 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolybdenum phosphide as an efficient electrocatalyst for the hydrogen evolution reaction†
Peng
Xiao
a,
Mahasin Alam
Sk
a,
Larissa
Thia
cd,
Xiaoming
Ge
b,
Rern Jern
Lim
a,
Jing-Yuan
Wang
c,
Kok Hwa
Lim
a and
Xin
Wang
*a
aSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 50 Nanyang Avenue, 639798, Singapore. E-mail: WangXin@ntu.edu.sg; Fax: +65 67947553
bInstitute of Materials Research and Engineering (IMRE), Agency of Science, Technology, and Research (A*STAR), 3 Research Link, Singapore 117602, Singapore
cResidues and Resource Reclamation Centre, Nanyang Technological University, Singapore
dInterdisciplinary Graduate School, Nanyang Technological University, 50 Nanyang Avenue Block S2 – B3a – 01, Singapore 639798, Singapore
First published on 29th May 2014
Abstract
Electrochemical production of hydrogen from water has been directed to the search for non-noble metal based and earth-abundant catalysts. In this work, we propose a novel cost-effective catalyst, molybdenum phosphide that exhibits high activity towards the hydrogen evolution reaction (HER) in both acid and alkaline media even in bulk form. Comparative analysis of Mo, Mo3P and MoP as catalysts for HER clearly indicates that phosphorization can potentially modify the properties of the metal and different degrees of phosphorization lead to distinct activities and stabilities. Theoretical calculations by density functional theory also show that a simple phosphorization of molybdenum to form MoP introduces a good ‘H delivery’ system which attains nearly zero binding to H at a certain H coverage. With the combination of experimental results and theoretical calculations, this work has enlightened a new way of exploring cost-effective catalysts for HER.
Broader contextHydrogen is currently pursued as a potential energy source and the electrochemical water splitting process has been proposed as a promising and clean means for large scale hydrogen production. Limited by the high cost and scarcity of noble metal catalysts, non-noble metal catalysts are being explored as possible alternatives, with a few successful cases to date, e.g. MoS2. However, the synthesis of these reported catalysts typically involve sophisticated methodologies in order to achieve desired nanostructures. Herein, we report a non-noble metal catalyst for the hydrogen evolution reaction (HER), molybdenum phosphide. Its synthesis can be realized via a facile two-step-sintering method. Our experimental results show that even in its bulk form, molybdenum phosphide exhibits high performance in both acidic and alkaline conditions. Furthermore, the observed performance of the bulk MoP was also comparable to that of the aforementioned nanostructured catalysts. Theoretical calculations reveal that phosphorization can potentially modify the properties of the metal and different degrees of phosphorization lead to distinct activities and stabilities. |
Introduction
Hydrogen production through electrochemical water-splitting has been extensively pursued as it could potentially achieve sustainable fuel production and the electricity required could be obtained from renewable energy.1 Noble metals, e.g. Pt, demonstrate exceptional behavior with nearly zero overpotential in acidic media.2–4 However, their high cost and scarcity impede their widespread usage and direct our attention to earth-abundant metals or their compounds. Non-noble electrocatalysts can be categorized into two categories: (1) the derivatives of organic liganded metals (e.g. Ni, Mo, Fe), such as hydrogenases or metalloenzyme, the mimics of active site of hydrogenases,5,6 molybdenum-oxo,5 and pyrene-functionalized nickel complexes;7 (2) inorganic compounds of metals. MoS2,8–16 as a typical