 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of multivalent ions on renewable energy generation in reverse electrodialysis†
David A.
Vermaas
ab,
Joost
Veerman
c,
Michel
Saakes
a and
Kitty
Nijmeijer
*b
aWetsus, Centre of Excellence for Sustainable Water Technology, P.O. Box 1113, 8900 CC, Leeuwarden, The Netherlands
bMembrane Science & Technology, University of Twente, MESA+ Institute for Nanotechnology, P.O. Box 217, 7500 AE, Enschede, The Netherlands. E-mail: d.c.nijmeijer@utwente.nl; Tel: +31 53 489 4185
cREDstack B.V., Pieter Zeemanstraat 6, 8606 JR, Sneek, The Netherlands
First published on 10th January 2014
Abstract
Renewable energy can be generated when mixing seawater and river water. This energy is captured in reverse electrodialysis (RED) using ion exchange membranes. Although natural sources of seawater and river water are composed of a mixture of monovalent and multivalent ions, laboratory research on RED is generally performed with artificial solutions of sodium chloride. This research demonstrates that the presence of magnesium- and sulphate ions in feed solutions with NaCl has a major effect on the obtained open circuit voltage and power density for three different membrane types. When using a mixture with a molar fraction of 10% MgSO4 and 90% of NaCl in both feed waters, the experimentally obtained power density in steady state decreases from 29% to 50% compared to the case where the feed solutions contain only NaCl as a salt. This effect is among others explained by the transport of Mg2+ and SO42− against their concentration gradient, as is elaborated in a theoretical framework and which is justified by experimental data. Non-stationary cases, where feed water is switched from a NaCl solution to a mixture of NaCl and MgSO4, show that the voltage response time is in the order of tens of minutes up to several hours, due to ion exchange between the membranes and the feed water. The knowledge gained from electrochemical measurements under stationary and non-stationary conditions and a novel technique to monitor the ion transport inside cation exchange membranes can be used to improve the obtained power density in practical applications of RED using natural feed water.
Broader contextThis paper describes the energy generation from waters that contain a mixture of monovalent and multivalent ions in different concentrations, which resembles the practical application of renewable energy generation from mixing natural seawater and river water, or brine and seawater or fresh water. Although the fraction of multivalent ions (such as Mg2+ and SO42−) is relatively small compared to that of monovalent ions (such as Na+ and Cl−) under environmental conditions, their impact on the power production is significant, as demonstrated in this paper. The theoretically available energy has been determined, which is relevant to all technologies that extract energy from salinity gradients, and the decrease in voltage and power density has been evaluated theoretically and experimentally for reverse electrodialysis. Furthermore, the remarkable large sensitivity of the power density to the fraction of multivalent ions has been unravelled from a (theoretical and experimental) analysis of the mass transport of monovalent and multivalent ions. These results imply new insights into the potential of salinity gradient power and strategies to maintain high power densities in reverse electrodialysis. |
Introduction
Renewable energy can be generated from mixing salt water, such as seawater, and fresh water, e.g. river water, using reverse electrodialysis (RED).1–5 This available energy from salinity differences can be captured using ion exchange membranes, which are selective for cations (cation exchange membranes, CEMs) or anions (anion exchange membranes, AEMs). The salinity difference induces a potential difference over the membranes when seawater is supplied at one side and river water is supplied at the other side of the membrane. Stacking the CEMs and AEMs alternately, with seawater and river water supplied in between, accumulates the voltage. When electrodes are introduced at the outer ends of the membrane pile, this voltage can be used to generate a redox reaction,6,7 or for capacitive storage,8,9 which enables an electrical current when an external circuit is connected.The potential to generate power from the mixing of seawater and river water is huge, as approximately 2 TW can be generated theoretically from the global river water runoff.10,11 This is close to the current worldwide electricity demand.12 Additionally, salinity gradient power can be generated by mixing brine (e.g., from salt water lakes) and seawater or fresh water.13 For artificial feed water and laboratory conditions, high energy efficiencies (>50%)3,14 and high gross power densities (>2 W m−2)15 can be obtained. However, the obtained power and energy efficiency decrease rapidly when natural feed water sources are used. A recent study on RED using natural feed water for 25 days indicated that the power density decreased to approximately 50% of the initial power at a timescale of several hours.16 This result implies that artificial feed water composed of pure NaCl solutions under laboratory conditions is not representative for natural feed water, although the dominant dissolved species in natural feed water is NaCl.
The decrease in power density as observed in previous research when using natural feed waters was among others attributed to organic foulants.16 Additionally, the presence of multivalent ions was identified to be a limiting factor for the obtained power density.16 Organic foulants, such as humic acids, are known to shield the charged groups of the ion exchange membranes and consequently lower the selectivity and increase the membrane resistance.17,18 The negative impact of multivalent ions on the obtained power density in RED is less well known.
Only very limited research is done on the effect of other salts than NaCl, such as MgSO4, on the obtained power in RED.19,20 Post et al.19 indicated that MgSO4 in the feed water reduces the stack voltage and increases the stack resistance compared to solutions with NaCl only.19 Lacey et al.21 indicated that multivalent ions such as iron, borates and silicates may shield the membrane charge irreversibly, which would decrease the membrane permselectivity and increase the membrane resistance. Quantitative results on the obtained power density and the typical timescale of the effects of multivalent ions are lacking, while they may have a major impact on the power density in practical applications of reverse electrodialysis.
Previous research on multivalent ions in electrodialysis (ED) cannot be used a priori for RED, as some effects may work similarly (e.g. effect on membrane resistance) but other effects work differently in ED to those in RED (e.g. direction of ion transport). In order to identify and understand the negative effect of multivalent ions on the power density in RED, the stationary and non-stationary effects of monovalent ions, multivalent ions and mixtures thereof need to be investigated systematically. Such research will explain the cause of the rapid decrease of approximately 50% in power density observed when using natural feed waters instead of artificial NaCl feed solutions.16
This paper unravels the effect of the presence of MgSO4 in feed waters on the RED performance and explains the underlying mechanisms. The use of MgSO4 provides an indication of the effects of multivalent ions in feed water in general, as the concentrations of Mg2+ and SO42− are typically highest after Na+ and Cl− in natural feed water.16 The concentration of MgSO4 in the feed water is systematically varied and the RED performance in terms of voltage, resistance and power density is analyzed. Additionally, the effect of the type of membrane and the distribution of MgSO4 in the membranes using SEM-EDX is investigated. The results show how RED can be operated using feed mixtures of NaCl and MgSO4, as a representative for natural feed waters, while maintaining a high power density.
Theory
Electromotive force
The supply of waters with different salinity on either side of an ion exchange membrane creates a voltage over that membrane due to the Donnan potential at the membrane–water interface. This voltage is given by the Nernst equation, corrected for the activity of the feed waters and the apparent permselectivity of the non-perfect membrane:22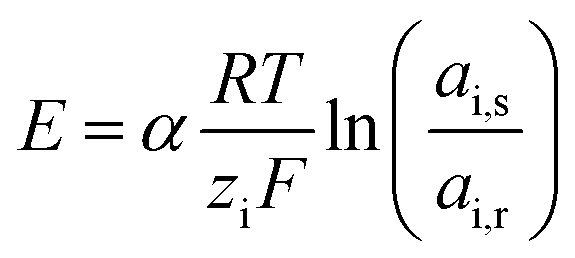 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), and ai is the activity of ionic species i in the feed water (M). The subscripts s and r indicate seawater or river water, respectively. Eqn (1) shows that the salinity ratio of monovalent ions (e.g. Na+, Cl−) creates twice the voltage created by the same salinity ratio of divalent ions (e.g. Mg2+, SO42−), assuming that the activities and apparent membrane permselectivity remain constant.
485 C mol−1), and ai is the activity of ionic species i in the feed water (M). The subscripts s and r indicate seawater or river water, respectively. Eqn (1) shows that the salinity ratio of monovalent ions (e.g. Na+, Cl−) creates twice the voltage created by the same salinity ratio of divalent ions (e.g. Mg2+, SO42−), assuming that the activities and apparent membrane permselectivity remain constant.
