discusses the transfer of protons through the AEM via negatively charged species (phosphate anions) and compared the use of CMI-7000 CEM and AMI-7001 AEM (both from Membrane International, USA). A number of key themes, apparent on review of the literature regarding MFCs containing AEMs [a small proportion of the total MFC literature], will now be discussed.
Many studies indicate that MFCs containing (dense) IEMs have superior performances (e.g. power density outputs‡ and coulombic efficiencies [CE]) and more stable performances with time422n when they contain AEMs (as opposed to CEMs). Major reasons that are put forward for this include:
(a) The lower O2 permeability of the AEMs, especially compared to Nafion® perfluorinated CEMs, where reduced O2 crossover from the cathode to the anode is beneficial in maintaining an anaerobic environment at the anode (for sustaining high CEs etc.);
(b) Reduced “pH splitting” effects leading to smaller pH gradients across the membrane with AEM-based systems: “pH splitting” is the undesirable lowering of the anode chamber pH and a raising of the pH at the cathode (common with CEM-based systems);
(c) “Reduced” [or different] membrane fouling characteristics;
(d) Reduced cathode resistances;
(e) Reduced cation-derived precipitates on the cathode catalyst;
(f) The higher ionic conductivities (lower internal ohmic resistances§) of the AEMs (vs. CEMs) in MFCs.
The latter point (f) above is an important contrast to the scenario of AAEM vs. PEMs in (abiotic) chemical fuel cells where H+ and OH− ions are the main conducting ions and where the OH− anions have the lower intrinsic mobilities (Table 1). However, IEMs in MFCs are predominantly in ionic forms that are not OH− and H+; the ions present depend on the nature of the anolyte and catholyte media and buffers (if present). If you compare Na+ to Cl−, the anion now has the higher mobility. Early generation (non-phase segregated) AAEMs generally have higher IECs compared to PEMs to offset the lower mobility OH− ions in APEFCs compared to H+ ions in PEMFCs. The combination of these factors (anion having higher mobility and AEMs having higher IECs) is why the conductivities of AEMs are now higher than CEMs when they are compared in MFCs (cf. the same IEMs used in abiotic fuel cells).
However, a major drawback in the use of AEMs is enhanced substrate (e.g. acetate) crossover from the anode to the cathode, which can lead to mixed potentials at the cathode and parasitic internal currents. If the system in question involves a catholyte solution (i.e. the O2 is dissolved as part of an aerated electrolyte), then changing this frequently can mitigate against this substrate crossover effect (if this is realistic in a real world application?). The loss of metabolites when using an AEM (especially at low external resistances) can lead to voltage losses over longer operational periods.422i Membrane deformation (often in the opposite direction to that seen with the use of CEMs) has also been witnessed, which led to an inferior performance with the use of an AEM;422l this was due to the trapping of water and gas between the AEM and cathode. This latter problem can be rectified by using a stainless steel mesh on the anode side to push the membrane onto the cathode.
The internal resistances§ of MFCs containing dense ion-exchange membranes (AEMs and CEMs) can also be higher compared to MFCs containing porous membranes or with membrane-less (e.g. single chamber) MFCs, though the lack of IEM can lead to higher O2 and substrate crossover. For example, a membrane-free MFC containing a cloth-cathode assembly (containing a GoreTex® cloth which enhances proton transport and O2 diffusion to the cathode) had a superior performance compared to MFCs containing a membrane–cathode assembly involving either an AEM or a CEM;422m The preparation of MFCs using the cloth-cathode assembly method was also less time consuming and is also claimed to be more optimal for scale-up.
Various types of AEMs have been evaluated in MFCs and these include AMI-7001 (the most commonly encountered),422c,d,f,g,p,q AEMs by Ralex,422b,i,j Tokuyama Neosepta types (this study compares Neosepta AFN, AM-1, and ACS AEMs and probes the interface resistances between the AEM and low buffer [low ionic strength] electrolyte),422k pore-filled type AEMs,422e and Chinese AEM types by Tianwei422n and Qianqiu Group (Zhejiang).422a,m
A selected case study is the recent comparison of an in-house synthesised QA poly(ether ether ketones) [QA-PEEK] AEM to AMI-7001 in a single chamber MFC.422h The “hydroxide conducting” QA-PEEK AEM ([allegedly] 0.2 μm thick, IEC = 1.39 meq. g−1) outperformed the AMI-7001 (450 μm thickness, IEC = 1.6 meq. g−1, gel polystyrene-divinylbenzene chemistry) with lower O2 crossover, despite higher substrate crossover; peak power densities of 60 W m−3 and 45 W m−3 and CE = 66 and 51%, respectively, were obtained at 30 °C. The MFCs contained PTFE wet-proofed Vulcan XC-72 covered carbon cloth anodes and carbon-cloth-based air-breathing cathodes that additionally contained a Pt catalyst (loading = 0.5 mgPt cm−2 geometric) and the anodes were inoculated with Anna University domestic wastewater in a phosphate buffer medium (pH = 7.3–7.6 and chemical oxygen demand = 400–600 mg dm−3). The AMI-7001 visibly degraded after 250 d of testing, unlike the QA-PEEK AEM.
A second case study422i is where a Ralex AM(H)-PES AEM (QA polyethylene/polyester based, unspecified thickness and IEC in the report but normally supplied with < 450 μm dry thickness and <750 μm fully hydrated thickness) similarly outperformed a Nafion® CEM in comparable flat-plate dual-chamber Shewanella putrefaciens (single species) inoculated MFCs at 27 °C; “non-catalysed” graphite plates were used for both the anode and cathode (maximum voltage and power density of 0.729 V and 57.8 mW m−2 for the AEM vs. 0.676 V and 39.2 mW m−2 for Nafion® [power densities normalised to anode geometric areas]).
A number of the above studies422m,n,q compared the performances of MFCs containing AEMs vs. CEMs.¶ This includes the original study by Logan et al.,422q which compared the performances of 2-chambered MFCs containing either AMI-7001 as an AEM or CMI-7000 as a CEM (450 μm thickness, IEC = 1.4 meq. g−1, gel polystyrene-divinylbenzene type). A research priority should be more in-depth comparisons between identical MFC set-ups containing thinner CEMs and AEMs of identical thicknesses, identical IECs and similar chemistries: i.e. the same backbone chemistries and just where the cationic (anion-exchange) and anionic (cation-exchange) head-groups are the variable. Aspects that should be studied, including for fundamental investigations, include:
(a) In situ beginning-of-life and longer term performances (including data on internal resistances [specifically including internal ohmic resistances]§ and current and power outputs [with clearly defined normalisation]‡);
(b) In situ and ex situ durabilities (i.e. changes in the IEM chemistry, IEC, conductivity, mechanical stability, and membrane (bio)fouling with time);
(c) Effects of the different membranes on the nature of the biofilms and microbial consortia in the various zones of the biotic chambers during in situ MFC testing;
(d) In situ and ex situ studies into the stability and longer-term performances of the electrodes (catalysts) including those at the cathode (e.g. cathode fouling).
There should also be concerted efforts into the development of cheap and thin (i.e. low area resistance especially in MFC-relevant anion forms) AEMs that are specifically tailored for MFC-related applications. The AEMs should maintain low O2 permeabilities, exhibit reduced substrate crossovers (to the cathode), and yield optimised biological outcomes including reduced performance losses due to biofouling (positively charged [polycationic] AEMs would selectively inhibit adhesion or fouling with positively charged bacteria [the opposite to when CEMs are used]). More research aimed at unambiguously confirming the nature of the in situ ion movement through the AEM (including exactly what ions are involved [OH−, PO43−, CO32−etc.]) is also justified.
Variant systems related to MFCs
MDC investigations are varied and include studies that have looked into: spatially decoupling the anode and cathode,433c hydraulically connecting multiple MDCs,433b hydraulically connecting an MDC to an osmotic MFC (MOFCs434 contain a Forward Osmosis [FO] membrane that can, on their own, allow desalinated water recovery along with power generation with superior energy recoveries compared to MFCs containing AEMs or CEMs),433e scaling up MDCs to litre scale capacities,433h developing MDC stacks (for faster desalination),432,433k developing hydrid desalination and electrolysis systems for water desalination and H2 generation,433l developing hybrid microbial electrolysis desalination and chemical production cells for desalination as well as acid and alkali production (this type of system also contains a bipolar membrane between the anode and chemical production chamber [the AEM separates the chemical production chamber and the desalination chamber]),432,433i packing MDCs with ion-exchange resins,433a pH control by using electrolyte circulation,433j and AEM biofouling.433d
Summary and concluding remarks
Alkaline anion-exchange membranes (AAEMs) are being developed for application in electrochemical devices such as alkaline polymer electrolyte fuel cells (APEFC). As with the more well-known proton-exchange membrane fuel cells (PEMFCs), APEFCs can be operated with Pt-based catalysts. However, it should be noted that even though the oxygen reduction reaction is slightly less of a problem at high pHs, the hydrogen oxidation reaction kinetics is poorer with Pt catalysts in alkali than in acid. In contrast to PEMFCs, the anode of APEFCs tends to produce larger performance limitations than the cathode. The main rationale for the development of devices such as APEFCs is the promise of the ability to use a broad range of non-precious metal (non-Pt-group-metals) catalysts.The conductivities of AAEMs in the OH− form can be as high as H+ conduction in proton-exchange membranes (PEMs such as Nafion®) when the AAEMs are well hydrated, which is the situation in electrolysers containing polymer electrolytes. However, the conductivities of AAEMs are much more sensitive to hydration levels and drop rapidly when exposed to lower humidity environments (e.g. as found in APEFCs). The development of AAEMs that retain conductivity at lower hydration levels is a research priority. If OH− conductivities are being measured, it is essential to totally exclude CO2 from each stage of the experiment otherwise conductivities will be under estimated (as a mixture of anions [OH−/CO32−/HCO3−] will be present). To aid in inter-laboratory comparisons, HCO3− conductivities should also be reported.
The biggest research challenge for devices with high pH environments is to develop chemically stable AAEMs and anion-exchange ionomers (AEIs), especially when less than fully hydrated. Most studies have focussed only on the alkali stabilities (especially of the cationic [anion-exchange] head-groups), as this is seen as the biggest problem. However, the stability to in situ generated peroxy species (and resulting radicals) needs to be explored in much more detail as not much is known to date regarding AAEMs in APEFCs etc. (in contrast to PEMs in PEMFCs). Even though a polymer backbone is stable in alkali, it may not be alkali stable once it is functionalised with cationic side-groups. Studies of the alkali stabilities of AAEMs and AEIs must consider the synergistic effects of the cation head-groups and the backbone. Stability studies should always use spectroscopic evidence alongside secondary evidence such as changes in ion-exchange capacities with ageing time; the relative changes in quaternary and non-quaternary exchange capacities should also be studied.
Despite all of this, AAEMs containing simple (low molecular weight) quaternary ammonium cationic head-groups (such as R–N+Me3) may well be stable enough in the OH− forms for some applications if they are kept fully hydrated. This will be especially true for AAEMs that possess phase segregated morphologies, where location of the cationic head-groups in the hydrophilic channels/clusters will maximise the chances that they remain suitably hydrated. It is vital that AAEMs/AEIs are developed with long term stabilities in the OH− form at temperatures >80 °C (especially when less that fully hydrated) for application in APEFCs. Operation at such elevated temperatures (and/or at high current densities) is suspected to lead to an enhanced or intrinsic tolerance to CO2 in the cathode air supplies. An alternative approach may be to deliberately direct the system to utilise CO32− (but not HCO3−) conduction as AAEMs are generally much more stable in the CO32− forms.
The use of alternative fuels in APEFCs is a mixed bag. The use of alcohols appears not to be viable without the addition of Na/KOH into the aqueous fuel supplies, which then begs the question: why use polymer electrolytes in such fuel cells? On the other hand, the use of non-carbon fuel options may have more application, especially sodium borohydride for niche (military) applications and hydrazine hydrate (not anhydrous hydrazine) for more main-stream markets.
Stability to OH− (or CO32−) anions is also required if the AAEMs are to be used in alkaline water electrolysers. However, such electrolysers have the advantage over APEFCs in that the AAEMs and AEIs can be kept well hydrated to maximise stability and conductivity. An aspiration is to develop alkaline polymer electrolyte electrolysers that generate H2 from pure water (no added alkali or acid). Electrolysis devices that are aimed at CO2 utilisation are also of increasing interest.
For application in redox flow batteries (RFBs), the anion-exchange membranes (AEMs) need to instead be stable to the redox species present (e.g. vanadium species in all-vanadium RFBs [VRFBs]) as well as to low pHs if acidic electrolytes are present. The hope is that AEMs selectively reject vanadium cations in VRFBs to reduce vanadium species crossover. A reduction in water crossover is also considered to be important with various RFBs and the use of AEMs should be investigated with regards to this technological requirement.
When applied to electrochemical devices such as reverse electrodialysis (RED) cells and microbial fuel cells (MFCs), the alkali stabilities of AEMs are almost irrelevant (as the membranes are not exposed to high pH environments). However, the conductivities are lower with ions that are typically present in these devices (e.g. Cl− and not OH−). The research challenge here is to develop thinner membranes (to counteract the lower conductivities with non-OH− anions) but where there is no reduction in other properties (e.g. no increase in O2 permeability in microbial systems or no lowering of permselectivity or mechanical properties in RED cells). As the AEMs are in environments with less extreme pHs, then chemistries that are not stable in alkali (such as imidazolium, phosphonium, and pyridinium cationic head-groups) may be applicable. This selection of a wider range of AEM chemistries may well be advantageous when applied to devices where fouling (including bio-fouling) may be an issue and needs to be minimised.
It is clear that a single AEM will not be optimal for all applications and AEMs will need to be specifically tailored for the job at hand. However, there is a significant chemical and materials space that can be probed and utilised for the development of tailored AEMs/AEIs.
Statement of author contributions
John Varcoe is lead author who wrote the bulk of the sections on membrane chemistries and the sections on biological systems and batteries. He also coordinated all efforts and integrated all contributions into the manuscript in a consistent manner. Plamen Atanassov provided the information on fuels that are alternative to hydrogen for use in fuel cells (such as hydrazine). Dario Dekel led the discussion (and provided an industrial perspective) on the application on AAEMs in hydrogen-based fuel cells. Andrew Herring, Tongwen Xu and Lin Zhuang all provided significant contributions on membrane chemistry, especially the critically important phase segregated systems. Lin Zhuang also provided input on alternative fuels and electrolysers. Michael Hickner wrote the section on redox flow batteries. Paul Kohl wrote the section on hybrid anion- and cation-exchange membrane systems. Anthony Kucernak wrote the key sections on issues with carbon and carbon-free supports. He gave some input into various catalyst chemistries. William Mustain wrote the sections on carbonate cycle fuel cells and electrolysers. Kitty Nijmeijer wrote the section on Reverse Electrodialysis Cells. Keith Scott led on the sections on hydrogen-based electrolysers.Abbreviations
This lists the subject-specific acronyms used in the main text for quick reference. This table does not list commonly used chemical symbols and nomenclature (e.g. NMR, TEM etc.) that are well known to the general chemistry audience. Acronyms in italics are carefully defined in the preamble at the start of the article.| AAEM | Alkaline anion-exchange membrane |
| AB | Ammonia borane |
| ABCO | 1-Azabicyclo[2.2.2]octane (quinuclidine) |
| ADFFC | Alkaline direct formate fuel cell |
| AEI | Anion-exchange ionomer |
| AEM | Anion-exchange membrane |
| AEMFC | Anion-exchange membrane fuel cell [≡ APEFC] |
| AFC | Alkaline fuel cell (taken here as the fuel cell type containing aqueous K/NaOH electrolytes and not a AAEM) |
| AMFC | Alkaline membrane fuel cell [≡ APEFC] |
| APEE | Alkaline polymer electrolyte electrolyser |
| APEFC | Alkaline polymer electrolyte fuel cell |
| APM | Alkali-doped poly(benzimidazole) |
| CCM | Catalysed coated membrane |
| CDU | Carbon dioxide utilisation |
| CE | Coulombic efficiency |
| CEM | Cation-exchange membrane |
| CNT | Carbon nanotube |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DAFC | Direct alcohol fuel cell |
| DBHFC | Direct borohydride fuel cell |
| DEFC | Direct ethanol fuel cell |
| DEGFC | Direct ethylene glycol fuel cell |
| DHFC | Direct hydrazine fuel cell |
| DMFC | Direct methanol fuel cell |
| EDL | Electrical double layer |
| EG | Ethylene glycol |
| EOR | Ethanol oxidation reaction |
| FO | Forward osmosis |
| HEM | Hydroxide-exchange membrane |
| HER | Hydrogen evolution reaction |
| HOR | Hydrogen oxidation reaction |
| IEC | Ion-exchange capacity |
| IEM | Ion-exchange membrane |
| MCCC | Microbial carbon capture cells |
| MDC | Microbial desalination cell |
| MEA | Membrane electrode assembly |
| MEC | Microbial electrolysis cell |
| MFC | Microbial fuel cell |
| MOFC | Microbial osmotic fuel cell |
| MOH | Metal (normally alkali metal) hydroxide |
| MOR | Methanol oxidation reaction |
| MRC | Microbial reverse electrodialysis cell |
| ORR | Oxygen reduction reaction |
| OER | Oxygen evolution reaction |
| PBI | Poly(benzimidazole) |
| PECH | Poly(epichlorohydrin) |
| PEEK | Poly(ether ether ketone) |
| PEM | Proton-exchange membrane |
| PEMFC | Proton-exchange membrane fuel cell |
| PFSA | Perfluoro sulfonic acid |
| PGM | Platinum-group metal |
| PPO | Poly(phenylene oxide) |
| PRO | Pressure retarded osmosis |
| PTFE | Poly(tetrafluoroethylene) |
| PVA | Poly(vinyl alcohol) |
| QA | Quaternary ammonium |
| QAPS | Quaternary ammonium poly(sulfone) |
| RED | Reverse electrodialysis |
| RFB | Redox flow battery |
| RFC | Regenerative fuel cell |
| RH | Relative humidity |
| RO | Reverse osmosis |
| ROMP | Ring opening metathesis polymerisation |
| RTCFC | Room temperature carbonate fuel cell |
| SAFC | Solid alkaline fuel cell [≡ APEFC] |
| SGE | Salinity gradient energy |
| SPE | Solid polymer electrolyte |
| SPEEK | Sulfonated poly(ether ether ketone) |
| URFC | Unitized regenerative fuel cell |
| VRFB | Vanadium redox flow battery |
Acknowledgements
We wish to thank the following funders and sponsors of the workshop on “Anion-exchange membranes for energy generation technologies” held at the University of Surrey on 25th – 26th July 2013:1 The UK's Engineering and Physical Sciences Research Council (EPSRC grants EP/I004882/1, EP/H019480/1 and EP/H025340/1), the University of Surrey's Institute of Advanced Studies (especially Mirela Dumic for her extensive assistance),1 the Research Council UK's Energy Programme's Supergen Hydrogen and Fuel Cell Research Hub (EPSRC grant EP/J016454/1), Royal Society of Chemistry publishing, Caltest Instruments Ltd. (UK), Alvatek Ltd. (UK), and Solartron Analytical UK (Ametek). We also thank the US Army Research Office for funding from a Multidisciplinary University Research Initiative (MURI contract W911NF-10-1-0520). The following members of the local organising committee are also thanked: Dr Simon Poynton, Dr Cathryn Hancock, Dr Ai Lien Ong, Sam Murphy, Sarah Mallinson, Jessica Whitehead and Lucy Howes.References
- http://www.ias.surrey.ac.uk/workshops/membranes/ .