inorganic compound for hydrodesulfurization,17 was discovered to possess biomimetic active sites as hydrogenases.18,19 Selenide,20,21 nickel molybdenum nitride,22 carbide,23–26 nickel phosphide,27,28 and first-row of transitional metal dichalcogenides29 are reported to be promising electrocatalysts towards the hydrogen evolution reaction (HER). By comparison, those cost-effective catalysts as the potential substitutes for Pt are either functionalized by organic ligands, e.g. [NiFe]hydrogenase or inorganics e.g. MoS2, Ni2P. In a typical ‘volcano plot’ of hydrogen adsorption energy of common metals (M = Nb, Mo, Ni, Pt, Au, Ag, etc.),30–32 it is revealed that strong metal–hydrogen (Mo, Ni, etc.) bonds could impede hydrogen release from the active sites, compared to Pt group metals, e.g. Pd and Pt, which excludes the non-noble metals (e.g. Ni, Mo) as suitable candidates to catalyze the hydrogen evolution reaction (HER). However, a subsequent study suggested that inorganic compounds of the non-noble metals can modify the metal–hydrogen bond strength and achieve a Pt-resembling Gibbs free energy for hydrogen evolution in an acidic environment. The study of MoS2 and Ni2P has successfully underpinned this theoretical calculation.18,33 Inspired by this, herein, we report a molybdenum compound, MoP as a highly efficient catalyst for HER both in acidic and alkaline media. Molybdenum phosphide is synthesized though facile sintering of molybdenum and phosphorus precursors assisted by citric acid. Unlike MoS2 which shows very poor activity in bulk form, MoP still exhibits high electrocatalytic activity in bulk form. Furthermore, it was also experimentally demonstrated that going from metal Mo, Mo3P to MoP, different degree of phosphorization results in distinct performance and different stability. Theoretically, we employed density functional theory (DFT) calculations to elucidate the underlying reasons behind the distinct performance by comparing the hydrogen chemisorption process and Gibbs free energy of Mo, Mo3P and MoP.Results and discussion
Mo3P and MoP were synthesized at 800 °C and 650 °C respectively via a two-step sintering method (Experimental section, ESI†), whilst metal Mo was obtained by reducing MoO3 in H2 at 850 °C. Their corresponding XRD patterns are shown in Fig. 1. The peaks are well indexed to the standard XRD profiles and no major impurities are detected. Metal Mo, in a cubic pack (ICSD code: 643962) belongs to Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (229). After the incorporation of phosphorus to form Mo3P, it evolves into a tetragonal structure. Its XRD pattern confirms its I
m (229). After the incorporation of phosphorus to form Mo3P, it evolves into a tetragonal structure. Its XRD pattern confirms its I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2m (121) space group in Fig. 1b, with the Mo atom being either 2 or 4-coordinated by P atoms. Further phosphorization leads to MoP, in an hexagonal structure (P
2m (121) space group in Fig. 1b, with the Mo atom being either 2 or 4-coordinated by P atoms. Further phosphorization leads to MoP, in an hexagonal structure (P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) m2), with Mo 6-coordinated by P atoms, as shown in Fig. 1c. A visualized structure change is presented in Scheme 1.
m2), with Mo 6-coordinated by P atoms, as shown in Fig. 1c. A visualized structure change is presented in Scheme 1.
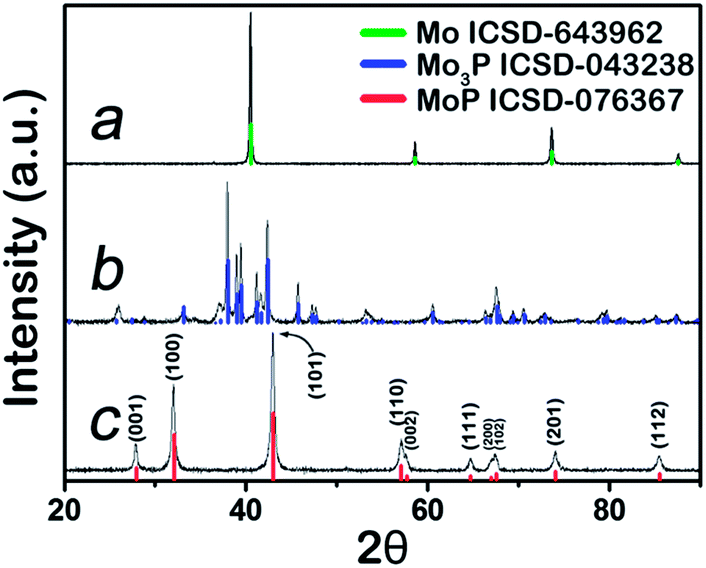 | ||
| Fig. 1 XRD patterns of (a) Mo; (b) Mo3P; (c) MoP. | ||
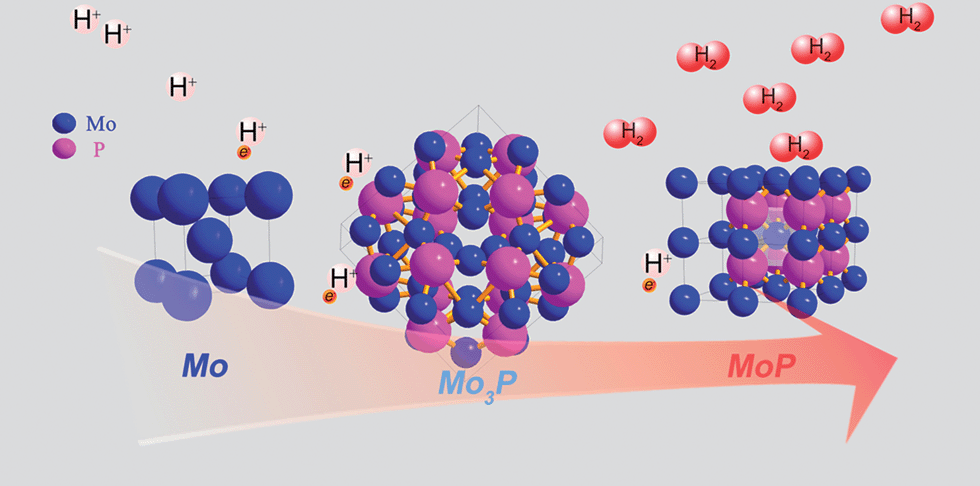 | ||
| Scheme 1 Schematic graph to show the structural evolution upon phosphorization. | ||
