 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Short-range Li diffusion vs. long-range ionic conduction in nanocrystalline lithium peroxide Li2O2—the discharge product in lithium-air batteries†
A.
Dunst
*a,
V.
Epp
*ab,
I.
Hanzu
ab,
S. A.
Freunberger
a and
M.
Wilkening
*ab
aChristian Doppler Laboratory for Lithium Batteries, Institute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: andreas.dunst@tugraz.at; vikror.epp@tugraz.at; wilkening@tugraz.at
bDFG Research Unit 1277, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
First published on 15th May 2014
Abstract
Understanding charge carrier transport in Li2O2, the storage material in the non-aqueous Li-O2 battery, is key to the development of this high-energy battery. Here, we studied ionic transport properties and Li self-diffusion in nanocrystalline Li2O2 by conductivity and temperature variable 7Li NMR spectroscopy. Nanostructured Li2O2, characterized by a mean crystallite size of less than 50 nm as estimated from X-ray diffraction peak broadening, was prepared by high-energy ball milling of microcrystalline lithium peroxide with μm sized crystallites. At room temperature the overall conductivity σ of the microcrystalline reference sample turned out to be very low (3.4 × 10−13 S cm−1) which is in agreement with results from temperature-variable 7Li NMR line shape measurements. Ball-milling, however, leads to an increase of σ by approximately two orders of magnitude (1.1 × 10−10 S cm−1); correspondingly, the activation energy decreases from 0.89 eV to 0.82 eV. The electronic contribution σeon, however, is in the order of 9 × 10−12 S cm−1 which makes less than 10% of the total value. Interestingly, 7Li NMR lines of nano-Li2O2 undergo pronounced heterogeneous motional narrowing which manifests in a two-component line shape emerging with increasing temperatures. Most likely, the enhancement in σ can be traced back to the generation of a spin reservoir with highly mobile Li ions; these are expected to reside in the nearest neighbourhood of defects generated or near the structurally disordered and defect-rich interfacial regions formed during mechanical treatment.
Broader contextElectrochemical energy storage is considered as the key technology to enable the shift from fossil to renewable energies. Among the few alternatives to Li-ion batteries that theoretically exceed significantly their specific energy, the non-aqueous Li-O2 battery has attracted intense interest. It relies on the reversible formation of lithium peroxide in the positive electrode, 2Li+ + O2 + 2e− ⇌ Li2O2. Major challenges, however, face its practical realization. One is poor charge transport in Li2O2 that is widely recognized as a major factor limiting its achievable amount on the conductive substrate during discharge and the rate and overpotentials at which it can be formed and decomposed during cycling. So far, charge transport in Li2O2 is only poorly understood. To shed light on Li ion dynamics in Li2O2 it needs to be investigated in a complementary way by both macroscopic as well as microscopic techniques. In particular, Li NMR relaxometry offers an atomic-scale access to information on the elementary steps of ion hopping. Such results will build a first basis to understand the complex interplay of morphology, defect density and ion transport in Li2O2. They may help to improve the key metrics of the Li-O2 battery, such as capacity, rate and efficiency. |
I. Introduction
The development of new energy storage systems is a vital topic in materials science. In particular, the lithium-air battery causes tremendous interest not only from the part of scientific community but also from an industrial point of view.1–5 The theoretical specific energy of non-aqueous lithium air batteries is ca. 3500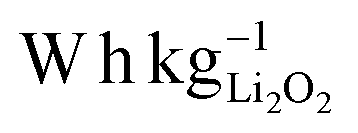 based on the mass of active materials alone. Therefore, it significantly exceeds that of lithium-ion batteries with conventional insertion materials.6–10 Potential key applications, particularly in the long term, include electric vehicles as well as the storage of electricity from intermittent sources such as wind, solar, and tidal.
based on the mass of active materials alone. Therefore, it significantly exceeds that of lithium-ion batteries with conventional insertion materials.6–10 Potential key applications, particularly in the long term, include electric vehicles as well as the storage of electricity from intermittent sources such as wind, solar, and tidal.
On discharge, at the positive electrode O2 from the atmosphere enters the porous electrode, is dissolved in the non-aqueous electrolyte within the pores, and is reduced at the electrode surface forming solid lithium peroxide. Li2O2 can be (re-)oxidized upon charging.1,7,8,10 The achievable amount of Li2O2 that can reversibly be formed upon discharge inside the porous electrode governs the reversible capacity. While the principal possibility to electrochemically form and decompose Li2O2 in non-aqueous electrolytes is well established, many challenges remain towards practical application.1,2,6–8,11,12 These include stability issues of the electrolyte and the porous electrode that lead to side reactions and compromised cycleability.13–19 Typical side reaction products at the cathode are Li2CO3, Li alkyl carbonates and Li carboxylates.13,17,20,21
Another major challenge is how to form a large amount of solid, insulating Li2O2 by O2 reduction in the conductive porous substrate and to effectively oxidize it on charging.22–26 On discharge, formation of a film of insulating Li2O2 limits the film thickness to a few nm before electron tunnelling or bulk conduction becomes ineffective.23,24,27 This is because of the poor conductivity estimated for pure Li2O2 that is significantly lower than that of other battery materials. In many cases particle growth has been observed that typically leads to particles in the range of 200 to 400 nm composed of a few 10 nm large primary crystallites; this suggests additional transport mechanisms to be in place.5,28 On charging, a two-stage process has been disclosed involving Li+ removal near the surface and bulk Li2O2 oxidation at low and high overpotentials, respectively.24,25,29 This requires Li motion in the surface region of Li2O2 and electron transport from the Li2O2 to the solid electron conducting surface. Electron transport may be relatively easy for Li2O2 directly in contact with the surface, but becomes difficult for the majority of Li2O2 more remote from the surface. When we consider issues such as the reversibility of Li peroxide accumulation and removal, it can be seen that transport in the solid state, that is, in the bulk, plays a major role. In general, enhanced Li diffusion in the bulk may assist in the electrochemical formation as well as the resorption and dissolution of Li peroxide. Considering recent studies, both electron and ion transport in Li2O2 are shown to be critical for the operation of the Li-air battery and improvements require their understanding on a microscopic level.5,26,30,31 In particular, it has been shown recently that finely crystalline or amorphous Li2O2 decomposes at lower voltages than large particles. The exact reasons, however, have not been identified yet.5,31
In addition to these recent findings, there is a lively debate on charge transport properties in Li2O2 in general; several theoretical studies have been published until now23,24,32–35 including also a very recent one on amorphous Li2O2 by Siegel and co-workers.36 As yet, only Maier and co-workers37 have investigated electron and ion transport in microcrystalline Li2O2experimentally by ac impedance spectroscopy and dc measurements; they showed that electronic conduction is governed by hole polarons, and ionic conduction is presumably mediated by negatively charged lithium vacancies; the crystal structure of Li2O2 (space group P63/mmc) is shown in Fig. 1. To the best of our knowledge, there is, so far, no investigation available reporting on activation energies of the elementary hopping processes of individual Li ions. Such information can be made available by nuclear magnetic resonance (NMR) spectroscopy.
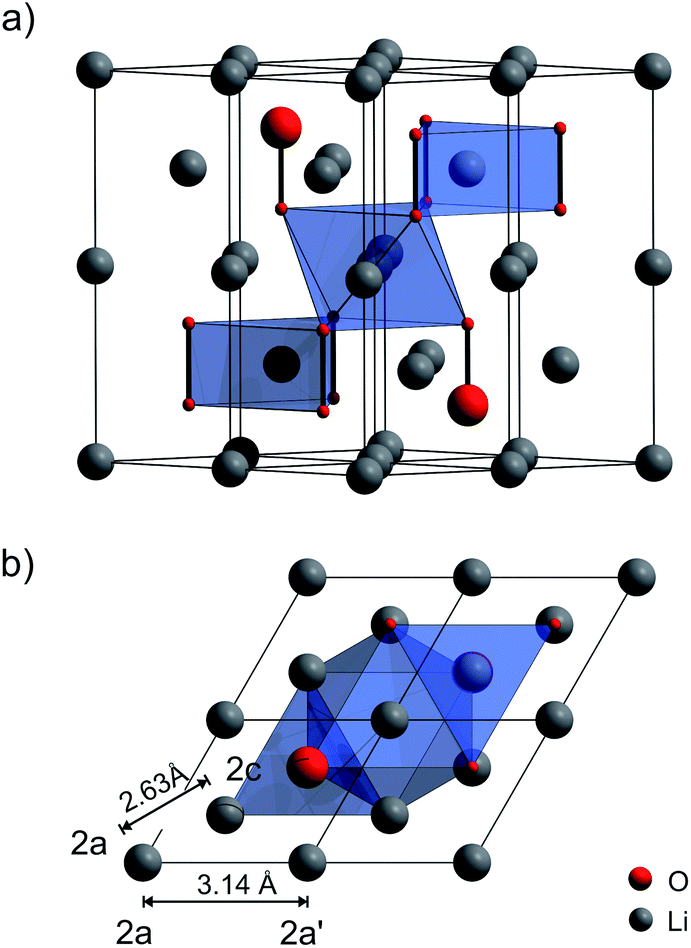 | ||
| Fig. 1 Crystal structure of Li2O2 (hexagonal space group P63/mmc), see also ref. 24. (a) 2 × 2 × 1 expansion of the unit cell. The covalently bonded O2 dimers are arranged in an alternating ABAB stacking. There are two unique Li sites indicated by polyhedra. The trigonal prismatic Li is in the same layer as the peroxide anions. The octahedrally coordinated one resides between the peroxide layers. (b) View along the z-axis showing the Li–Li distances of the two crystallographically inequivalent Li sites 2a and 2c. | ||
The present work is aimed at understanding both short-range and long-range Li ion transport in nanocrystalline Li2O2, that is, a nanostructured sample with crystallites having a mean diameter of less than 50 nm which is comparable to that found in electrochemically formed Li2O2. The samples investigated have been prepared ex situ via a top-down approach: chemically synthesised microcrystalline lithium peroxide was mechanically treated in a high-energy planetary ball mill. The starting material was obtained in high purity by dehydration of a mixture of LiOH and H2O2. Although composed of crystallites with the same mean diameter, a nanocrystalline sample prepared by ball-milling is not directly comparable with the discharge product formed in a lithium-air battery. Here, however, we considered ball milling as an appropriate means to resemble the electrochemically formed Li2O2 as closely as possible with respect to both crystallite size and defects. From the point of view of structural disorder, the sample investigated might serve well to study the influence of structural disorder on ion transport properties. As has been shown also for other Li ion conductors, the transport properties of ball milled oxides may also resemble those of completely amorphous materials reflecting the upper limit of a disordered oxide or peroxide.
In the present study, long-range as well as short-range Li ion dynamics have been probed by temperature-variable conductivity spectroscopy and 7Li NMR relaxometry. The overall conductivity σdc of the microcrystalline sample turned out to be very low which is in agreement with previous results as well as with 7Li NMR line shape measurements presented here. Ball-milling, however, leads to an increase of σdc, being characteristic for long-range ion transport, by approximately two orders of magnitude; correspondingly, the activation energy decreases. Similarly, also short-range Li ion dynamics are influenced by high-energy ball milling. In general, a large volume fraction of interfacial regions is formed and structural disorder is introduced by mechanical treatment. The defects generated are anticipated to largely influence both ionic transport over long distances as well as the elementary steps of ion hopping to which NMR is extremely sensitive when carried out at sufficiently low temperatures.