- (a) K.-D. Kreuer, Chem. Mater., 2014, 26, 361 CrossRef CAS; (b) M. A. Hickner, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1727 CrossRef CAS PubMed; (c) Y.-J. Wang, J. Qiao, R. Baker and J. Zhang, Chem. Soc. Rev., 2013, 42, 5768 RSC; (d) A. Brouzgou, A. Podias and P. Tsiakaras, J. Appl. Electrochem., 2013, 43, 119 CrossRef CAS; (e) J. Pan, C. Chen, L. Zhuang and J. Lu, Acc. Chem. Res., 2012, 45, 473 CrossRef CAS PubMed; (f) H. Zhang and P. K. Shen, Chem. Soc. Rev., 2012, 41, 2382 RSC; (g) H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780 CrossRef CAS PubMed; (h) G. Couture, A. Alaaeddine, F. Boschet and B. Ameduri, Prog. Polym. Sci., 2011, 1521 CrossRef CAS PubMed; (i) G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1 CrossRef CAS PubMed; (j) Y. A. Elabd and M. A. Hickner, Macromolecules, 2011, 44, 1 CrossRef CAS; (k) R. Zeng and J. R. Varcoe, Recent Pat. Chem. Eng., 2011, 4, 93 CAS; (l) E. H. Yu, U. Krewer and K. Scott, Energies, 2010, 3, 1499 CrossRef CAS PubMed; (m) V. V. Shevchenko and M. A. Gumennaya, Theor. Exp. Chem., 2010, 46, 139 CrossRef CAS; (n) D. Tang, J. Pan, S. Lu, L. Zhuang and J. Lu, Sci. China: Chem., 2010, 53, 357 CrossRef CAS PubMed; (o) E. Antolini and E. R. Gonzalez, J. Power Sources, 2010, 195, 3431 CrossRef CAS PubMed; (p) A. Serov and C. Kwak, Appl. Catal., B, 2010, 98, 1 CrossRef CAS PubMed; (q) F. Bidault, D. J. L. Brett, P. H. Middleton and N. P. Brandon, J. Power Sources, 2009, 187, 39 CrossRef CAS PubMed; (r) J. R. Varcoe, J. P. Kizewski, D. M. Halepoto, S. D. Poynton, R. C. T. Slade and F. Zhao, in Encyclopedia of Electrochemical Power Sources, ed. C. Garche, P. Dyer, Z. Moseley, D. R. Ogumi and B. Scrosati, Elsevier, Amsterdam, 2009, vol. 2, pp. 329–343 Search PubMed; (s) J. R. Varcoe, S. D. Poynton and R. C. T. Slade, in Handbook of Fuel Cells - Fundamentals, Technology and Applications, ed. W. Vielstich, H. Yokokawa and H. A. Gasteiger, John Wiley and Sons, Ltd., 2009, vol. 5, ch. 21, pp. 322–333 Search PubMed; (t) R. C. T. Slade and J. R. Varcoe, Fuel Cells, 2005, 5, 187 CrossRef PubMed; (u) T. Xu, J. Membr. Sci., 2005, 263, 1 CrossRef CAS PubMed.
- S. Marini, P. Salvi, P. Nelli, R. Pesenti, M. Villa, M. Berrettoni, G. Zangari and Y. Kiros, Electrochim. Acta, 2012, 82, 384 CrossRef CAS PubMed.
- (a) W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Adv. Funct. Mater., 2013, 23, 970 CrossRef CAS PubMed; (b) B. Schwenzer, J. Zhang, S. Kim, L. Li and Z. Yang, ChemSusChem, 2011, 4, 1388 CrossRef CAS PubMed; (c) X. Li, H. Zhang, Z. Mai, H. Zhang and I. Vankelecom, Energy Environ. Sci., 2011, 4, 1147 RSC; (d) M. Skyllas-Kazacos, M. H. Chakrabarti, S. A. Hajimolana, F. S. Mjalli and M. Saleem, J. Electrochem. Soc., 2011, 158, R55 CrossRef CAS PubMed ; also ref. 2a above.
- (a) B. E. Logan and M. Elimelech, Nature, 2012, 488, 313 CrossRef CAS PubMed; (b) G. Z. Ramon, B. J. Feinberg and E. M. V. Hoek, Energy Environ. Sci., 2011, 4, 4423 RSC.
- (a) J. X. Leong, W. R. W. Daud, M. Ghasemi, K. B. Liew and M. Ismail, Renewable Sustainable Energy Rev., 2013, 575 CrossRef CAS PubMed; (b) V. B. Oliveria, M. Simões, L. F. Melo and A. M. F. R. Pinto, Biochem. Eng. J., 2013, 73, 53 CrossRef PubMed; (c) Y. Yang, G. Sun and M. Xu, J. Chem. Technol. Biotechnol., 2011, 86, 625 CrossRef CAS PubMed ; also ref. 5a above.
- W. Gellet, J. Schumacher, M. Kesmez, D. Le and S. D. Minteer, J. Electrochem. Soc., 2010, 157, B557 CrossRef PubMed.
- (a) X. Li, Q. Liu, Y. Yu and Y. Meng, J. Mater. Chem. A, 2013, 1, 4324 RSC; (b) A. Jasti and V. K. Shahi, J. Mater. Chem. A, 2013, 1, 6134 RSC; (c) D. Chen, M. A. Hickner, S. Wang, J. Pan, M. Xiao and Y. Meng, Int. J. Hydrogen Energy, 2012, 37, 16168 CrossRef CAS PubMed; (d) M. Tanaka, M. Koike, K. Miyatake and M. Watanabe, Polym. Chem., 2011, 2, 99 RSC; (e) J. Fang and P. K. Shen, J. Membr. Sci., 2006, 285, 317 CrossRef CAS PubMed.
- (a) X. Li, Y. Yu and Y. Meng, ACS Appl. Mater. Interfaces, 2013, 5, 1414 CrossRef CAS PubMed; (b) D. W. Seo, Y. D. Lim, M. A. Hossain, S. H. Lee, H. C. Lee, H. H. Jang, S. Y. Choi and W. G. Kim, Int. J. Hydrogen Energy, 2013, 38, 579 CrossRef CAS PubMed; (c) D. W. Seo, M. A. Hossain, D. H. Lee, Y. D. Lim, S. H. Lee, H. C. Lee, T. W. Hong and W. G. Kim, Electrochim. Acta, 2012, 86, 360 CrossRef CAS PubMed; (d) Q. Zhang, Q. Zhang, J. Wang, S. Zhang and S. Li, Polymer, 2010, 5407 CrossRef CAS PubMed; (e) J. Zhou, M. Ünlü, J. A. Vega and P. A. Kohl, J. Power Sources, 2009, 190, 285 CrossRef CAS PubMed; (f) D. Xing, S. Zhang, C. Yin, C. Yan and X. Jian, Mater. Sci. Eng., B, 2009, 157, 1 CrossRef CAS PubMed; (g) L. Li and Y. Wang, J. Membr. Sci., 2005, 262, 1 CrossRef CAS PubMed; (h) J. Kerres, A. Ullrich and M. Hein, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2874 CrossRef CAS PubMed.
- (a) X. Yan, S. Gu, G. He, X. Wu and J. Benziger, J. Power Sources, 2014, 250, 90 CrossRef CAS PubMed; (b) J. Han, H. Peng, J. Pan, L. Wei, G. Li, C. Chen, l. Xiao, J. Lu and L. Zhuang, ACS Appl. Mater. Interfaces, 2013, 5, 13405 CrossRef CAS PubMed; (c) Z. Liu, X. Li, K. Shen, P. Feng, Y. Zhang, X. Xu, W. Hu, Z. Jiang, B. Liu and M. D. Guiver, J. Mater. Chem. A, 2013, 1, 6481 RSC; (d) X. Lin, Y. Liu, S. D. Poynton, A. Ong, J. R. Varcoe, L. Wu, Y. Li, X. Liang, Q. Li and T. Xu, J. Power Sources, 2013, 233, 259 CrossRef CAS PubMed; (e) Z. Liu, X. Zhu, G. Wang, X. Hou and D. Liu, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1632 CAS; (f) K. Shen, J. Pang, S. Feng, Y. Wang and Z. Jiang, J. Membr. Sci., 2013, 440, 20 CrossRef CAS PubMed; (g) X. Yan, G. He, X. Wu and J. Benziger, J. Membr. Sci., 2013, 429, 13 CrossRef CAS PubMed; (h) A. Jasti, S. Prakash and V. K. Shahi, J. Membr. Sci., 2013, 428, 470 CrossRef CAS PubMed; (i) J. Wang, J. Wang and S. Zhang, J. Membr. Sci., 2012, 415–416, 205 CrossRef CAS PubMed; (j) H. Zhang and Z. Zhou, Solid State Ionics, 2008, 179, 1296 CrossRef CAS PubMed.
- X. Yan, S. Gu, G. He, X. Wu, W. Zheng and X. Ruan, J. Membr. Sci., 2014, 466, 220 CrossRef CAS PubMed.
- (a) G. Wang, Y. Weng, J. Zhao, D. Chu, D. Xie and R. Chen, Polym. Adv. Technol., 2010, 21, 554 CAS; (b) G. Wang, Y. Weng, D. Chu, D. Xie and R. Chen, J. Membr. Sci., 2009, 326, 4 CrossRef CAS PubMed.
- (a) W. Wang, S. Wang, W. Li, X. Xie and Y. Lv, Int. J. Hydrogen Energy, 2013, 38, 11045 CrossRef CAS PubMed; (b) C. Li, S. Wang, W. Wang, X. Xie, Y. Lv and C. Deng, Int. J. Hydrogen Energy, 2013, 38, 11038 CrossRef CAS PubMed; (c) Q. Hu, Y. Shang, Y. Wang, M. Xu, S. Wang, X. Xie, Y. Liu, H. Zhang, J. Wang and Z. Mao, Int. J. Hydrogen Energy, 2012, 37, 12659 CrossRef CAS PubMed.
- (a) N. T. Rebek, Y. Li and D. M. Knauss, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1770 CrossRef PubMed; (b) A. Katzfuß, V. Gogel, L. Jörissen and J. Kerres, J. Membr. Sci., 2013, 425–426, 131 CrossRef PubMed; (c) X. Lin, C. Wu, Y. Wu and T. Xu, J. Appl. Polym. Sci., 2012, 123, 3644 CrossRef CAS PubMed; (d) S. Gu, J. Skovgard and Y. Yan, ChemSusChem, 2012, 5, 843 CrossRef CAS PubMed; (e) A. Ong, S. Saad, R. Lan, R. J. Goodfellow and S. Tao, J. Power Sources, 2011, 196, 8272 CrossRef CAS PubMed; (f) Y. Wu, C. Wu, J. R. Varcoe, S. D. Poynton, T. Xu and Y. Fu, J. Power Sources, 2010, 195, 3069 CrossRef CAS PubMed; (g) L. Wu, T. Xu, D. Wu and X. Zheng, J. Membr. Sci., 2008, 310, 577 CrossRef CAS PubMed; (h) L. Wu, T. Xu and W. Yang, J. Membr. Sci., 2006, 286, 185 CrossRef CAS PubMed; (i) T. Xu and F. F. Zha, J. Membr. Sci., 2002, 199, 203 CrossRef CAS.
- (a) R. Janarthanan, J. L. Horan, B. R. Caire, Z. C. Zieger, Y. Yang, X. Zuo, M. W. Liberatore, M. R. Hibbs and A. M. Herring, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1743 CrossRef CAS PubMed; (b) C. Fujimoto, D.-S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423–424, 438 CrossRef CAS PubMed; (c) E. E. Switzer, T. S. Olson, A. K. Datye, P. Atanassov, M. R. Hibbs, C. Fujimoto and C. J. Cornelius, Electrochim. Acta, 2010, 55, 3404 CrossRef CAS PubMed.
- (a) M. A. Vandiver, J. L. Horan, Y. Yang, E. T. Tansey, S. Seifert, M. W. Liberatore and A. M. Herring, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1761 CrossRef CAS PubMed; (b) D. S. Kim, C. H. Fujimoto, M. R. Hibbs, A. Labouriau, Y.-K. Choe and Y. S. Kim, J. Polym. Sci., Part B: Polym. Phys., 2013, 46, 7826 CAS; (c) D. S. Kim, C. H. Fujimoto, M. R. Hibbs, A. Labouriau, Y.-K. Choe and Y. S. Kim, Macromolecules, 2013, 46, 7826 CrossRef CAS; (d) X. Kong, K. Wadhwa, J. G. Verkade and K. Schmidt-Rohr, Macromolecules, 2009, 42, 1659 CrossRef CAS.
- A. Bosnjakovic, M. Danilczuk, S. Schlick, P. N. Xiong, G. M. Haugen and S. J. Hamrock, J. Membr. Sci., 2014, 467, 136 CrossRef CAS PubMed.
- (a) D. Henkensmeier, H. Cho, M. Brela, A. Michalak, A. Dyck, W. Germer, N. M. H. Duong, J. H. Jang, H.-J. Kim, N.-S. Woo and T.-H. Lim, Int. J. Hydrogen Energy, 2014, 39, 2842 CrossRef CAS PubMed; (b) H.-J. Lee, J. Choi, J. Y. Han, H.-J. Kim, Y.-E. Sung, H. Kim, D. Henkensmeier, E. A. Cho, J. H. Jang and S. J. Yoo, Polym. Bull., 2013, 70, 2619 CrossRef CAS; (c) L. An, L. Zeng and T. S. Zhao, Int. J. Hydrogen Energy, 2013, 38, 10602 CrossRef CAS PubMed; (d) O. D. Thomas, K. J. W. Y. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, J. Am. Chem. Soc., 2012, 134, 10753 CrossRef CAS PubMed; (e) Z. Xia, S. Yuan, G. Jiang, X. Guo, J. Fang, L. Liu, J. Qiao and J. Yin, J. Membr. Sci., 2012, 390–391, 152 CrossRef CAS PubMed; (f) D. Henkensmeier, H.-R. Cho, H.-J. Kim, C. N. Kirchner, J. Leppin, A. Dyke, J. H. Jang, E. A. Cho, S.-W. Nam and T.-H. Lim, Polym. Degrad. Stab., 2012, 97, 264 CrossRef CAS PubMed; (g) H. Luo, G. Vaivars, B. Agboola, S. Mu and M. Mathe, Solid State Ionics, 2012, 208, 52 CrossRef CAS PubMed; (h) O. D. Thomas, K. J. W. Y. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, Polym. Chem., 2011, 2, 1641 RSC; (i) H. Hou, G. Sun, R. He, B. Sun, W. Jin, H. Liu and Q. Xin, Int. J. Hydrogen Energy, 2008, 33, 7172 CAS; (j) B. Xing and O. Savadogo, Electrochem. Commun., 2000, 2, 697 CrossRef CAS.
- (a) C. C. Yang, J. Appl. Electrochem., 2012, 42, 305 CrossRef CAS PubMed; (b) T. Y. Guo, Q. H. Zeng, C. H. Zhao, Q. L. Liu, A. M. Zhu and I. Broadwell, J. Membr. Sci., 2011, 371, 268 CrossRef CAS PubMed; (c) C. Sollogoub, A. Guinault, C. Bonnebat, M. Bennjima, L. Akrour, J. F. Fauvarque and L. Ogier, J. Membr. Sci., 2009, 335, 37 CrossRef CAS PubMed; (d) D. Stoica, F. Alloin, S. Marais, D. Langevin, C. Chappey and P. Judeinstein, J. Phys. Chem. B, 2008, 112, 12338 CrossRef CAS PubMed; (e) D. Stoica, L. Ogier, L. Akrour, F. Alloin and J. F. Fauvarque, Electrochim. Acta, 2007, 53, 1596 CrossRef CAS PubMed; (f) E. Agel, J. Bouet and J. F. Fauvarque, J. Power Sources, 2001, 101, 267 CrossRef CAS.
- A. M. Maes, T. P. Pandey, M. A. Vandiver, L. K. Lundquist, Y. Yang, J. L. Horan, A. Krosovsky, M. W. Liberatore, S. Seifert and A. M. Herring, Electrochim. Acta, 2013, 110, 260 CrossRef CAS PubMed.
- (a) M. Zhang, H. K. Kim, E. Chalkova, F. Mark, S. L. Lvov and T. C. M. Chung, Macromolecules, 2011, 44, 5937 CrossRef CAS; (b) H. A. Kostalik IV, T. J. Clark, N. J. Robertson, P. F. Mutolo, J. M. Longo, H. D. Abruña and G. W. Coates, Macromolecules, 2010, 43, 7147 CrossRef; (c) N. J. Robertson, H. A. Kostalik IV, T. J. Clark, P. F. Mutolo, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 3400 CrossRef CAS PubMed.
- T. J. Clarke, N. J. Robertson, H. A. Kostalik IV, E. B. Lobkovsky, P. F. Mutolo, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 12888 CrossRef PubMed.
- (a) W. Lu, Z.-G. Shao, G. Zhang, Y. Zhao and B. Yi, J. Power Sources, 2014, 248, 905 CrossRef CAS PubMed; (b) T.-H. Tsai, A. M. Maes, M. A. Vandiver, C. Versek, S. Seifert, M. Tuominen, M. W. Liberatore, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1751 CrossRef CAS PubMed; (c) J. Zhou, J. Guo, D. Chu and R. Chen, J. Power Sources, 2012, 219, 272 CrossRef CAS PubMed.
- C. Chen, A. R. Hess, A. R. Jones, X. Liu, G. D. Barber, T. E. Mallouk and H. R. Allcock, Macromolecules, 2012, 45, 1182 CrossRef CAS.
- (a) L. Gubler, Adv. Energy Mater., 2014, 4, 1300827 Search PubMed; (b) H. Koshikawa, K. Yoshimura, W. Sinnananchi, T. Yamaki, M. Asano, K. Yamamoto, S. Yamaguchi, H. Tanaka and Y. Maekawa, Macromol. Chem. Phys., 2013, 214, 1756 CrossRef CAS PubMed; (c) T. A. Sharazi, J. Y. Sohn, Y. M. Lee and M. D. Guiver, J. Membr. Sci., 2013, 441, 148 CrossRef PubMed; (d) J. P. Kizewski, N. H. Mudri and J. R. Varcoe, Radiat. Phys. Chem., 2013, 89, 64 CrossRef CAS PubMed; (e) M. Mamlouk, J. A. Horsfall, C. Williams and K. Scott, Int. J. Hydrogen Energy, 2012, 37, 11912 CrossRef CAS PubMed; (f) B.-S. Ko, J.-Y. Sohn and J. Shin, Polymer, 2012, 53, 4652 CrossRef CAS PubMed; (g) L. Ajis, A. A. Nazli, M. A. Abd Malik, M. D. Khairul Zaman and Y. Muhd Zu Azhan, Adv. Mater. Res., 2012, 476–478, 636 Search PubMed; (h) J. Fang, Y. Yang, X. Lu, M. Ye, W. Li and Y. Zhang, Int. J. Hydrogen Energy, 2012, 37, 594 CrossRef CAS PubMed; (i) J. R. Varcoe, R. C. T. Slade, E. Lam How Yee, S. D. Poynton, D. J. Driscoll and D. C. Apperley, Chem. Mater., 2007, 19, 2686 CrossRef CAS; (j) T. N. Danks, R. C. T. Slade and J. R. Varcoe, J. Mater. Chem., 2003, 13, 712 RSC; (k) B. L. Svarfvar, K. B. Ekman, M. J. Sundell and J. H. Nasman, Polym. Adv. Technol., 1996, 7, 839 CrossRef CAS.