Uphill transport
However, transport is more complex when a mixture of monovalent and multivalent ions is present at both sides of an ion exchange membrane.23–26 In that case, ions are exchanged through the membrane, in order to obtain equilibrium in chemical potential at both sides of the membrane.23 This can be illustrated using an exemplary system with NaCl and MgSO4 at both sides of a cation exchange membrane (CEM), in the same ratio but at different concentrations, as illustrated in Fig. 1.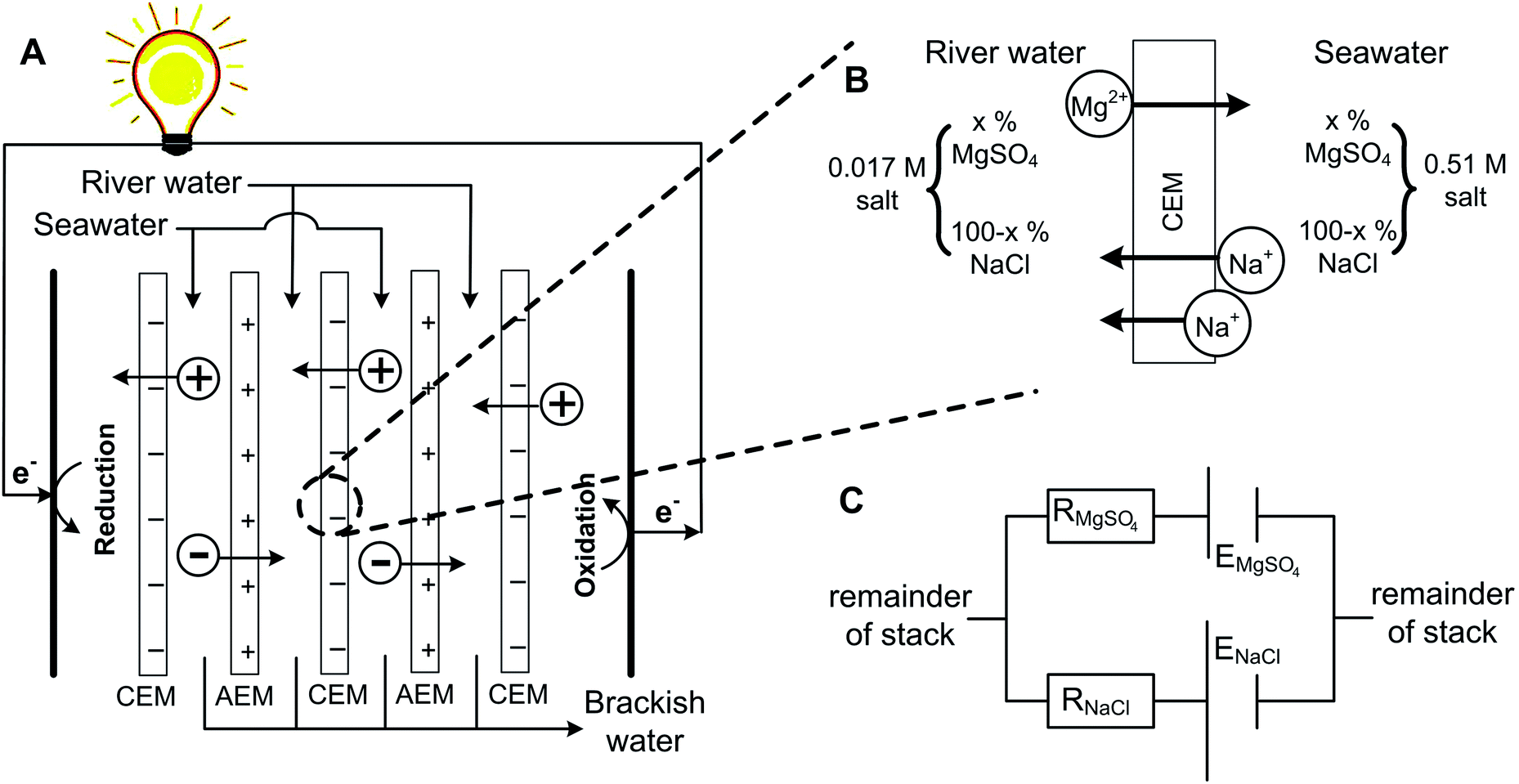 | ||
| Fig. 1 Principle of RED (A), with a zoomed section of a CEM showing the principle of uphill transport (B). The equivalent electrical circuit of this membrane is presented at the right bottom (C). | ||
Because of the valence of the ion, the electromotive force related to the difference in [Na+] is larger than that of [Mg2+] (see eqn (1)). Hence, Mg2+ is transported from the dilute solution to the concentrated solution in exchange for double the amount of Na+ in the opposite direction (Fig. 1B), to obey electroneutrality at both sides of the membrane. The transport of Mg2+ against the concentration gradient is described in the literature as the uphill transport.19,23
This system can be illustrated in an electrical circuit, having two power sources with an unequal voltage (Fig. 1C) in parallel. The source with the lowest voltage will be loaded, at the cost of the source with the highest voltage. The membrane resistances for Mg2+ and Na+-ions, which are illustrated in the electrical circuit as an electrical resistance, slow down the uphill transport of Mg2+.
The uphill transport of Mg2+ sacrifices the salinity difference of the monovalent species, in this case Na+, while at the same time no net charge transport is obtained. As a consequence, the presence of multivalent ions causes an irreversible loss in the available energy from monovalent ions.19 A similar process occurs at the anion exchange membrane (AEM) for Cl− and SO42−.
The uphill transport stops when the salinity differences are such that the electromotive forces of both ionic species are equal. This can be best defined considering the case in which the feed waters are not refreshed (i.e., a batch process). Using eqn (1) and assuming that the apparent permselectivity is equal for both ionic species, this stationary situation is reached when the ion activities at either side of the membrane obey:
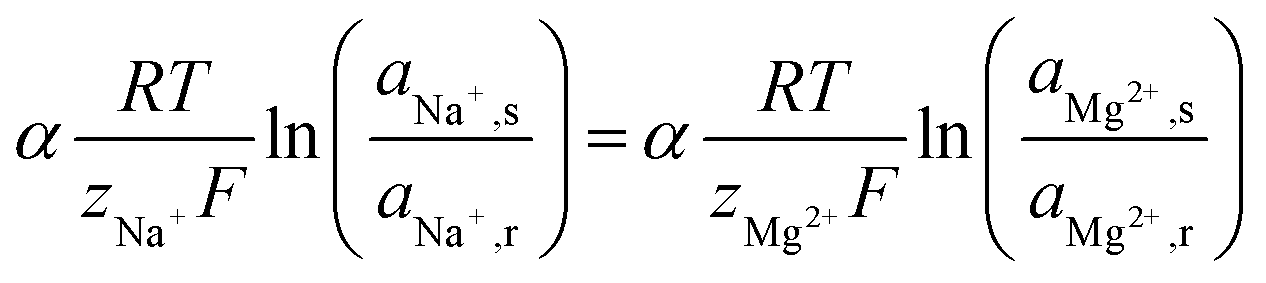 | (2) |
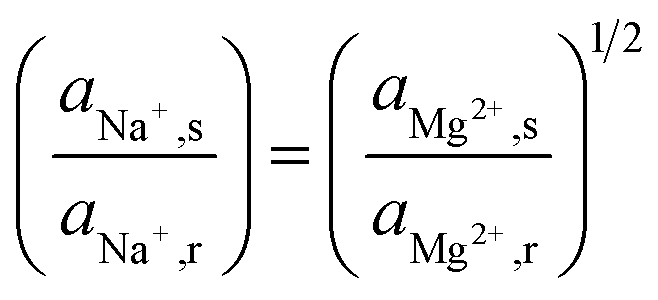 | (3) |
The molar transport of Mg2+, in exchange for 2Na+, can then be calculated as a function of the initial feed water concentrations and the activity coefficients. When the changes in feed water concentration are regarded small, the activity coefficients γ (-) can be assumed constant, and the molar transport of Mg2+ can be solved from:
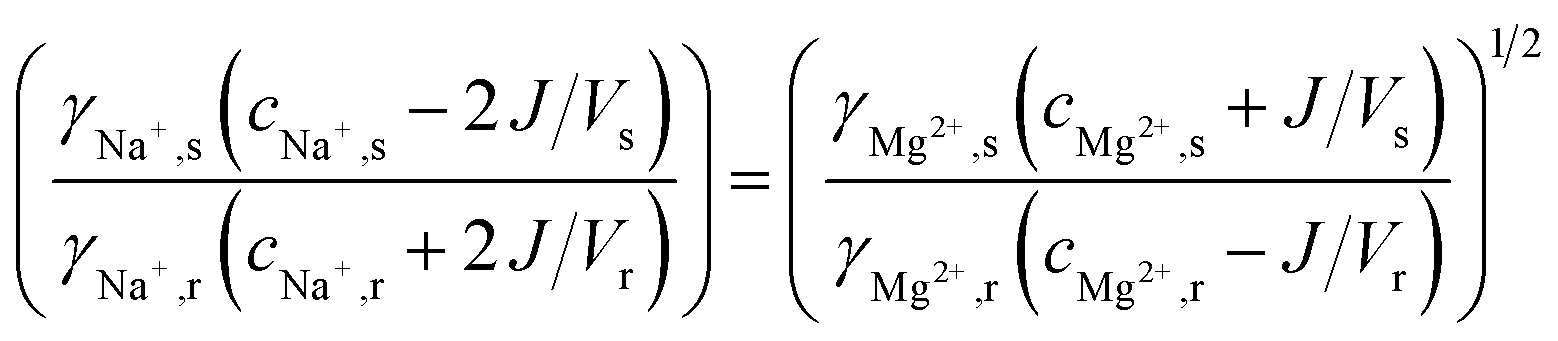 | (4) |
Theoretical energy and voltage
The available energy from mixing solutions with different concentrations can be derived from the Gibbs free energy of mixing.27 When using mixtures of salts, the contribution of each ionic species to the available energy can be calculated individually considering the molar fraction of that specific salt only. Such a calculation has been performed for mixtures of the monovalent salt NaCl and the multivalent salt MgSO4, disregarding uphill transport. In addition, the available energy is calculated for the concentrations after uphill transport, as are derived from the equilibrium condition in eqn (3). These calculations of the available energy are shown in Fig. 2A as a function of the molar salt fraction of MgSO4 in both the seawater and river water feed. Fig. 2B considers the open circuit voltage over a perfectly selective membrane, when uphill transport is completed, as a function of the fraction of MgSO4 in both feed waters. The voltage without considering uphill transport cannot be given, as the voltage due to the monovalent species conflicts that of the multivalent species. In addition, Fig. 2B shows the cases in which MgSO4 is only present in the seawater feed or river water feed.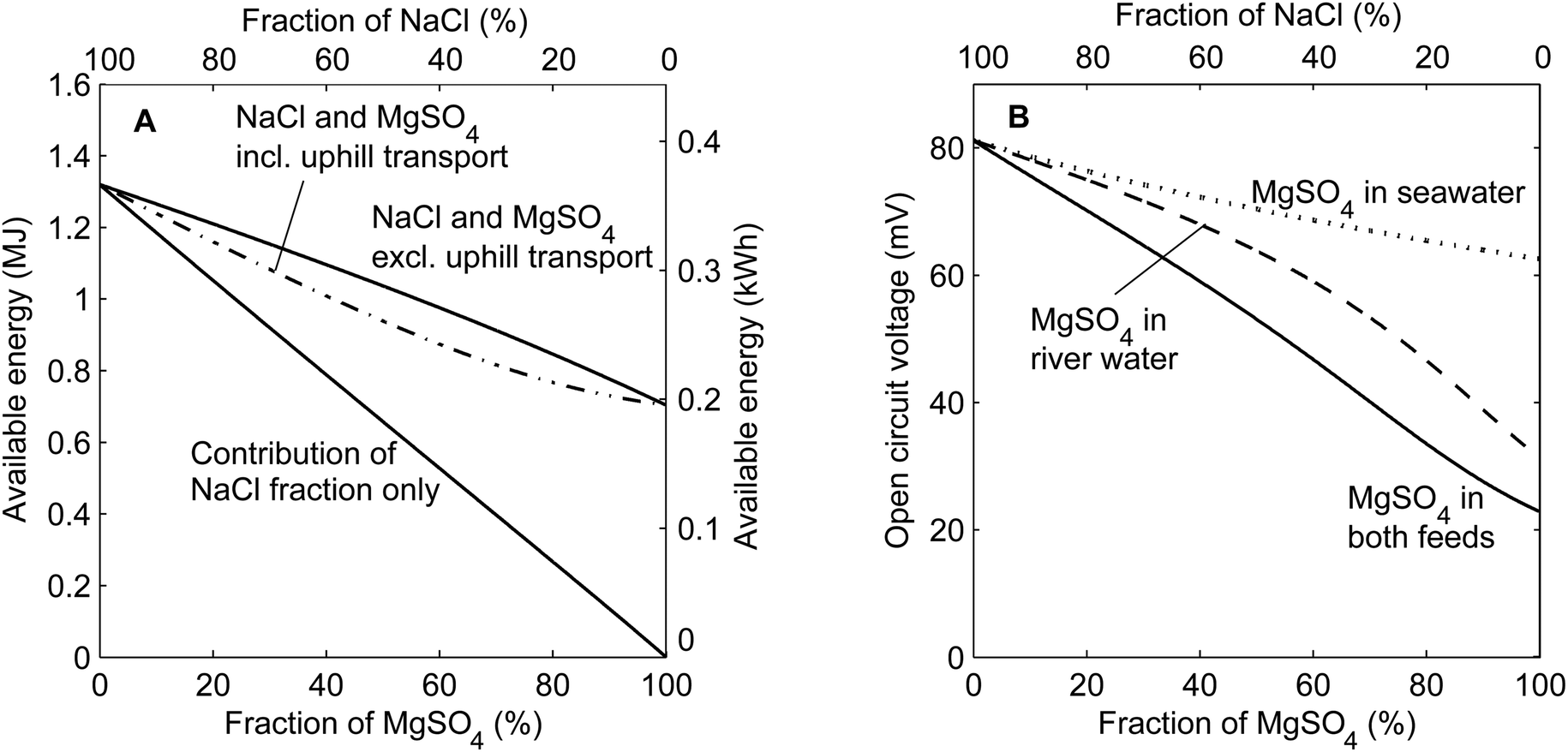 | ||