II. Experimental
A. Preparation, structural and thermal characterization of lithium peroxide
Lithium hydroxide monohydrate (Sigma Aldrich) was dissolved in water (dest.) and an equimolar aqueous solution of hydrogen peroxide 30% (Merck) was added under an Ar atmosphere. The solution was stirred under an argon atmosphere at ambient conditions. The initially produced lithium hydroperoxide LiOOH was dried in vacuo and subsequently treated at 493 K for 12 h yielding microcrystalline lithium peroxide Li2O2 of high purity. To prepare nanocrystalline Li2O2, the source material micro-Li2O2 was mechanically treated in a high-energy ball mill (Fritsch Pulverisette 7 (premium line)). A grinding beaker made of stabilized ZrO2 (45 mL) and 140 milling balls (ZrO2, 5 mm in diameter) were employed; the microcrystalline starting sample was milled for 180 min at a rotational speed of 600 rpm. The complete synthesis was carried out under an Ar atmosphere.From then on, the samples prepared were stored in an argon-filled glove box. They were characterized by X-ray powder diffraction (XRD) carried out at room temperature with a Bruker D8 Advance diffractometer operating in Bragg–Brentano geometry with Cu-Kα radiation at 40 kV. Diffractograms were analyzed by Rietveld refinement with the programs FULLPROF and X'PertHighScorePlus (Panalytical). To determine decomposition temperatures we carried out thermogravimetric (TG) and differential scanning calorimetry (DSC) measurements; for simultaneous thermal analysis a 449C Jupiter (Netzsch) was used. The heating rate was 10 K min−1 from 293 K to 773 K; experiments were carried out with a helium gas stream of 50 mL min−1.
B. Characterization of ion transport and Li diffusivity
In order to estimate the ionic and electronic contributions to the total conductivity, potentiostatic polarization measurements were carried out on the gold-sputtered Li2O2 pellets which were pressed under a protective Ar atmosphere. The pellets were placed inside air-tight Swagelok-type electrochemical cells in order to protect them during measurements from air and humidity. The polarization experiments were carried out at 293 K with a VMP3 instrument from Biologic that was equipped with a low-current option and controlled by EC-lab software. Polarization measurements were carried out with a constant voltage U = 5 V. The current I was recorded as a function of time t.
Prior to the measurements, the samples were fire-sealed in evacuated glass NMR tubes of 5 mm in diameter and ca. 3 cm in length. The measurements were carried out at temperatures ranging from 133 K to 604 K. Temperature regulation and monitoring were carried out with a Eurotherm temperature controller in combination with a type T thermocouple.
7Li NMR spectra were obtained after Fourier transformation of the free induction decays (FIDs) recorded by a single pulse experiment using recycle delays of up to 60 s at the lowest accessible temperatures. 7Li NMR spin–lattice relaxation (SLR) rates R1 in the laboratory frame of reference were acquired by means of the classical saturation recovery pulse sequence 10 × π/2 − td − π/2 − acquisition (acq.)38–42 The initial ten π/2 pulses in rapid succession are used to destroy any longitudinal magnetization Mz. Immediately after this saturation comb, the recovery of Mz is recorded as a function of delay time td at different temperatures. In addition, 7Li NMR SLR rates have also been recorded in the rotating frame of reference (SLRρ). For this purpose, the spin-locking technique π/2p(tlock) − acq.38,43–48 was employed. The corresponding locking frequency was chosen to be ω1/2π = 28 kHz with the duration of the locking pulse tlock varying from 100 μs to 100 ms. The recycle delay was set to at least 5 × R1−1 ≡ 5 × T1 in order to guarantee full longitudinal relaxation between each scan. Both R1 and R1ρ were obtained by parameterizing the resulting magnetic transients Mz(td) and Mϱ(tlock), respectively, by stretched exponentials: Mz(td) ∝ 1 − exp(−(td/T1ϱ)γ) and Mϱ(tlock) ∝ exp(−(tlock/T1ϱ)γϱ), respectively.
In addition, 7Li NMR stimulated echoes49–53 have been recorded by utilizing the Jeener–Broekaert54 pulse sequence as a function of mixing time tm (10 μs ≤ tm ≤ 100 s) at a fixed preparation time tp: (90°)x − tp − (45°)y − tm − (45°) − t − acquisition. Note that SAE NMR, that is, two-time correlation spectroscopy, takes advantage of interactions between the quadrupole moment of the nucleus and the electric field gradient (EFG) tensor at the nuclear site.55 The technique gives access to single-particle correlation functions, so-called 7Li sin–sin hopping correlation functions. In general, the results from stimulated echo measurements are comparable to those from SLRρ NMR measurements.53 The difference is that Li SAE NMR, being comparable to Li exchange NMR,56–60 is sensitive to even slower Li diffusion processes.
III. Results and discussion
A. X-ray diffraction, chemical and thermal analysis
The purity of the lithium peroxide prepared was checked by X-ray powder diffraction (see Fig. 2), energy dispersive X-ray diffraction (EDX), infrared (IR) spectroscopy and inductively coupled plasma mass spectrometry (ICP-MS). The powder diffractogram obtained for microcrystalline Li2O2 can be fully indexed with literature 2θ values. We could not detect any significant amounts of crystalline impurity phases. Fortunately, the same is true for the ball-milled material which was treated for 3 h in a planetary mill. It has also been characterized by XRD, EDX, IR spectroscopy and ICP-MS (see also the ESI†). As expected, the XRD peaks have broadened due to size effects and strain generated, but neither any mechanochemically induced phases were formed nor any traces of crystalline impurities, such as abrasion of ZrO2, show up. Negligible abrasion is also corroborated by ICP-MS (see ESI†). IR spectroscopy (see Fig. S1†) reveals a negligible amount of X-ray amorphous lithium carbonate which is most likely due to post-contamination. The impurity level concerning metal ions is fortunately very low as has been verified by EDX (see Fig. S2†) and particularly by ICP-MS (see Table S1 in the ESI†). Altogether, this is a perfect starting condition which helps us to interpret both results from NMR line shape measurements and relaxometry as well as conductivity spectroscopy.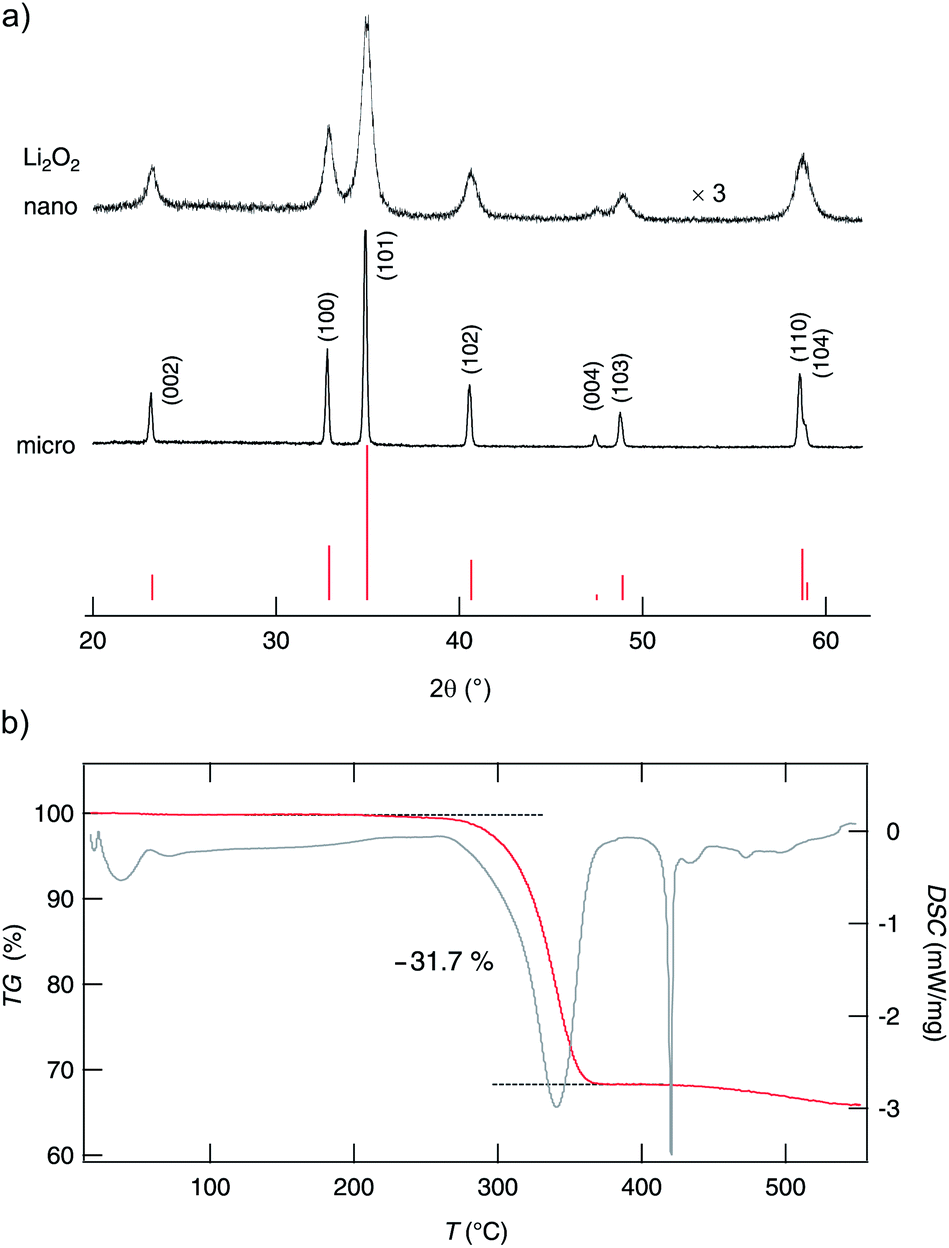 | ||
| Fig. 2 (a) X-ray powder diffractograms of micro- and nanocrystalline lithium peroxide. Miller indices (hkl) indicate the reflections of Li2O2. Vertical bars in the lower part of the figure illustrate peak positions and relative intensities of pure lithium peroxide. Compared to the pattern of micro-Li2O2 that of nano-Li2O2 is magnified by a factor of three. (b) TG (red line) and DSC measurements (grey line) of nano-Li2O2; above ca. 500 K Li2O2 starts to decompose. A weight loss of ca. 32% points to the release of oxygen leaving over Li2O. | ||
The mean crystallite size of micro-Li2O2, which is our source material, lies in the μm range as can be seen from the sharp XRD reflections. From the extent of peak broadening we can roughly estimate the mean diameter 〈d〉 of the crystallites. The well-known equation introduced by Scherrer yields 〈d〉 ≈ 25 nm. This is in good agreement with other oxides which had been treated in high-energy ball mills to produce nanocrystalline ceramics; usually the crystallite sizes are much less than 50 nm and centre about 10 to 20 nm.61
Unfortunately, nanocrystalline Li2O2 decomposes at elevated temperatures. Our TG/DSC measurements (see Fig. 2) reveal that oxygen is released at temperatures significantly larger than 493 K. The TG curve is very similar to that reported in ref. 37. Thus, NMR (and conductivity) measurements (see below) had to be restricted to this limit. Their interpretation needs careful consideration of the thermal stability of nano-Li2O2, also with respect to grain growth and healing of defects initially introduced.
B. Long-range transport: conductivity measurements and potentiostatic polarization experiments
![[small sigma, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e111.gif) , of nanocrystalline Li2O2 are shown which were recorded during heating the sample from 353 K up to 533 K. They reveal distinct dc plateaus referring to σ′(ν → 0) ≡ σdc. At lower T, a dispersive regime shows up which can be associated with local, i.e., short-ranged (forward–backward), jump processes. In many cases, a Jonscher power law, σ′ ∝ νκ, is appropriate to account for the frequency dependence of σ′ in this frequency range. The deviation from σdc in the low-frequency regime is because of polarization effects of the ion-blocking electrodes applied. σ′ in the regime of electrode polarization (EP) often shows a slight change in slope in this frequency range as indicated in Fig. 3(a); the arrow points to the EP flank of the isotherm recorded at 533 K.
, of nanocrystalline Li2O2 are shown which were recorded during heating the sample from 353 K up to 533 K. They reveal distinct dc plateaus referring to σ′(ν → 0) ≡ σdc. At lower T, a dispersive regime shows up which can be associated with local, i.e., short-ranged (forward–backward), jump processes. In many cases, a Jonscher power law, σ′ ∝ νκ, is appropriate to account for the frequency dependence of σ′ in this frequency range. The deviation from σdc in the low-frequency regime is because of polarization effects of the ion-blocking electrodes applied. σ′ in the regime of electrode polarization (EP) often shows a slight change in slope in this frequency range as indicated in Fig. 3(a); the arrow points to the EP flank of the isotherm recorded at 533 K.