- (a) Z. Jiang and Z.-J. Jiang, J. Membr. Sci., 2014, 456, 84 CrossRef PubMed; (b) J. Hu, C. Zhang, L. Jiang, S. Fang, X. Zhang, X. Wang and Y. Meng, J. Power Sources, 2014, 248, 831 CrossRef CAS PubMed; (c) N. Follain, S. Roualdès, S. Marais, J. Frugier, M. Reinholdt and J. Durand, J. Phys. Chem. C, 2012, 116, 8510 CrossRef CAS; (d) C. Zhang, J. Hu, X. Wang, H. Toyoda, M. Nagatsu, X. Zhang and Y. Meng, J. Power Sources, 2012, 198, 112 CrossRef CAS PubMed; (e) C. Zhang, J. Hu, Y. Meng, M. Nagatsu and H. Toyoda, Chem. Commun., 2011, 47, 10230 RSC; (f) C. Zhang, J. Hu, J. Cong, Y. Zhao, W. Shen, H. Toyoda, M. Nagatsu and Y. Meng, J. Power Sources, 2011, 196, 5386 CrossRef CAS PubMed; (g) J. Hu, Y. Meng, C. Zhang and S. Fang, Thin Solid Films, 2011, 519, 2155 CrossRef CAS PubMed; (h) M. Sudoh, S. Niimi, N. Takaota and M. Watanabe, ECS Trans., 2010, 25, 61 CAS; (i) K. Matsuoka, S. Chiba, Y. Iriyama, T. Abe, M. Matsuoka, K. Kikuchi and Z. Ogumi, Thin Solid Films, 2008, 516, 3309 CrossRef CAS PubMed; (j) M. Schieda, S. Roualdès, J. Durand, A. Martinent and D. Marsacq, Desalination, 2006, 199, 286 CrossRef CAS PubMed.
- (a) D.-H. Kim, J.-H. Park, S.-J. Seo, J.-S. Park, S. Jung, Y. S. Kang, J.-H. Choi and M.-S. Kang, J. Membr. Sci., 2013, 447, 80 CrossRef CAS PubMed; (b) S.-H. Park, Y.-W. Choi, C.-S. Kim and S. B. Park, J. Solid State Chem., 2013, 17, 1247 CAS; (c) H. Jung, H. Ohashi, T. Tamaki and T. Yamaguchi, Chem. Lett., 2013, 42, 14 CrossRef CAS; (d) H. Zhang, H. Ohashi, T. Tamaki and T. Yamaguchi, J. Phys. Chem. C, 2012, 116, 7650 CrossRef CAS; (e) H. Jung, K. Fujii, T. Tamaki, H. Ohashi, T. Ito and T. Yamaguchi, J. Membr. Sci., 2011, 373, 107 CrossRef CAS PubMed; (f) X. Zhang, S. W. Tay, Z. Liu and L. Hong, J. Power Sources, 2011, 196, 5494 CrossRef CAS PubMed; (g) A. K. Pandey, R. F. Childs, M. West, J. N. A. Lott, B. E. McCarry and J. M. Dickson, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 807 CrossRef CAS.
- (a) A. M. Park, F. E. Turley, R. J. Wycisk and P. N. Pintauro, Macromolecules, 2014, 47, 227 CrossRef CAS; (b) S. Roddecha, Z. Dong, Y. Wu and M. Anthamatten, J. Membr. Sci., 2012, 389, 478 CrossRef CAS PubMed.
- (a) G. Li, J. Pan, J. Han, C. Chen, J. Lu and L. Zhuang, J. Mater. Chem. A, 2013, 1, 12497 RSC; (b) Y. Zhao, J. Pan, H. Yu, D. Yang, J. Li, L. Zhuang, Z. Shao and B. Yi, Int. J. Hydrogen Energy, 2013, 38, 1983 CrossRef CAS PubMed; (c) Y. Zhao, H. Yu, D. Yang, J. Li, Z. Shao and B. Yi, J. Power Sources, 2013, 221, 247 CrossRef CAS PubMed; (d) Y.-C. Cao, K. Scott and X. Wang, Int. J. Hydrogen Energy, 2012, 37, 12688 CrossRef CAS PubMed; (e) F. Zhang, H. Zhang, J. Ren and C. Qu, J. Mater. Chem., 2010, 20, 8139 RSC.
- (a) T. Zhou, J. Zhang, J. Jingfu, G. Jiang, J. Zhang and J. Qiao, Synth. Met., 2013, 167, 43 CrossRef CAS PubMed; (b) L. Zeng, T. S. Zhao and Y. S. Li, Int. J. Hydrogen Energy, 2012, 37, 18425 CrossRef CAS PubMed; (c) G. Merle, S. S. Hosseiny, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2012, 409–410, 191 CrossRef CAS PubMed; (d) Y. Wu, J. Luo, L. Yao, C. Wu, F. Mao and T. Xu, J. Membr. Sci., 2012, 399–400, 16 CrossRef CAS PubMed; (e) C.-C. Yang, S.-S. Chiu, S.-C. Kuo and T. H. Liou, J. Power Sources, 2012, 199, 37 CrossRef CAS PubMed; (f) J. Fu, J. Qiao, X. Wang, J. Ma and T. Okada, Synth. Met., 2010, 160, 193 CrossRef CAS PubMed; (g) Y. Xiao, J. Fang, Q. H. Zeng and Q. L. Liu, J. Membr. Sci., 2008, 311, 319 CrossRef PubMed; (h) C.-C. Yang, S.-J. Chiu and W.-C. Chien, J. Power Sources, 2006, 162, 21 CrossRef CAS PubMed; (i) T. Sata, K. Kawamura and K. Matsusaki, J. Membr. Sci., 2001, 181, 167 CrossRef CAS.
- O. I. Deavin, S. Murphy, A. Ong, S. D. Poynton, R. Zeng, H. Herman and J. R. Varcoe, Energy Environ. Sci., 2012, 5, 8584 CAS.
- (a) S. Maurya, S.-H. Shin, M.-K. Kim, S.-H. Yun and S.-H. Moon, J. Membr. Sci., 2013, 443, 28 CrossRef CAS PubMed; (b) X. Wang, M. Li, B. T. Golding, M. Sadeghi, Y. Cao, E. H. Yu and K. Scott, Int. J. Hydrogen Energy, 2011, 36, 10022 CrossRef CAS PubMed; (c) M. Faraj, E. Elia, M. Boccia, A. Filpi, A. Pucci and F. Ciardelli, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3437 CrossRef CAS PubMed; (d) A. K. Pandey, A. Goswami, D. Sen, S. Mazumder and R. F. Childs, J. Membr. Sci., 2003, 217, 117 CrossRef CAS; (e) B. Bauer, H. Strathmann and F. Effenberger, Desalination, 1990, 79, 125 CrossRef CAS.
- (a) J. Zhou, K. Joseph, J. M. Ahlfield, D.-Y. Park and P. A. Kohl, J. Electrochem. Soc., 2013, 160, F573 CrossRef CAS PubMed; (b) C. G. Arges, L. Wang, J. Parrondo and V. Ramani, J. Electrochem. Soc., 2013, 160, F1258 CrossRef CAS PubMed.
- (a) M. A. Hossain, Y. Lim, S. Lee, H. Jang, S. Choi, Y. Jeon, S. Lee, H. Ju and W. G. Kim, Solid State Ionics, 2014, 262, 754 CrossRef CAS PubMed; (b) J. Wang, S. Gu, R. B. Kaspar, B. Zhang and Y. Yan, ChemSusChem, 2013, 6, 2079 CrossRef CAS PubMed; (c) O. M. M. Page, S. D. Poynton, S. Murphy, A. Ong, D. M. Hillman, C. A. Hancock, M. G. Hale, D. C. Apperley and J. R. Varcoe, RSC Adv., 2013, 3, 579 RSC; (d) B. Lin, H. Dong, Y. Li, Z. Si, F. Gu and F. Yan, Chem. Mater., 2013, 25, 1858 CrossRef CAS; (e) X. Lin, J. R. Varcoe, S. D. Poynton, X. Liang, A. Ong, J. Ran, Y. Li and T. Xu, J. Mater. Chem. A, 2013, 1, 7262 RSC; (f) D. Chen and M. A. Hickner, ACS Appl. Mater. Interfaces, 2012, 4, 5775 CrossRef CAS PubMed; (g) J. Ran, L. Wu, J. R. Varcoe, A. Ong, S. D. Poynton and T. Xu, J. Membr. Sci., 2012, 415–416, 242 CrossRef CAS PubMed; (h) B. Qiu, B. Lin, Z. Si, L. Qiu, F. Chu, J. Zhao and F. Yan, J. Power Sources, 2012, 217, 329 CrossRef CAS PubMed; (i) B. S. Aitken, C. F. Buitrago, J. D. Heffley, M. Lee, H. W. Gibson, K. I. Winey and K. B. Wagener, Macromolecules, 2012, 45, 681 CrossRef CAS; (j) B. Qiu, B. Lin, L. Qiu and F. Yan, J. Mater. Chem., 2012, 22, 1040 RSC; (k) B. Lin, L. Qiu, B. Qiu, Y. Peng and F. Yan, Macromolecules, 2011, 44, 9642 CrossRef CAS; (l) Y. Ye and Y. A. Elabd, Macromolecules, 2011, 44, 8494 CrossRef CAS; (m) F. Zhang, H. Zhang and C. Qu, J. Mater. Chem., 2011, 21, 12744 RSC; (n) W. Li, J. Fang, M. Lv, C. Chen, X. Chi, Y. Yang and Y. Zhang, J. Mater. Chem., 2011, 21, 11340 RSC; (o) B. Lin, L. Qiu, J. Lu and F. Yan, Chem. Mater., 2010, 22, 6718 CrossRef CAS; (p) M. Guo, J. Fang, H. Xu, W. Li, X. Lu, C. Lan and K. Li, J. Membr. Sci., 2010, 362, 97 CrossRef CAS PubMed.
- X. Lin, X. Liang, S. D. Poynton, J. R. Varcoe, A. Ong, J. Ran, Y. Li, Q. Li and T. Xu, J. Membr. Sci., 2013, 443, 193 CrossRef CAS PubMed.
- L.-C. Jheng, S. L.-C. Hsu, B.-Y. Lin and Y.-L. Hsu, J. Membr. Sci., 2014, 460, 160 CrossRef CAS PubMed.
- (a) J. Miyake, K. Fukasawa, M. Watanabe and K. Miyatake, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 383 CrossRef CAS PubMed; (b) X. Li, Y. Yu, Q. Luiu and Y. Meng, Int. J. Hydrogen Energy, 2013, 38, 11067 CrossRef CAS PubMed; (c) L. I. Şanli and S. A. Gürsel, J. Appl. Polym. Sci., 2011, 120, 2313 CrossRef PubMed; (d) Y. Li and T. Xu, Sep. Purif. Tech., 2008, 61, 430 CrossRef CAS PubMed; (e) A. Huang, C. Xia, C. Xiao and L. Zhuang, J. Appl. Polym. Sci., 2006, 100, 2248 CrossRef CAS PubMed; (f) A. Huang, C. Xiao and L. Zhuang, J. Appl. Polym. Sci., 2005, 96, 2146 CrossRef CAS PubMed.
- (a) S. D. Sajjad, Y. Hong and F. Liu, Polym. Adv. Technol., 2014, 25, 108 CrossRef CAS PubMed; (b) C. H. Zhao, Y. Gong, Q. L. Liu, Q. G. Zhang and A. M. Zhu, Int. J. Hydrogen Energy, 2012, 37, 11383 CrossRef CAS PubMed; (c) X. Lin, L. Wu, Y. Liu, A. Ong, S. D. Poynton, J. R. Varcoe and T. Xu, J. Power Sources, 2012, 217, 373 CrossRef CAS PubMed; (d) C. Qu, H. Zhuang, F. Zhang and B. Liu, J. Mater. Chem., 2012, 22, 8203 RSC; (e) D. S. Kim, A. Labouriau, M. D. Guiver and Y. S. Kim, Chem. Mater., 2011, 23, 3795 CrossRef CAS; (f) M. Li, L. Yang, S. Fang and S. Dong, J. Membr. Sci., 2011, 366, 245 CrossRef CAS PubMed; (g) J. Wang, S. Li and S. Zhang, Macromolecules, 2010, 43, 3890 CrossRef CAS; (h) Q. Zhang, S. Li and S. Zhang, Chem. Commun., 2010, 46, 7495 RSC.
- (a) L. Jiang, X. Lin, J. Ran, C. Li, L. Wu and T. Xu, Chin. J. Chem., 2012, 30, 2241 CrossRef CAS PubMed; (b) S. Gu, R. Cai and Y. Yan, Chem. Commun., 2011, 47, 2856 RSC; (c) K. K. Stokes, J. A. Orlicki and F. L. Beyer, Polym. Chem., 2011, 2, 80 RSC; (d) C. G. Arges, S. Kulkarni, A. Baranek, K.-J. Pan, M.-S. Jung, D. Patton, K. A. Mauritz and V. Ramani, ECS Trans., 2010, 33, 1903 CAS; (e) S. Gu, R. Cai, T. Luo, K. Jensen, C. Contreras and Y. Yan, ChemSusChem, 2010, 3, 555 CrossRef CAS PubMed; (f) S. Gu, R. Cai, T. Luo, Z. Chen, M. Sun, Y. Liu, G. He and Y. Yan, Angew. Chem., Int. Ed., 2009, 48, 6499 CrossRef CAS PubMed; (g) M. Wada and S. Higashizaki, J. Chem. Soc., Chem. Commun., 1984, 482 RSC.
- K. J. T. Noonan, K. M. Hugar, H. A. Kostalik IV, E. B. Lobkovsky, H. A. Abruña and G. W. Coates, J. Am. Chem. Soc., 2012, 134, 18161 CrossRef CAS PubMed.
- B. Zhang, S. Gu, J. Wang, Y. Liu, A. M. Herring and Y. Yan, RSC Adv., 2012, 2, 12683 RSC.
- (a) Y. Wang, A. Rapakousiou and D. Astruc, Macromolecules, 2014, 47, 3767 CrossRef CAS; (b) M. L. Disabb-Miller, Y. Zha, A. J. DeCarlo, M. Pawar, G. N. Tew and M. A. Hickner, Macromolecules, 2013, 46, 9279 CrossRef CAS; (c) P. Y. Xu, C. H. Zhao and Q. L. Liu, J. Appl. Polym. Sci., 2013, 130, 1172 CrossRef PubMed; (d) Y. Zha, M. L. Disabb-Miller, Z. D. Johnson, M. A. Hickner and G. N. Tew, J. Am. Chem. Soc., 2012, 134, 4493 CrossRef CAS PubMed.
- (a) Z. Siroma, S. Watanabe, K. Yasuda, K. Fukuta and H. Yanagi, J. Electrochem. Soc., 2011, 158, B682 CrossRef CAS PubMed; (b) H. Yanagi and K. Fukuta, ECS Trans., 2008, 16, 257 CAS.
- Y.-L. S. Tse, H. N. Sarode, G. E. Lindberg, T. A. Witten, A. M. Herring and G. A. Voth, J. Phys. Chem. C, 2014, 118, 845 CAS.
- V. Heagu, I. Bunia and I. Plesca, Polym. Degrad. Stab., 2000, 70, 463 CrossRef.
- (a) M. R. Hibbs, M. A. Hickner, T. M. Alam, S. K. McIntyre, C. H. Fujimoto and C. J. Cornelius, Chem. Mater., 2008, 20, 2566 CrossRef CAS; (b) C. Zhang, J. Hu, M. Nagatsu, Y. Meng, W. Shen, H. Toyoda and X. Shu, Plasma Processes Polym., 2011, 8, 1024 CrossRef CAS PubMed; (c) R. C. T. Slade and J. R. Varcoe, Solid State Ionics, 2005, 176, 585 CrossRef CAS PubMed.
- (a) E. N. Komkova, D. F. Stamatialis, H. Strathmann and M. Wessling, J. Membr. Sci., 2004, 244, 25 CrossRef CAS PubMed; (b) A. A. Zagorodni, D. L. Kotova and V. F. Selemenev, React. Funct. Polym., 2002, 53, 157 CrossRef CAS.
- F. Karas, J. Hnát, M. Paidar, J. Schauer and K. Bouzek, Int. J. Hydrogen Energy, 2014, 39, 5054 CrossRef CAS PubMed.
- D. Y. Park, P. A. Kohl and H. W. Beckham, J. Phys. Chem. C, 2013, 117, 15468 CAS.
- N. Li, Y. Leng, M. A. Hickner and C.-Y. Wang, J. Am. Chem. Soc., 2013, 135, 10124 CrossRef CAS PubMed.
- (a) H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem. C, 2012, 116, 9419 CrossRef CAS; (b) J. B. Edson, C. S. Macomber, B. S. Pivovar and J. M. Boncella, J. Membr. Sci., 2012, 399–400, 49–59 CrossRef CAS PubMed; (c) S. Chempath, J. M. Boncella, L. R. Pratt, N. Henson and B. S. Pivovar, J. Phys. Chem. C, 2010, 114, 11977 CrossRef CAS; (d) S. Chempath, B. R. Einsla, L. R. Pratt, C. S. Macomber, J. M. Boncella, J. A. Rau and B. S. Pivovar, J. Phys. Chem. C, 2008, 112, 3179 CrossRef CAS; (e) C. S. Macomber, J. M. Boncella, B. S. Pivovar and J. A. Rau, J. Therm. Anal. Calorim., 2008, 93, 225 CrossRef CAS.
- J. R. Varcoe, Phys. Chem. Chem. Phys., 2007, 9, 1479 RSC.
- J. Yan and M. A. Hickner, Macromolecules, 2010, 43, 2349 CrossRef CAS.
- (a) W. Lu, Z.-G. Shao, G. Zhang, Y. Zhao, J. Li and B. Yi, Int. J. Hydrogen Energy, 2013, 38, 9285 CrossRef CAS PubMed; (b) P. Y. Xu, T. Y. Guo, C. H. Zhao, I. Broadwell, Q. G. Zhang and Q. L. Liu, J. Appl. Polym. Sci., 2013, 128, 3853 CrossRef PubMed; (c) F. Schönberger, J. Kerres, H. Dilger and E. Roduner, Phys. Chem. Chem. Phys., 2009, 11, 5782 RSC; (d) G. Hübner and E. Roduner, J. Mater. Chem., 1999, 9, 409 RSC.
- (a) M. P. Rodgers, L. J. Bonville, H. R. Kunz, D. K. Slattery and J. F. Fenton, Chem. Rev., 2012, 112, 6075 CrossRef CAS PubMed; (b) R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K.-I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904 CrossRef CAS PubMed.
- T. Sata, M. Tsujimoto, T. Yamaguchi and K. Matsusaki, J. Membr. Sci., 1996, 112, 161 CrossRef CAS.
- (a) M. R. Hibbs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1736 CrossRef CAS PubMed; (b) Z. Zhang, L. Wu, J. R. Varcoe, C. Li, A. Ong, S. D. Poynton and T. Xu, J. Mater. Chem. A, 2013, 1, 2595 RSC; (c) L. Zeng and T. S. Zhao, Electrochem. Commun., 2013, 34, 278 CrossRef CAS PubMed; (d) W. Lu, Z.-S. Shao, G. Zhang, J. Li, Y. Zhao and B. Yi, Solid State Ionics, 2013, 245–246, 8 CrossRef CAS PubMed; (e) J.-S. Park, S.-H. Park, S.-D. Yim, Y.-G. Yoon, W.-Y. Lee and C.-S. Kim, J. Power Sources, 2008, 178, 620 CrossRef CAS PubMed; (f) J.-S. Park, G.-G. Park, S.-H. Park, Y.-G. Yoon, C. S. Kim and W. Y. Lee, Macromol. Symp., 2007, 249–250, 174 CrossRef PubMed; (g) M. Tomoi, K. Yamaguchi, R. Ando, Y. Kantake, Y. Aosaki and H. Kubota, J. Appl. Polym. Sci., 1997, 64, 1161 CrossRef CAS.
- R. Janarthanan, S. K. Pilli, J. L. Horan, D. A. Gamarra, M. R. Hibbs and A. M. Herring, J. Electrochem. Soc., 2014, 161, F944 CrossRef CAS PubMed.
- C. Iojoiu, F. Chabert, M. Maréchal, N. El Kissi, J. Guindet and J.-Y. Sanchez, J. Power Sources, 2006, 153, 198 CrossRef CAS PubMed.
- U.-S. Hwang and J.-H. Choi, Sep. Purif. Technol., 2006, 48, 16 CrossRef CAS PubMed.
- http://www.sigmaaldrich.com/, USA website accessed 12 January 2014.
- http://www.sigmaaldrich.com/catalog/product/aldrich/197602, Safety Data Sheet.
- L. Liu, Q. Li, J. Dai, H. Wang, B. Jin and R. Bai, J. Membr. Sci., 2014, 453, 52 CrossRef CAS PubMed.