| Fig. 2 Theoretical available energy using 1 m3 of both feed waters (A) and open circuit voltage over one membrane (B) as a function of the molar salt fraction MgSO4 in the feed water. The available energy (A) corresponds to the case when MgSO4 is in both feed waters. The open circuit voltage (B) is also calculated for cases when MgSO4 is only present in the river water or in the seawater. The total salt concentrations for the inflowing feed water are 0.508 M and 0.017 M for all cases. The activity coefficients are derived from the modified three characteristic parameter correlation (TCPC) model.28 | ||
Fig. 2A shows that the available energy decreases when a larger fraction of NaCl is substituted for MgSO4, even when disregarding the loss due to uphill transport. The contribution of NaCl decreases practically linearly, proportional to the molar fraction of NaCl, due to the lower concentrations of NaCl. However, the contribution of MgSO4 cannot compensate for this decrease in available energy, due to the relatively low activity coefficients for MgSO4, which effectively limit the increase in entropy when mixing the feed waters. Hence, the total available energy of a mixture of NaCl and MgSO4 is lower than when using NaCl only. This effect is inevitably related to the use of MgSO4 as a salt and hence will also be relevant to other salinity gradient power technologies such as pressure retarded osmosis (PRO)27,29 and capacitive mixing (CAPMIX).9,30,31
The uphill transport of MgSO4, and the corresponding exchange for NaCl, further limits the available energy in RED. Because two moles of NaCl are exchanged for one mole of MgSO4, available energy is lost when uphill transport of MgSO4 occurs. This irreversible loss is largest for a fraction MgSO4 of approximately 60%, where it accounts for approximately 10% of the available energy. The maximum is found at this particular fraction due to a combination of large uphill transport J (eqn (4)) when both ionic species are present in significant fractions and the different contributions of NaCl and MgSO4 to the available energy.
The open circuit voltage (OCV) over each ion exchange membrane decreases relatively much more than the total available energy when increasing the fraction of MgSO4 in the feed water (Fig. 2B). When MgSO4 is mixed in both feed waters, the OCV decreases from approximately 80 mV for feed water without MgSO4 to little more than 20 mV for feed water with MgSO4 only. This decrease is much larger than that for the available energy, mainly due to the fact that the valence (zi) appears in the denominator in eqn (1). Hence, the voltage is halved for solutions with MgSO4 only compared to NaCl only even when equal activities would be considered.
Additionally, uphill transport plays a larger role in the OCV because the salinity ratio, which determines the OCV, is more sensitive to small changes in the river water concentrations than the available energy. Although it is impossible to calculate the voltage without uphill transport, the effect of the uphill transport is visible when considering the cases in which a fraction of MgSO4 is present in the seawater or in the river water only. As shown in the ESI,† the case with MgSO4 in the seawater only, at fractions <80%, has a minimum of uphill transport. The difference between this case and the other two cases provides an indication of the uphill transport effect on the open circuit voltage. Fig. 2B shows that uphill transport has substantial influence on the obtained voltage, and hence on the power density, while the effect is rather small for the available energy (Fig. 2A).