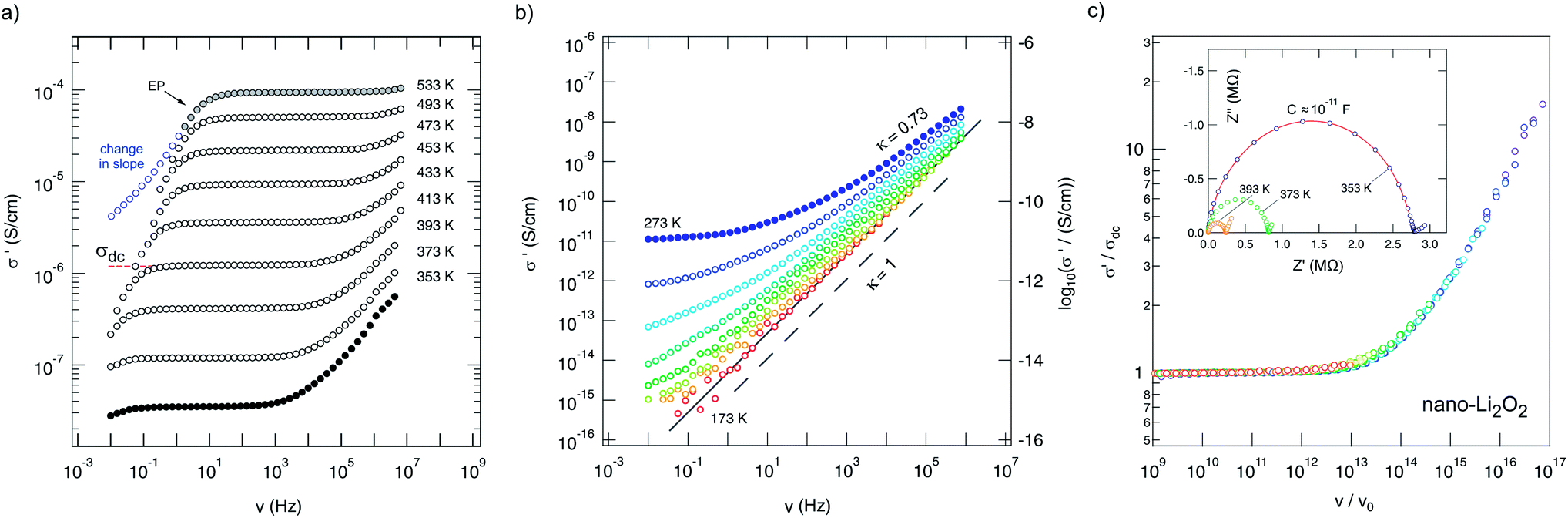 | ||
| Fig. 3 (a) Conductivity spectra of nanocrystalline Li2O2 prepared by high-energy ball milling. (b) Conductivity spectra recorded under the same conditions as in (a) but at lower T. κ (= 1 − α) is the Jonscher exponent; a value close to 1 (indicated by the dashed line) points to so-called NCL-type behaviour being frequently interpreted in terms of localized ion motions. The isotherms shown were recorded at 173 K, 183 K, 193 K, 213 K, 233 K, 253 K and 273 K. (c) Scaling of the isotherms shown in (a); the inset shows selected complex plane plots (−Z′′ vs. Z′). | ||
At lower T (Fig. 3(b)), the dispersive part starts to dominate the spectra and σ′ tends to obey the relation σ′ ∝ ν. This is the signature of nearly-constant-loss (NCL) behavior being typical for complex crystalline and structurally disordered ion conductors, see ref. 62 for an overview on this topic. The behavior σ′ ∝ ν implies a negligible frequency dependence of the dielectric loss, that is the imaginary part of the complex permittivity ε′′(ν) = σ′(ν)/(2πν) yielding σ′NCL(ν) = 2πνε′′NCL(ν) ≈ Aν1−α with α > 0 but close to 0 and A being almost independent of T: ε′′NCL(ν)2π ≈ Aν−α.
NCL behavior is frequently interpreted in terms of strictly localized ion hopping which can be illustrated as caged dynamics in a double-well potential, for example.62 The activation energy associated is anticipated to be very small. The electrical response of crystalline Li2O2 found at low T clearly suggests the presence of NCL-type (ion) dynamics, most probably because of the short distances of possible occupied and unoccupied (interstitial) Li sites within the crystal structure (see Fig. 1).
In contrast to such shallow activated processes, σdc is sensitive to long-range ion transport usually determined by much higher activation energies being the sum of both the migration enthalpy and the enthalpy needed to form, e.g., vacant Li sites. Siegel and co-coworkers24 clearly summarized the types of defects, their equilibrium formation energies and concentrations. As it turned out by first principle calculations, the dominant positively and negatively charged defects are hole polarons and Li vacancies, respectively. In single crystalline Li2O2, the formation energies have been calculated to range from 0.93 to 0.95 eV with concentrations in the order of 107 cm−3.24
In general, slow Li ion transport in oxides is characterized by activation energies being much larger than 0.6 eV.63–65σdc can either be read out from the distinct dc plateaus in Fig. 3(a) or determined by analyzing complex plane plots, which can be constructed when the imaginary part Z′′ of the impedance is plotted as a function of the real part, that is Z′. An ideal Debye process, governed by an exponential relaxation function, would give a semicircle whose origin coincides with the abscissa. Depressed semicircles, as found for many complex ion conductors showing structural disorder,66 directly correlate with the presence of a dispersive part of σ′ and point to a non-Debye response as well as so-called correlated motion.67,68 The single semicircles, observed for each temperature here, have to be regarded as a sum of both the response from the bulk and that from the grain boundaries. Electrode effects show up as spikes or tails in the Nyquist plots presented in Fig. 3(c). Analyzing −Z′′(Z′) with an appropriate equivalent circuit unit including a resistance and a constant phase element connected in parallel yields a (total) capacity C in the order of 10−11 F (inset of Fig. 3) for the main relaxation process observed. This indicates that, to a major extent, the electrical response, and thus σdc, reflects bulk ion conduction.69
The same can be deduced from permittivity spectra ε′(ν) shown in Fig. 4. While EP shows up once again at low frequencies and high temperatures, the permittivity found at higher frequencies (or lower temperatures) is well below 100 and indicates presumably bulk response. The dashed line in Fig. 4(b) illustrates the permittivity plateau leading to ε′(0) ≈ 30. This is in line with the value (ε′(0) ≈ 35) which has been reported for microcrystalline Li2O2 by Gerbig et al.37 Moreover, experiments with different electrodes (Ti or Pt) and ac conductivity measurements as a function of oxygen partial pressure37 did not show any indications that oxygen ions determine the overall conductivity. In an O2 atmosphere the incorporation of oxygen consumes electrons leading to the formation of holes that are quite localized corresponding to a superoxide ion O2− on O22− sites as electron spin resonance (EPR) spectra had indicated; moreover, the superoxide O2− anions seem to associate with lithium vacancies.37
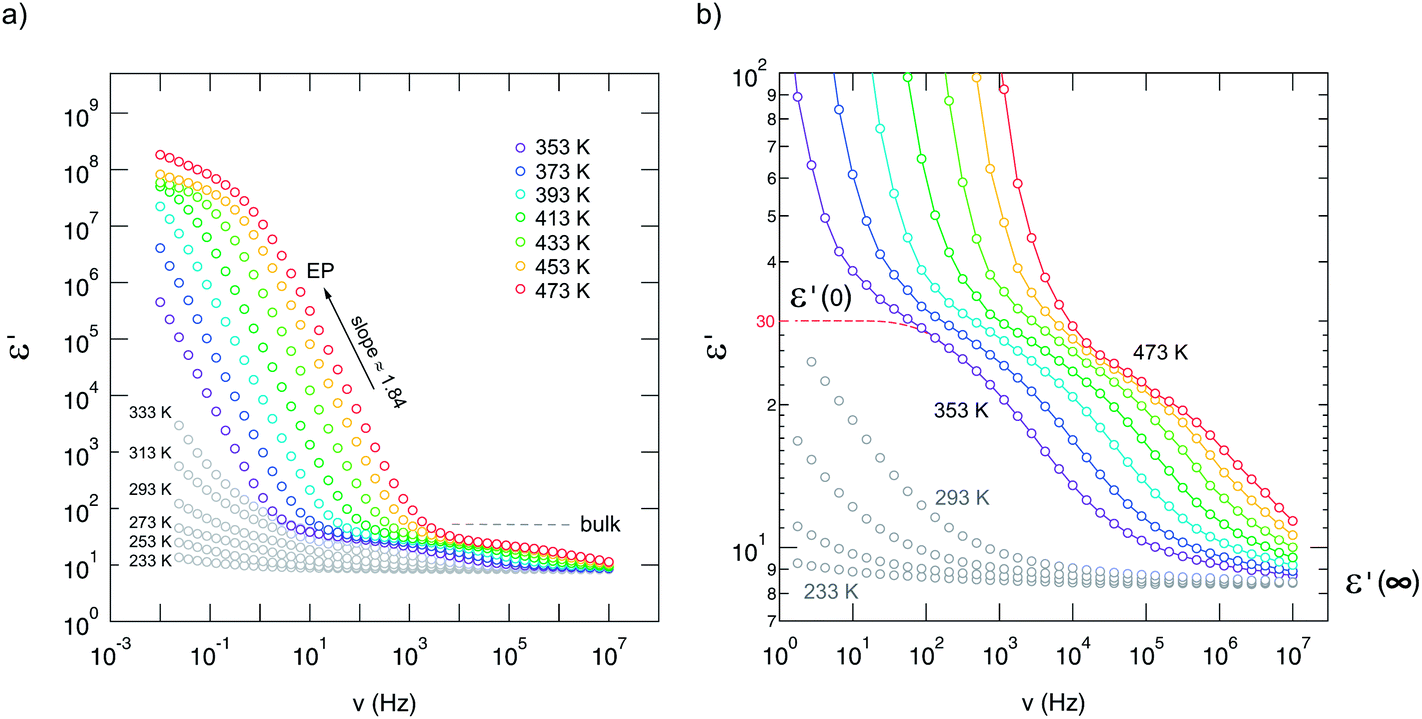 | ||
| Fig. 4 (a) Permittivity spectra of nanocrystalline Li2O2 recorded at the temperatures indicated. (b) Magnification of the spectra shown on the left side. Spectra have been recorded in steps of 20 K. ε(0) ≈ 30, which is in good agreement with that deduced for microcrystalline Li2O2, see ref. 37, indicates bulk response rather than large contributions from grain boundaries. | ||
It is interesting to note that ε′(0) decreases with increasing T although the temperature was still below the decomposition point. Since this process is irreversible, it might reflect the influence of defect healing due to soft sintering of the sample or reflect the smooth onset of Li2O release. According to Sidebottom,70 and similarly to Roling,71 with the help of the Debye model for dipolar relaxation the change in permittivity Δε = ε′(0) − ε′(∞) (see Fig. 4) can be related to the number fraction n′ of the mobile ions and the distance traversed in a single hop. Thus, a slight decrease of n′, associated with a decrease in ion conductivity σ′ (and consequently σ′(dc), as presented below), could explain the decrease observed. As pointed out by Ngai and Rendell,72 such a decrease could also be interpreted in terms of a change of the underlying motional correlation function. In a wider sense, both interpretations reflect a change of the defect chemistry37 in Li2O2 at elevated T because of the oxygen loss when the sample is exposed to a reducing nitrogen atmosphere.
Despite this slight irreversibility observed, the conductivity spectra σ′(ν) recorded up to 450 K (at least) can be scaled such that they collapse into a single master curve. This means that their shape does not vary significantly with increasing temperature. Here, ν0 (see Fig. 3c) is meant to be the ‘characteristic’ frequency at which σ′ = 2σdc holds. This scaling behavior roughly proves that a single (main) charge carrier process seems to be relevant up to 473 K.
Finally, in Fig. 5σdcT, determined from σ′(ν), is plotted for micro- and nanocrystalline Li2O2 as a function of inverse temperature. The arrows mark the conductivities recorded during the heating run and the cooling run, respectively. Heating up to 570 K has negligible influence on σdc of the micro-sample; a much larger influence, however, is observed for nanocrystalline Li2O2. σdcT of the nano-sample starts to deviate from the Arrhenius line drawn if the temperature was raised to 500 K. This temperature is in good agreement with that determined via TG/DSC (see above). Note that between each data point (σdcT) the equilibration time to reach a constant temperature was about 20 min. As mentioned above, this feature might be due to Li2O release and/or a kind of sintering of the sample. Grain growth and significant sintering, however, could not be observed by XRD.