- Y. Yang and P. Knauss, Proceedings of the 242nd American Chemical Society Meeting, Denver, USA, 2011, Abstract 241 Search PubMed.
- (a) J. Hu, D. Wan, W. Zhu, L. Huang, S. Tan, X. Cai and X. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 4720 CrossRef CAS PubMed; (b) A. H. N. Rao, S. Nam and T.-H. Kim, Int. J. Hydrogen Energy, 2014, 39, 5919 CrossRef CAS PubMed.
- C. G. Arges and V. Ramani, J. Electrochem. Soc., 2013, 160, F1006 CrossRef CAS PubMed.
- (a) W. Wang, S. Wang, X. Xie, Y. Lv and V. Ramani, Int. J. Hydrogen Energy, 2014 DOI:10.1016/j.ijhydene.2014.03.053; (b) W. Wang, S. Wang, X. Xie, Y. Lv and V. K. Ramani, J. Membr. Sci., 2014, 462, 112 CrossRef CAS PubMed.
- (a) Z. Si, L. Qiu, H. Dong, F. Gu, Y. Li and F. Yan, ACS Appl. Mater. Interfaces, 2014, 6, 4346 CrossRef CAS PubMed; (b) Z. Si, Z. Sun, F. Gu, L. Qiu and F. Yan, J. Mater. Chem. A, 2014, 2, 4413 RSC; (c) Y. Yang, J. Wang, J. Zheng, S. Li and S. Zhang, J. Membr. Sci., 2014, 467, 48 CrossRef CAS PubMed.
- F. Gu, H. Dong, Y. Li, Z. Si and F. Yan, Macromolecules, 2014, 47, 208 CrossRef CAS.
- H. Long and B. S. Pivovar, J. Phys. Chem. C, 2014, 118, 9880 CAS.
- S. C. Price, K. S. Williams and F. L. Beyer, ACS Macro Lett., 2014, 3, 160 CrossRef CAS.
- Y. Liu, J. Wang, Y. Yang, T. M. Brenner, S. Seifert, Y. Yan, M. W. Liberatore and A. M. Herring, J. Phys. Chem. C, 2014, 118, 15136 CAS.
- K. C. Lethesh, W. Dehaen and K. Binnemans, RSC Adv., 2014, 4, 4472 RSC.
- A. G. Wright and S. Holdcroft, ACS Macro Lett., 2014, 3, 444 CrossRef CAS.
- Y. Ye, K. K. Stokes, F. L. Beyer and Y. A. Elabd, J. Membr. Sci., 2013, 443, 93 CrossRef CAS PubMed.
- R. Schwesinger, R. Link, P. Wenzl, S. Kossek and M. Keller, Chem.–Eur. J., 2006, 12, 429 CrossRef PubMed.
- S. A. Nuñez and M. A. Hickner, ACS Macro Lett., 2013, 2, 49 CrossRef.
- A. Amel, L. Zhu, M. Hickner and Y. Ein-Eli, J. Electrochem. Soc., 2014, 161, F615 CrossRef CAS PubMed.
- (a) A. D. Mohanty, Y.-B. Lee, L. Zhu, M. A. Hickner and C. Bae, Macromolecules, 2014, 47, 1973 CrossRef CAS; (b) X. Li, G. Nie, J. Tao, W. Wu, L. Wang and S. Liao, ACS Appl. Mater. Interfaces, 2014, 6, 7585 CrossRef CAS PubMed.
- (a) Q. Li, L. Liu, Q. Miao, B. Jin and R. Bai, Chem. Commun., 2014, 50, 2791 RSC; (b) J. Pan, Y. Li, J. Han, G. Li, L. Tan, C. Chen, J. Lu and L. Zhuang, Energy Environ. Sci., 2013, 6, 2912 RSC.
- (a) C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490 CrossRef CAS PubMed; (b) Z. Zhao, F. Gong, S. Zhang and S. Li, J. Power Sources, 2012, 218, 368 CrossRef CAS PubMed.
- D. M. Hillman, S. H. Stephens, S. D. Poynton, S. Murphy, A. Ong and J. R. Varcoe, J. Mater. Chem. A, 2013, 1, 1018 CAS.
- J. P. Kizewski, N. H. Mudri, R. Zeng, S. D. Poynton, R. C. T. Slade and J. R. Varcoe, ECS Trans., 2010, 33, 27 CAS.
- D. Marx, A. Chandra and M. E. Tukerman, Chem. Rev., 2010, 110, 2174 CrossRef CAS PubMed.
- R. F. Silva, M. De Francesco and A. Pozio, J. Power Sources, 2004, 134, 18 CrossRef CAS PubMed.
- M. A. Vandiver, B. R. Caire, J. R. Carver, K. Waldrop, M. R. Hibbs, J. R. Varcoe, A. M. Herring and M. W. Liberatore, J. Electrochem. Soc., 2014, 161, H677 CrossRef CAS PubMed.
- T. Kimura and Y. Yamazaki, Electrochemistry, 2011, 79, 94 CrossRef CAS PubMed.
- Atkins' Physical Chemistry, ed. P. Atkins and J. de Paula, Oxford University Press, Oxford, 8th edn, Table 21.6, p. 019 Search PubMed.
- http://web.med.unsw.edu.au/phbsoft/mobility_listings.htm, accessed 27 September 2013.
- I. T. Lucas, S. Durand-Vidal, E. Dubois, J. Chevalet and P. Turq, J. Phys. Chem. C, 2007, 111, 18568 CAS.
- D. Chaturvedi and S. Ray, Monatshefte für Chemie, 2006, 137, 459 CrossRef CAS PubMed.
- M. G. Marino, J. P. Melchior, A. Wohlfarth and K. D. Kreuer, J. Membr. Sci., 2014, 464, 61 CrossRef CAS PubMed.
- H. N. Sarode, G. E. Lindberg, Y. Yang, L. Felberg, G. A. Voth and A. M. Herring, J. Phys. Chem. B, 2014, 118, 1363 CrossRef CAS PubMed.
- (a) M. Mamlouk, K. Scott, J. A. Horsfall and C. Williams, Int. J. Hydrogen Energy, 2011, 36, 7191 CrossRef CAS PubMed; (b) T. Soboleva, Z. Xie, Z. Shi, E. Tsang, T. Navessin and S. Holdcroft, J. Electroanal. Chem., 2008, 622, 145 CrossRef CAS PubMed; (c) S. Ma, Z. Siroma and H. Tanaka, J. Electrochem. Soc., 2006, 153, A2274 CrossRef CAS PubMed.
- (a) K. W. Han, K. K. Ko, K. Abu-Hakmeh, C. Bae, Y. J. Sohn and S. S. Jang, J. Phys. Chem. C, 2014, 118, 12577 CrossRef CAS; (b) A. M. Kiss, T. D. Myles, K. N. Grew, A. A. Peracchino, G. J. Nelson and W. K. S. Chiu, J. Electrochem. Soc., 2013, 160, F994 CrossRef CAS PubMed; (c) B. V. Merinov and W. A. Goddard III, J. Membr. Sci., 2013, 431, 79 CrossRef CAS PubMed; (d) G. A. Giffin, S. Lavina, G. Pace and V. Di Noto, J. Phys. Chem. C, 2012, 116, 23965 CrossRef CAS; (e) T. D. Myles, A. M. Kiss, K. N. Grew, A. A. Peracchino, G. J. Nelson and W. K. S. Chiu, J. Electrochem. Soc., 2011, 158, B790 CrossRef CAS PubMed; (f) K. N. Grew, X. Ren and D. Chu, Electrochem. Solid-State Lett., 2011, 14, B127 CrossRef CAS PubMed; (g) K. N. Grew, D. Chu and W. K. S. Chiu, J. Electrochem. Soc., 2010, 57, B1024 CrossRef PubMed; (h) K. N. Grew and W. K. S. Chiu, J. Electrochem. Soc., 2010, 157, B327 CrossRef CAS PubMed; (i) Y. S. Li, T. S. Zhao and W. W. Yang, Int. J. Hydrogen Energy, 2010, 35, 5656 CrossRef CAS PubMed.
- (a) S. Zhang, C. Li, X. Xie and F. Zhang, Int. J. Hydrogen Energy, 2014, 39, 13718 CrossRef CAS PubMed; (b) J. Wang, G. He, X. Wu, X. Yan, Y. Zhang, Y. Wang and L. Du, J. Membr. Sci., 2014, 459, 86 CrossRef CAS PubMed; (c) L. Wang and M. A. Hickner, Polym. Chem., 2014, 5, 2928 RSC.
- N. Li, M. D. Guiver and W. H. Binder, ChemSusChem, 2013, 6, 1376 CrossRef CAS PubMed.
- N. Li and M. D. Guiver, Macromolecules, 2014, 47, 2175 CrossRef CAS.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535 CrossRef CAS.
- M. E. Tuckerman, D. Marx and M. Parrinello, Nature, 2002, 417, 925 CrossRef CAS PubMed.
- J. Pan, S. Lu, Y. Li, A. Huang, L. Zhuang and J. Lu, Adv. Funct. Mater., 2010, 20, 312 CrossRef CAS PubMed.
- (a) G. L. Han, P. Y. Xu, K. Zhou, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Membr. Sci., 2014, 464, 72 CrossRef CAS PubMed; (b) H.-C. Lee, K.-L. Liu, L.-D. Tsai, J.-Y. Lai and C.-Y. Chao, RSC Adv., 2014, 4, 10944 RSC; (c) J. Ran, L. Wu, X. Lin, L. Jiang and T. Xu, RSC Adv., 2012, 2, 4250 RSC.
- T. J. Peckham and S. Holdcroft, Adv. Mater., 2010, 22, 4667 CrossRef CAS PubMed.
- (a) M. A. Hossain, Y. Lim, S. Lee, H. Jang, S. Choi, Y. Jeon, J. Lim and W. G. Kim, Int. J. Hydrogen Energy, 2014, 39, 2731 CrossRef CAS PubMed; (b) Y. Ye, S. Sharick, E. M. Davis, K. I. Winey and Y. A. Elabd, ACS Macro Lett., 2013, 2, 575 CrossRef CAS; (c) X. Wang, M. Goswami, R. Kumar, B. G. Sumpter and J. Mays, Soft Matter, 2012, 8, 3036 RSC; (d) Z. Zhao, J. Wang, S. Li and S. Zhang, J. Power Sources, 2011, 196, 4445 CrossRef CAS PubMed; (e) M. Tanaka, K. Fukasawa, E. Nishino, S. Yamaguchi, K. Yamada, H. Tanaka, B. Bae, K. Miyatake and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 10646 CrossRef CAS PubMed.
- M. L. Disabb-Miller, Z. D. Johnson and M. A. Hickner, Macromolecules, 2013, 46, 949 CrossRef CAS.
- S. C. Price, X. Ren, A. C. Jackson, Y. Ye, Y. A. Elabd and F. B. Beyer, Macromolecules, 2013, 46, 7332 CrossRef CAS.
- Q. Li, L. Liu, Q. Mao, B. Jin and R. Bai, Polym. Chem., 2014, 5, 2208 RSC.
- N. Li, Q. Zhang, C. Wang, Y. M. Lee and M. D. Guiver, Macromolecules, 2012, 45, 2411 CrossRef CAS.
- N. Li, T. Yan, Z. Li, T. Thurn-Albrecht and W. H. Binder, Energy Environ. Sci., 2012, 5, 7888 CAS.
- N. Li, L. Wang and M. A. Hickner, Chem. Commun., 2014, 50, 4092 RSC.
- J. Ran, L. Wu and T. Xu, Polym. Chem., 2013, 4, 4612 RSC.
- J. Pan, C. Chen, Y. Li, L. Wang, L. Tan, G. Li, X. Tang, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2014, 7, 354 CAS.
- S. Karaborni, K. Esselink, P. A. J. Hilbers, B. Smit, J. Karthäuser, N. M. Van Os and R. Zana, Science, 1994, 266, 254 CAS.
- http://www2.dupont.com/FuelCells/en_US/products/nafion.html , accessed February 2014 .
- K. Scott, E. H. Yu, G. Vlachogiannopoulos, M. Shivare and N. Duteanu, J. Power Sources, 2008, 175, 452 CrossRef CAS PubMed.
- (a) G. K. S. Prakash, F. C. Krause, F. A. Viva, S. R. Narayanan and G. A. Olah, J. Power Sources, 2011, 196, 7967 CrossRef CAS PubMed; (b) K. Fukuta, H. Inoue, Y. Chikashige and H. Yanagi, ECS Trans., 2010, 28, 221 CAS; (c) Y. S. Li, T. S. Zhao and Z. X. Liang, J. Power Sources, 2009, 190, 223 CrossRef CAS PubMed; (d) H. Bunazawa and Y. Yamazaki, J. Power Sources, 2008, 182, 48 CrossRef CAS PubMed.
- (a) M. S. Naughton, G. H. Gu, A. A. Moradia and P. J. A. Kenis, J. Power Sources, 2013, 242, 581 CrossRef CAS PubMed; (b) C. V. Rao and Y. Ishikawa, J. Phys. Chem. C, 2012, 116, 4340 CrossRef; (c) A. Santasalo-Aarnio, S. Hietala, T. Rauhala and T. Kallio, J. Power Sources, 2011, 196, 6153 CrossRef CAS PubMed; (d) C. Delacourt, P. L. Ridgway, J. B. Kerr and J. Newman, J. Electrochem. Soc., 2008, 155, B42 CrossRef CAS PubMed.
- (a) L. Sun, J. Guo, J. Zhou, Q. Xu, D. Chu and R. Chen, J. Power Sources, 2012, 202, 70 CrossRef CAS PubMed; (b) M. Ünlü, J. Zhou, I. Anestis-Richard and P. A. Kohl, ChemSusChem, 2011, 3, 1398 CrossRef PubMed; (c) F. Zhang, H. Zhang, C. Qu and J. Ren, J. Power Sources, 2011, 196, 3099 CrossRef CAS PubMed; (d) K. Miyazaki, N. Sugimura, K.-I. Kawakita, T. Abe, K. Nisho, H. Nakanishi, M. Matsuoka and Z. Ogumi, J. Electrochem. Soc., 2010, 157, A1153 CrossRef CAS PubMed; (e) M. Piana, M. Boccia, A. Filipi, E. Flammia, H. A. Miller, M. Orsini, F. Salusti, S. Santiccioli, F. Ciardelli and A. Pucci, J. Power Sources, 2010, 195, 5875 CrossRef CAS PubMed; (f) J. R. Varcoe, R. C. T. Slade and E. Lam How Yee, Chem. Commun., 2006, 1428 RSC.
- R. Zeng, J. Handsel, S. D. Poynton, A. J. Roberts, R. C. T. Slade, H. Herman, D. C. Apperley and J. R. Varcoe, Energy Environ. Sci., 2011, 4, 4925 CAS.
- D. Dekel, Alkaline membrane fuel cells, in Encyclopedia of Applied Electrochemistry, ed. R. Savinell, K.-I. Ota and G. Kreysa, Springer, Berlin/Heidelberg, SpringerReference number 303632 Search PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102 CrossRef CAS PubMed.
- M. Weissmann, C. Coutanceau, P. Brault and J.-M. Léger, Electrochem. Commun., 2007, 9, 1097 CrossRef CAS PubMed.
- (a) T. Lee, E. K. Jeon and B.-S. Kim, J. Mater. Chem. A, 2014, 2, 6167 RSC; (b) J. Ohyama, Y. Okata, N. Watabe, M. Katagiri, A. Nakamura, H. Arikawa, K.-I. Shimizu, T. Takeguchi, W. Ueda and A. Satsuma, J. Power Sources, 2014, 245, 998 CrossRef CAS PubMed; (c) Q. Wu, L. Jiang, Q. Tang, J. Liu, S. Wang and G. Sun, Electrochim. Acta, 2013, 91, 314 CrossRef CAS PubMed; (d) S. Gu, W. Sheng, R. Cai, S. M. Alia, S. Song, K. O. Jensen and Y. Yan, Chem. Commun., 2013, 49, 131 RSC; (e) N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443 CrossRef CAS PubMed; (f) Y. Fang, X. Yang, L. Wang and Y. Li, Electrochim. Acta, 2013, 90, 421 CrossRef CAS PubMed; (g) J. Xu, P. Gao and T. S. Zhao, Energy Environ. Sci., 2012, 5, 5333 RSC; (h) J. Sunarso, A. A. J. Torriero, W. Zhou, P. C. Howlett and M. Forsyth, J. Phys. Chem. C, 2012, 116, 5827 CrossRef CAS; (i) N. Ramaswamy and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 18015 CrossRef CAS; (j) J. Guo, A. Hsu, D. Chu and R. Chen, J. Phys. Chem. C, 2010, 114, 4324 CrossRef CAS; (k) T. S. Olson, S. Pylypenko, P. Atanassov, K. Asazawa, K. Yamada and H. Tanaka, J. Phys. Chem. C, 2010, 114, 5049 CrossRef CAS; (l) R. Chen, H. Li, D. Chu and G. Wang, J. Phys. Chem. C, 2009, 113, 20689 CrossRef CAS.
- B. S. Pivovar, AMFC Workshop Final Report, 2006, located at, http://www1.eere.energy.gov/hydrogenandfuelcells/pdfs/amfc_dec2006_workshop_report.pdf.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9 CrossRef CAS PubMed.
- D. S. Su and G. Sun, Angew. Chem., Int. Ed., 2011, 50, 11570 CrossRef CAS PubMed.
- (a) Q. Hu, G. Li, J. Pan, L. Tan, J. Lu and L. Zhuang, Int. J. Hydrogen Energy, 2013, 38, 16264 CrossRef CAS PubMed; (b) D. Dekel, ECS Trans., 2012, 50, 2051 CrossRef PubMed; (c) S. Lu, J. Pan, A. Huang, L. Zhuang and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20611 CrossRef CAS; (d) J. R. Varcoe, R. C. T. Slade, G. L. Wright and Y. Chen, J. Phys. Chem. B, 2006, 110, 21041 CrossRef CAS PubMed.
- C. A. Hancock, A. Ong, P. R. Slater and J. R. Varcoe, J. Mater. Chem. A, 2014, 2, 3047 CAS.
- J. S. Spendelow and A. Wieckowski, Phys. Chem. Chem. Phys., 2007, 9, 2654 RSC.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529 CrossRef CAS PubMed.
- L. Jiang, A. Hsu, D. Chu and R. Chen, Electrochim. Acta, 2010, 55, 4506 CrossRef CAS PubMed.
- M. Piana, S. Catanorchi and H. A. Gasteiger, ECS Trans., 2008, 16, 2045 CAS.
- H. Meng, F. Jaouen, E. Proietti, M. Lefèvre and J. P. Dodelet, Electrochem. Commun., 2009, 11, 1986 CrossRef CAS PubMed.
- Q. He, X. Yang, R. He, A. Bueno-López, H. Miller, X. Ren, W. Yang and B. E. Koel, J. Power Sources, 2012, 213, 169 CrossRef CAS PubMed.
- K. C. Neyerlin, W. Gu, J. Jorne and H. A. Gasteiger, J. Electrochem. Soc., 2007, 154, B631 CrossRef CAS PubMed.
- (a) R. Zeng, R. C. T. Slade and J. R. Varcoe, Electrochim. Acta, 2010, 56, 607 CrossRef CAS PubMed; (b) R. Zeng, S. D. Poynton, J. P. Kizewski, R. C. T. Slade and J. R. Varcoe, Electrochem. Commun., 2010, 12, 823 CrossRef CAS PubMed.
- P. Rheinländer, S. Henning, J. Herranz and H. A. Gasteiger, ECS Trans., 2012, 50, 2163 CrossRef PubMed.
- R. Jervis, N. Mansor, C. Gibbs, C. A. Murray, C. C. Tang, P. R. Shearing and D. J. L. Brett, J. Electrochem. Soc., 2014, 161, F458 CrossRef CAS PubMed.