In practical cases, the fraction of multivalent ions in seawater (typically 10%16) is often lower than that in river water, which makes the uphill transport extra relevant. The uphill transport as demonstrated for MgSO4 occurs similarly for other multivalent ions. As a consequence, scaling of poorly soluble multivalent salts (such as calcium phosphates) is possible at the seawater compartments, which was already observed in RED using natural feed water.16
The use of monovalent selective membranes reduces the disadvantageous influence of multivalent ions19 and hence increases the actual obtained voltage. A perfectly monovalent selective membrane would induce an open circuit voltage that is based on the difference in NaCl concentration only, i.e., approximately 80 mV for all fractions of MgSO4 in Fig. 2B. Although this would significantly improve the obtained voltage, and hence the power density, the existing monovalent selective membranes do still allow a substantial part of the multivalent ion transport.19,32
Power density
The much lower open circuit voltage, but only slightly lower available energy, suggests that a larger membrane area is required to capture an equal portion of the available energy when a fraction of MgSO4 is present in the feed water. In other words, the power density is expected to be lower when the fraction of multivalent ions is increased. The obtainable power density of a RED stack can be derived from the open circuit voltage and the electrical resistance of the stack:2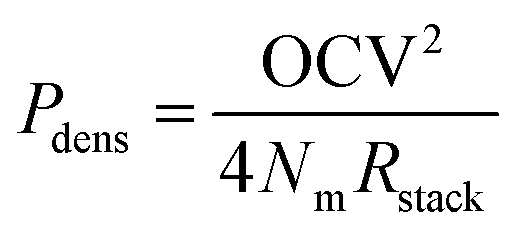 | (5) |
Although this theoretical framework helps to understand the effect of MgSO4, the actual open circuit voltage, electrical resistance and power density are more complex to calculate. For example, the equilibrium as described in eqn (4) assumes a batch mode and hence disregards the supply of new feed water. Therefore, the actually generated voltage, electrical resistance and obtained power in RED need to be investigated experimentally, which is described in the next section.
Experimental setup
Stack
Three RED stacks were built, each with different ion exchange membranes. One stack was composed of heterogeneous membranes (Ralex CMH/AMH, MEGA AS, Czech Republic), a second stack was composed of commercial homogeneous membranes (Neosepta CMX/AMX, Tokuyama, Japan) and a third stack was composed of a new type of homogeneous membranes (V1 CEM/V1 AEM, Fujifilm Europe, The Netherlands). The specifications of these membranes are listed in Table 1.| Membrane name | Membrane type | Thickness (wet) | Charge density35 | Apparent permselectivity (in 0.5 –0.1 M KCl) | Area resistance (in 0.5 M NaCl) |
|---|---|---|---|---|---|
| Ralex CMH/AMH | Heterogeneous | <725 μm | 5.5 M | >91% | <8.8 Ω cm2 |
| Neosepta CMX/AMX | Homogeneous | 155 μm | 8.4 M | 98% | 2.7 Ω cm2 |
| Fujifilm V1 | Homogeneous | 125 μm | — | 93% | 1.6 Ω cm2 |
These membranes are selected to observe the effect of MgSO4 for different types of membranes (heterogeneous and homogeneous) and possible difference in (uphill) ion transport due to the differences in membrane thickness and membrane resistance.
A woven net spacer of 200 micrometer thickness (Sefar 06-300/51, Switzerland) was used in between the ion exchange membranes to create alternately compartments for salt water and fresh water. The spacer geometry, gaskets and inflow geometry were described in previous studies.3,33 All stacks were confined between two endplates with embedded Ti–Ru/Ir mesh electrodes (MAGNETO Special Anodes B.V., The Netherlands) with a dimension of 10 cm by 10 cm.
The electrode rinse solution, composed of 0.05 M Fe(CN)63−/4− and 0.25 M NaCl, was circulated in the electrode compartments at a flow rate of 150 mL min−1 and kept at a static overpressure of 0.5 bar to ensure that the membranes are closely packed.
The stack with Fujifilm membranes was equipped with additional buffer compartments similarly to a previous study,34 created by a Neosepta CMX membrane between the membrane pile and the electrode compartment. This buffer compartment was circulated with 0.25 M NaCl. The buffer compartment was installed in this case to avoid leakage of the electrode rinse solution towards the feed water, which was no issue with the other membrane types.
To correct for the additional resistance of the electrode compartment and the possible buffer compartment, the resistance of an empty stack, composed of two electrodes, corresponding electrode compartments and possibly buffer compartments, was measured. The resistance of such a blank was used to correct the resistance of the measurement series, and allowed comparison of the different stacks.
Feed water
Salt and fresh water streams, composed of a mixture of dissolved NaCl (99.5% purity, ESCO, The Netherlands) and/or MgSO4, MgCl2 or Na2SO4 (at least 98% purity, VWR, The Netherlands), were fed into the RED stacks. The total salt concentration of the artificial seawater was 0.508 M in all cases, whereas the total salt concentration of the artificial river water was always 0.017 M. In other words, compared to artificial solutions with only NaCl, part of the NaCl was replaced with MgSO4, MgCl2 or Na2SO4, in order to maintain the molarity of the inflowing feed (and hence the conductivity, to a certain degree) constant.When considering mixtures of NaCl and MgSO4, a molar percentage of 0%, 5%, 10%, 25%, 50% or 100% of the total dissolved salt was accounted for by MgSO4. This percentage was equal for both feed water streams. The remaining amount of dissolved salt was NaCl. When considering the mixtures of NaCl and MgCl2 or Na2SO4, 10% of the dissolved salts was MgCl2 or Na2SO4 and the remaining 90% was NaCl.
All stacks were first fed with solutions containing 0.508 M NaCl and 0.017 M NaCl, to equilibrate the membranes with NaCl solutions. After at least one hour of operation, the feed water solutions were suddenly switched to one of the previously described mixtures. The stacks operated for at least 4 hours using these solutions before switching back to solutions with dissolved NaCl only. The effluent was sampled and effluent concentrations were measured using ion chromatography (IC) and inductively coupled plasma (ICP).
The flow rate of the feed water solutions was 60 mL min−1 in all cases, which is equivalent to a flow velocity of 1.3 cm s−1 and a Reynolds number of 5. The effluent was not re-cycled, i.e., new feed water was supplied continuously. The temperature of the feed waters was maintained at 25 ± 1.5 °C.
Electrochemical measurements
A chronopotentiometric series was applied (Ivium Technologies, The Netherlands), comprising a stage with no current (open circuit) and a stage with a current density of 10 A m−2. Both stages were applied for 60 seconds and repeated continuously. The stack resistance was calculated from the difference in voltage at these two stages, divided by the current density.Some measurements were duplicated to check the reproducibility. The steady voltages of these duplicates were typically within 3% of the original series.
In addition, the open circuit voltage over a single membrane (CEM or AEM, 10 cm × 10 cm) was measured, using a single membrane enveloped with only two compartments for feed water connected to an Ag/AgCl reference electrode (QIS, The Netherlands). The same feed water solutions as for the stacks were used for these individual membrane setups and were supplied at a flow rate of 12 mL min−1.
Membrane cross-sections
To study the ion transport through the membranes, the position of the ionic species in the membrane cross-sections was identified with energy dispersive X-ray spectra (EDX or EDS). Membrane cross-sections were made for all membranes, after 0, 2, 15, 30, 45 and 300 min of operation with feed water solutions in which 10% of the salt concentration was MgSO4 and 90% was NaCl.After this specified duration, the stacks were opened, the membranes were rinsed in demineralized water and directly frozen in liquid nitrogen to conserve the position of the ions in the membranes. The process to stop the stack operation, dismantle the stack and freeze the membranes took approximately 1 minute. Subsequently, the frozen membranes were broken and freeze dried (Edwards, United Kingdom).