Field-emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM) were applied to examine its morphology. FE-SEM graphs in Fig. 2a show that the as-synthesized MoP and Mo3P (Fig. S3†) are in bulk form due to the calcination at high temperature. The high-resolution TEM (HR-TEM) graph exhibits a well-arrayed (001) plane of MoP with a plane distance of 0.32 nm (Fig. 2b), which is correlated with the peak at 28° in the XRD characterization. The energy dispersive X-ray spectroscopy (EDS) results confirm the compositions of MoP and Mo3P (Fig. S4†), which is quite consistent with the chemical formulas, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 3:1 for MoP, Mo3P respectively. To elucidate the valence states of individual elements of MoP, X-ray photoelectron spectroscopy (XPS) experiments were conducted. Fig. 3a and b show the XPS profiles of Mo 3d and P 2p of the as-synthesized MoP. Deconvolution of the spectra indicates two doublets at 235.28 eV/232.08 eV (Mo6+ 3d3/2/3d5/2) and 231.9 eV/228.8 eV (Mo4+ 3d3/2/3d5/2) in Fig. 3a, which can be assigned to high oxidation state of Mo (MoO3 and MoO2).34,35 We believe that the oxidation only happens on the surface. To validate our hypothesis, we characterize the aged MoP (Fig. S5†) by XRD, which still manifests the well-crystallized MoP and does not indicate any discernible peaks of the oxides, implying a mere surface-oxidation (less than 10 nm) that could only be detected by XPS. The rest of the XPS profile depicted in green (Fig. 3a) is ascribed to MoP, represented by a 231.2 eV/228.0 eV doublet, which agrees well with previous reports.36,37 The profile of P 2p also exhibits a peak with high binding energy (peak 1 in Fig. 3b) which could be attributed to PO43− or P2O5 (ref. 36) caused by oxidation. To identify the rest peaks for MoP, we further compared the XPS profile of the as-prepared MoP sample after testing with the aged sample. As shown in Fig. 3d, the P 2p profile of the aged sample does not display the low-binding-energy peaks as we observe in Fig. 3b. On the contrary, it manifests even stronger peaks in the low-binding-energy region for the sample after testing (Fig. S6†). Considering the above, we assign the doublet (130.1/129.2 eV for peak 2/peak 3) to low valence of P, MoP in this case.
:1 and 3:1 for MoP, Mo3P respectively. To elucidate the valence states of individual elements of MoP, X-ray photoelectron spectroscopy (XPS) experiments were conducted. Fig. 3a and b show the XPS profiles of Mo 3d and P 2p of the as-synthesized MoP. Deconvolution of the spectra indicates two doublets at 235.28 eV/232.08 eV (Mo6+ 3d3/2/3d5/2) and 231.9 eV/228.8 eV (Mo4+ 3d3/2/3d5/2) in Fig. 3a, which can be assigned to high oxidation state of Mo (MoO3 and MoO2).34,35 We believe that the oxidation only happens on the surface. To validate our hypothesis, we characterize the aged MoP (Fig. S5†) by XRD, which still manifests the well-crystallized MoP and does not indicate any discernible peaks of the oxides, implying a mere surface-oxidation (less than 10 nm) that could only be detected by XPS. The rest of the XPS profile depicted in green (Fig. 3a) is ascribed to MoP, represented by a 231.2 eV/228.0 eV doublet, which agrees well with previous reports.36,37 The profile of P 2p also exhibits a peak with high binding energy (peak 1 in Fig. 3b) which could be attributed to PO43− or P2O5 (ref. 36) caused by oxidation. To identify the rest peaks for MoP, we further compared the XPS profile of the as-prepared MoP sample after testing with the aged sample. As shown in Fig. 3d, the P 2p profile of the aged sample does not display the low-binding-energy peaks as we observe in Fig. 3b. On the contrary, it manifests even stronger peaks in the low-binding-energy region for the sample after testing (Fig. S6†). Considering the above, we assign the doublet (130.1/129.2 eV for peak 2/peak 3) to low valence of P, MoP in this case.