- W. Sheng, A. P. Bivens, M. Myint, Z. Zhuang, R. V. Forest, Q. Fang, J. G. Cheng and Y. Yan, Energy Environ. Sci., 2014, 7, 1719 CAS.
- (a) I. Gunasekara, M. Lee, D. Abbott and S. Mukerjee, ECS Electrochem. Lett., 2012, 1, F16 CrossRef CAS PubMed; (b) A. E. S. Sleightholme and A. R. Kucernak, Electrochim. Acta, 2011, 56, 4396 CrossRef CAS PubMed; (c) A. E. S. Sleightholme, J. R. Varcoe and A. R. Kucernak, Electrochem. Commun., 2008, 10, 151 CrossRef CAS PubMed; (d) A. R. Kucernak and E. Toyoda, Electrochem. Commun., 2008, 10, 1728 CrossRef CAS PubMed; (e) J. Jiang and A. R. Kucernak, J. Electroanal. Chem., 2004, 567, 123 CrossRef CAS PubMed.
- A. Ong, D. K. Whelligan, M. L. Fox and J. R. Varcoe, Phys. Chem. Chem. Phys., 2013, 15, 18992 RSC.
- A. Ong, D. K. Whelligan, S. Murphy, J. R. Varcoe, manuscript in preparation.
- T. J. Schmidt, U. A. Paulus, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 508, 41 CrossRef CAS.
- A. Anastasopoulos, J. C. Davis, L. Hannah, B. E. Hayden, C. E. Lee, C. Milhano, C. Mormiche and L. Offin, ChemSusChem, 2013, 6, 1973 CrossRef CAS PubMed.
- W. Lu, Z.-G. Shao, G. Zhang, Y. Zhao, J. Li and B. Yi, Int. J. Hydrogen Energy, 2013, 38, 9285 CrossRef CAS PubMed.
- D. Konopka, M. A. Johnson, M. Errico, P. Bahrami and C. A. Hays, Electrochem. Solid-State Lett., 2012, 15, B17 CrossRef CAS PubMed.
- A. J. Lemke, A. W. O'Toole, R. S. Phillips and E. T. Eisenbraun, J. Power Sources, 2014, 256, 319 CrossRef CAS PubMed.
- M. H. Robson, K. Artyushkova, W. Patterson, P. Atanassov and M. R. Hibbs, Electrocatalysis, 2014, 5, 148 CrossRef CAS PubMed.
- S. D. Poynton, R. C. T. Slade, W. E. Mustain, T. J. Omasta, R. Escudero-Cid, P. Ocón and J. R. Varcoe, J. Mater. Chem. A, 2014, 2, 5124 CAS.
- (a) G. F. McLean, T. Niet, S. Prince-Richard and N. Djilali, Int. J. Hydrogen Energy, 2002, 27, 507 CrossRef CAS; (b) A. J. Appleby and F. R. Foulkes, Fuel Cell Handbook, Van Nostrand Reinhold, New York, 1989 Search PubMed.
- Y. Wang, L. Li, L. Hu, L. Zhuang, J. Lu and B. Xu, Electrochem. Commun., 2003, 5, 662 CrossRef CAS.
- L. A. Adams, S. D. Poynton, C. Tamain, R. C. T. Slade and J. R. Varcoe, ChemSusChem, 2008, 1, 79 CrossRef CAS PubMed.
- M. Ünlü, J. Zhou and P. A. Kohl, Electrochem. Solid-State Lett., 2009, 12, B27 CrossRef PubMed.
- (a) M. Carmo, G. Doubek, R. C. Sekol, M. Linardi and A. D. Taylor, J. Power Sources, 2013, 230, 169 CrossRef CAS PubMed; (b) Q. He and X. Ren, J. Power Sources, 2012, 220, 373 CrossRef CAS PubMed; (c) X. Wang, J. P. McClure and P. S. Fedkiw, Electrochim. Acta, 2012, 79, 126 CrossRef CAS PubMed.
- Y. S. Kim, US DOE Hydrogen and fuel cells program and vehicle technologies program annual merit review, 2011, http://www.hydrogen.energy.gov/pdfs/review11/fc043_kim_2011_o.pdf.
- M. J. Jung, C. G. Arges and V. Ramani, J. Mater. Chem., 2011, 21, 6158 RSC.
- Y. Luo, J. Guo, C. Wang and D. Chu, Macromol. Chem. Phys., 2011, 212, 2094 CrossRef CAS PubMed.
- S. D. Poynton, J. P. Kizewski, R. C. T. Slade and J. R. Varcoe, Solid State Ionics, 2010, 181, 219 CrossRef CAS PubMed.
- K. Fukuta, “Electrolyte Materials for AMFCs and AMFC Performance” at the AMFC Workshop held on 8th May 2011 in Washington (USA).
- H. Zhang, H. Ohashi, T. Tamaki and T. Yamaguchi, J. Phys. Chem. C, 2013, 117, 16791 CAS.
- K. Jiao, P. He, Q. Du and Y. Yin, Int. J. Hydrogen Energy, 2014, 39, 5981 CrossRef CAS PubMed.
- E. M. Sommer, L. S. Martins, J. V. C. Vargas, J. E. F. C. Gardolinski, J. C. Ordonez and C. E. B. Marino, J. Power Sources, 2012, 213, 16 CrossRef CAS PubMed.
- (a) E. H. Yu, X. Wang, U. Krewer, L. Li and K. Scott, Energy Environ. Sci., 2012, 5, 5668 RSC; (b) L. An, T. S. Zhao, Y. Li and Q. Wu, Energy Environ. Sci., 2012, 5, 7536 RSC.
- (a) C. Jin, Y. Song and Z. Chen, Electrochim. Acta, 2009, 54, 4136 CrossRef CAS PubMed; (b) Y. Chen, L. Zhuang and J. Lu, Chin. J. Catal., 2007, 28, 870 CrossRef CAS; (c) A. V. Tripković, K. D. Popović and J. D. Lović, Electrochim. Acta, 2001, 46, 3163 CrossRef.
- R. R. Adzic, M. L. Avramovivic and A. V. Tripković, Electrochim. Acta, 1984, 29, 1353 CrossRef CAS.
- (a) J. Zhang, C. Guo, L. Zhang and C. M. Li, Chem. Commun., 2013, 49, 6334 RSC; (b) S. Maheswari, P. Sridhar and S. Pitchmani, Fuel Cells, 2012, 12, 963 CrossRef CAS PubMed; (c) G. Dong, M. Huang and L. Guan, Phys. Chem. Chem. Phys., 2012, 14, 2557 RSC; (d) M. Zhiani, H. A. Gasteiger, M. Piana and S. Catanorchi, Int. J. Hydrogen Energy, 2011, 36, 5110 CrossRef CAS PubMed; (e) S. Song, Y. Wang, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2008, 78, 381 CrossRef CAS PubMed; (f) N. Park, T. Shiraishi, K. Kamisugi, Y. Hara, K. Iizuka, T. Kado and S. Hayase, J. Appl. Electrochem., 2008, 38, 371 CrossRef CAS; (g) H. Meng, M. Wu, X. X. Hu, M. Nie, Z. D. Wei and P. K. Shen, Fuel Cells, 2006, 6, 447 CrossRef CAS PubMed; (h) E. H. Yu, K. Scott and R. W. Reeve, Fuel Cells, 2003, 3, 169 CrossRef PubMed.
- (a) M. Saito, S. Tsuzuki, K. Hayamizu and T. Okada, J. Phys. Chem. B, 2006, 110, 24410 CrossRef CAS PubMed; (b) J. Ling and O. Savadogo, J. Electrochem. Soc., 2004, 151, A1604 CrossRef CAS PubMed; (c) X. Ren, T. E. Springer and S. Gottesfeld, J. Electrochem. Soc., 2000, 147, 92 CrossRef CAS PubMed.
- C. Xu and T. S. Zhao, J. Power Sources, 2007, 168, 143 CrossRef CAS PubMed.
- C. Weinzierl and U. Krewer, J. Power Sources, 2014, 268, 911 CrossRef CAS PubMed.
- J. Divisek, US Pat. 20030049509, 2003; M. Stefener and J. Müller, US Pat. 20050069757, 2005.
- K. Matsuoka, Y. Iriyama, T. Abe, M. Matsuoka and Z. Ogumi, J. Power Sources, 2005, 150, 27 CrossRef CAS PubMed.
- T. F. Fuller and D. J. Wheeler, US Pat. 6068941 A, 2000.
- (a) J. Liu, J. Ye, C. Xu, S. P. Jiang and Y. Tong, J. Power Sources, 2008, 177, 67 CrossRef CAS PubMed; (b) J. Ye, J. Liu, C. Xu, S. P. Jiang and Y. Tong, Electrochem. Commun., 2007, 9, 2760 CrossRef CAS PubMed; (c) M. E. P. Markiewicz, D. M. Hebert and S. H. Bergens, J. Power Sources, 2006, 161, 761 CrossRef CAS PubMed.
- J. R. Varcoe, R. C. T. Slade, E. Lam How Yee, S. D. Poynton and R. C. T. Slade, J. Power Sources, 2007, 173, 194 CrossRef CAS PubMed.
- G. A. Koscher and K. Kordesch, J. Solid State Electrochem., 2003, 7, 632 CrossRef CAS.
- (a) Y. Zhang, J. Fang, Y. Wu, H. Xu, X. Chi, W. Li, Y. Yang, G. Yan and Y. Zhuang, J. Colloid Interface Sci., 2012, 381, 59 CrossRef CAS PubMed; (b) C.-C. Yang, J. Appl. Electrochem., 2012, 42, 305 CrossRef CAS PubMed; (c) H. Kim, J. Zhou, M. Ünlü, I. Anestis-Richard, K. Joseph and P. A. Kohl, Electrochim. Acta, 2011, 56, 3085 CrossRef CAS PubMed; (d) J.-H. Kim, H.-K. Kim, K.-T. Hwang and J.-Y. Lee, Int. J. Hydrogen Energy, 2010, 35, 768 CrossRef CAS PubMed; (e) J. Kim, T. Momma and T. Osaka, J. Power Sources, 2009, 189, 999 CrossRef CAS PubMed; (f) E. H. Yu and K. Scott, J. Appl. Electrochem., 2005, 35, 91 CrossRef CAS PubMed; (g) E. H. Yu and K. Scott, Electrochem. Commun., 2004, 6, 361 CrossRef CAS PubMed.
- R. W. Verjulio, J. Santander, N. Sabaté, J. P. Esquivel, N. Torres-Herrero, A. Habrioux and N. Alonso-Vante, Int. J. Hydrogen Energy, 2014, 39, 5406 CrossRef CAS PubMed.
- A. V. Tripković, K. D. Popović, B. N. Grgur, B. Blizanac, P. N. Ross and N. M. Marković, Electrochim. Acta, 2002, 47, 3707 CrossRef.
- K. L. Nagashree and M. F. Ahmed, Synth. Met., 2008, 158, 610 CrossRef CAS PubMed.
- N. Spinner and W. E. Mustain, Electrochim. Acta, 2011, 56, 5656 CrossRef CAS PubMed.
- M. Ünlü, D. Abbott, N. Ramaswamy, X. Ren, S. Mukerjee and P. A. Kohl, J. Electrochem. Soc., 2011, 158, B1423 CrossRef PubMed.
- (a) P. Joghee, S. Pylypenko, K. Wood, G. Bender and R. O'Hayre, ChemSusChem, 2014, 7, 1854 CrossRef CAS PubMed; (b) J. Wang, R. Shi, X. Guo, J. Xin, J. Zhao, C. Song, L. Wang and J. Zhang, Int. J. Hydrogen Energy, 2014, 123, 309 CAS; (c) E. E. Switzer, T. S. Olson, A. K. Datye, P. Atanassov, M. R. Hibbs and C. J. Cornelius, Electrochim. Acta, 2009, 54, 989 CrossRef CAS PubMed; (d) C. Nishihara and T. Okada, J. Electroanal. Chem., 2005, 577, 355 CrossRef CAS PubMed; (e) P. V. Samant, C. M. Rangel, M. H. Romero, J. B. Fernades and J. L. Figueiredo, J. Power Sources, 2005, 151, 79 CrossRef CAS PubMed; (f) X. Zhang, K.-Y. Tsang and K.-Y. Chan, J. Electroanal. Chem., 2004, 573, 1 CrossRef CAS.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183 CrossRef CAS PubMed.
- V. Bambagioni, C. Bianchini, A. Marchionni, J. Filippi, F. Vizza, J. Teddy, P. Serp and M. Zhiani, J. Power Sources, 2009, 190, 241 CrossRef CAS PubMed.
- (a) D. H. Nagaraju and V. Lakshminarayanan, J. Phys. Chem. C, 2009, 113, 14922 CrossRef CAS; (b) K. A. Assiongbon and D. Roy, Surf. Sci., 2005, 594, 99 CrossRef CAS PubMed; (c) Z. Borkowska, A. Tymosiak-Zielinska and R. Nowakowski, Electrochim. Acta, 2004, 49, 2613 CrossRef CAS PubMed.
- M. G. Hosseini, M. Abdolmaleki and S. Ashrafpoor, J. Appl. Electrochem., 2012, 42, 153 CrossRef CAS; M. A. Abdel Rahim, R. M. Abdel Hameed and M. W. Khalil, J. Power Sources, 2004, 134, 160 CrossRef PubMed; J. Taraszewska and G. Roslonek, J. Electroanal. Chem., 1994, 364, 209 CrossRef.
- G. G. W. Lee, J. Leddy and S. D. Minteer, Chem. Commun., 2012, 48, 11972 RSC.
- (a) E. Antolini and J. Perez, Int. J. Hydrogen Energy, 2011, 36, 15752 CrossRef CAS PubMed; (b) R. N. Singh, T. Sharma, A. Singh, D. Anindita, D. Mishra and S. K. Tiwari, Electrochim. Acta, 2008, 53, 2322 CrossRef CAS PubMed.
- (a) J. Zhou, M. Ünlü, I. Anestis-Richard and P. A. Kohl, J. Membr. Sci., 2010, 350, 286 CrossRef CAS PubMed; (b) H. Hou, G. Sun, R. He, B. Sun, W. Jin, H. Liu and Q. Xin, Int. J. Hydrogen Energy, 2008, 33, 7172 CAS; (c) E. H. Yu and K. Scott, J. Power Sources, 2004, 137, 248 CrossRef CAS PubMed.
- T. M. Alam and M. R. Hibbs, Macromolecules, 2014, 47, 1073 CrossRef CAS.
- (a) A. Jasti and V. K. Shahi, RSC Adv., 2014, 4, 19238 RSC; (b) C. Zhao, W. Ma, W. Sun and H. Na, J. Appl. Polym. Sci., 2014, 131, 40256 Search PubMed; (c) A. H. N. Rao, H.-J. Kim, S. Nam and T.-H. Kim, Polymer, 2013, 54, 6918 CrossRef CAS PubMed; (d) M. M. Nasef, N. A. Zubirm, A. F. Ismail, M. Khayet, K. Z. M. Dahlan, H. Saidi, R. Rohani, T. I. S. Ngah and N. A. Sulaiman, J. Membr. Sci., 2006, 268, 96 CrossRef CAS PubMed.
- (a) G. K. Goswami, R. Nandan, B. K. Barman and K. K. Nanda, J. Mater. Chem. A, 2013, 1, 3133 RSC; (b) A. Santasalo-Aarnio, S. Tuomi, K. Jalkanen, K. Kontturi and T. Kallio, Electrochim. Acta, 2013, 87, 730 CrossRef CAS PubMed; (c) H. Bunazawa and Y. Yamazaki, J. Power Sources, 2009, 190, 210 CrossRef CAS PubMed.
- H. Deng, J. Chen, K. Jiao and X. Huang, Int. J. Heat Mass Transfer, 2014, 7, 376 CrossRef PubMed.
- A. Katzfuß, S. Poynton, J. Varcoe, V. Gogel, U. Storr and J. Kerres, J. Membr. Sci., 2014, 465, 129 CrossRef PubMed.
- T. S. Zhao, Y. S. Li and S. Y. Shen, Front. Energy Power Eng. Chin., 2010, 4, 443 CrossRef.
- (a) S. Zinoviev, F. Müller-Langer, P. Das, N. Bertero, P. Fornasiero, M. Kaltschmitt, G. Centi and S. Miertus, ChemSusChem, 2010, 3, 1106 CrossRef CAS PubMed; (b) K. David and A. J. Ragauskas, Energy Environ. Sci., 2010, 3, 1182 RSC.
- S. Y. Shen, T. S. Zhao and Q. X. Wu, Int. J. Hydrogen Energy, 2012, 37, 575 CrossRef CAS PubMed.
- V. Rao, Hariyanto, C. Cremers and U. Stimming, Fuel Cells, 2007, 7, 417 CrossRef CAS PubMed.
- (a) S. Sun, Z. Jusys and R. J. Behm, J. Power Sources, 2013, 231, 122 CrossRef CAS PubMed; (b) E. Antolini, J. Power Sources, 2007, 170, 1 CrossRef CAS PubMed.
- (a) P. A. Christensen, S. W. M. Jones and A. Hamnett, J. Phys. Chem. C, 2012, 116, 24681 CrossRef CAS; (b) Y. Bai, J. Wu, J. Xi, J. Wang, W. Zhu, L. Chen and X. Qiu, Electrochem. Commun., 2005, 7, 1087 CrossRef CAS PubMed; (c) C. Su and P. K. Shen, J. Power Sources, 2005, 142, 27 CrossRef PubMed.
- (a) E. Antolini, Energy Environ. Sci., 2009, 2, 915 RSC; (b) G. Cui, S. Song, P. K. Shen, A. Kowal and C. Bianchini, J. Phys. Chem. C, 2009, 113, 15639 CrossRef CAS; (c) Z. X. Liang, T. S. Zhao, J. B. Xu and L. D. Zhu, Electrochim. Acta, 2009, 54, 2203 CrossRef CAS PubMed; (d) Q. He, W. Chen, S. Mukerjee, S. Chen and F. Laufek, J. Power Sources, 2009, 187, 298 CrossRef CAS PubMed; (e) F. P. Hu, Z. Wang, Y. Li, C. Li, X. Zhang and P. K. Shen, J. Power Sources, 2008, 177, 61 CrossRef CAS PubMed; (f) C. Xu, H. Wang, P. K. Shen and S. P. Jiang, Adv. Mater., 2007, 19, 4256 CrossRef CAS PubMed; (g) H. Wang, C. Xu, F. Cheng and S. Jiang, Electrochem. Commun., 2007, 9, 1212 CrossRef CAS PubMed; (h) J. Bagchi and S. K. Bhattacharya, Transition Met. Chem., 2007, 32, 47 CrossRef CAS PubMed.
- T. Sheng, W.-F. Lin, C. Hardacre and P. Hu, J. Phys. Chem. C, 2014, 118, 5762 CAS.
- Y. S. Li and T. S. Zhao, Int. J. Hydrogen Energy, 2012, 37, 4413 CrossRef CAS PubMed.
- C. Xu, Z. Tian, P. K. Shen and S. P. Jiang, Electrochim. Acta, 2008, 53, 2610 CrossRef CAS PubMed.
- F. Hu, C. Chen, Z. Wang, G. Wei and P. K. Shen, Electrochim. Acta, 2006, 52, 1087 CrossRef CAS PubMed.
- P. K. Shen and C. Xu, Electrochem. Commun., 2006, 8, 184 CrossRef CAS PubMed.
- A. N. Geraldes, D. F. da Silva, E. S. Pino, J. C. M. da Silva, R. F. B. de Souza, P. Hammer, E. V. Spinancé, A. O. Neto, M. Linardi and M. C. dos Santos, Electrochim. Acta, 2013, 111, 455 CrossRef CAS PubMed.
- A. Dutta and J. Datta, J. Phys. Chem. C, 2012, 116, 25677 CAS.
- S. Shen, T. S. Zhao, J. Xu and Y. Li, Energy Environ. Sci., 2011, 4, 1428 CAS.
- L. Ma, H. He, A. Hsu and R. Chen, J. Power Sources, 2013, 241, 696 CrossRef CAS PubMed.