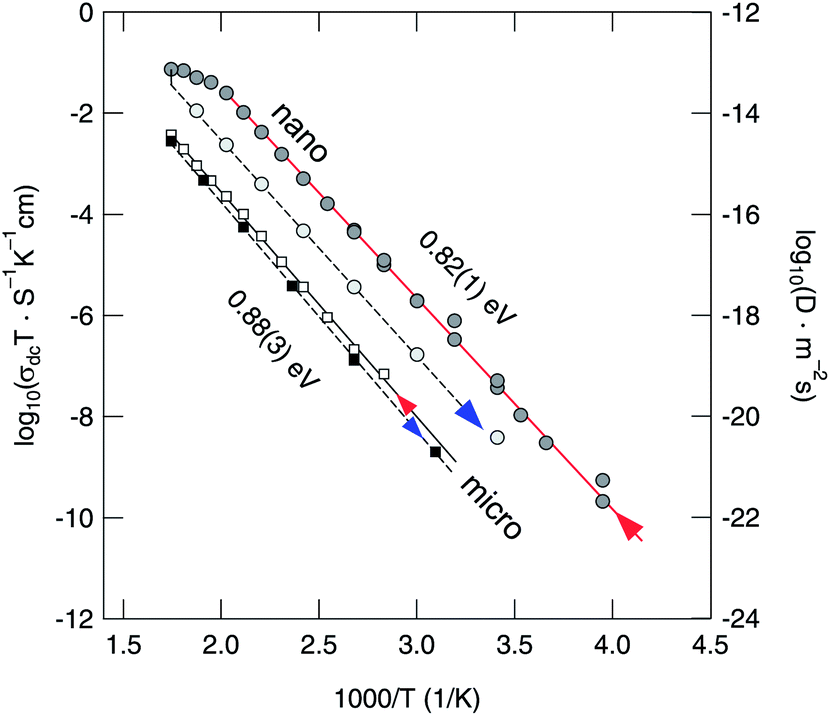 | ||
| Fig. 5 Arrhenius plot of the total conductivity of nanocrystalline and microcrystalline Li2O2 measured under a flow of dry nitrogen. The arrows denote the heating and cooling runs, respectively. The activation energies refer to the heating runs. During cooling we obtain 0.90(7) eV (micro) and 0.83(7) eV (nano). To obtain the right axis, σdc values have been roughly converted into solid-state diffusion coefficients D via the Nernst–Einstein equation. | ||
Most importantly, when comparing the initial conductivities of micro- and nanocrystalline Li2O2 (heating runs), that of nano-Li2O2 is by more than two orders of magnitude larger than that of micro-Li2O2. At room temperature we have σdc,nano ≈ 1.35 × 10−10 S cm−1 which corresponds – when the Nernst–Einstein equation is applied – to an overall solid-state diffusion coefficient in the order of 4 × 10−20 m2 s−1. In accordance with the increase of σdc, the corresponding activation energy for overall (long-range) charge carrier transport is reduced from Ea = 0.88 eV to 0.82 eV. This result reflects the influence of crystallite size and structural disorder introduced via ball-milling on macroscopic properties such as overall conductivity, which is the sum of both the electronic and ionic contributions. For comparison, Maier and co-workers, who measured ionic (ion) and electronic (eon) conductivities of microcrystalline Li2O2, found Ea,ion = 0.95(5) eV (423–473 K) and Ea,eon = 1.3(1) eV (423–473 K), respectively.37 The bulk ion conductivity σion,micro of 10−10 to 10−9 S cm−1 recorded at 373 K is in perfect agreement with our value of 2 × 10−9 S cm−1 (373 K) keeping in mind that the latter represents the total conductivity.
For Li2O2 which was treated for 1 h in a Pulverisette 5 mill, Gerbig et al. reported an activation energy being reduced by ΔEa = 0.14 eV compared to that of microcrystalline Li2O2. This result agrees well with our value of 0.82 eV. In order to estimate any electronic contributions to the total value of σdc, we carried out preliminary potentiostatic polarization measurements at room temperature.
The nanocrystalline sample exhibited normal behavior and the current dropped exponentially (see Fig. 6). It has to be noticed that the time needed for stabilization of the current is very long, a fact that indicates a very low conductivity, and thus a very low mobility of the ionic species. Unfortunately, no clear plateau was reached during the experiment. Nevertheless, the electronic conductivity of nano-Li2O2 is estimated to be approximately 9 × 10−12 S cm−1, that is less than 10% of the total conductivity measured at room temperature (≈1.1 × 10−10 S cm−1).
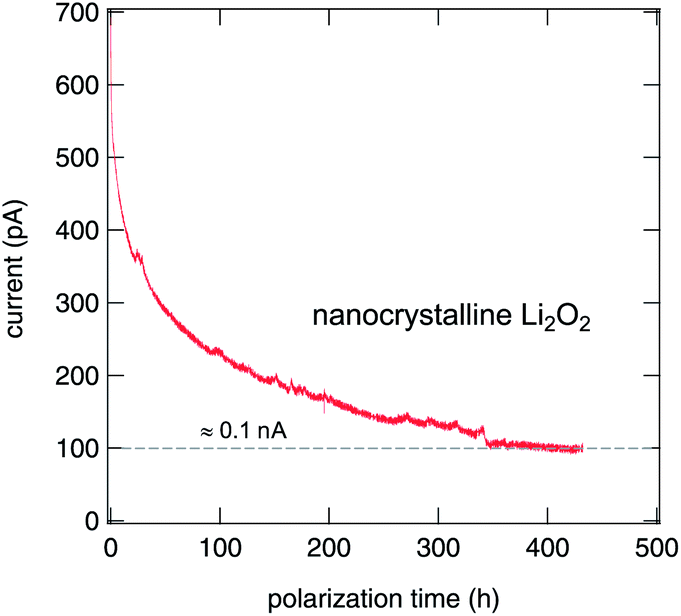 | ||
| Fig. 6 Polarization curve (dc) of a nanocrystalline lithium peroxide prepared by high-energy ball milling. The electric contact to the pressed-pellet was realized by plasma gold sputtering. The voltage applied across the pellet was 0.5 V. The almost constant current which is reached at long polarization time, that is the (quasi) stationary state, is slightly below 100 pA. | ||
In the case of our microcrystalline sample, the conductivity was so low that it was impossible to measure any current at the same polarization voltage (0.5 V) that was used for the nanocrystalline material. Consequently, the voltage applied was increased tenfold, up to 5 V. Even in this case, it was not possible to see any unequivocal exponential decrease of the current. It has to be noticed that by being submitted to such high voltages, the electrodes will no longer be blocking and there will be no accumulation at the electrode since there is an ongoing chemical reaction. Remarkably, even under such harsh conditions micro-Li2O2 does not show any traces of damage during 1000 hours of the experiment. The conductivity of this sample seems to be extremely low at room temperature. This makes it impossible to measure it in such a direct way with our VMP3 instrument, which is so far limited to measurements at ambient temperature.
Maier and co-workers performed similar measurements but at higher T; they were able to exactly determine both σion and σeon of microcrystalline lithium peroxide.37 If we roughly extrapolate their data on micro-Li2O2 towards room temperature we get σion/σeon ≈ 1000. The same can be anticipated from our experiments. For our ball-milled sample, however, we found σion/σeon ≈ 10. This trend is in qualitative agreement with the result reported for nano-Li2O2, which was, however, only softly milled in a Pulverisette 5 planetary mill for just 1 hour: Gerbig et al. reported on an increase of σion by half an order and σeon by one order of magnitude.37 As is well known, the milling conditions, such as the type of mill, the duration of milling, the atmosphere, the ball-to-powder ratio, etc., can drastically influence the structural properties and defect chemistry of the material obtained.
Irrespective of the exact influence of the milling conditions, this comparison reveals that during milling σeon increases more than σion does, leading to a smaller σion/σeon ratio. On the basis of EPR, see ref. 37, the increase of σeon (measured in an oxygen atmosphere) of a ball-milled sample has been associated with an increasing concentration of superoxide O2− species on peroxide sites corresponding to electron holes.
Before we look in detail on Li jump rates and activation energies deduced from NMR, we shall compare the conductivity results with other ion conductors. Taking into account the electronic contribution to the total conductivity, ion dynamics in nanocrystalline Li2O2 turned out to be relatively poor. Values of σion in the order of 10−10 S cm−1 and σeon in the order of 10−12 S cm−1 are below those being desired for a lithium-oxygen battery. Preferably, ionic conductivities should be in the order of 10−5 to 10−3 S cm−1; electronic ones even two orders of magnitude higher. For comparison, the record holder in terms of extremely good ion conductivity is Li10GeP2S12 with 10−3 S cm−1 at room temperature,75 followed by garnet-type oxides42,76 (10−3 S cm−1 at approximately 300 K) or Li-bearing argyrodites.77 Activation energies range from 0.35 to 0.25 eV. The electronic conductivity of nanocrystalline Li2O2 prepared by ball-milling is still lower than that of LiFePO4 (10−6 S cm−1, 0.6 eV (ref. 37 and 78)) that may serve here as a yardstick for a mixed conductor with practical battery applications. In the case of LiFePO4, carbon coating and nanosizing are necessary to prepare an electrode material able to work as a cathode in lithium-ion batteries. Similar conductive coating, however, cannot be realized on the forming/disappearing Li2O2; the electronic conductivity values not to be rate limiting would have to be again several orders of magnitude higher. This might be achieved by further doping of the material as well as further introduction of structural disorder.36 Taking advantage of non-trivial nano-size effects would lead to the question how does ion conductivity depend on the size of the crystallites? This would require a systematic study which is beyond the scope of the present investigation focussing on the elementary steps of hopping in ball-milled Li2O2.