 | ||
| Fig. 2 Morphology of MoP (a) FE-SEM and (b) TEM graph. | ||
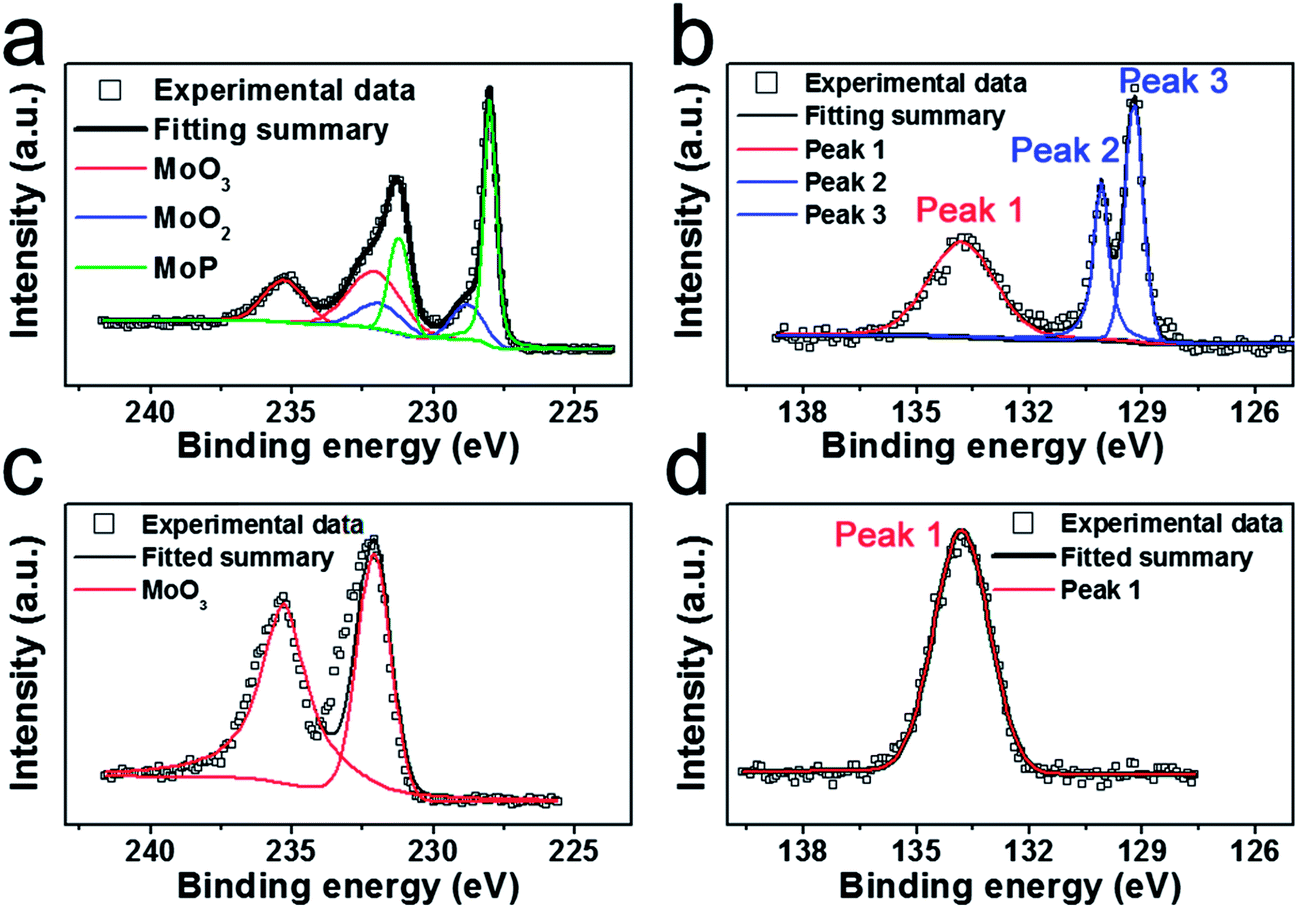 | ||
| Fig. 3 X-ray photoelectron spectra (XPS) of the as-synthesized MoP (a) Mo 3d and (b) P 2p; aged MoP (c) Mo 3d and (d) P 2p. | ||
In a three-electrode configuration, the catalytic activity towards HER was investigated in acid and alkaline media. Fig. 4 shows the performance of Mo, Mo3P and MoP in acidic medium; Pt/C was also tested as a comparison. The polarization curve and Tafel slope ∼30 mV dec−1 of Pt/C presented in Fig. 4a and b are consistent with previous studies.38 After being drop-cast on a glassy carbon electrode (which exhibits negligible activity towards HER), to achieve a current density of 10 mA cm−2, metal Mo requires a potential of up to 0.5 V vs. RHE with an onset potential of ∼0.3 V vs. RHE, and Mo3P is found to perform similarly as shown by the green line, indicating that metal Mo itself is not a good catalyst for HER and its phosphide Mo3P is not suitable for HER either. In contrast, MoP shows a rather high performance in 0.5 M H2SO4. The onset of the hydrogen evolution reaction can be observed at around 50 mV vs. RHE and the current density reaches 30 mA cm−2 at a potential of 0.18 V vs. RHE. To clarify these differences in performance, from the perspective of an elementary step, i.e. hydrogen chemisorption, we use Gibbs free energy (ΔGH°) as the descriptor to assess the binding strength of catalysts to H (e.g. Mo and MoP) and evaluate the catalysts (detailed DFT calculations can be found in the ESI†). Metal Mo exhibits strong binding to H, indicated by the highly negative binding energy (ΔEH) in Table S1† when H coverage reaches 1/4 of a monolayer (ML, one hydrogen adsorbed on 2 × 2 slab). This agrees well with the previous report.18 Calculation of the Mo terminated surface on (001)-MoP also shows it has similar or even stronger binding to H which excludes Mo as the active site. Nevertheless, the investigation of P sites implies that P has played a crucial role by acting as a ‘hydrogen deliverer’. As shown in Table S2,† ΔGH° changes from −0.36 eV to 0.54 eV when H coverage increases from 1/4 ML to full coverage, indicating that