- Y. Li and Y. He, RSC Adv., 2014, 4, 16879 RSC.
- (a) R. B. de Lima and H. Varela, Gold Bull., 2008, 41, 15 CrossRef CAS; (b) C. Xu, H. Yu, J. Rong, S. P. Jiang and Y. Liu, Electrochem. Commun., 2007, 9, 2009 CrossRef CAS PubMed; (c) S. S. Gupta and J. Datta, J. Power Sources, 2005, 145, 124 CrossRef PubMed.
- M. R. Tarasevich, Z. R. Karichev, V. A. Bogdanovskaya, E. N. Lubnin and A. V. Kapustin, Electrochem. Commun., 2005, 7, 141 CrossRef CAS PubMed.
- H. Hou, G. Sun, R. He, Z. Wu and B. Sun, J. Power Sources, 2008, 182, 95 CrossRef CAS PubMed.
- L. An, T. S. Zhao, Q. X. Wu and L. Zeng, Int. J. Hydrogen Energy, 2012, 37, 14536 CrossRef CAS PubMed.
- C. Bianchini, V. Bambagioni, J. Filippi, A. Marchionni, F. Vizza, P. Bert and A. Tampucci, Electrochem. Commun., 2009, 11, 1077 CrossRef CAS PubMed.
- J. B. Xu, T. S. Zhao, Y. S. Li and W. W. Yang, Int. J. Hydrogen Energy, 2010, 35, 9693 CrossRef CAS PubMed.
- L. An and T. S. Zhao, Int. J. Hydrogen Energy, 2011, 36, 9994 CrossRef CAS PubMed.
- (a) L. An, T. S. Zhao, S. Y. Shen, Q. X. Wu and R. Chen, Int. J. Hydrogen Energy, 2010, 35, 4329 CrossRef CAS PubMed; (b) K. Miyazaki, N. Sugimura, K. Matsuoka, Y. Iriyama, T. Abe, M. Matsuoka and Z. Ogumi, J. Power Sources, 2008, 178, 683 CrossRef CAS PubMed; (c) L. Demarconnay, S. Brimaud, C. Coutanceau and J.-M. Léger, J. Electroanal. Chem., 2007, 601, 169 CrossRef CAS PubMed; (d) C. Coutanceau, L. Demarconnay, C. Lamy and J.-M. Léger, J. Power Sources, 2006, 156, 14 CrossRef CAS PubMed.
- (a) K. Matsuoka, Y. Iriyama, T. Abe, M. Matsuoka and Z. Ogumi, J. Electrochem. Soc., 2005, 182, A729 CrossRef PubMed; (b) K. Matsuoka, M. Inaba, Y. Iriyama, T. Abe, Z. Ogumi and M. Matsuoka, Fuel Cells, 2002, 2, 35 CrossRef CAS.
- K. Matsuoka, Y. Iriyama, T. Abe, M. Matsuoka and Z. Ogumi, Electrochim. Acta, 2005, 51, 1085 CrossRef CAS PubMed.
- C. Cremers, D. Bayer, J. Meier, S. Berenger, B. Kintzel, M. Joos and J. Tübke, ECS Trans., 2010, 25, 27 CAS.
- A. Serov, U. Martinez and P. Atanassov, Electrochem. Commun., 2013, 34, 185 CrossRef CAS PubMed.
- (a) J. Qi, L. Xin, D. J. Chadderdon, Y. Qiu, Y. Jiang, N. Benipal, C. Liang and W. Li, Appl. Catal., B, 2014, 154–155, 360 CrossRef CAS PubMed; (b) Z. Wang, L. Xin, X. Zhao, Y. Qiu, Z. Zhang, O. A. Baturina and W. Li, Renewable Energy, 2014, 62, 556 CrossRef CAS PubMed; (c) Z. Zhang, L. Xin, J. Qi, D. J. Chadderdon and W. Li, Appl. Catal., B, 2013, 136–137, 29 CAS; (d) Z. Zhang, L. Xin and W. Li, Int. J. Hydrogen Energy, 2012, 37, 9393 CrossRef CAS PubMed; (e) Z. Zhang, L. Xin and W. Li, Appl. Catal., B, 2012, 119–120, 40 CrossRef CAS PubMed; (f) A. Ilie, M. Simões, S. Baranton, C. Coutanceau and S. Martemianov, J. Power Sources, 2011, 196, 4965 CrossRef CAS PubMed.
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542 CAS.
- R. L. Arechederra and S. D. Minteer, Fuel Cells, 2009, 9, 63 CrossRef CAS PubMed.
- (a) V. L. Oliveira, C. Morais, K. Servat, T. W. Napporn, G. Tremiliosa-Filho and K. B. Kokoh, Electrochim. Acta, 2014, 117, 255 CrossRef CAS PubMed; (b) L. Roquet, E. M. Belgsir, J.-M. Léger and C. Lamy, Electrochim. Acta, 1994, 39, 2387 CrossRef CAS.
- M. Simões, S. Baranton and C. Coutanceau, Appl. Catal., B, 2010, 93, 354 CrossRef PubMed.
- J.-H. Zhang, Y.-J. Liang, N. Li, Z.-Y. Li, C.-W. Xu and S. P. Jiang, Electrochim. Acta, 2012, 59, 156 CrossRef CAS PubMed.
- D. J. Jeffrey and G. A. Camara, Electrochem. Commun., 2010, 12, 1129 CrossRef PubMed.
- A. Zalineeva, A. Serov, M. Padilla, U. Martinez, K. Artyushkova, S. Baranton, C. Coutanceau and P. B. Atanassov, J. Am. Chem. Soc., 2014, 136, 3937 CrossRef CAS PubMed.
- http://www.superiorideas.org/projects/fuel-cells .
- (a) L. Zeng, Z. K. Tang and T. S. Zhao, Appl. Energy, 2014, 115, 405 CrossRef CAS PubMed; (b) A. M. Bartrom, J. Ta, T. Q. Nguyen, J. Her, A. Donovan and J. L. Haan, J. Power Sources, 2013, 229, 234 CrossRef CAS PubMed; (c) A. M. Bartrom and J. L. Haan, J. Power Sources, 2012, 214, 68 CrossRef CAS PubMed.
- (a) R. Pathak and S. Basu, Electrochim. Acta, 2013, 113, 42 CrossRef CAS PubMed; (b) J. Chen, C. X. Zhao, M. M. Zhi, K. Wang, L. Deng and G. Xu, Electrochim. Acta, 2012, 66, 133 CrossRef CAS PubMed; (c) L. An, T. S. Zhao, S. Y. Shen, Q. X. Wu and R. Chen, J. Power Sources, 2011, 196, 186 CrossRef CAS PubMed; (d) N. Fujiwara, S.-I. Yamazaki, Z. Siroma, T. Ioroi, H. Senoh and K. Yasuda, Electrochem. Commun., 2009, 11, 390 CrossRef CAS PubMed.
- (a) R. Lan and S. W. Tao, J. Power Sources, 2011, 196, 5021 CrossRef CAS PubMed; (b) A. N. Rollinson, J. Jones, V. Dupont and M. V. Twigg, Energy Environ. Sci., 2011, 4, 1216 RSC; (c) R. Lan, S. W. Tao and J. T. S. Irvine, Energy Environ. Sci., 2010, 3, 438 RSC.
- K. Xu, S. J. Lao, H. Y. Qin, B. H. Liu and Z. P. Li, J. Power Sources, 2010, 195, 5606 CrossRef CAS PubMed.
- N. V. Rees and R. G. Compton, Energy Environ. Sci., 2011, 4, 1255 CAS.
- (a) G. E. Evans and K. V. Kordesch, Science, 1967, 158, 1148 CAS; (b) A. Kunugi, Z. Takehara and S. Yoshizawa, Denki Kagaku, 1965, 33, 211 CAS; (c) S. Karp and L. Meites, J. Am. Chem. Soc., 1962, 84, 906 CrossRef CAS.
- S. Takahashi, S. Higuchi, R. Fujii and Y. Miyake, Report of Governmental Industrial Research Institute, Osaka (Japan), 1974, vol. 346 Search PubMed.
- K. Yamada, K. Yasuda, N. Fujiwara, Z. Siroma, H. Tanaka, Y. Miyazaki and T. Kobayashi, Electrochem. Commun., 2003, 5, 892 CrossRef CAS PubMed.
- K. Asazawa, K. Yamada, H. Tanaka, A. Oka, M. Taniguchi and T. Kobayshi, Angew. Chem., Int. Ed., 2007, 46, 8024 CrossRef CAS PubMed.
- (a) T. Sakamoto, K. Asazawa, J. Sanabria-Chinchilla, U. Martinez, B. Halevi, P. Antanassov, P. Strasser and H. Tanaka, J. Power Sources, 2014, 247, 605 CrossRef CAS PubMed; (b) T. Sakamoto, K. Asazawa, U. Martinez, B. Halevi, T. Susuki, S. Arai, D. Matsumura, Y. Nishihata, P. Atanassov and H. Tanaka, J. Power Sources, 2013, 234, 252 CrossRef CAS PubMed.
- H. Tanaka, K. Asazawa, T. Sakamoto, T. Kato, M. Kai, S. Yamaguchi, K. Yamada and H. Fujikawa, ECS Trans., 2008, 16, 459 CAS.
- W. X. Yin, Z. P. Li, J. K. Zhu and H. Y. Qin, J. Power Sources, 2008, 182, 520 CrossRef CAS PubMed.
- K. Asazawa, K. Yamada, H. Tanaka, M. Taniguchi and K. Oguro, J. Power Sources, 2009, 191, 362 CrossRef CAS PubMed.
- (a) U. Martinez, K. Asazawa, B. Halevi, A. Falase, B. Kiefer, A. Serov, M. Padilla, T. Olson, A. Datye, H. Tanaka and P. Atanassov, Phys. Chem. Chem. Phys., 2012, 14, 5512 RSC; (b) T. Sakamoto, K. Asazawa, K. Yamada and H. Tanaka, Catal. Today, 2011, 164, 181 CrossRef CAS PubMed.
- G. Karim-Nezhad, R. Jafarloo and P. S. Dorraji, Electrochim. Acta, 2009, 54, 5721 CrossRef CAS PubMed.
- H. Gao, Y. Wang, F. Xiao, C. B. Chung and H. Duan, J. Phys. Chem. C, 2012, 116, 7719 CAS.
- S. J. Lao, H. Y. Qin, L. Q. Ye, B. H. Liu and Z. P. Li, J. Power Sources, 2010, 195, 4135 CrossRef CAS PubMed.
- (a) M. H. M. T. Assumpção, S. G. da Silva, R. F. B. de Souza, G. S. Buzzo, E. V. Spinacé, A. O. Neto and J. C. M. Silva, Int. J. Hydrogen Energy, 2014, 39, 5148 CrossRef PubMed; (b) R. Lan, J. T. S. Irvine and S. W. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482 CrossRef CAS PubMed; (c) S. Suzuki, H. Muroyama, T. Matsui and K. Eguchi, J. Power Sources, 2012, 208, 257 CrossRef CAS PubMed; (d) R. Lan and S. W. Tao, Electrochem. Solid-State Lett., 2010, 13, B83 CrossRef CAS PubMed.
- K. R. Lee, D. Song, S. B. Park and J.-I. Han, RSC Adv., 2014, 4, 5638 RSC.
- (a) E. Bertin, C. Roy, S. Garbarino, D. Guay, J. Solla-Gullón, F. J. Vidal-Iglesias and J. M. Feliu, Electrochem. Commun., 2012, 22, 197 CrossRef CAS PubMed; (b) B. K. Boggs and G. G. Botte, Electrochim. Acta, 2010, 55, 5287 CrossRef CAS PubMed; (c) K. Yao and Y. F. Cheng, Int. J. Hydrogen Energy, 2008, 33, 6681 CrossRef CAS PubMed; (d) L. Zhou, Y. F. Cheng and M. Amrein, J. Power Sources, 2008, 177, 50 CrossRef CAS PubMed; (e) F. J. Vidal-Iglesias, J. Solla-Gullón, V. Montiel, J. M. Feliu and A. Aldaz, J. Power Sources, 2007, 171, 448 CrossRef CAS PubMed; (f) V. Rosca and M. T. M. Koper, Phys. Chem. Chem. Phys., 2006, 8, 2513 RSC; (g) F. J. Vidal-Iglesias, J. Solla-Gullón, J. M. Feliu, H. Baltruschat and A. Aldaz, J. Electroanal. Chem., 2006, 588, 331 CrossRef CAS PubMed; (h) F. J. Vidal-Iglesias, J. Solla-Gullón, J. M. Pérez and A. Aldaz, Electrochem. Commun., 2006, 8, 102 CrossRef CAS PubMed; (i) K. Endo, Y. Katayama and T. Miura, Electrochim. Acta, 2005, 50, 2181 CrossRef CAS PubMed; (j) A. C. A. de Vooys, M. T. M. Koper, R. A. Van Santen and J. A. R. Van Veen, J. Electroanal. Chem., 2001, 506, 127 CrossRef CAS.
- W. Peng, L. Xiao, B. Huang, L. Zhuang and J. Lu, J. Phys. Chem. C, 2011, 115, 23050 CAS.
- T. Hejze, J. O. Besenhard, K. Kordesch, M. Cifrain and R. R. Aronsson, J. Power Sources, 2008, 176, 490 CrossRef CAS PubMed.
- R. Halseid, P. J. S. Vie and R. Tunold, J. Power Sources, 2006, 154, 343 CrossRef CAS PubMed.
- T. Lopes, D. S. Kim, Y. S. Kim and F. H. Garzon, J. Electrochem. Soc., 2012, 159, B265 CrossRef CAS PubMed.
- I. Merino-Jiménez, C. Ponce de León, A. A. Shah and F. C. Walsh, J. Power Sources, 2012, 219, 339 CrossRef PubMed.
- A. Serov, A. Aziznia, P. H. Benhangi, K. Artyushkova, P. Atanassov and E. Gyenge, J. Mater. Chem. A, 2013, 1, 14384 CAS.
- C. Ponce de León, F. C. Walsh, C. J. Patrissi, M. G. Medeiros, R. R. Bessette, R. W. Reeve, J. B. Lakeman, A. Rose and D. Browning, Electrochem. Commun., 2008, 10, 1610 CrossRef PubMed.
- H. Cheng, K. Scott, K. V. Lovell, J. A. Horsfall and S. C. Waring, J. Membr. Sci., 2007, 288, 168 CrossRef CAS PubMed.
- (a) B. Šljukić, D. M. F. Santos and C. A. C. Sequeira, J. Electroanal. Chem., 2013, 694, 77 CrossRef PubMed; (b) M. Chatenet, F. Micoud, I. Roche, E. Chainet and J. Vondrák, Electrochim. Acta, 2006, 51, 5452 CrossRef CAS PubMed; (c) R. X. Feng, H. Dong, Y. D. Wang, X. P. Ai, Y. L. Cao and H. X. Yang, Electrochem. Commun., 2005, 7, 449 CrossRef CAS PubMed.
- (a) C. Celik, F. G. Boyaci San and H. I. Sarac, Int. J. Hydrogen Energy, 2010, 35, 8678 CrossRef CAS PubMed; (b) R. Jamard, A. Latour, J. Salomon, P. Capron and A. Martinent-Beaumont, J. Power Sources, 2008, 176, 287 CrossRef CAS PubMed; (c) U. B. Demirci, Electrochim. Acta, 2007, 52, 5119 CrossRef CAS PubMed.
- B. H. Liu, Z. P. Li, J. K. Zhu and S. Suda, J. Power Sources, 2008, 183, 151 CrossRef CAS PubMed.
- J.-H. Wee, J. Power Sources, 2006, 155, 329 CrossRef CAS PubMed.
- (a) F. H. B. Lima, A. M. Pasqualeti, M. B. M. Concha, M. Chatenet and E. A. Ticianelli, Electrochim. Acta, 2012, 84, 202 CrossRef CAS PubMed; (b) M. Chatenet, F. H. B. Lima and E. A. Ticianelli, J. Electrochem. Soc., 2010, 157, B697 CrossRef CAS PubMed; (c) D. A. Finkelstein, N. Da Mota, J. L. Cohen and H. D. Abuña, J. Phys. Chem. C, 2009, 13, 19700 CrossRef.
- D. A. Finkelstein, C. D. Letcher, D. J. Jones, L. M. Sandberg, D. J. Watts and H. D. Abuña, J. Phys. Chem. C, 2013, 117, 1571 CAS.
- (a) M. H. Atwan, D. O. Northwood and E. L. Gyenge, Int. J. Hydrogen Energy, 2007, 32, 3116 CrossRef CAS PubMed; (b) E. Sanli, H. Çelikkan, B. Z. Uysal and M. L. Aksu, Int. J. Hydrogen Energy, 2006, 31, 1920 CrossRef CAS PubMed.
- (a) G. Behmenyar and A. N. Akin, J. Power Sources, 2014, 249, 239 CrossRef CAS PubMed; (b) M. Simões, S. Baranton and C. Coutanceau, J. Phys. Chem. C, 2009, 113, 13369 CrossRef.
- G. Rostamikia and M. J. Janik, Energy Environ. Sci., 2010, 3, 1262 CAS.
- (a) C.-C. Huang, Y.-L. Liu, W.-H. Pan, C.-M. Chang, C.-M. Shih, H.-Y. Chu, C.-H. Chien, C.-H. Juan and S. J. Lue, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1779 CrossRef CAS PubMed; (b) N. Duteanu, G. Vlachogiannopoulos, M. R. Shivhare, E. H. Yu and K. Scott, J. Appl. Electrochem., 2007, 37, 1085 CrossRef CAS.
- C. Qu, H. Zhang, F. Zhang and B. Liu, J. Mater. Chem., 2012, 22, 8203 RSC.
- L. Gu, N. Luo and G. H. Miley, J. Power Sources, 2007, 173, 77 CrossRef CAS PubMed.
- Y. Wang, P. He and H. Zhou, Energy Environ. Sci., 2010, 3, 1515 CAS.
- X. Yang, Y. Liu, S. Li, X. Wei, L. Wang and Y. Chen, Sci. Report., 2012, 2, 567 Search PubMed.
- H. Qin, Z. Liu, Y. Guo and Z. Li, Int. J. Hydrogen Energy, 2010, 35, 2868 CrossRef CAS PubMed.
- (a) L. C. Nagle and J. F. Rohan, J. Electrochem. Soc., 2011, 158, B772 CrossRef CAS PubMed; (b) U. B. Demirci and P. Miele, Energy Environ. Sci., 2009, 2, 627 RSC; (c) X.-B. Zhang, J.-M. Yan, S. Han, H. Shioyama, K. Yasuda, N. Kuriyama and Q. Xu, J. Power Sources, 2008, 182, 515 CrossRef CAS PubMed; (d) X.-B. Zhang, S. Han, J.-M. Yan, M. Chandra, H. Shioyama, K. Yasuda, N. Kuriyama, T. Kobayashi and Q. Xu, J. Power Sources, 2007, 168, 167 CrossRef CAS PubMed; (e) C. Yao, H. Yang, L. Zhuang, X. Ai, Y. Cao and J. Lu, J. Power Sources, 2007, 165, 125 CrossRef CAS PubMed.
- T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116 CrossRef CAS PubMed.
- X.-B. Zhang, S. Han, J.-M. Yan, H. Shioyama, N. Kuriyama, T. Kobayashi and Q. Xu, Int. J. Hydrogen Energy, 2009, 34, 174 CrossRef CAS PubMed.
- (a) J. A. Vega, S. Smith and W. E. Mustain, J. Electrochem. Soc., 2011, 158, B349 CrossRef CAS PubMed; (b) J. A. Vega, C. Chartier and W. E. Mustain, J. Power Sources, 2010, 195, 7176 CrossRef CAS PubMed; (c) J. A. Vega and W. E. Mustain, Electrochim. Acta, 2010, 55, 1638 CrossRef CAS PubMed.
- I. Gunasekara, M. Lee, D. Abbott and S. Mukerjee, ECS Electrochem. Lett., 2012, 1, F16 CrossRef CAS PubMed.