C. Short-range motions: NMR spectra and relaxation rates
Temperature-variable 7Li NMR spectra, when recorded under static, i.e., non-rotating conditions, are sensitive to Li ion hopping processes.79–83 In Fig. 77Li NMR spectra of both micro- and nano-crystalline Li2O2 are shown. Although 7Li is a quadrupole nucleus, due to symmetry reasons there is no electric field gradient present at the Li sites which would lead to a so-called quadruple powder pattern.73 At sufficiently low temperature the NMR line is dipolarly broadened and its shape is similar to a Gaussian. For comparison, the corresponding rigid-lattice Li NMR spectrum of Li2O, crystallizing with cubic symmetry, looks very similar.74,84 At temperatures below ambience, the Li hopping rate 1/τ is low and does not affect the spectra. With increasing temperature, however, 1/τ increases and causes line narrowing. If 1/τ reaches the value of the NMR line width, which is 17.5 kHz at 290 K, homonuclear dipole–dipole interactions governing the line in the rigid lattice regime become increasingly averaged. In the present case, the so-called motional narrowing clearly proceeds in a heterogenous way which means that a narrow NMR line emerges at elevated T and superimposes the broad one.74,84,85 The spectrum of micro-Li2O2 reveals that this process starts at around 482 K; it can be even better recognized when the spectrum recorded at 604 K is regarded.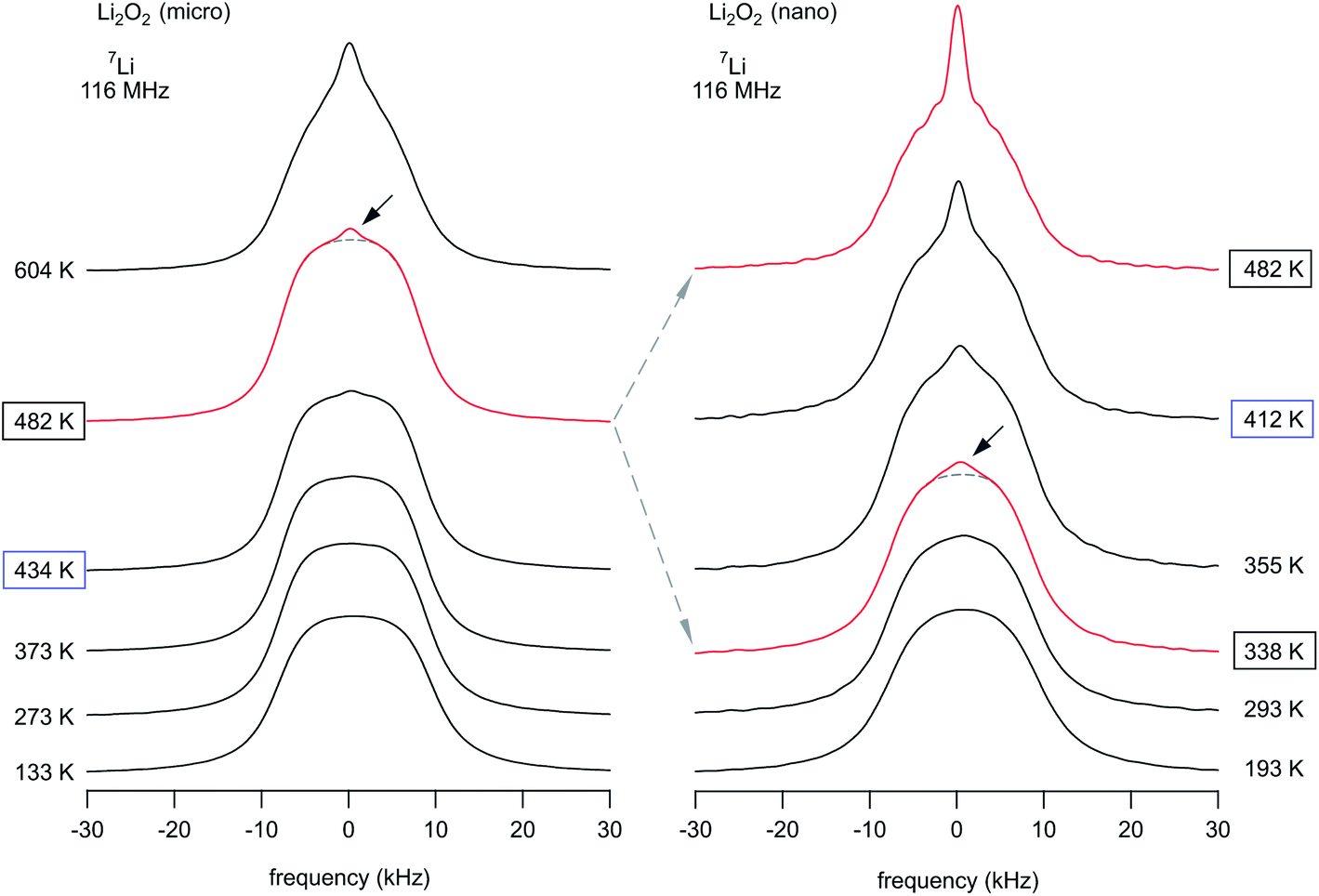 | ||
| Fig. 7 Static 7Li NMR spectra of microcrystalline (left) and nanocrystalline (right) Li2O2 recorded at 116 MHz and at the temperatures indicated. The arrows indicate a motionally narrowed NMR line superimposing the broad line, being characteristic for Li ions in microcrystalline Li2O2. Since the electric field gradient at the Li sites in Li2O2 vanishes due to symmetry reasons, 7Li NMR spectra of Li2O2 are composed of a single line (the ‘central’ line) only, see also ref. 73. The same situation was found, e.g., for Li2O with perfect cubic symmetry.74 | ||
Importantly, in the case of nanocrystalline Li2O2 the same process occurs, however, it starts at significantly lower temperatures. For comparison, at 355 K the two dynamically different spin reservoirs can be well differentiated from each other while the corresponding spectra of the microcrystalline sample still show the broad Gaussian-shaped line. Regarding previous studies on nanocrystalline Li2O by Heitjans and co-workers,85,86 such two-component NMR line shapes have been interpreted in terms of (mobile) Li spins residing in or near the large volume fraction of interfacial regions generated during milling.74,85 This assignment can also be adapted to explain the spectra of nano-Li2O2, which is anticipated to be characterized by a large number fraction of defects. Since the feature of a two-component line shape did also show up for micro-Li2O2 at elevated temperatures, the emergence of a narrow NMR line is either connected to thermally induced defects, formed also in micro-Li2O2, or to an intrinsic heterogeneous Li+ dynamics in the peroxide.
It is worth noting that the heterogenous narrowing shown in Fig. 7 for nanocrystalline Li2O2 is fully reversible. The irreversible decomposition into Li2O, which smoothly starts above 500 K according to TG/DSC and which could also have its microscopic (atomic-scale) infancy at 473 K, cannot be used to explain the two-component behavior observed. Here, we believe that its origin is almost completely related to the mechanically induced defect-rich structure formed. The area fraction Af of the narrow NMR component as well as the evolution of the widths of the two NMR lines of nano-Li2O2 are shown in Fig. 8.
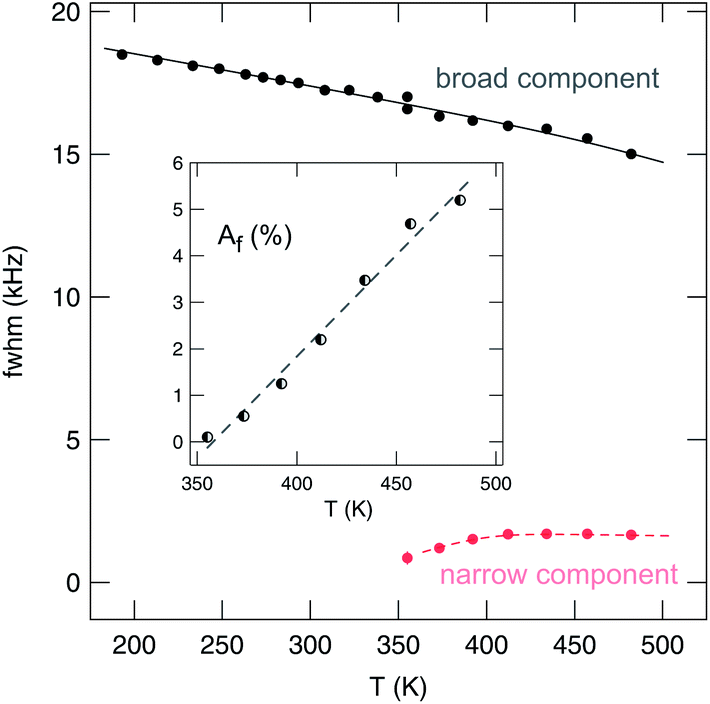 | ||
| Fig. 8 Line widths (fwhm, full width at half maximum) of the two NMR components visible in the 7Li NMR spectra shown in Fig. 7. Af denotes the area fraction, which also equals the number fraction, of fast diffusing Li spins in nanocrystalline lithium peroxide prepared by high-energy ball milling. | ||
Starting from approximately 18 kHz the broad component slightly decreases. The motionally narrowed NMR line shows up at elevated T; its width is 1.7(1) kHz. Af reaches 5% at 450 K, thus, indicating that only a small number fraction of Li ions is involved in the fast diffusion process. Af corresponds to the area fraction of that component of the FIDs which after Fourier transformation yields a narrowed NMR line. In Fig. 9, two FIDs of a spin-locking R1ρ experiment (28 kHz) are shown, which have been recorded at quite different temperatures, viz. 233 K and 455 K. While in both cases the fast decaying part corresponds to the broad Gaussian obtained after Fourier transformation, only the FID recorded at 455 K reveals an additional intensity slowly decaying with acquisition time, see also ref. 84. Before we briefly discuss the separate analysis of this slowly decaying part in the frame of a spin-locking NMR experiment, which is certainly limited because of the low intensity of this contribution, we firstly present the rates R1 and R1ρ, which can be deduced from the FIDs when the whole area under the curve is evaluated as a function of waiting or locking time.
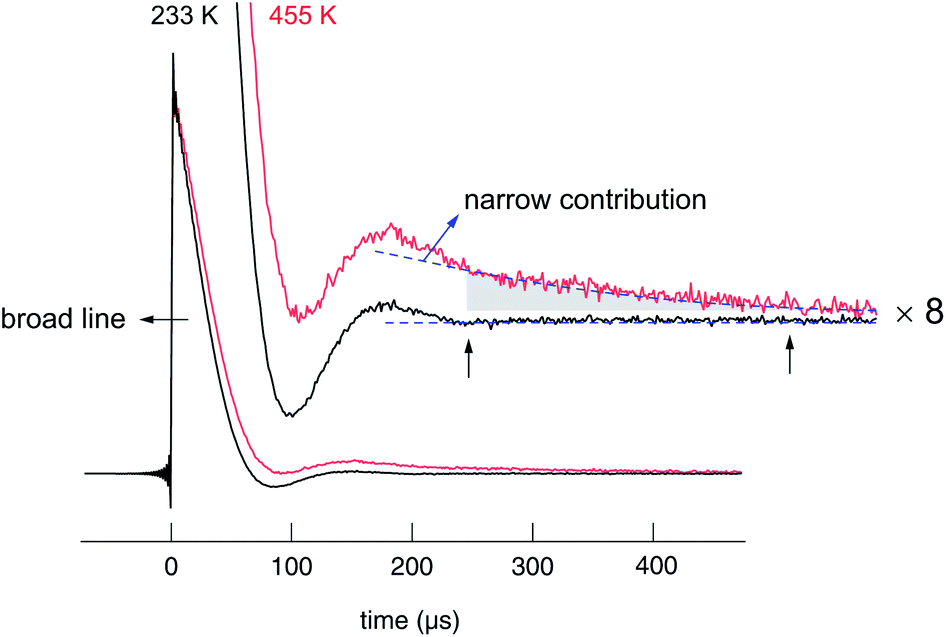 | ||
| Fig. 9 7Li NMR (transversal) transients of nanocrystalline Li2O2 recorded by the spin-lock technique at 28 kHz and a Larmor frequency of 116 MHz. In contrast to the signal recorded at 233 K, the FID obtained at elevated T reveals a slowly decaying intensity, which is responsible for the motionally narrowed NMR line visible in the Fourier transforms in Fig. 7. | ||
As an example, in Fig. 10 selected magnetization transients of our spin-lock experiments are shown. They quantify the transversal decay of Mϱ(tlock) according to Mϱ(tlock) ∝ exp(−(tlock/T1ϱ)γϱ). γϱ(T) is shown in Fig. 11. Over the whole temperature range the transients can be best fitted with stretched exponentials; a separation of the two spin reservoirs discussed above, e.g., by the use of two single exponentials, is hardly possible because of the low number fraction of fast relaxing spins. Therefore, the rates shown in Fig. 11, which gives an overview of the NMR rates (and stretching exponents γ) of the nano-Li2O2 measured, represent those ions which are not directly responsible for the motionally narrowed NMR line detected. The observation that the R1ρ transients can only be fitted with stretched exponentials rather than with a single exponential might be connected to the fact that non-exponential curves can be expected for 7Li; this can be due to intrinsic as well as extrinsic reasons: (i) as a spin-3/2 nucleus 7Li interacts with electric field gradients being associated with defects introduced, (ii) the interaction with paramagnetic impurities may also lead to such a behavior, and/or (iii) independent of such extrinsic influences the underlying hopping correlation function could be of non-exponential shape from the outset. If not being an intrinsic feature of a single ion hopping process in nanocrystalline Li2O2, the latter could also be the result of heterogeneous dynamics, i.e., the overall dynamics observed is the result of a superposition of many jump processes each being describable by single exponential motional correlation functions. The latter might be corroborated by the observation that at least γ of our R1ρ transients varies with temperature. This points to a shift of Li dynamics in favor of an overall (non-exponential) diffusion process. Such a process increasingly may become governed by correlated motion either due to structural disorder or repulsive (Coulomb) interactions of the jumping ions.