P could bond hydrogen at low coverage whilst desorb H at high coverage. This enables P to behave like a ‘hydrogen deliverer’, resembling the S-edges in MoS2.39 Interestingly, when we applied the same method to calculate the binding energy of H on (001) of Mo3P, where a four-P-site slab is chosen, the binding energy ΔEH becomes positive (Table S3†) with the initial two H adsorbed, indicating unfavorable binding of H on the P sites of Mo3P. This is in accordance with the experimental results.
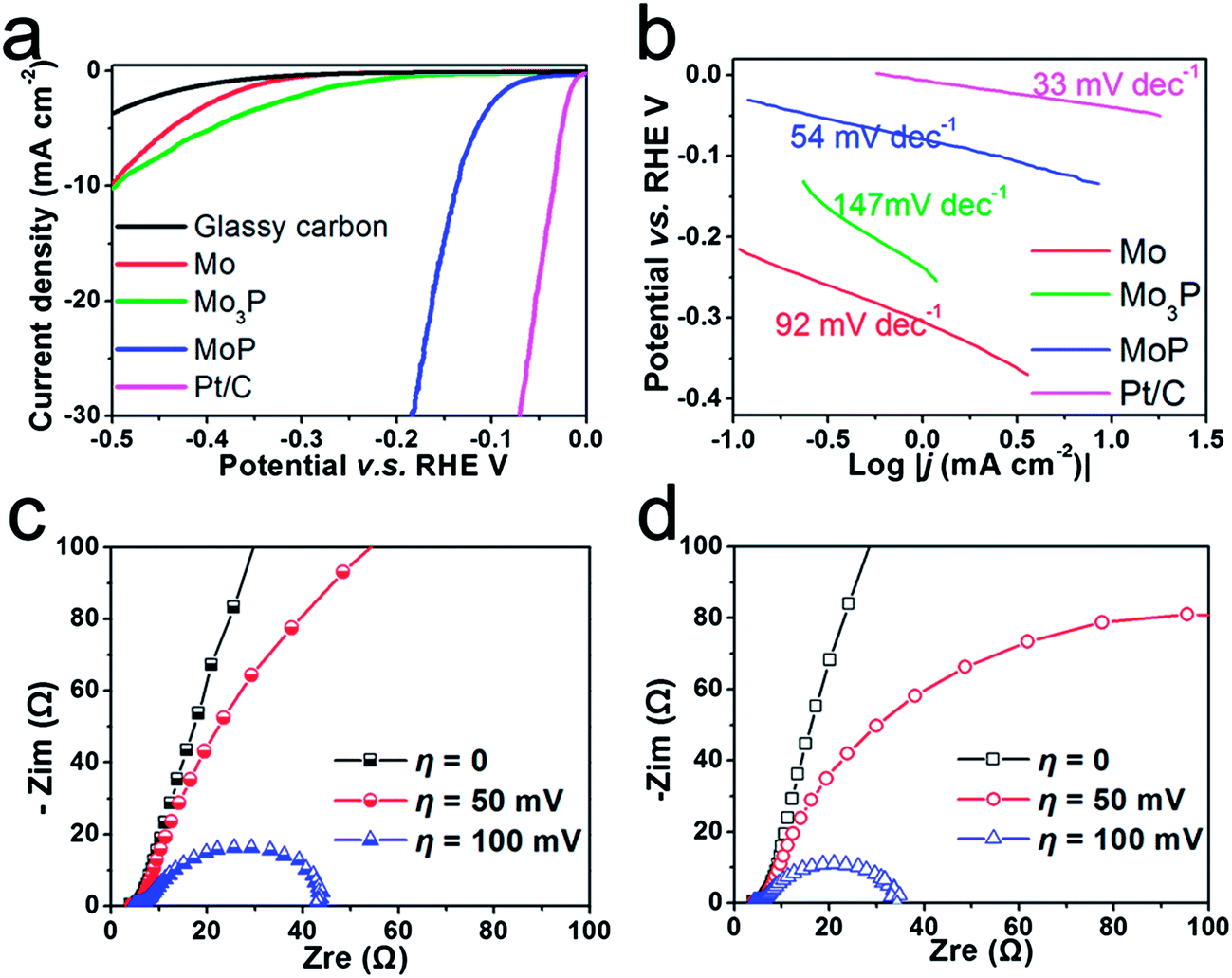 | ||
| Fig. 4 (a) Polarization curves of Mo, Mo3P and MoP in 0.5 M H2SO4, scan rate: 2 mV s−1; (b) Tafel plots in 0.5 M H2SO4; electrochemical impedance spectra of MoP: (c) in 0.5 M H2SO4 and (d) in 1 M KOH. | ||
After electrochemical testing, the catalyst composition was again examined by XPS. Compared to the as-synthesized MoP, the XPS profile of the post-test catalysts displays a similar profile to Mo 3d and P 2p as shown in Fig. S6† except for the decreased content of MoO3 and MoO2. A previous study of Mo2C by Hu et al.23 has proved that MoO3 and MoO2 are not efficient catalysts, which further suggests the high performance could be attributed to MoP.
Tafel plots of potential V–log|j (current density)| could be interpreted by the Volmer–Tafel or Heyrovsky mechanism in the classical two-electron-reaction model for cathodic HER.24 The Tafel slope of 92 mV dec−1 (Fig. 4b) for metal Mo suggests a typical Volmer–Tafel route with the Volmer step as the rate-determining step. After phosphorization, the plot exhibits slopes of 147 mV dec−1 and 54 mV dec−1 for Mo3P and MoP respectively. Here, the Tafel slope of 54 mV dec−1 implies that hydrogen evolution on MoP undergoes a Heyrovsky mechanism which is still different from the route noble metals, e.g. Pt, are subjected to. Based on the Langmuir Isotherm model,40 a fast discharging reaction (Volmer reaction) rate under low coverage (<0.1) of adsorbed hydrogen (Had) leads to ∼30 mV dec−1 and the Tafel slope of 54 mV dec−1 is most possibly caused by a large coverage of Had (>0.6). The large coverage of H is also rendered by theoretical calculation that zero ΔGH° could be achieved at a certain