- (a) B. Connors and B. Ramson, J. Physiol., 1984, 355, 619 CAS; (b) E. Nightingale, J. Phys. Chem., 1959, 63, 1381 CrossRef CAS.
- C. M. Lang, K. Kim and P. A. Kohl, Electrochem. Solid-State Lett., 2006, 9, A545 CrossRef CAS PubMed.
- (a) N. Spinner and W. E. Mustain, J. Electrochem. Soc., 2013, 160, F1275 CrossRef CAS PubMed; (b) N. Spinner and W. E. Mustain, J. Electrochem. Soc., 2012, 159, E187 CrossRef CAS PubMed; (c) J. A. Vega, N. Spinner, M. Catanese and W. E. Mustain, J. Electrochem. Soc., 2012, 159, B19 CrossRef CAS PubMed; (d) J. A. Vega, S. Shrestha, M. Ignatowich and W. E. Mustain, J. Electrochem. Soc., 2012, 159, B12 CrossRef CAS PubMed; (e) H. W. Pennline, E. J. Granite, D. R. Luebke, J. R. Kitchin, J. Landon and L. M. Weiland, Fuel, 2010, 89, 1307 CrossRef CAS PubMed.
- I. Gunasekara, M. Lee, D. Abbott and S. Mukerjee, ECS Electrochem. Lett., 2012, 1, F16 CrossRef CAS PubMed.
- C. A. Hancock, A. Ong and J. R. Varcoe, RSC Adv., 2014, 4, 30035 RSC.
- (a) I. Gunasekara, D. Abbott and S. Mukerjee, Proceedings of the 221st Electrochemical Society Meeting, Seattle USA, 2012, Abstract 228 Search PubMed; (b) M. S. Naughton, F. R. Brushett and P. A. Kenis, J. Power Sources, 2011, 196, 1762 CrossRef CAS PubMed.
- (a) M. Stoukides, J. Appl. Electrochem., 1995, 25, 899 CrossRef CAS; (b) Y. Amenomiya, V. Birss, M. Goledzinowski, J. Galuszka and A. Sanger, Catal. Rev.: Sci. Eng., 1990, 32, 163 CrossRef CAS.
- J. S. Spendelow, J. D. Goodpaster, P. J. A. Kenis and A. Wieckowski, J. Phys. Chem. B, 2006, 110, 9545 CrossRef CAS PubMed.
- (a) S. Mafé and P. Ramírez, Acta Polym., 1997, 48, 234 CrossRef PubMed; (b) P. Ramírez, S. Mafé, J. A. Manzanares and J. Pellicer, J. Electroanal. Chem., 1996, 404, 187 CrossRef; (c) R. Simons, J. Membr. Sci., 1993, 78, 13 CrossRef CAS; (d) P. Ramírez, V. M. Aguilella, J. A. Manzanares and S. Mafé, J. Membr. Sci., 1992, 73, 191 CrossRef; (e) R. Simons, Electrochim. Acta, 1985, 30, 275 CrossRef CAS; (f) R. Simons and G. Khanarian, J. Membr. Biol., 1978, 38, 11 CrossRef CAS.
- (a) A. Iizuka, K. Hashimoto, H. Nagasawa, Y. Yanagisawa and A. Yamasaki, Sep. Purif. Technol., 2012, 101, 49 CrossRef CAS PubMed; (b) M. D. Eisaman, L. Alvarado, D. Larner, P. Wang and K. A. Littau, Energy Environ. Sci., 2011, 4, 4031 RSC; (c) M. D. Eisaman, L. Alvarado, D. Larner, P. Wang, B. Garg and K. A. Littau, Energy Environ. Sci., 2011, 4, 1319 RSC.
- (a) M. Ünlü, J. Zhou and P. A. Kohl, Angew. Chem., Int. Ed., 2010, 49, 1299 CrossRef PubMed; (b) M. Ünlü, J. Zhou and P. A. Kohl, J. Electrochem. Soc., 2010, 157, B1391 CrossRef PubMed; (c) M. Ünlü, J. Zhou and P. A. Kohl, J. Phys. Chem. C, 2009, 113, 11416 CrossRef.
- M. Ünlü, J. Zhou and P. A. Kohl, Fuel Cells, 2010, 10, 54 Search PubMed.
- (a) I. D. Raistrick, Electrochim. Acta, 1990, 35, 1579 CrossRef CAS; (b) T. E. Springer, T. A. Zawodzinski, M. S. Wilson and S. Gottesfeld, J. Electrochem. Soc., 1996, 143, 587 CrossRef CAS PubMed.
- J. M. Spurgeon, M. G. Walter, J. Zhou, P. A. Kohl and N. S. Lewis, Energy Environ. Sci., 2011, 4, 1172 Search PubMed.
- (a) A. Heller, H. J. Lewerenz and B. Miller, J. Am. Chem. Soc., 1981, 103, 200 CrossRef CAS; (b) M. Szklarczyk and J. O. M. Bockris, J. Phys. Chem., 1984, 88, 1808 CrossRef CAS.
- B. D. Alexander, P. J. Kulesza, L. Rutkowska and J. Augustynski, J. Mater. Chem., 2008, 18, 2298 RSC.
- A. L. Dicks, J. Power Sources, 2006, 156, 128 CrossRef CAS PubMed.
- (a) J. P. Meyers and R. M. Darling, J. Electrochem. Soc., 2006, 153, A1432 CrossRef CAS PubMed; (b) C. A. Reiser, L. Bregoli, T. W. Patterson, J. S. Yi, J. D. L. Yang, M. L. Perry and T. D. Jarvi, Electrochem. Solid-State Lett., 2005, 8, A273 CrossRef CAS PubMed.
- W. W. Jacques, US Pat., 555511, 1896.
- M. L. Perry and T. F. Fuller, J. Electrochem. Soc., 2002, 149, S59 CrossRef CAS PubMed.
- (a) D. Cao, Y. Sun and G. Wang, J. Power Sources, 2007, 167, 250 CrossRef CAS PubMed; (b) S. Zecevic, E. M. Patton and P. Parhami, Carbon, 2004, 42, 1983 CrossRef CAS PubMed.
- K. Tomantschger, R. Findlay, M. Hanson, K. Kordesch and S. Srinivasan, J. Power Sources, 1992, 39, 21 CrossRef CAS.
- B. K. Kakati, D. Sathiyamoorthy and A. Verma, Int. J. Hydrogen Energy, 2010, 35, 4185 CrossRef CAS PubMed.
- H. Kinoshita, S. Yonezawa, J.-H. Kim, M. Kawai, M. Takashima and T. Tsukatani, J. Power Sources, 2008, 183, 464 CrossRef CAS PubMed.
- (a) F. Bidault, D. J. L. Brett, P. H. Middleton, N. Abson and N. P. Brandon, Int. J. Hydrogen Energy, 2010, 35, 1783 CrossRef CAS PubMed; (b) F. Bidault, D. J. L. Brett, P. H. Middleton, N. Abson and N. P. Brandon, Int. J. Hydrogen Energy, 2009, 34, 6799 CrossRef CAS PubMed.
- F. Bidault and A. Kucernak, J. Power Sources, 2010, 195, 2549 CrossRef CAS PubMed.
- (a) A. Kucernak, F. Bidault and G. Smith, Electrochim. Acta, 2012, 82, 284 CrossRef CAS PubMed; (b) F. Bidault and A. Kucernak, J. Power Sources, 2011, 196, 4950 CrossRef CAS PubMed.
- S. M. Alia, K. Duong, T. Liu, K. Jensen and Y. Yan, ChemSusChem, 2012, 5, 1619 CrossRef CAS PubMed.
- A. Luzzi, L. Bonadio and M. McCann, in Pursuit of the Future. 25 years of IEA research towards the realisation of Hydrogen Energy Systems, IEA-HIA Report, 2004, ISBN 0-9752270-0-9 Search PubMed.
- W. Kreuter and H. Hofmann, Int. J. Hydrogen Energy, 1998, 23, 661 CrossRef CAS.
- Electrochemical hydrogen technologies, Electrochemical production and combustion of hydrogen, ed. H. Wendt, Elsevier, Amsterdam, 1990 Search PubMed.
- S.-E. Jang and H. Kim, J. Am. Chem. Soc., 2010, 132, 14700 CrossRef CAS PubMed.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865 RSC.
- P. Millet, F. Andolfatto and R. Durand, Int. J. Hydrogen Energy, 1996, 21, 87 CrossRef CAS.
- J. W. Vogt, Materials for electrochemical cell separators, NASA Report NAS CR72493, NASA Lewis Research Center, 1968 Search PubMed.
- L. H. Baetsle and D. Huys, J. Inorg. Nucl. Chem., 1968, 30, 639 CrossRef CAS.
- P. Vermeiren, W. Adriansens, J. P. Moreels and R. Leysen, Hydrogen Power: Theoretical and Engineering Solutions, in Proceedings of the HYPOTHESIS II Symposium, ed. T. O. Saetre, Grimstad (Norway), 1997, p. 179 Search PubMed.
- J. Xu, G.-P. Sheng, H.-W. Luo, W.-W. Li, L.-F. Wang and H.-Q. Yu, Water Res., 2012, 46, 1817 CrossRef CAS PubMed.
- M. B. Rodrigues, A. S. Paleta, J. A. R. Marquez and O. Soalorza, Int. J. Electrochem. Sci., 2009, 4, 43 Search PubMed.
- D. Pletcher and X. Li, Int. J. Hydrogen Energy, 2011, 36, 15089 CrossRef CAS PubMed.
- V. Bambagioni, M. Bevilacqua, C. Bianchini, J. Filippi, A. Lavacchi, A. Marchionni, F. Vizza and P. K. Shen, ChemSusChem, 2010, 3, 851 CrossRef CAS PubMed.
- S. H. Ahn, B.-S. Lee, I. Choi, S. J. Yoo, H.-J. Kim, E. Cho, D. Henkensmeier, S. W. Nam, S.-K. Kim and J. H. Jang, Appl. Catal., B, 2014, 154–155, 197 CrossRef CAS PubMed.
- Y. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C.-Y. Wang, J. Am. Chem. Soc., 2012, 134, 9054 CrossRef CAS PubMed.
- X. Li, F. C. Walsh and D. Pletcher, Phys. Chem. Chem. Phys., 2011, 13, 1162 RSC.
- D. Pletcher, X. Li and S. Wang, Int. J. Hydrogen Energy, 2012, 37, 7429 CrossRef CAS PubMed.
- X. Wu and K. Scott, J. Mater. Chem., 2011, 21, 12344 RSC.
- X. Wu and K. Scott, J. Power Sources, 2012, 214, 124 CrossRef CAS PubMed.
- X. Wu and K. Scott, J. Power Sources, 2012, 206, 14 CrossRef CAS PubMed.
- C. C. Pavel, F. Cecconi, C. Emiliani, S. Santiccioli, A. Scaffidi, S. Catanorchi and M. Comotti, Angew. Chem., Int. Ed., 2014, 53, 1378 CrossRef CAS PubMed.
- L. Xiao, S. Zhang, J. Pan, C. Yan, M. He, L. Zhuang and J. Lu, Energy Environ. Sci., 2012, 5, 7869 CAS.
- J. Parrondo, C. G. Arges, M. Niedzwiecki, E. B. Anderson, K. E. Ayres and V. Ramani, RSC Adv., 2014, 4, 9875 RSC.
- F. Mitlitsky, B. Myers and A. H. Weisberg, Enegy & Fuels, 1998, 12, 56 CAS.
- T. Ioroi, N. Kitazawa, K. Yasuda, Y. Yamamoto and H. Takenaka, J. Appl. Electrochem., 2001, 31, 1179 CrossRef CAS.
- P. Hosseini Benhangi, A. Alfantazi and E. Gyenge, Electrochim. Acta, 2014, 123, 42 CrossRef PubMed.
- X. Wu, K. Scott, F. Xie and N. Alford, J. Power Sources, 2014, 246, 225 CrossRef CAS PubMed.
- H. B. Suffredini, J. L. Cerne, F. C. Crnkovic, S. A. S. Machado and L. A. Avaca, Int. J. Hydrogen Energy, 2000, 25, 415 CrossRef CAS.
- X. Tang, L. Xiao, C. Yang, J. Lu and L. Zhuang, Int. J. Hydrogen Energy, 2014, 39, 3055 CrossRef CAS PubMed.
- W. Hu and J.-Y. Lee, Int. J. Hydrogen Energy, 1998, 23, 253 CrossRef CAS.
- W.-X. Chen, Int. J. Hydrogen Energy, 2001, 26, 603 CrossRef CAS.
- A. C. Tavares and S. Trasatti, Electrochim. Acta, 2000, 45, 4195 CrossRef CAS.
- H. Obayashi and T. Kudo, Mater. Res. Bull., 1978, 13, 1409 CrossRef CAS.
- Lodon Metal Exchange, https://www.lme.com/metals/non-ferrous/nickel/, accessed 22 November 2013.
- G. E. Stoner and P. J. Moran, Proceedings of the Symposium on Industrial Water Electrolysis, Vancouver, Canada, 1978, p. 169 Search PubMed.
- C. Ballleux, A. Damien and A. Montet, Int. J. Hydrogen Energy, 1983, 8, 529 CrossRef.
- Batelle Geneva Research Centre Technical Note: Plasma-sprayed inorganic membranes as diaphragms for electrolysis, 1978.
- A. C. C. Tseung and S. Jasem, Electrochim. Acta, 1977, 22, 31 CrossRef CAS.
- P. Rasiyah and A. C. C. Tseung, Adv. Hydrogen Energy, 1982, 1982(3), 383 Search PubMed.
- M. Prigent, L. J. Mas and F. C. Verillon, Proceedings of the Symposium on Industrial Water Electrolysis, Vancouver, Canada, 1978, p. 234 Search PubMed.
- V. K. V. P. Srirapu, C. S. Sharma, R. Awasthi, R. N. Singh and A. S. K. Sinha, Phys. Chem. Chem. Phys., 2014, 16, 7385 RSC.
- H. S. Horowitz, J. M. Longo and H. H. Horowitz, J. Electrochem. Soc., 1983, 130, 1851 CrossRef CAS PubMed.
- A. Simon, T. Fujioka, W. E. Price and L. D. Nghiem, Sep. Purif. Technol., 2014, 127, 70 CrossRef CAS PubMed.
- W. E. Mustain, N. S. Spinner and J. A. Vega, WIPO Pat. Application WO/2012/061728, submitted November 2011.
- (a) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (b) A. Goeppert, M. Czaun, G. K. S. Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833 RSC; (c) M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43 RSC; (d) http://co2chem.co.uk/, accessed 4 October 2013.
- H. He, W. Li, M. Zhong, D. Konkolewicz, D. Wu, K. Yaccato, T. Rappold, G. Sugar, N. E. David and K. Matyjaszewski, Energy Environ. Sci., 2013, 6, 488 CAS.
- (a) E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112 RSC; (b) C. Coestentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423 RSC; N. S. Spinner, J. A. Vega and W. E. Mustain, Catal. Sci. Technol., 2012, 2, 19 Search PubMed.
- http://www.eon.com/en/media/news-detail.jsp?id=10738%26year=2011 .
- http://theotcinvestor.com/mantra-venture-group-mvtg-moves-to-commercialize-carbon-dioxide-reduction-technology-1196/ .
- H.-R. Jhong, S. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191 CrossRef PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jamamillo, Energy Environ. Sci., 2012, 5, 7050 CAS.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504 CrossRef CAS PubMed.
- (a) E. Ruiz-Trejo and J. T. S. Irvine, Solid State Ionics, 2013, 252, 157 CrossRef CAS PubMed; (b) K. Xie, Y. Zhang, G. Meng and J. T. S. Irvine, Energy Environ. Sci., 2011, 4, 2218 RSC.
- M. A. Rahman, X. Wang and C. Wen, J. Electrochem. Soc., 2013, 160, A1759 CrossRef CAS PubMed.
- Q. Wu, L. Jiang, L. Qi, L. Yuan, E. Wang and G. Sun, Electrochim. Acta, 2014, 123, 167 CrossRef CAS PubMed.
- A. A. Mohamad, N. S. Mohamed, M. Z. A. Yahya, R. Othman, S. Ramesh, Y. Alias and A. K. Arof, Solid State Ionics, 2003, 156, 171 CrossRef CAS.
- http://bestar.lbl.gov/bli5/files/2012/06/Imanishi_session3.pdf, accessed 22 January 2014.
- (a) N. Vassal, E. Salmon and J. F. Fauvarque, Electrochim. Acta, 2000, 45, 1527 CrossRef CAS; (b) S. Guinot, E. Salmon, J. F. Penneau and J. F. Fauvarque, Electrochim. Acta, 1998, 43, 1163 CrossRef CAS; (c) J. F. Fauvarque, S. Guinot, N. Bouzir, E. Salmon and J. F. Penneau, Electrochim. Acta, 1995, 40, 2449 CrossRef CAS.
- (a) C.-C. Yang, S.-J. Lin and S.-T. Hsu, J. Power Sources, 2003, 122, 210 CrossRef CAS; (b) C.-C. Yang, J. Power Sources, 2002, 109, 22 CrossRef CAS.
- Z. Zhang, C. Zuo, Z. Liu, Y. Yu, Y. Zuo and Y. Song, J. Power Sources, 2014, 251, 470 CrossRef CAS PubMed.
- N. Fujiwara, M. Yao, Z. Siroma, H. Senoh, T. Ioroi and K. Yasuda, J. Power Sources, 2011, 196, 808 CrossRef CAS PubMed.
- P. Alotto, M. Guarnieri and F. Moro, Renewable Sustainable Energy Rev., 2014, 29, 325 CrossRef CAS PubMed.
- A. Z. Weber, M. M. Mench, J. P. Meyers, P. N. Ross, J. T. Gostick and Q. H. Liu, J. Appl. Electrochem., 2011, 41, 1137 CrossRef CAS.
- S. C. Chieng, M. Kazacos and M. Skyllas-Kazacos, J. Membr. Sci., 1992, 75, 81 CrossRef CAS.
- (a) D.-H. Kim, S.-J. Seo, M.-J. Lee, J.-S. Park, S.-H. Moon, Y. S. Kang, Y.-W. Choi and M.-S. Kang, J. Membr. Sci., 2014, 454, 44 CrossRef CAS PubMed; (b) S. Maurya, S.-H. Shin, K.-W. Sung and S.-H. Moon, J. Power Sources, 2014, 255, 325 CrossRef CAS PubMed; (c) A. A. Shinkle, A. E. S. Sleightholme, L. D. Griffith, L. T. Thompson and C. W. Monroe, J. Power Sources, 2012, 206, 490 CrossRef CAS PubMed.
- Q. Luo, L. Li, W. Wang, Z. Nie, X. Wei, B. Li, B. Chen, Z. Yang and V. Sprenkle, ChemSusChem, 2013, 6, 268 CrossRef CAS PubMed.
- (a) D. Chen and M. A. Hickner, Phys. Chem. Chem. Phys., 2013, 15, 11299 RSC; (b) S. Kim, T. Tighe, B. Schwenzer, J. Yan, J. Zhang, J. Liu, Z. Yang and M. A. Hickner, J. Appl. Electrochem., 2011, 41, 1201 CrossRef CAS.
- D. Chen, S. Kim, L. Li, G. Yang and M. A. Hickner, RSC Adv., 2012, 2, 8087 RSC.
- M. S. J. Jung, J. Parrondo, C. G. Arges and V. Ramani, J. Mater. Chem. A, 2013, 1, 10458 CAS.
- K. D. Kreuer, J. Membr. Sci., 2001, 185, 29 CrossRef CAS.
- (a) B. Pivovar, Y. Yang and E. L. Cussler, J. Membr. Sci., 1999, 154, 155 CrossRef CAS; (b) M. A. Hickner and B. Pivovar, Fuel Cells, 2005, 5, 213 CrossRef CAS PubMed.
- D. Chen, S. Kim, V. Sprenkle and M. A. Hickner, J. Power Sources, 2013, 231, 301 CrossRef CAS PubMed.