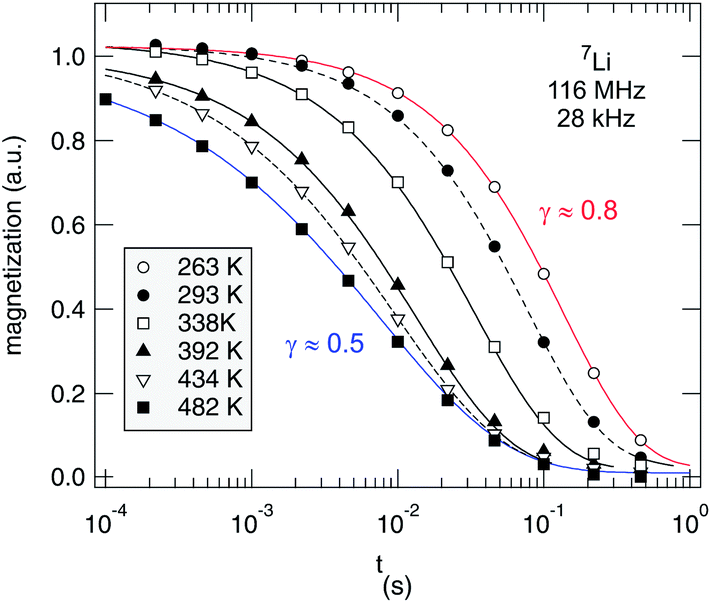 | ||
| Fig. 10 Temperature-variable 7Li NMR R1ρ transients recorded in the rotating frame using a spin-locking frequency of 28 kHz. Solid and dashed lines show fits with stretched exponentials. The stretching exponents γϱ are shown as a function of T in Fig. 10. | ||
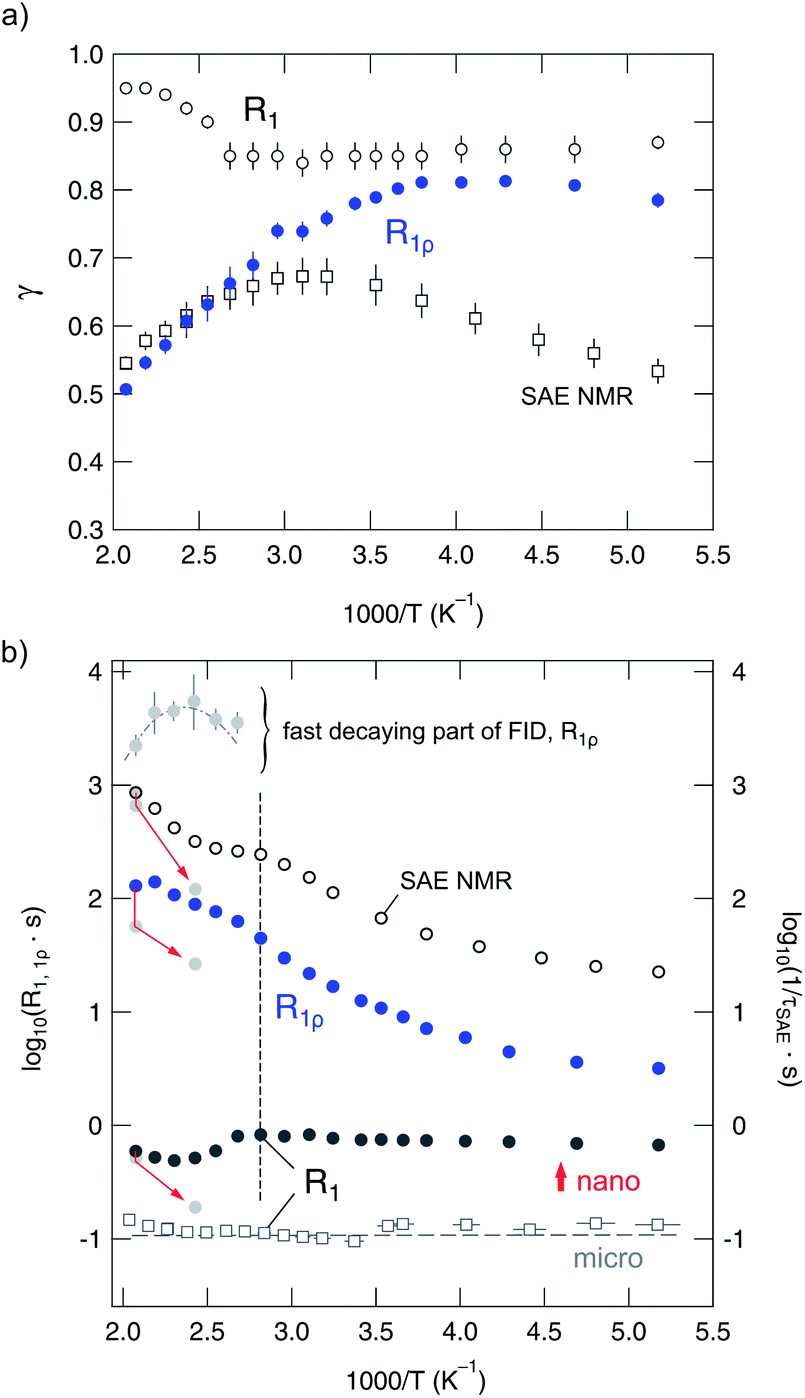 | ||
| Fig. 11 (a) Stretching factors γ(ρ) used to parameterize the various magnetization transients (R1, R1ρ) and echo decay curves (SAE NMR) of nano-Li2O2. (b) Arrhenius plot of the 7Li NMR relaxation rates of nanocrystalline Li2O2 measured. The rates shown were obtained by analysing the magnetization transients and echo damping curves with stretched exponentials. The data were acquired at 116 MHz and at a locking frequency of ω1/2π = 28 kHz. For comparison, the R1 rates of microcrystalline Li2O2 are also shown. The arrows point to those rates which were recorded during cooling. | ||
Compared to the corresponding γ values of microcrystalline Li2O2, which vary, over the temperature range covered, from 0.84 to 0.72 (R1) and from 0.64 to 0.44 (R1ρ), the results indicate that ball-milling shifts γ of R1 towards values closer to one rather than affecting the stretching exponents in the opposite direction. At least, this is valid for the so-called non-diffusive temperature regime which in the case of R1ρ becomes apparent below 200 K. For comparison, in the temperature regime covered here, the R1 rates are clearly not induced by translational Li diffusion; even up to 500 K only a non-diffusive background rate is detected which does only weakly depend on T. This can be easily explained by the different sensitivities of the two methods to Li diffusivity.40,87 While R1ρ measurements do probe Li dynamics with jump rates in the order of the locking frequency, that is, 28 kHz in our case, R1 is, however, sensitive to jumps with rates on the MHz time-scale. The latter is defined by the Larmor frequency itself.
It is worth mentioning that the R1 rates apparently show a shallow local minimum around 430 K. In the same temperature regime, steps in R1ρ(1/T) (and τ−1SAE(1/T)) show up (see Fig. 12). These might be related to variations in the diffusion mechanism and/or slight structural changes. The observations will be discussed in more detail below. When analyzing the low-T flanks of diffusion-induced NMR rate peaks, we expect activation energies being much smaller than those probed by conductivity spectroscopy in the dc limit. This is simply due to the fact that in the low-T limit, NMR relaxometry is in general sensitive to the local jump process rather than to long-range ion transport. This has been recently outlined by our group on a fluorine anion conductor.88 Here, we use the results from NMR to make comparisons with recent theoretical predictions about local hopping barriers.
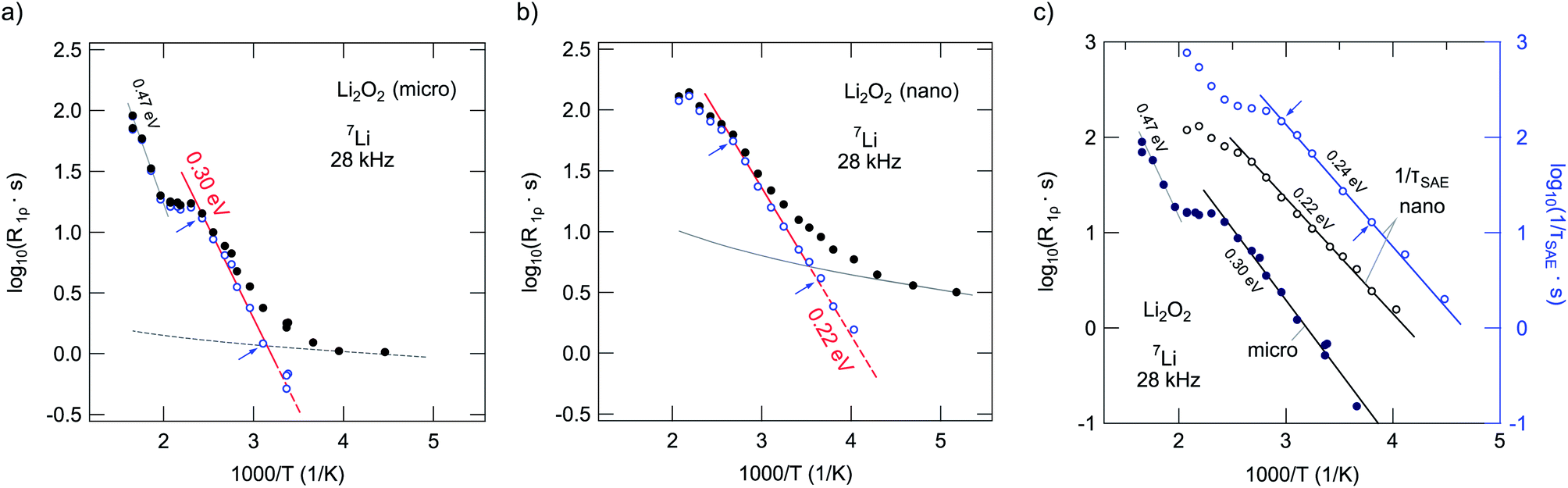 | ||
| Fig. 12 (a) and (b) 7Li spin–lattice relaxation rates (116 MHz, 28 kHz locking frequency) of micro- and nanocrystalline Li2O2 as a function of inverse temperature. The dashed lines correspond to power laws characterizing the non-diffusive background contributions to the overall rates. The correction procedure to obtain SLR rates mainly induced by diffusion is described in the text; solid lines represent Arrhenius fits, the activation energies obtained are indicated. Arrows mark the fitting ranges. (c) Comparison of the 7Li SLR NMR rates recorded in the rotating frame of reference with those obtained from SAE NMR for nano-Li2O2. | ||
For this purpose and for further analysis of our data, it is important to emphasize that the poor ion conductivity probed expectedly manifests itself in the observation that only the rate R1ρ is able to probe Li dynamics below 500 K. This also holds for the SAE NMR measurements, which reveal a diffusion-induced contribution at elevated temperatures. To separate the influence of non-diffusive and weaker-than-activated background effects on the (purely) diffusion-induced rates R1ρ and τ−1SAE, we parameterized the low-temperature background rates with appropriate power laws R1ρ ∝ Tβ. An extrapolation to higher temperatures allowed for calculating the desired diffusion-induced R1ρ,diff rates (see unfilled symbols in Fig. 12(a) and (b)). After this correction procedure, R1ρ,diff(1/T) of microcrystalline Li2O2 reveals a low-temperature flank pointing to a (low-T) activation energy of 0.30 eV (Fig. 12(a)). This is in good agreement with calculated hopping barriers for negatively charged Li vacancies (0.33–0.39 eV) by Radin et al.24 and Hummelshøj et al.32,33 The calculations of Radin and Siegel reveal that the lowest energy pathway (0.33 eV) corresponds to migration between adjacent octahedral sites.24
At elevated temperatures, that is at 430 K, the NMR rates reach a plateau value. If not associated with slight microstructural changes of the material, which are not detectable via XRD, it might be related to a shallow local rate maximum. Since no substantial NMR line narrowing shows up, this, however, can only be an indication of localized, i.e., strictly short-ranged Li motions between adjacent energy minima in the structure of the peroxide. As mentioned above, such localized motions appear as ε′′ ≠ f(ν) (or σ′ ∝ ν) in conductivity spectroscopy. With increasing temperature, the rates increase further and follow an Arrhenius line with an activation energy of 0.47 eV (Fig. 12(a)). Interestingly, this value is in agreement with that proposed for the binding or interaction energy between hole polarons and lithium vacancies (0.47 eV).34 As we will see below, a somewhat smaller activation energy was found by SAE NMR in this region (0.41 eV), which is in agreement with the first-principle calculations of Radin and Siegel for nearest neighbor hole polaron hopping (0.42 eV).24 For comparison, the model used by Luntz et al., for example, points to 0.38 eV.27 Hence, it cannot be excluded that also polaron dynamics can be indirectly seen by 7Li NMR. When comparing these values we have to keep in mind, however, that the involvement of trigonal prismatic sites in Li dynamics leads to a barrier of 0.39 eV.24
Even after a generous background correction, the corresponding rates of the nanocrystalline sample are shifted towards larger values (see Fig. 12(b)) and follow an Arrhenius law as determined by a smaller activation energy of only 0.22 eV. As in the case of the microcrystalline sample, a discontinuity is observed. It shows up at 400 K and is most likely of the same origin as discussed for the non-treated sample. For comparison, in the case of τ−1SAE of nano-Li2O2 (see Fig. 12(c)) it is shifted towards even lower temperatures (360 K). The activation energy turned out to be slightly larger than that probed via the spin-lock NMR technique. Altogether, the shift of the relaxation and decay rates observed for the sample mechanically treated indicates enhanced local Li hopping as compared to the starting material, which has also been observed for nanocrystalline Li2O74,85 and other ion conductors prepared by milling.63,79,89–92 As in the case of R1 of micro-Li2O2, above 400 K the rates τ−1SAE increase further; the corresponding activation energy is approximately 0.42 eV (as already mentioned above).