coverage of H on P sites from 2/4 ML to 3/4 ML. In comparison with metal Mo, we can conclude that a simple phosphorization could completely change the reaction route. By extrapolating the Tafel plot to an overpotential of 0 V, the exchange current density can be extracted (Table 1). It is found that MoP exhibits a value of 3.4 × 10−2 mA cm−2, ∼70 times higher than that of metal Mo (4.9 × 10−4 mA cm−2) and also outperforms MoS2 that has been reported as a promising catalyst for HER.13,20 As a non-noble catalyst in bulk form, MoP has achieved comparably competitive performance to Ni2P,27 and is even superior to MoB, Mo2C,23 Mo2C/CNT25 at a similar loading for HER in acidic medium.
| b (mV dec−1) | j o (mA cm−2) | |
|---|---|---|
| Mo | 92 | 4.9 × 10−4 |
| Mo3P | 147 | — |
| MoP in 0.5 M H2SO4 | 54 | 3.4 × 10−2 |
| MoP in 1 M KOH | 48 | 4.6 × 10−2 |
| Pt/C | 33 | 0.63 |
Few catalysts can exhibit good activity and stability under both acidic and alkaline conditions. e.g. Ni2P was found to deteriorate rapidly in alkaline medium.27 Herein we further examined the activity of MoP in both acidic and alkaline conditions using an electrochemical impedance spectroscopy (EIS) technique. Bode plots of MoP (Fig. S7†) suggest a one-time-constant process in both acidic and alkaline media and Nyquist plots show a depressed arc intercepted by the x-axis in Fig. 4c and d. The high-frequency intersection with the x-axis represents ohmic resistance, mainly arising from the electrolyte and all contact resistances. To decouple the ohmic resistance from the polarization resistance, we applied a model of ohmic resistance (Rs) in series with a module, where the polarization resistance (Rct) is in parallel with a constant phase element (CPE). As summarized in Table S4,† MoP performs slightly better in an alkaline medium than in an acidic medium, as indicated by a smaller Rct at an overpotential of 50 mV and 100 mV. The polarization curve in Fig. 5a agrees with the EIS results. At low current density, MoP in an alkaline medium outperforms that in an acidic medium, and exhibits a nearly overlapping polarization curve in the high-current region, suggesting a good performance in alkaline medium.