The EDX analysis (JEOL, JSM 6010LA, United States) was performed over at least two cross-sectional lines of each sample, each comprising at least 10 scan points. The average atomic percentages of the detected elements of Na, Cl, Mg and S were calculated. More details on the EDX analysis are provided in the ESI.†
Results and discussion
The results will first comprise the stationary RED performance with different NaCl–MgSO4 mixtures, which is representative for large-scale operations with steady feed water conditions. Subsequently, the transition from one feed to another feed is presented, which resembles the cases where e.g. tides cause fluctuating feed water concentrations.Stationary open circuit voltage
The open circuit voltages (OCV) for steady operation are shown in Fig. 3 for stacks with Ralex, Neosepta or Fujifilm membranes, as a function of the percentage of MgSO4 to the total dissolved salt of both feed water streams.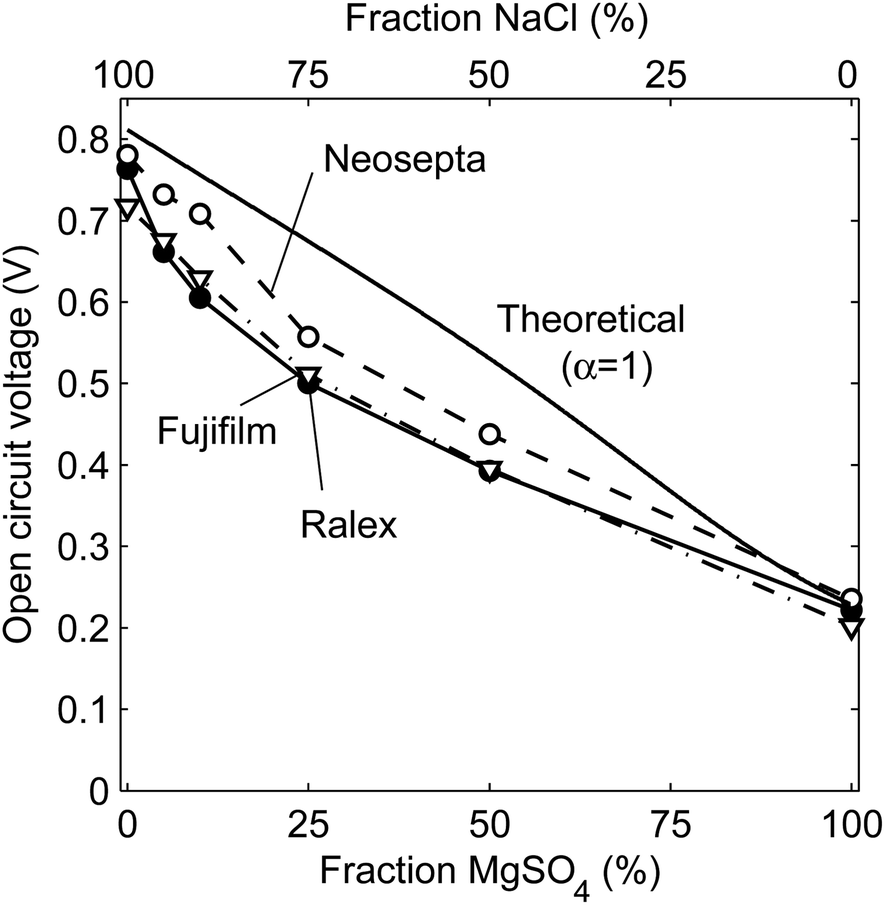 | ||
| Fig. 3 Open circuit voltage versus the molar fraction of MgSO4 of the total amount of dissolved salt in the feed water. The experimental results are obtained with Ralex, Neosepta or Fujifilm membranes. The presented values are averaged based on a stationary data series of at least 1 hour. Error bars at twice the standard deviations of this series are too small to be visible. The voltage for the Neosepta membranes at 100% MgSO4 is slightly higher (2%) than theoretical, which is due to a small deviation in the theoretical activity (which includes estimated activity coefficients) with respect to the actual activity. | ||
The open circuit voltage decreases when more MgSO4, relative to the total amount of dissolved salt, is present in the feed water (Fig. 3). This decreasing trend was expected, as demonstrated already in Fig. 2B. The open circuit voltage is highest for the stack with Neosepta membranes, due to the high charge density of these membranes35,36 and the high permselectivity according to its specifications (Table 1).
The open circuit voltage is close to the theoretical voltage for perfectly selective membranes in the case of 0 or 100% MgSO4, which indicates that the membranes are highly selective and co-ions are excluded in these cases. The exclusion of co-ions is dependent on the electrical force on these ions, which is proportional to the product of the Donnan potential and the ion charge. In other words, co-ions are optimally excluded when a high Donnan potential is available and all co-ions have a high valence, as all co-ions experience the maximum electric force expelling them from the double layer in this case. The relatively high selectivity for 0% MgSO4 is well known and originates from the high Donnan potentials that exclude the co-ions effectively from the double layer when using monovalent ions only. On the other hand, when using multivalent ions only, the high ionic charges (2+ or 2−) exclude the co-ions from the double layer, which is visible as a good agreement between the theoretical and experimental values at 100% MgSO4.
However, the experimental OCV is lower than the theoretical one when mixtures of monovalent and multivalent ions are used. This is explained from the combination of relatively low Donnan potentials (due to the presence of multivalent ions) and relatively low valence of the monovalent co-ions, which increases the permeation of co-ions through the ion exchange membranes.24 In this case, more monovalent cations will permeate through the AEMs and more monovalent anions through CEMs, compared to solutions with purely monovalent or multivalent ions. In other words, the permeation of co-ions through the non-ideal membranes is an increasing issue when using a mixture of monovalent and multivalent ions. This co-ion transport is not taken into account for the theoretical curve, as the theory assumes a perfect 100% selective membrane under all conditions (α = 1). The actual apparent permselectivity is lower when using mixtures of monovalent and multivalent ions, which results in a lower open circuit voltage than the theoretical equivalents. This effect is significant, and even more pronounced in the power density which is dependent on the OCV squared (eqn (5)).
Stationary power density
The area resistance and obtained power density for steady operation are shown in Fig. 4 for stacks with Ralex, Neosepta or Fujifilm membranes, as a function of the percentage of MgSO4 to the total dissolved salt of both feed water streams. | ||
| Fig. 4 Ohmic resistance (A) and power density (B) versus the molar fraction of MgSO4 of the total amount of dissolved salt in the feed water. The experimental results are obtained with Ralex, Neosepta or Fujifilm membranes. The presented values are averaged based on a stationary data series of at least 1 hour, and twice the standard deviation is shown as error bar. Most error bars are too small to be visible. | ||
The ohmic resistance increases slightly with increasing MgSO4 fraction for all membrane types (Fig. 4A), due to higher membrane resistance as well as slightly lower conductivity of the feed water. A higher electrical resistance in the case of mixtures with MgSO4 was observed before19 and can be explained from the slightly lower diffusion coefficients of Mg2+ and SO42− compared to Na+ and Cl−. Additionally, steric hindrance of the larger (hydrated) Mg2+ and SO42− ions possibly plays a role.37 Nevertheless, the increase in ohmic resistance is less pronounced than the decrease in open circuit voltage when increasing the fraction of MgSO4. The non-ohmic resistance on the other hand does not change significantly (not shown), as this contribution is mainly dependent on the hydrodynamics of the feed water.38–40
The decreasing OCV and to a lesser extent the increasing ohmic resistance are reflected in the obtained power density (Fig. 4B). Because the power density is proportional to the OCV squared, the power density decreases rapidly when increasing the fraction of MgSO4 in the feed water. When the salt in the feed water comprises only 10% MgSO4, which is most representative for typical seawater and river water, the power density decreases with 29% (Fujifilm), 37% (Neosepta) and even 50% (Ralex) compared to pure NaCl as feed. This large decrease in power density emphasizes the importance of the open circuit voltage when using mixtures of NaCl and MgSO4.