Let us point out that the difference between R1ρ and τ−1SAE of nano-Li2O2 likely reflects the increased sensitivity of SAE NMR to very slow Li exchange processes.55,87,93–99 These might be found in the crystalline cores of the nanostructured sample. Indeed, the shape of the curve τ−1SAE(1/T) resembles that of R1ρ(1/T) of micro-Li2O2. In the present case, we tend to interpret the SAE decay rates as dipolar relaxation rates rather than taking them as real jump rates. The latter has been found for a variety of materials with Li migration pathways being formed by multiple electrically inequivalent Li sites.87 Because of both the small activation energy found by SAE NMR and the low pre-exponential factor associated, we thus suspect that dipole–dipole interactions strongly influence the spin-alignment echo damping and hence govern the decay rates obtained. Furthermore, at the lowest temperatures τ−1SAE seems to be increasingly influenced by spin-diffusion, which hampers the detection of echo damping that is (purely) induced by particle diffusion.
Mechanical treatment has also an effect on the background rates measured by the various NMR techniques applied. For both R1 and R1ρ we observed an increase of the non-diffusion induced rates by one order of magnitude. Several phenomena could serve as origins for this observation such as (i) strictly localized motions, as characterized by an NCL effect (vide supra), (ii) coupling of spins to unpaired electrons, or (iii) a change of lattice vibrations. The increasing concentration of electron holes observed by Gerbig and co-workers37 might be useful to explain such differences observed for ball-milled Li2O2.
Taken together, enhanced long-range ion transport, as evidenced by conductivity spectroscopy, is also accompanied by increased short-range motions of all of the 7Li NMR spins. This is reflected by decreased activation energies and increased (diffusion-induced) overall relaxation rates. Importantly, the latter shows increased Li ion dynamics even in the bulk of Li2O2 nanocrystallites. To detail the fast 7Li spin reservoir as seen via NMR line shapes, let us reconsider the two-component line shape and the associated FIDs of nanocrystalline Li2O2 at elevated T in a quantitative way. If we integrate only the slowly decaying part of the FID, it should be possible to separate the ions which are responsible for the narrowed line from those reflected by the broad one.
In Fig. 9 the two vertically drawn arrows indicate the integration range that we used to construct the corresponding temperature-variable magnetization transients. For comparison, in Fig. 11(b) the SLR rates obtained are shown as grey circles in the upper part of the Arrhenius diagram. Their temperature dependence might be interpreted as a shallow maximum since the rates indeed start to decrease with increasing temperature. With the maximum condition valid for a R1ρ(1/T)-peak, ω1τc ≈ 0.5, we obtained a motional correlation rate of τc−1 = 3.5 × 105 s−1 at 473 K. Within a factor of two the correlation time τc is expected to be equal to the Li residence time between two successful jumps. Assuming a mean jump distance of only 2.5 Å, the Einstein–Smoluchowski equation for 3D uncorrelated motion100 yields a self-diffusion coefficient D in the order of D(473 K) = 1 × 10−14 m2 s−1. For comparison with conductivity spectroscopy, a diffusion coefficient within this order is in good agreement with 2.1 × 10−5 S cm−1 as probed at the same temperature. This agreement points out that ion transport to which σdc is sensitive seems to be mainly governed by the fast Li sub-ensemble detected by NMR. This Li reservoir could be a spatially local one near or in the surface-influenced interfacial regions of the nm sized, structurally disordered Li2O2 crystallites.74,85 It might be comparable to that expected for Li ion surface diffusion.
For comparison with other Li ion conductors, both active materials and solid electrolytes, a diffusion coefficient in the order of D(473 K) = 1 × 10−14 m2 s−1 reflects rather poor ion diffusivity. Considering recent examples that have been investigated by NMR, Li-bearing argyrodite, Li6PSe5Br, shows values as high as 10−12 m2 s−1 at 263 K;83 Li diffusion in the anode material LiC6 is in the order of 10−15 m2 s−1 at 300 K, while we have 4 × 10−20 m2 s−1 in the case of nano-Li2O2 (see Fig. 5).82 In addition, the well-known garnet-type electrolyte, Al-doped Li7Zr2La3O12, shows a D value of 10−14 m2 s−1 at ambient temperature rather than at 473 K; the corresponding activation energy is much lower and turned out to be 0.35 eV.42 Interestingly, quite recently, Siegel and co-workers36 calculated the ionic conductivity for amorphous Li2O2 and found enhanced dynamics characterized by 10−7 S cm−1. This is three orders of magnitude larger than that for nanocrystalline Li2O2 prepared by ball milling.
Considering electronic properties, quite recently, Geng et al.101 have employed first-principles density functional theory calculations to study the properties of gain boundaries in Li2O2. They report on electronic conduction channels in polycrystalline Li2O2 opened up by stable grain boundaries. Considering also space charge effects, which are known to influence both ionic and electronic charge carrier transport,102–105 it underpins the important role of interfaces to be considered when dynamic properties shall be engineered, see also ref. 5.
IV. Summary and conclusions
Charge carrier transport and Li ion dynamics of nanocrystalline lithium peroxide were investigated by the complementary techniques of conductivity and NMR spectroscopy. In agreement with the recent study by Gerbig et al. translational Li ion dynamics as well as electron transport turned out to be rather poor in microcrystalline Li2O2. After high-energy ball milling, however, the overall conductivity has been increased by more than two orders of magnitude and reaches 1.1 × 10−10 S cm−1 at room temperature. Potentiostatic polarization measurements reveal a discernable electronic contribution (approximately 10%) to the total conductivity. Compared to the non-milled starting material, the activation energy decreased from 0.88 eV to 0.82 eV.While at very low temperatures the nearly constant loss phenomenon leaves its fingerprints in the frequency dependence of the conductivity σ′, above 250 K the 7Li NMR relaxometry measurements on nano-Li2O2 point to sort-range motions with an activation energy of approximately 0.22 eV. In a similar way, this is also observed for microcrystalline lithium peroxide, the activation energy, however, is somewhat larger (0.30 eV). Most importantly, well below the decomposition range of nanocrystalline Li2O2, the static 7Li NMR line shapes are composed of two components. While the broad one is identical to that observed for micro Li2O2, the motionally narrowed line represents a sub-ensemble of relatively fast Li ions. Analyzing this component in the frame of a T1ρ NMR experiment – to the best of our knowledge such a separation has not been reported so far for R1ρ rates – we were able to roughly estimate the corresponding Li jump rate at 473 K and, hence, the self-diffusion coefficient. The latter, which is in the order of 10−14 m2 s−1, corresponds well with the dc conductivity probed at the same temperature.
In summary, enhancement of charge carrier dynamics in nanocrystalline Li2O2 is most likely related to various defects and strain that high-energy ball milling can introduce. Defects are introduced in both the bulk and the surface-influenced regions of the nanocrystallites. In many cases the large volume fraction of interfacial regions formed during the mechanical impact show structural disorder facilitating ion transport over short and long distances. Although the enhancement is easily measurable, compared to other oxidic lithium ion conductors it turned out to be much lower. It seems that ball-milling has a larger influence on σeon than on σion; at room temperature we found σion/σeon ≈ 10, which is much smaller than the ratio estimated for the microcrystalline counterpart.
Author contributions
The authors were equally involved in experimental work, data analysis and project planning. All authors have approved the final version of the manuscript.Acknowledgements
We thank our colleagues at the TU Graz for valuable discussions. Financial support from the Austrian Federal Ministry of Economy, Family and Youth, and the Austrian National Foundation for Research, Technology and Development is greatly appreciated. Furthermore, we thank the Deutsche Forschungsgemeinschaft (DFG) for financial support within the DFG Research Unit 1277, grant no. WI3600/2-1 and 4-1, and the DFG Prority Program 1415, grant no. WI3600/5-1.References
- S. H. Oh, R. Black, E. Pomerantseva, J.-H. Lee and L. F. Nazar, Nat. Chem., 2012, 4, 1004 CrossRef CAS PubMed.
- K. M. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 1, 143 Search PubMed.
- Y.-C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 750 CAS.
- F. Li, H. Kitaura and H. Zhou, Energy Environ. Sci., 2013, 6, 2302 CAS.
- B. D. Adams, C. Radtke, R. Black, M. L. Trudeau, K. Zaghib and L. F. Nazar, Energy Environ. Sci., 2013, 6, 1772 CAS.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287 CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef CAS PubMed.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854 CAS.
- J. Read, J. Electrochem. Soc., 2002, 149, A1190 CrossRef CAS PubMed.
- T. Ogasawara, A. Débart, M. Holzapfel, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 1390 CrossRef CAS PubMed.
- L. Zhong, R. R. Mitchell, Y. Liu, B. M. Gallant, C. V. Thompson, J. Y. Huang, S. X. Mao and Y. Shao-Horn, Nano Lett., 2013, 13, 2209 CrossRef CAS PubMed.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193 CrossRef CAS.
- S. A. Freunberger, Y. Chen, Z. Peng, J. M. Griffin, L. J. Hardwick, F. Bardé, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040 CrossRef CAS PubMed.
- F. Mizuno, S. Nakanishi, Y. Kotani, S. Yokoishi and H. Iba, Electrochemistry, 2010, 78, 403 CrossRef CAS PubMed.
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, G. Girishkumar and A. C. Luntz, J. Phys. Chem. Lett., 2011, 2, 1161 CrossRef CAS.
- W. Xu, J. Hu, M. H. Engelhard, S. A. Towne, J. S. Hardy, J. Xiao, J. Feng, M. Y. Hu, J. Zhang and F. Ding, et al. , J. Power Sources, 2012, 215, 240 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2013, 135, 494 CrossRef CAS PubMed.
- W. Xu, K. Xu, V. V. Viswanathan, S. A. Towne, J. S. Hardy, J. Xiao, Z. Nie, D. Hu, D. Wang and J.-G. Zhang, J. Power Sources, 2011, 196, 9631 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng, Y. Chen, Z. Liu and P. G. Bruce, Nat. Mater., 2013, 12, 1050 CrossRef CAS PubMed.
- S. A. Freunberger, Y. Chen, N. E. Drewett, L. J. Hardwick, F. Bardé and P. G. Bruce, Angew. Chem., Inter. Ed., 2011, 50, 8609 CrossRef CAS PubMed.
- B. D. McCloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshøj, J. K. Nørskov and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 997 CrossRef CAS.
- J. S. Hummelshoj, A. C. Luntz and J. K. Norskov, J. Phys. Chem., 2013, 138, 034703 CrossRef CAS PubMed.