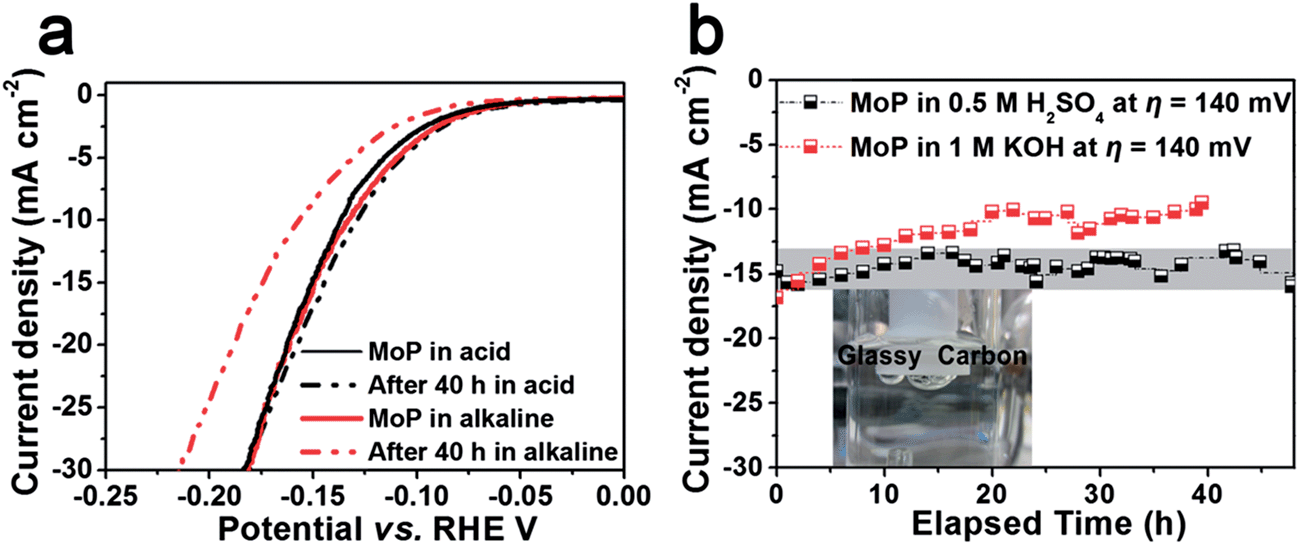 | ||
| Fig. 5 (a) The polarization curve before and after durability tests, scan rate: 2 mV s−1; (b) chronoamperometric electrolysis. | ||
Durability is a key factor in evaluating catalyst performance. Long-term stability is investigated both in 1 M KOH and 0.5 M H2SO4. Unfortunately, slow corrosion happens in 1 M KOH as shown in the amperometric plot (Fig. 5b) and the performance degrades to ca. 60% of its initial value in 40 h (Fig. 5a). However, chronoamperometric electrolysis provides evidence of the excellent stability of MoP in 0.5 M H2SO4 at an applied potential of 0.14 mV vs. RHE in Fig. 5b, and no performance degradation was observed after electrolysis for more than 40 h. The polarization curve after electrolysis shows the slightly enhanced performance in the region of low current density, which could be ascribed to surface activation in cathodic polarization. In contrast, Mo and Mo3P suffer from severe performance degradation in both acidic and alkaline media. Inspection of the morphology after the stability test shows no difference compared to the one before the test (Fig. S8†) in a panoramic view.
Mo is widely known as a chemically unstable metal in acidic media, and with insufficient phosphorization, the degradation of current density was observed even in the second scan of the polarization curve of Mo3P (not shown), implying that corrosion occurs. Stability is commonly correlated with structure. As shown in Scheme 1, the Mo atom is 6-coordinated by P atom in MoP in contrast to 2 or 4 P-atoms bonded to Mo in Mo3P, which could probably be related to the stability in acidic or alkaline media, and suggests the significance of phosphorization.
Conclusions
Distinct performance was observed from a traditional metal Mo phosphide compound. Our results show catalysts having a higher degree of phosphorization as in MoP results in better performance compared to a lower degree of phosphorization or lack thereof as in Mo3P and Mo respectively. Performance in this case is determined based on polarization, Tafel analysis in the low-current region, and stability tests by chronoamperometric electrolysis.DFT calculations verified that the active site should be ascribed to P atoms, which achieve a nearly zero Gibbs free energy. The results show that P atoms have a role analogous to the S atoms in MoS2, which are responsible for creating a high numbers of edges for HER.19 In summary, we have demonstrated that facile phosphorization can transform metal Mo, a poor catalyst, into MoP, an active and stable catalyst for HER. Similar to organic-ligand-functionalized metals, e.g. nickel complexes,7 molybdenum-oxo,5 this strategy could be easily extended to other earth-abundant and inexpensive metals (e.g. Ni, Fe, etc.).