- D. Chen, M. A. Hickner, E. Agar and E. C. Kumbur, Electrochem. Commun., 2013, 26, 37 CrossRef CAS PubMed.
- S.-G. Park, N.-S. Kwak, C. W. Hwang, H.-M. Park and T. S. Hwang, J. Membr. Sci., 2012, 423, 429 CrossRef PubMed.
- S.-J. Seo, B.-C. Kim, K.-W. Sung, J. Shim, J.-D. Jeon, K.-H. Shin, S.-H. Shin, S.-H. Yun, J.-Y. Lee and S.-H. Moon, J. Membr. Sci., 2013, 428, 17 CrossRef CAS PubMed.
- D. Chen, M. A. Hickner, E. Agar and E. C. Kumbur, ACS Appl. Mater. Interfaces, 2013, 5, 7559 CAS.
- D. Chen, M. A. Hickner, E. Agar and E. C. Kumbur, J. Membr. Sci., 2013, 437, 108 CrossRef CAS PubMed.
- (a) C. W. Hwang, H.-M. Park, C. M. Oh, T. S. Hwang, J. Shim and C.-S. Jin, J. Membr. Sci., 2014, 468, 98 CrossRef CAS PubMed; (b) F. Zhang, H. Zhang and C. Qu, ChemSusChem, 2013, 6, 2290 CrossRef CAS PubMed; (c) S. H. Zhang, B. G. Zhang, D. B. Xing and X. G. Jian, J. Mater. Chem. A, 2013, 1, 12246 RSC; (d) J. Y. Qiu, M. Y. Li, J. F. Ni, M. L. Zhai, J. Peng, L. Xu, H. H. Zhou, J. Q. Li and G. S. Wei, J. Membr. Sci., 2007, 297, 174 CrossRef CAS PubMed; (e) G. J. Hwang and H. Ohya, J. Membr. Sci., 1997, 132, 55 CrossRef CAS; (f) T. Mohammadi and M. Skyllas-Kazacos, J. Power Sources, 1996, 63, 179 CrossRef CAS.
- (a) J. Fang, H. Xu, X. Wei, M. Guo, X. Lu, C. Lan, Y. Zhang, Y. Liu and T. Peng, Polym. Adv. Technol., 2013, 24, 168 CrossRef CAS PubMed; (b) Y. Wang, J. Qiu, J. Peng, L. Xu, J. Li and M. Zhai, J. Membr. Sci., 2011, 376, 70 CrossRef CAS PubMed.
- (a) S. Zhang, B. Zhang, G. Zhao and X. Jian, J. Mater. Chem. A, 2014, 2, 3083 RSC; (b) S.-G. Park, N.-S. Kwak, C. W. Hwang, H.-M. Park and T. S. Hwang, J. Membr. Sci., 2012, 423, 429 CrossRef PubMed; (c) F. Zhang, H. Zhang and C. Qu, J. Phys. Chem. B, 2012, 116, 9016 CrossRef CAS PubMed; (d) S. Zhang, C. Yin, D. Xing, D. Yang and X. Kiang, J. Membr. Sci., 2010, 363, 243 CrossRef CAS PubMed.
- D. S. Aaron, Q. Liu, Z. Tang, G. M. Grim, A. B. Papandrew, A. Turhan, T. A. Zawodzinski and M. M. Mench, J. Power Sources, 2012, 206, 450 CrossRef CAS PubMed.
- S. Yun, J. Parrondo and V. Ramani, J. Mater. Chem. A, 2014, 2, 6605 CAS.
- G. L. Wick and W. R. Schmitt, Mar. Technol. Soc. J., 1977, 11, 16 Search PubMed.
- J. W. Post, J. Veerman, H. V. M. Hamelers, G. J. W. Euverink, S. J. Metz, K. Nijmeijer and C. J. N. Buisman, J. Membr. Sci., 2007, 288, 218 CrossRef CAS PubMed.
- S. Loeb, J. Membr. Sci., 1976, 1, 49 CrossRef.
- R. E. Pattle, Nature, 1954, 174, 660 CrossRef CAS.
- D. A. Vermaas, S. Bajracharya, B. B. Sales, M. Saakes, B. Hamelers and K. Nijmeijer, Energy Eniron. Sci., 2013, 6, 643 RSC.
- J. N. Wienstein and F. B. Leitz, Science, 1976, 191, 557 Search PubMed.
- R. E. Lacey, Ocean Eng., 1980, 7, 1 CrossRef CAS.
- R. Audinos, J. Power Sources, 1983, 10, 203 CrossRef CAS.
- P. Długolęcki, A. Gambier, K. Nijmeijer and M. Wessling, Environ. Sci. Technol., 2009, 43, 6888 CrossRef.
- J. W. Post, H. V. M. Hamelers and C. J. N. Buisman, Environ. Sci. Technol., 2008, 42, 5785 CrossRef CAS.
- J. Veerman, M. Saakes, S. J. Metz and G. J. Harmsen, J. Membr. Sci., 2009, 327, 136 CrossRef CAS PubMed.
- M. Turek and B. Bandura, Desalination, 2007, 205, 67 CrossRef CAS PubMed.
- (a) G. M. Geise, M. A. Hickner and B. E. Logan, ACS Macro Lett., 2013, 2, 814 CrossRef CAS; (b) M. C. Hatzell and B. E. Logan, J. Membr. Sci., 2013, 446, 449 CrossRef CAS PubMed; (c) X. Luo, X. Cao, Y. Mo, K. Xiao, X. Zhang, P. Liang and X. Huang, Electrochem. Commun., 2012, 19, 25 CrossRef CAS PubMed.
- E. Brauns, Desalin. Water Treat., 2010, 13, 53 CrossRef CAS.
- W. Li, B. Krantz, E. R. Cornelissen, J. W. Post, A. R. D. Verliefde and C. Y. Tang, Appl. Energ., 2013, 104, 592 CrossRef CAS PubMed.
- J. Veerman, R. M. de Jong, M. Saakes, S. J. Metz and G. J. Harmsen, J. Membr. Sci., 2009, 343, 7 CrossRef CAS PubMed.
- M. C. Hatzell, I. Ivanov, R. D. Cusick, X. Zhu and B. E. Logan, Phys. Chem. Chem. Phys., 2014, 16, 1632 RSC.
- P. Długołęcki, K. Nijmeijer, S. Metz and M. Wessling, J. Membr. Sci., 2008, 319, 214 CrossRef PubMed.
- E. Güler, R. Elizen, D. Vermaas, M. Saakes and K. Nijmeijer, J. Membr. Sci., 2013, 446, 266 CrossRef PubMed.
- E. Güler, Y. Zhang, M. Saakes and K. Nijmeijer, ChemSusChem, 2012, 5, 2262 CrossRef PubMed.
- G. M. Geise, M. A. Hickner and B. E. Logan, ACS Appl. Mater. Interf., 2013, 5, 10294 CrossRef CAS PubMed.
- K. Kwon, S. J. Lee, L. Li, C. Han and D. Kim, Int. J. Energ. Res., 2013, 38, 530 CrossRef PubMed.
- P. Długołęcki, J. Dąbrowska, K. Nijmeijer and M. Wessling, J. Membr. Sci., 2010, 347, 101 CrossRef PubMed.
- D. A. Vermaas, M. Saakes and K. Nijmeijer, J. Membr. Sci., 2011, 385–386, 234 CrossRef PubMed.
- E. Güler, R. Elizen, M. Saakes and K. Nijmeijer, J. Membr. Sci., 2014, 455, 254 CrossRef PubMed.
- E. Güler, R. Elizen, M. Saakes and K. Nijmeijer, J. Membr. Sci., 2014, 458, 136 CrossRef PubMed.
- J. W. Post, H. V. M. Hamelers and C. J. N. Buisman, J. Membr. Sci., 2009, 330, 65 CrossRef CAS PubMed.
- D. A. Vermaas, J. Veerman, M. Saakes and K. Nijmeijer, Energy Environ. Sci., 2014, 7, 1434 CAS.
- D. A. Vermaas, D. Kunteng, M. Saakes and K. Nijmeijer, Water Res., 2013, 47, 1289 CrossRef CAS PubMed.
- D. A. Vermaas, D. Kunteng, J. Veerman, M. Saakes and K. Nijmeijer, Environ. Sci. Technol., 2014, 48, 3065 CrossRef CAS PubMed.
- E. Guler, W. van Baak, M. Saakes and K. Nijmeijer, J. Membr. Sci., 2014, 455, 254 CrossRef PubMed.
- (a) X. Zhang, F. Zhu, L. Chen, Q. Zhao and G. Tao, Bioresour. Technol., 2013, 146, 161 CrossRef CAS PubMed; (b) I. Singh and A. Chandra, Bioresour. Technol., 2013, 142, 77 CrossRef CAS PubMed; (c) X. Xia, J. C. Tokash, F. Zhang, P. Liang, X. Huang and B. E. Logan, Environ. Sci. Technol., 2013, 47, 2085 CrossRef CAS PubMed; (d) X. Zhang, J. Shi, P. Liang, J. Wei, X. Huang, C. Zhang and B. E. Logan, Bioresour. Technol., 2013, 142, 109 CrossRef CAS PubMed; (e) J. M. Gohil and D. G. Karamanev, J. Power Sources, 2013, 243, 603 CrossRef CAS PubMed; (f) Y. Ahn and B. E. Logan, Bioresour. Technol., 2013, 132, 436 CrossRef CAS PubMed; (g) S. Haddadi, E. Elbeshbishy and H.-S. Lee, Bioresour. Technol., 2013, 142, 562 CrossRef CAS PubMed; (h) E. Mahendiravarman and D. Sangeetha, Int. J. Hydrogen Energy, 2013, 38, 2471 CrossRef CAS PubMed; (i) S. Pandit, S. Ghosh, M. M. Ghangrekar and D. Das, Int. J. Hydrogen Energy, 2012, 37, 9383 CrossRef CAS PubMed; (j) S. Pandit, A. Sengupta, S. Kale and D. Das, Bioresour. Technol., 2011, 102, 2736 CrossRef CAS PubMed; (k) E. Ji, H. Moon, J. Piao, P. T. Ha, J. An, D. Kim, J.-J. Woo, Y. Lee, S.-H. Moon, B. E. Rittmann and I. S. Chang, Biosens. Bioelectron., 2011, 26, 3266 CrossRef CAS PubMed; (l) X. Zhang, S. Cheng, X. Huang and B. E. Logan, Biosens. Bioelectron., 2010, 25, 1825 CrossRef CAS PubMed; (m) L. Zhuang, C. Feng, S. Zhou, Y. Li and Y. Wang, Process Biochem., 2010, 45, 929 CrossRef CAS PubMed; (n) Y. Mo, P. Liang, X. Huang, H. Wang and X. Cao, J. Chem. Technol. Biotechnol., 2009, 84, 1767 CrossRef CAS PubMed; (o) G. J. la O’, J. J. Walish and E. J. Crumlin, US Pat. Application 2009/0087690 A1, 2009; (p) Y. Zuo, S. Cheng and B. E. Logan, Environ. Sci. Technol., 2008, 42, 6967 CrossRef CAS; (q) J. R. Kim, S. Cheng, S.-E. Oh and B. E. Logan, Environ. Sci. Technol., 2007, 41, 1004 CrossRef CAS ; also reviews in ref. 6 above.
- H. Wang and Z. J. Ren, Biotechnol. Adv., 2013, 31, 1796 CrossRef CAS PubMed.
- (a) B. E. Logan and K. Rabaey, Science, 2012, 337, 686 CrossRef CAS PubMed; (b) O. Lefebvre, A. Uzabiaga, I. S. Chang, B.-H. Kim and H. Y. Ng, Appl. Microbiol. Biotechnol., 2011, 89, 259 CrossRef CAS PubMed; (c) Y. Qiao, S.-J. Bao and C. M. Li, Energy Environ. Sci., 2010, 3, 544 RSC; (d) D. R. Lovley, Nat. Rev. Microbiol., 2006, 4, 497 CrossRef CAS PubMed.
- K. B. Liew, W. R. W. Daud, M. Ghasemi, J. X. Leong, S. S. Lim and M. Ismail, Int. J. Hydrogen Energy, 2014, 39, 4870 CrossRef PubMed.
- (a) E. Martin, B. Tartakovsky and O. Savadogo, Electrochim. Acta, 2011, 58, 58 CrossRef CAS PubMed; (b) M. Lu, S. Kharkwal, H. Y. Ng and S. F. Y. Li, Biosens. Bioelectron., 2011, 26, 4728 CrossRef CAS PubMed; (c) I. Roche and K. Scott, J. Appl. Electrochem., 2009, 39, 197 CrossRef CAS.
- (a) X. Shi, Y. Feng, X. Wang, H. Lee, J. Liu, Y. Qu, W. He, S. M. S. Kumar and N. Ren, Bioresour. Technol., 2012, 108, 89 CrossRef CAS PubMed; (b) B. Erable, N. Duteanu, S. M. S. Kumar, Y. Feng, M. M. Ghangrekar and K. Scott, Electrochem. Commun., 2009, 11, 1547 CrossRef CAS PubMed.
- (a) C. Wu, X. Liu, W. Li, G. Sheng, G. Zang, Y. Cheng, N. Shen, Y. Yang and H. Yu, Appl. Energy, 2012, 98, 594 CrossRef CAS PubMed; (b) O. Schaetzle, F. Barrière and U. Schröder, Energy Environ. Sci., 2009, 2, 96 RSC.
- X. Cao, X. Huang, P. Liang, N. Boon, M. Fan, L. Zhang and X. Zhang, Energy Environ. Sci, 2009, 2, 498 CAS.
- Y. Zhang and I. Angelidaki, Water Res., 2014, 56, 11 CrossRef CAS PubMed.
- (a) J. An and H.-S. Lee, RSC Adv., 2013, 3, 14021 RSC; (b) E. Ribot-Llobet, J.-Y. Nam, J. C. Tokash, A. Guisasola and B. E. Logan, Int. J. Hydrogen Energy, 2013, 38, 2951 CrossRef CAS PubMed; (c) T. H. J. A. Sleutels, H. V. M. Hamelers, R. A. Rozendal and C. J. N. Buisman, Int. J. Hydrogen Energy, 2009, 34, 3612 CrossRef CAS PubMed; (d) S. Cheng and B. E. Logan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18871 CrossRef CAS PubMed.
- Y. Kim and B. E. Logan, Desalination, 2013, 308, 122 CrossRef CAS PubMed.
- (a) K. Zuo, L. Yuan, J. Wei, P. Liang and X. Huang, Bioresour. Technol., 2013, 146, 637 CrossRef CAS PubMed; (b) Y. Qu, Y. Feng, J. Liu, X. Shi, Q. Yang, J. Lv and B. E. Logan, Desalination, 2013, 317, 17 CrossRef CAS PubMed; (c) Q. Ping and Z. He, Bioresour. Technol., 2013, 144, 304 CrossRef CAS PubMed; (d) Q. Ping, B. Cohen, C. Dosoretz and Z. He, Desalination, 2013, 325, 48 CrossRef CAS PubMed; (e) B. Zhang and Z. He, J. Membr. Sci., 2013, 441, 18 CrossRef CAS PubMed; (f) K. S. Brastad and Z. He, Desalination, 2013, 309, 32 CrossRef CAS PubMed; (g) H. Luo, P. Xu, P. E. Jenkins and Z. Ren, J. Membr. Sci., 2012, 409–410, 16 CrossRef CAS PubMed; (h) K. S. Jacobson, D. M. Drew and Z. He, Environ. Sci. Technol., 2011, 45, 4652 CrossRef CAS PubMed; (i) S. Chen, G. Liu, R. Zhang, B. Qin and Y. Luo, Environ. Sci. Technol., 2012, 46, 2467 CrossRef CAS PubMed; (j) Y. Qu, Y. Feng, X. Wang, J. Liu, J. Lv, W. He and B. E. Logan, Bioresour. Technol., 2012, 106, 89 CrossRef CAS PubMed; (k) X. Chen, X. Xia, P. Liang, X. Cao, H. Sun and X. Huang, Environ. Sci. Technol., 2011, 45, 2465 CrossRef CAS PubMed; (l) M. Mehanna, P. D. Kiely, D. F. Call and B. E. Logan, Environ. Sci. Technol., 2010, 44, 9578 CrossRef CAS PubMed; (m) M. Mehanna, T. Saito, J. Yan, M. Hickner, X. Cao, X. Huang and B. E. Logan, Energy Environ. Sci., 2010, 3, 1114 RSC; (n) X. Cao, X. Huang, P. Liang, K. Xiao, Y. Zhou, X. Zhang and B. E. Logan, Environ. Sci. Technol., 2009, 43, 7148 CrossRef CAS ; also ref. 5a above.
- C. M. Werner, B. E. Logan, P. E. Saikaly and G. L. Amy, J. Membr. Sci., 2013, 428, 116 CrossRef CAS PubMed.
- (a) R. D. Cusick, M. Hatzell, F. Zhang and B. E. Logan, Environ. Sci. Technol., 2013, 47, 14518 CrossRef CAS PubMed; (b) X. Luo, J.-Y. Nam, F. Zhang, X. Zhang, P. Liang, X. Huang and B. E. Logan, Bioresour. Technol., 2013, 140, 399 CrossRef CAS PubMed; (c) X. Zhu, M. C. Hatzell, R. D. Cusick and B. E. Logan, Electrochem. Commun., 2013, 31, 52 CrossRef CAS PubMed; (d) R. D. Cusick, Y. Kim and B. E. Logan, Science, 2012, 335, 1474 CrossRef CAS PubMed; (e) Y. Kim and B. E. Logan, Environ. Sci. Technol., 2011, 45, 5834 CrossRef CAS PubMed; (f) Y. Kim and B. E. Logan, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16176 CrossRef CAS PubMed ; also ref. 5a above.
- (a) T. Zhang, H. Nie, T. S. Bain, H. Lu, M. Cui, O. L. Snoeyenbos-West, A. E. Franks, K. P. Nevin, T. P. Russell and D. R. Lovley, Energy Environ. Sci., 2013, 6, 217 RSC; (b) Y. Gong, A. Ebrahim, A. M. Feist, M. Embree, T. Zhang, D. Lovley and K. Zengler, Environ. Sci. Technol., 2013, 47, 568 CrossRef CAS PubMed; (c) S. Pandit, B. K. Nayak and D. Das, Bioresour. Technol., 2012, 107, 97 CrossRef CAS PubMed.
- H. Li and J. C. Liao, Energy Environ. Sci., 2013, 6, 2892 CAS.
- (a) X. Zhu and B. E. Logan, Bioresour. Technol., 2014, 159, 24 CrossRef CAS PubMed; (b) X. Zhu, M. C. Hatzell and B. E. Logan, Environ. Sci. Technol. Lett., 2014, 1, 231 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: This gives the biographies for all authors of this perspective. See DOI: 10.1039/c4ee01303d |
| ‡ Currents and power densities can be normalised to various areas (such as geometric or “projected” anode, cathode, or membrane areas) and volumes (such as anode chamber volume or total MFC volume). These are not always well defined in reported W m−2 or W m−3 values in the MFC-related literature. A recommendation is to either unambiguously define the normalisation being used or report the absolute currents and powers alongside detailed listing of relevant areas and volume of the system (and its most relevant components). The latter will facilitate easier comparisons between the wide diversity of different systems (configurations) reported in the literature, which currently use a wide variety of normalisations. |
| § Care should be taken when referring to internal resistances, which should be unambiguously defined. MFC studies often refer to internal resistances as meaning the total internal resistances (sum of ohmic + anodic charge transfer + cathodic charge transfer + contact + interfacial resistances + mass transport resistances etc.). However, chemical fuel cell (and related devices) studies often refer to internal resistances as meaning the internal ohmic resistances. |
| ¶ The properties of the membranes being used are not always unambiguously reported. As a minimum, we recommend that the following is reported: thickness (in a well-defined hydration state), IEC, and the chemical nature of the membrane. |
| This journal is © The Royal Society of Chemistry 2014 |