- V. Viswanathan, K. S. Thygesen, J. S. Hummelshhøj and G. G. J. K. Nørskov, J. Chem. Phys., 2011, 135, 214704 CrossRef CAS PubMed.
- M. D. Radin and D. J. Siegel, Energy Environ. Sci., 2013, 6, 2370 CAS.
- S. Kang, Y. Mo, S. P. Ong and G. Ceder, Chem. Mater., 2013, 25, 3328 CrossRef CAS.
- Y. Chen, S. A. Freunberger, Z. Peng, O. Fontaine and P. G. Bruce, Nat. Chem., 2013, 5, 489 CrossRef CAS PubMed.
- A. C. Luntz, V. Viswanathan, J. Voss, J. B. Varley, J. K. Nørskov, R. Scheffler and A. Speidel, J. Phys. Chem. Lett., 2013, 4, 3494 CrossRef CAS.
- R. R. Mitchel, B. M. Gallant, Y. Shao-Horn and C. V. Thompson, J. Phys. Chem. Lett., 2013, 4, 1060 CrossRef.
- Y.-C. Lu and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 4, 93 CrossRef.
- V. Timoshevskii, Z. Feng, K. H. Bevan, J. Goodenough and K. Zaghib, Appl. Phys. Lett., 2013, 103, 073901 CrossRef PubMed.
- D. Zhai, H.-H. Wang, J. Yang, K. C. Lau, K. Li, L. A. Curtiss and K. Amine, J. Am. Chem. Soc., 2013, 135, 15364 CrossRef CAS PubMed.
- J. S. Hummelshøj, J. Blomqvist, S. Datta, T. Vegge, J. Rossmeisl, K. S. Thygesen, A. C. Luntz, K. W. Jacobsen and J. K. Nørskov, J. Chem. Phys., 2010, 132, 071101 CrossRef PubMed.
- J. Chen, J. S. Hummelshøj, K. S. Thygesen, J. S. G. Myrdal, J. K. Nørskov and T. Vegge, Catal. Today, 2011, 165, 2 CrossRef CAS PubMed.
- S. P. Ong, Y. Mo and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 081105 CrossRef.
- M. D. Radin, J. F. Rodriguez, F. Tian and D. J. Siegel, J. Am. Chem. Soc., 2012, 134, 1093 CrossRef CAS PubMed.
- F. Tian, M. D. Radin and D. J. Siegel, Chem. Mater., 2014 Search PubMed , in press.
- O. Gerbig, R. Merkle and J. Maier, Adv. Mater., 2013, 25, 3129 CrossRef CAS PubMed.
- E. Fukushima and S. B. W. Roeder, Experimental Pulse NMR: A Nuts and Bolts Approach, Addison-Wesley Pub. Co., Advanced Book Program, Reading, Mass., 1981 Search PubMed.
- P. Heitjans, A. Schirmer, and S. Indris, in Diffusion in Condensed Matter, ed. P. Heitjans and J. Kärger, Springer, 2005, ch. 9, p. 367 Search PubMed.
- A. Kuhn, S. Narayanan, L. Spencer, G. R. Goward, V. Thangadurai and M. Wilkening, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 94302 CrossRef.
- A. Kuhn, J.-Y. Choi, L. Robben, F. Tietz, M. Wilkening and P. Heitjans, Z. Phys. Chem., 2012, 226, 525 CrossRef CAS.
- H. Buschmann, J. Dölle, S. Berendts, A. Kuhn, P. Bottke, M. Wilkening, P. Heitjans, A. Senyshyn, H. Ehrenberg and A. Lotnyk, et al. , Phys. Chem. Chem. Phys., 2011, 13, 19378 RSC.
- D. Ailion and C. P. Slichter, Phys. Rev. Lett., 1964, 12, 168 CrossRef CAS.
- C. P. Slichter and D. Ailion, Phys. Rev., 1964, 135, A1099 CrossRef.
- D. C. Ailion and C. P. Slichter, Phys. Rev., 1965, 137, A235 CrossRef.
- D. C. Look and I. J. Lowe, J. Chem. Phys., 1966, 44, 2995 CrossRef CAS PubMed.
- T. J. Rowland and F. Y. Fradin, Phys. Rev., 1969, 182, 760 CrossRef CAS.
- D. Wolf, Phys. Rev. B: Condens. Matter Mater. Phys., 1974, 10, 2724 CrossRef CAS.
- X.-P. Tang and Y. Wu, J. Magn. Reson., 1998, 133, 155 CrossRef CAS.
- R. Böhmer, J. Magn. Reson., 2000, 147, 78 CrossRef PubMed.
- F. Qi, G. Diezemann, H. Böhm, J. Lambert and R. Böhmer, J. Magn. Reson., 2004, 169, 225 CrossRef CAS PubMed.
- F. Qi, T. Jörg and R. Böhmer, Solid State Nucl. Magn. Reson., 2002, 22, 484 CrossRef CAS PubMed.
- M. Wilkening and P. Heitjans, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 024311 CrossRef.
- J. Jeener and P. Broekaert, Phys. Rev., 1967, 157, 232 CrossRef CAS.
- R. Böhmer, K. Jeffrey and M. Vogel, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 50, 87 CrossRef PubMed.
- P. Bottke, D. Freude and M. Wilkening, J. Phys. Chem. C, 2013, 117, 8114 CAS.
- L. S. Cahill, R. Chapman, J. Britten and G. Goward, J. Phys. Chem. B, 2006, 110, 7171 CrossRef CAS PubMed.
- Z. Xu and J. Stebbins, Science, 1995, 270, 1332 CAS.
- C. Grey and N. Dupré, Chem. Rev., 2004, 104, 4493 CrossRef CAS.
- J. Cabana, N. Dupré, G. Rousse, C. P. Grey and M. R. Palacin, Solid State Ionics, 2005, 176, 2205 CrossRef CAS PubMed.
- S. Indris, D. Bork and P. Heitjans, J. Mater. Synth. Process., 2000, 8, 245 CrossRef CAS.
- K. Funke, C. Cramer and D. Wilmer, in Diffusion in Condensed Matter – Methods, Materials, Models, ed. P. Heitjans and J. Kärger, Springer, Berlin, 2nd edn, 2005, ch. 21, pp. 857–893 Search PubMed.
- P. Heitjans, M. Masoud, A. Feldhoff and M. Wilkening, Faraday Discuss., 2007, 134, 67 RSC.
- B. Ruprecht, M. Wilkening, R. Uecker and P. Heitjans, Phys. Chem. Chem. Phys., 2012, 14, 11974 RSC.
- B. Ruprecht, H. Billetter, U. Ruschewitz and M. Wilkening, J. Phys.: Condens. Matter, 2010, 22, 245901 CrossRef CAS PubMed.
- D. L. Sidebottom, Rev. Mod. Phys., 2009, 81, 999 CrossRef.
- K. Funke, Prog. Solid State Chem., 1993, 22, 111 CrossRef CAS.
- A. Bunde, W. Dieterich, P. Maass and M. Meyer, in Diffusion in Condensed Matter – Methods, Materials, Models, ed. P. Heitjans and J. Kärger, Springer, Berlin, 2nd edn, 2005, ch. 20, pp. 813–856 Search PubMed.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater, 1990, 2, 132 CrossRef CAS.
- D. L. Sidebottom, Phys. Rev. Lett., 1999, 82, 3653 CrossRef CAS.
- B. Roling, Solid State Ionics, 1998, 105, 185 CrossRef CAS.
- K. Ngai and R. W. Rendell, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 9393 CrossRef CAS.
- M. Leskes, N. Drewett, L. Hardwick, P. Bruce, G. Goward and C. Grey, Angew. Chem., Int. Ed., 2012, 51, 8560 CrossRef CAS PubMed.
- M. Wilkening, S. Indris and P. Heitjans, Phys. Chem. Chem. Phys., 2003, 5, 2225 RSC.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama and K. Kawamoto, et al. , Nat. Mater., 2011, 10, 682 CrossRef CAS PubMed.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778 CrossRef CAS PubMed.
- H.-J. Deiseroth, S.-T. Kong, H. Eckert, J. Vannahme, C. Reiner, T. Zaiss and M. Schlosser, Angew. Chem., Int. Ed., 2008, 47, 755 CrossRef CAS PubMed.
- R. Amin, J. Maier, P. Balaya, D. P. Chen and C. T. Lin, Solid State Ionics, 2008, 179, 1683 CrossRef CAS PubMed.
- M. Wilkening, V. Epp, A. Feldhoff and P. Heitjans, J. Phys. Chem. C, 2008, 112, 9291 CAS.
- M. Wilkening, W. Iwaniak, J. Heine, V. Epp, A. Kleinert, M. Behrens, G. Nuspl, W. Bensch and P. Heitjans, Phys. Chem. Chem. Phys., 2007, 9, 6199 RSC.
- S. Narayanan, V. Epp, M. Wilkening and V. Thangadurai, RSC Adv., 2012, 2, 2553 RSC.
- J. Langer, V. Epp, P. Heitjans, F. A. Mautner and M. Wilkening, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 094304 CrossRef.
- V. Epp, z. Gün, H.-J. Deiseroth and M. Wilkening, J. Phys. Chem. Lett., 2013, 4, 2118 CrossRef CAS.
- P. Heitjans and M. Wilkening, Mater. Res. Bull., 2009, 34, 915 CrossRef CAS.
- S. Indris and P. Heitjans, J. Non-Cryst. Solids, 2002, 307, 555 CrossRef.
- P. Heitjans, A. Schirmer and S. Indris, in Diffusion in Condensed Matter – Methods, Materials, Models, ed. P. Heitjans and J. Kärger, Springer, Berlin, 2nd edn, 2005b, ch. 9, pp. 369–415 Search PubMed.
- M. Wilkening and P. Heitjans, ChemPhysChem, 2012, 13, 53 CrossRef CAS PubMed.
- F. Preishuber-Pflügl, P. Bottke, B. Bitschnau and M. Wilkening, Phys. Chem. Chem. Phys., 2014 Search PubMed , in press.
- D. Bork and P. Heitjans, J. Phys. Chem. B, 1998, 102, 7303 CrossRef CAS.
- D. Bork and P. Heitjans, J. Phys. Chem. B, 2001, 105, 9162 CrossRef CAS.
- M. Wilkening, D. Bork, S. Indris and P. Heitjans, Phys. Chem. Chem. Phys., 2002, 4, 3246 RSC.
- M. Masoud and P. Heitjans, Defect Diffus. Forum, 2005, 237–240, 1016 CrossRef CAS.
- X.-P. Tang, R. Busch, W. Johnson and Y. Wu, Phys. Rev. Lett., 1998, 81, 5358 CrossRef CAS.
- X.-P. Tang, U. Geyer, R. Busch, W. Johnson and Y. Wu, Nature, 1999, 402, 160 CrossRef CAS PubMed.
- R. Böhmer, T. Jörg, F. Qi and A. Titze, Chem. Phys. Lett., 2000, 316, 419 CrossRef.
- M. Wilkening, W. Küchler and P. Heitjans, Phys. Rev. Lett., 2006, 97, 065901 CrossRef CAS.
- M. Wilkening and P. Heitjans, J. Phys.: Condens. Matter, 2006, 18, 9849 CrossRef CAS.
- M. Wilkening, C. Lyness, A. R. Armstrong and P. G. Bruce, J. Phys. Chem. C, 2009, 113, 4741 CAS.
- M. Wilkening, J. Heine, C. Lyness, A. R. Armstrong and P. G. Bruce, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 064302 CrossRef.
- H. Mehrer, Diffusion in Solids, Springer, Berlin, 2006 Search PubMed.
- W. T. Geng, B. L. He and T. Ohno, J. Phys. Chem. C, 2013, 117, 25222 CAS.
- J. Maier, Prog. Solid State Chem., 1995, 23, 171 CrossRef CAS.
- J. Maier, Nat. Mater., 2005, 4, 805 CrossRef CAS.
- N. Sata, K. Eberman, K. Eberl and J. Maier, Nature, 2000, 408, 946 CrossRef CAS PubMed.
- W. Puin, S. Rodewald, R. Ramlau, P. Heitjans and J. Maier, Solid State Ionics, 2000, 131, 159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ee00496e |
| This journal is © The Royal Society of Chemistry 2014 |