Membrane voltage
To further understand the behavior of NaCl–MgSO4 mixtures, the individual effect of Mg2+ and SO42− is investigated separately using mixtures with MgCl2 or Na2SO4 next to NaCl. The obtained open circuit voltages using single membranes (CEM or AEM) are shown in Fig. 5.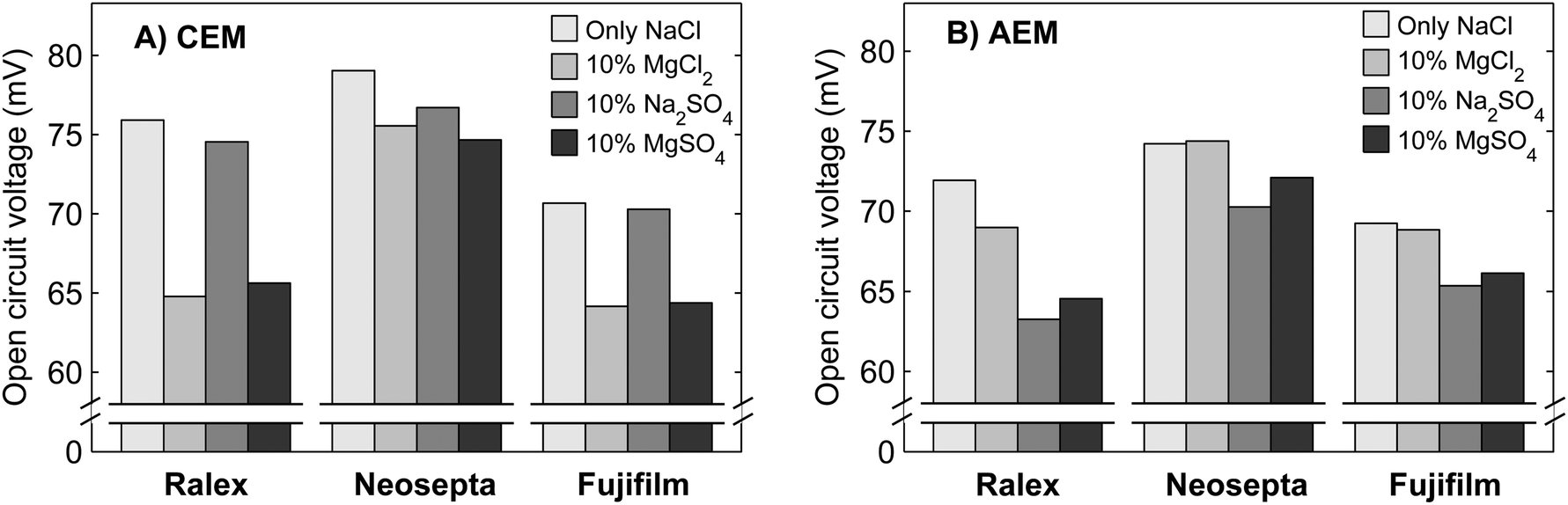 | ||
| Fig. 5 Open circuit voltage for single cation exchange membranes (A) and anion exchange membranes (B) when using feed water composed of NaCl only or 10% MgCl2, Na2SO4 or MgSO4 and 90% NaCl. The values of the obtained voltages have typically a precision of 1 mV. | ||
Fig. 5 shows that the type of cation (Mg2+ or Na+) only affects the voltage over the CEMs significantly, while the type of anion (SO42− or Cl−) only influences the voltages over the AEMs significantly. This is a direct effect of the charge selectivity of the ion exchange membranes.
Ralex membranes show the largest decrease in OCV when comparing 100% NaCl to 10% MgSO4/90% NaCl, while these membranes feature the largest membrane resistance. Apparently, the uphill transport is not strongly influenced by the membrane resistance. This can be explained from the concept of the double layer at the membrane–water interface. A very small flux of ions through the membrane (due to uphill transport) is sufficient to bring the concentration in the thin double layer to equilibrium with the chemical potential and determine the voltage over the membrane. Therefore, the membrane resistance does not play a significant role for this ion transport in the stationary case.
The reason that Ralex membranes demonstrate even largest decreases in OCV when mixtures of monovalent and multivalent ions are present is that these membranes have a relatively low fixed charge density.35 Therefore, these membranes feature a relatively low permselectivity, especially when mixtures of monovalent and multivalent ions are used. As explained before, the combination of relatively low Donnan potentials due to the presence of multivalent ions and monovalent co-ions increases the permeation of co-ions through the ion exchange membranes.24
Both homogeneous membrane types (Neosepta and Fujifilm) show only a minor decrease in OCV due to the presence of multivalent counter-ions when compared to Ralex membranes (Fig. 5). The Neosepta membranes have a higher fixed charge density than the Ralex membranes (Table 1).35 The charge density of Fujifilm membranes is not specified, but it is expected to be more or less similar to that of the homogeneous Neosepta membranes.
The presence of Mg2+ generally induces a larger decrease in membrane voltage for CEMs (Fig. 5A) than the presence of SO42− does for AEMs (Fig. 5B). This may be due to the formation of ion pairs of sodium and sulphate, NaSO4−, which is effectively a monovalent ionic species.41 Hence, the OCV for a stack with CEMs and AEMs will decrease less pronounced when SO42− is present than when Mg2+ is present.
Mass transport
The ionic transport rate, among others, of the uphill transport, can be analyzed when considering the salinity ratio (seawater to river water) of individual ionic species in the effluent. In the influent, all ions have a salinity ratio of 30:1 between the seawater and river water feed. The ratio of each ionic species in the effluent, when using 10% MgSO4 in both feed waters, is shown in Fig. 6 as a function of the current density, for the stack with Neosepta membranes. Similar results are obtained for stacks with Ralex or Fujifilm membranes (see ESI†).
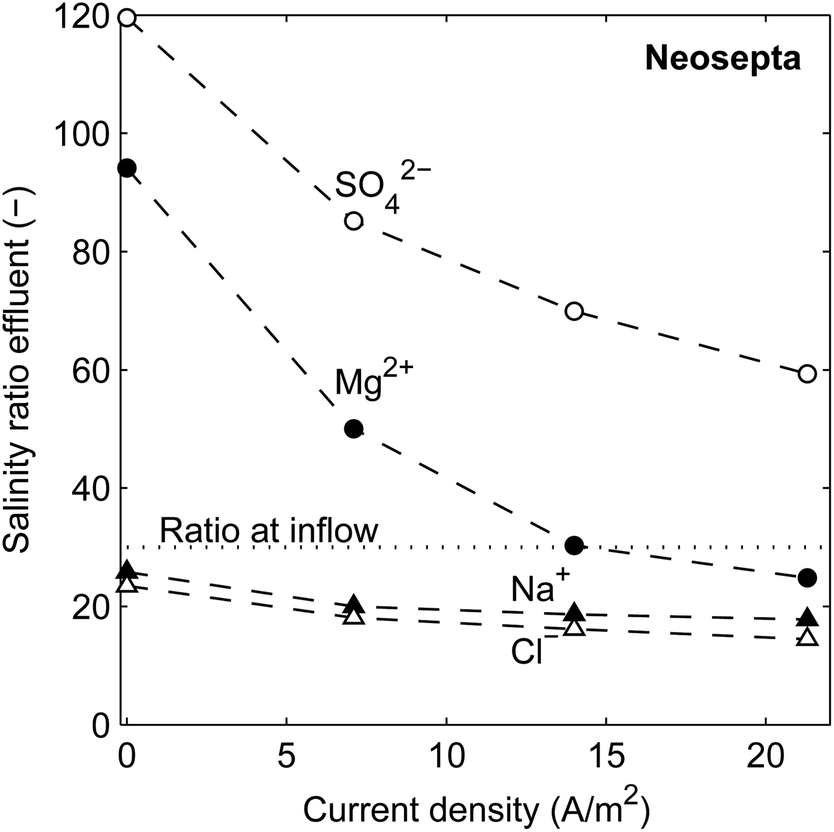 | ||
| Fig. 6 Ratio of Na+, Cl−, Mg2+ and SO42− between the seawater effluent and the river water effluent, as a function of the current density, for the stack with Neosepta membranes. | ||
Fig. 6 shows that the ratio for all considered ions in the effluent decreases for increasing current density, as a higher ionic current directly implies a larger ionic flux from seawater to river water. In other words, a part of the available energy is used. However, also when no electrical current is generated, the concentration ratio for the monovalent ions in the effluent is already significantly lower than 30. This is explained from the uphill transport of multivalent ions, in exchange for monovalent ions.
This uphill transport of multivalent ions is proven from the concentration ratio for Mg2+ and SO42−, which is significantly higher than 30 when no current density is generated. The concentrations of SO42− are more pronounced than that for Mg2+, due to a lower resistance of the AEM compared to the CEM and the higher diffusion coefficient of the used anionic species than that of the cationic species.42 Although the high concentration ratios of the multivalent ions clearly indicate uphill transport, the concentration ratios for multivalent ions are expected to be even higher based on the theoretical framework (eqn (3)). This discrepancy is also observed in previous research,19 and justifies the concept that the concentrations in the double layer (which determine the OCV) adapt quickly while the effluent concentrations are dominated by the concentrations in the bulk fluids. The continuous feed water flow prevents that the concentrations in the bulk fluids equilibrate with these in the double layer.
The concentration ratios for multivalent ions decrease sharply when the current density increases. This is caused by a decrease in voltage over each membrane, due to an ohmic and non-ohmic voltage drop. Such a decrease in voltage reduces the electromotive force for uphill transport of multivalent ions at a higher current density. As a consequence, this effect shifts the optimum current density to a higher value for a system with a mixture of monovalent and multivalent ions.