Acknowledgements
Financial support from the academic research fund AcRF tier 1 (M4011020 RG8/12) Ministry of Education, Singapore and competitive research program (2009 NRF-CRP 001-032), National Research Foundation, Singapore. The support by the Singapore National Research Foundation under its Campus for Research Excellence And Technological Enterprise (CREATE) programme is also acknowledged.References
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- R. Cammack, Nature, 1999, 397, 214–215 CrossRef CAS PubMed.
- A. Volbeda and J. C. Fontecilla-Camps, Dalton Trans., 2003, 4030–4038 RSC.
- S. Shima, O. Pilak, S. Vogt, M. Schick, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer and U. Ermler, Science, 2008, 321, 572–575 CrossRef CAS PubMed.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS PubMed.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371–1374 CrossRef CAS PubMed.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878–3888 CAS.
- I. Hatay, P. Y. Ge, H. Vrubel, X. Hu and H. H. Girault, Energy Environ. Sci., 2011, 4, 4246–4251 CAS.
- H. Vrubel, D. Merki and X. Hu, Energy Environ. Sci., 2012, 5, 6136–6144 CAS.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- Z. Chen, D. Cummins, B. N. Reinecke, E. Clark, M. K. Sunkara and T. F. Jaramillo, Nano Lett., 2011, 11, 4168–4175 CrossRef CAS PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262–1267 RSC.
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916–1923 CrossRef CAS.
- R. R. Chianelli, M. H. Siadati, M. P. De la Rosa, G. Berhault, J. P. Wilcoxon, R. Bearden and B. L. Abrams, Catal. Rev. Sci. Eng., 2006, 48, 1–41 CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- D. Kong, H. Wang, J. J. Cha, M. Pasta, K. J. Koski, J. Yao and Y. Cui, Nano Lett., 2013, 13, 1341–1347 CrossRef CAS PubMed.
- H. Wang, D. Kong, P. Johanes, J. J. Cha, G. Zheng, K. Yan, N. Liu and Y. Cui, Nano Lett., 2013, 13, 3426–3433 CrossRef CAS PubMed.
- W.-F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- H. Vrubel and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703–12706 CrossRef CAS PubMed.
- W.-F. Chen, S. Iyer, S. Iyer, K. Sasaki, C.-H. Wang, Y. Zhu, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2013, 6, 1818–1826 CAS.
- W.-f. Chen, C. H. Wang, K. Sasaki, N. Marinkovic, W. Xu, J. T. Muckerman, Y. Zhu and R. R. Adzic, Energy Environ. Sci., 2013, 6, 943–951 CAS.
- L. Liao, S. Wang, J. Xiao, X. Bian, Y. Zhang, M. D. Scanlon, X. Hu, Y. Tang, B. Liu and H. H. Girault, Energy Environ. Sci., 2014, 7, 387–392 CAS.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- L. Feng, H. Vrubel, M. Bensimon and X. Hu, Phys. Chem. Chem. Phys., 2014, 16, 5917–5921 RSC.
- D. Kong, J. J. Cha, H. Wang, H. R. Lee and Y. Cui, Energy Environ. Sci., 2013, 6, 3553–3558 CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef PubMed.
- W. Schmickler and S. Trasatti, J. Electrochem. Soc., 2006, 153, L31–L32 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2006, 153, L33 CrossRef PubMed.
- P. Liu and J. A. Rodriguez, J. Am. Chem. Soc., 2005, 127, 14871–14878 CrossRef CAS PubMed.
- X. Zhao, M. Cao, B. Liu, Y. Tian and C. Hu, J. Mater. Chem., 2012, 22, 13334–13340 RSC.
- O. G. Marin Flores and S. Ha, Appl. Catal., A, 2009, 352, 124–132 CrossRef CAS PubMed.
- J. Bai, X. Li, A. Wang, R. Prins and Y. Wang, J. Catal., 2012, 287, 161–169 CrossRef CAS PubMed.
- D. C. Phillips, S. J. Sawhill, R. Self and M. E. Bussell, J. Catal., 2002, 207, 266–273 CrossRef CAS.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- A. B. Laursen, S. Kegnaes, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577–5591 CAS.
- J. G. N. Thomas, Trans. Faraday Soc., 1961, 57, 1603–1611 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ee00957f |
| This journal is © The Royal Society of Chemistry 2014 |