Non-stationary performance
The previous results discussed stationary performance, i.e., when feed water with the same concentrations is fed for a long time. Non-stationary cases, for example when feed water with monovalent ions only is suddenly replaced by feed water with mono- and multivalent ions, allow the mass transport mechanisms to be analyzed in the membrane, double layer and feed water bulk. The power density as a function of time after such feed water change is shown for all stacks in Fig. 7. | ||
| Fig. 7 Power density as a function of time, for stacks with (A) Ralex membranes, (B) Neosepta membranes and (C) Fujifilm membranes. At time = 0 h, the feed water source is switched from NaCl solutions to salt solutions with a certain percentage of MgSO4 (as indicated in the graphs) and the remaining percentage of NaCl. Small deviations in power density are observed for all stacks with 5% MgSO4 and the Neosepta stack with 10% MgSO4, which are due to temporary entrapment of air in the feed water. | ||
In all cases, the steady power densities for the stack with heterogeneous Ralex membranes are lower than for the stacks with homogeneous membranes, especially when mixtures of NaCl and MgSO4 are used, as already demonstrated in Fig. 3. However, the non-stationary power density in Fig. 7 reveals that the stack with Ralex membranes responds slower than the stack with Neosepta membranes and this is even more pronounced when compared to the stack with Fujifilm membranes. When using a mixture with 10% MgSO4, the power density does not reach a constant value after 4 hours of operation in the case of Ralex membranes, while for Fujifilm membranes the power density is rather constant after 0.5 h already. These differences in response time are also observed when the feed water is switched back to feed water with NaCl only (not shown).
The decrease in power density and the corresponding response time match qualitatively and quantitatively very well with previous experiments with real natural feed water, in which Ralex membranes were used and approximately 10% of the ions is multivalent.16,43 Comparing this practical case with the present experiments with Ralex membranes and 10% MgSO4, the power density decreases in both cases by approximately 50% in a typical timescale of hours. Therefore, this artificial mixture of monovalent (90%) and multivalent (10%) ions is considered to be representative for at least the first hours of operation of RED with natural feeds.
Membrane cross-sections
The slow response of the power in the case of mixtures with MgSO4 is the consequence of the limitations in ion exchange between the membrane and the double layer and that between the double layer and the diffusive boundary layer. The double layer, which is as thin as only a few nm, experiences hardly any ion transport from the bulk fluid by convection and hence is strongly influenced by the ionic composition of the membranes. The Ralex membranes are much thicker than the homogeneous membranes, and therefore have the largest NaCl buffer when switched to a solution with a fraction of MgSO4. This process suggests a gradual change of the counter-ions within the membrane interior over time. Fig. 8 shows the percentage of magnesium elements relative to the total counter-ions (magnesium + sodium) in cross-sections of the cation exchange membranes of Ralex, Neosepta and Fujifilm, for different durations of operation on feed water with 10% MgSO4–90% NaCl.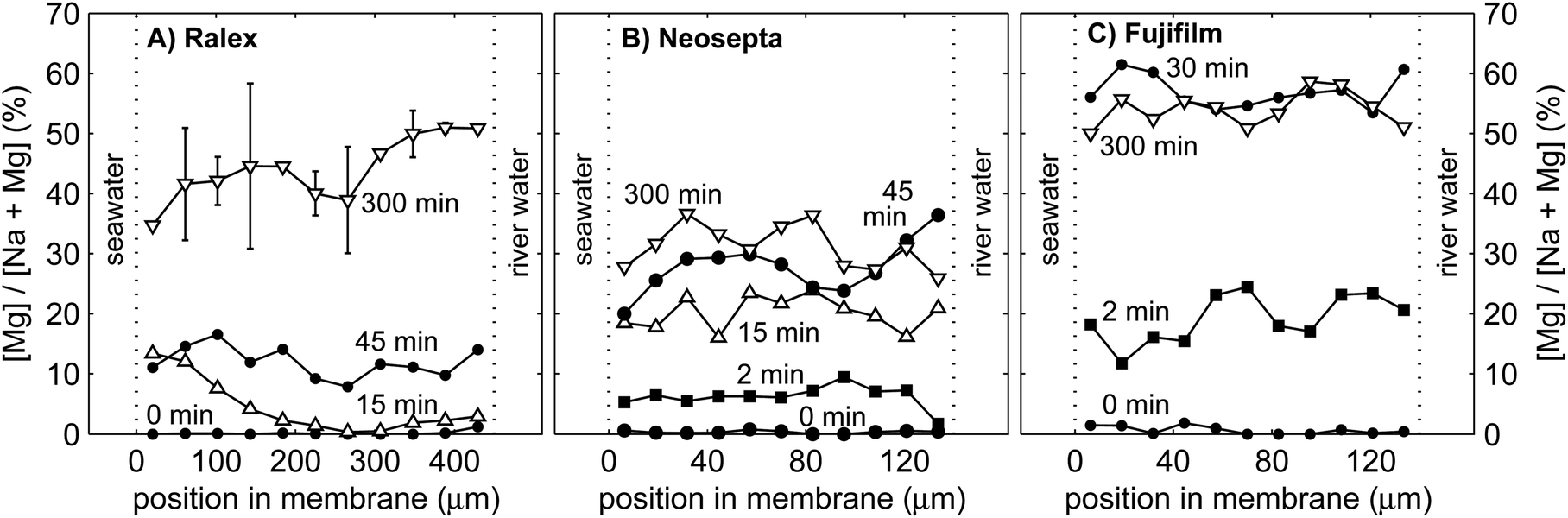 | ||
| Fig. 8 Relative contribution of magnesium as a counter-ion in Ralex (A), Neosepta (B) and Fujifilm (C) cation exchange membranes, as a function of the position in the membrane cross-section. The elements are experimentally detected with EDX. The left sides of each graph correspond to the membrane side that faced the seawater compartment, while the right side faced the river water compartment. The standard error is indicated by the error bars for Ralex 300 min. | ||
Fig. 8 shows that the interior of the cation exchange membranes does not contain any magnesium before switching to feed water with MgSO4 (i.e. at 0 min) within the accuracy of the measurement. The contribution of magnesium strongly increased to 30% to 60% of the total amount of counter-ions after 300 minutes of operation with feed water with 10% MgSO4. The relatively large fraction of magnesium in the membrane material is explained from its high valence (z = 2 for Mg2+) compared to Na+ (z = 1). Therefore, Mg2+ binds stronger to the negative fixed membrane charges than Na+ does. This results in a relatively large fraction of magnesium elements in the membrane after 300 min. The strong affinity for Mg2+ of the CEMs explains why the ohmic resistance is most sensitive to only small fractions of MgSO4, as observed already in Fig. 3B.
When considering the Ralex membrane in more detail, the gradient in the relative concentration of magnesium is directed from the seawater compartment towards the river water compartment after 15 min, while it is opposite after 300 min of operation (Fig. 8A). The exact shape of this curve cannot be determined due to the limited accuracy, but the contributions of MgSO4 at the seawater side are significantly different from those at the river water side. Both observations are expected based on the theoretical framework. Because the ionic current is directed from the seawater compartment towards the river water compartment, magnesium enters the membrane initially from the seawater side, which explains the profile at 15 min. When steady concentration profiles are established in the membrane (i.e., at 300 min), the concentration gradient of multivalent ions should be directed towards the highest concentration,24 in line with the uphill transport of multivalent ions. Such a concentration profile was already predicted by Higa et al.,24 and is, to the best of our knowledge, for the first time experimentally observed, in this case in the Ralex membrane. These details in concentration profiles cannot be distinguished for the other membrane types, probably due to ion exchange in these thinner membranes when dismantling the stack and preparing the membrane samples for EDX analysis. Hence, no conclusions can be drawn from the gradient in each cross-section for the Neosepta and Fujifilm membranes. The much larger thickness of the Ralex membranes better conserves the concentration gradients as representative during the experiment. Future experiments using (thick) membranes that can be frozen directly after operation will provide more details on the exact shape of the concentration profiles inside the membranes, but are beyond the scope of this paper.
The relative concentration of Mg2+ increases much faster in Fujifilm CEMs and (in less degree) in Neosepta CEMs compared to Ralex CEMs. The contribution of magnesium more than doubles between 45 min and 300 min for Ralex membranes (Fig. 8A), while it increases only slightly for Neosepta membranes in this period (Fig. 8B). For Fujifilm membranes (Fig. 8C), the relative concentration of magnesium does not even change significantly anymore after the first 30 min. The transition time to establish a steady concentration profile in the membrane is expected to be proportional to the ion exchange capacity and the membrane thickness. The Ralex membranes offer a slightly higher ion exchange capacity and are much thicker than Neosepta membranes35 and therefore provide the largest source of Na+, which is in agreement with Fig. 8. Although the ion exchange capacity of Fujifilm membranes is not specified, the fast stabilization of the concentration profile predicts a low ion exchange capacity of these membranes. However, an exact value cannot be inferred, as micro-heterogeneity in the membranes complicates the uniform release of monovalent ions from the membrane interior.44
During the non-stationary period, the double layer is supplied with Na+ ions transported out of the membrane in exchange for Mg2+ transported into the membrane, which effectively reduces the fraction of multivalent ions in the double layer. Hence, the voltage over the CEMs is temporarily higher compared to steady operation on feed water with a fraction of MgSO4. The same effect occurs for AEMs, where Cl− is slowly released in exchange for SO42− (not shown due to the less reliable detection of sulfur). This slow release of monovalent ions towards the double layer also explains the slow transition in power density as observed in Fig. 7, in particular for Ralex membranes.
Outlook
The effect of multivalent ions on the obtained power density should be considered for practical applications of RED. As indicated in Fig. 2B, the voltage is mostly affected by the concentration in the river water side. It was already known that the (monovalent) salinity45 and fouling potential16 are important factors that need to be considered for selection of the feed water sources. The present research emphasizes that the contribution of multivalent ions in the feed water, in particular in the diluted stream, should be considered as well.Additionally, the observed slow response in power density (Fig. 7) suggests a strategy that reduces the impact of multivalent ions: the slow ion supply from the membrane interior predicts that the response can be accelerated at a higher current density, and that the response can be decelerated when the current density is periodically reversed. The latter behavior is recently observed for RED stacks that operate using natural feed water; a stack with periodic feed water switch (which induces a switch of the current direction) showed a slower decrease in power density than a stack with continuous feed water supply.43 A fast response, which may occur at high current densities in the same direction, can be useful when applying a treatment with NaCl (brine) to flush the multivalent ions from the membranes. Such treatment should be applied as short as possible, to limit the volume of feed water required for such cleaning. In this way, the gradual ion exchange between monovalent and multivalent ions in the membrane can be used beneficially. Hence, such an operational strategy can be applied to improve the power density when RED is used for power production from natural feed water streams.
Conclusions
Energy generation from mixing seawater and river water is mimicked representatively using artificial mixtures of salts with monovalent ions (e.g., NaCl) and salts with multivalent ions (e.g. MgSO4). The global potential for salinity gradient power, as quantified by the available energy per water volume, decreases only slightly for increasing fractions of MgSO4, while the obtained voltage and power density decrease drastically. For a mixture with a molar fraction of 10% MgSO4 and 90% of NaCl in both feed waters, the experimentally obtained power density in steady state decreases between 29% and 50%, depending on the membrane type, compared to the case where the feed solutions contain only NaCl as a salt.This decrease in power density is mainly due to a lower open circuit voltage and only slightly due to higher electrical resistance. This lower open circuit voltage is theoretically predicted due to the transport of Mg2+ and SO42− against their concentration gradient. This uphill transport of multivalent ions is driven by the difference in voltage as generated by a monovalent species and a multivalent species. In addition, the permeation of co-ions through the non-ideal membranes is an increasing issue when using a mixture of monovalent and multivalent ions. This effect is more pronounced for heterogeneous membranes than for homogeneous membrane types.
Non-stationary cases, where feed water is switched from NaCl solutions to mixtures of NaCl and MgSO4, show that the voltage response time is in the order of tens of minutes up to several hours, due to ion exchange between the membranes and the feed water. The voltage remains temporarily higher than in its steady state due to the gradual release of monovalent ions from the membrane interior in exchange for multivalent ions, as is visible in the concentration profiles in membrane cross-sections. This knowledge opens the possibility to improve the power density in practical applications of RED using varying concentrations of feed water.
Acknowledgements
This work was performed in the cooperation framework of Wetsus, Centre of Excellence for Sustainable Water Technology (http://www.wetsus.nl). Wetsus is co-funded by the Dutch Ministry of Economic Affairs and Ministry of Infrastructure and Environment, the European Union Regional Development Fund, the Province of Fryslân and the Northern Netherlands Provinces. The authors are thankful for the support of the participants of the research theme ‘Blue Energy’. In addition, the authors thank Fujifilm Europe B.V. for supplying their membranes and Karst Wierenga and Steven Koffijberg for performing experiments.References
- R. E. Pattle, Nature, 1954, 174, 660 CrossRef CAS.
- P. E. Długołęcki, A. Gambier, K. Nijmeijer and M. Wessling, Environ. Sci. Technol., 2009, 43, 6888–6894 CrossRef.
- J. W. Post, H. V. M. Hamelers and C. J. N. Buisman, Environ. Sci. Technol., 2008, 42, 5785–5790 CrossRef CAS.
- B. E. Logan and M. Elimelech, Nature, 2012, 488, 313–319 CrossRef CAS PubMed.
- G. Z. Ramon, B. J. Feinberg and E. M. V. Hoek, Energy Environ. Sci., 2011, 4, 4423–4434 CAS.
- J. Veerman, M. Saakes, S. Metz and G. Harmsen, J. Appl. Electrochem., 2010, 40, 1461–1474 CrossRef CAS.
- O. Scialdone, C. Guarisco, S. Grispo, A. D. Angelo and A. Galia, J. Electroanal. Chem., 2012, 681, 66–75 CrossRef CAS PubMed.
- D. A. Vermaas, S. Bajracharya, B. B. Sales, M. Saakes, B. Hamelers and K. Nijmeijer, Energy Environ. Sci., 2013, 6, 643–651 CAS.
- O. S. Burheim, F. Liu, B. B. Sales, O. Schaetzle, C. J. N. Buisman and H. V. M. Hamelers, J. Phys. Chem. C, 2012, 116, 19203–19210 CAS.
- G. L. Wick, Energy, 1978, 3, 95–100 CrossRef CAS.
- J. D. Isaacs and R. J. Seymour, Int. J. Environ. Stud., 1973, 4, 201–205 CrossRef.
- EIA, International energy outlook 2011, U.S. Department of Energy, Washington, D.C., 2011 Search PubMed.
- M. Tedesco, A. Cipollina, A. Tamburini, W. van Baak and G. Micale, Desalin. Water Treat., 2012, 49, 404–424 CrossRef CAS.
- D. A. Vermaas, J. Veerman, N. Y. Yip, M. Elimelech, M. Saakes and K. Nijmeijer, ACS Sustainable Chem. Eng., 2013, 1, 1295–1302 CrossRef CAS.
- D. A. Vermaas, M. Saakes and K. Nijmeijer, Environ. Sci. Technol., 2011, 45, 7089–7095 CrossRef CAS PubMed.
- D. A. Vermaas, D. Kunteng, M. Saakes and K. Nijmeijer, Water Res., 2013, 47, 1289–1298 CrossRef CAS PubMed.
- V. D. Grebenyuk, R. D. Chebotareva, S. Peters and V. Linkov, Desalination, 1998, 115, 313–329 CrossRef CAS.
- V. Lindstrand, A.-S. Jönsson and G. Sundström, Desalination, 2000, 130, 73–84 CrossRef CAS.
- J. W. Post, H. V. M. Hamelers and C. J. N. Buisman, J. Membr. Sci., 2009, 330, 65–72 CrossRef CAS PubMed.
- G. D. Mehta, J. Membr. Sci., 1982, 11, 107–120 CrossRef CAS.
- R. E. Lacey, Ocean Eng., 1980, 7, 1–47 CrossRef CAS.
- J. Veerman, M. Saakes, S. J. Metz and G. J. Harmsen, Environ. Sci. Technol., 2010, 44, 9207–9212 CrossRef CAS PubMed.
- M. Higa, A. Tanioka and K. Miyasaka, J. Membr. Sci., 1988, 37, 251–266 CrossRef CAS.
- M. Higa, A. Tanioka and K. Miyasaka, J. Membr. Sci., 1990, 49, 145–169 CrossRef CAS.
- M. Tasaka, R. Kiyono and D.-S. Yoo, J. Phys. Chem. B, 1999, 103, 173–177 CrossRef CAS.
- M. Higa and A. Kira, J. Phys. Chem., 1992, 96, 9518–9523 CrossRef CAS.
- N. Y. Yip and M. Elimelech, Environ. Sci. Technol., 2012, 46, 5230–5239 CrossRef CAS PubMed.
- X. Ge, X. Wang, M. Zhang and S. Seetharaman, J. Chem. Eng. Data, 2007, 52, 538–547 CrossRef CAS.
- S. Chou, R. Wang and A. G. Fane, J. Membr. Sci., 2013, 448, 44–54 CrossRef CAS PubMed.
- D. Brogioli, Phys. Rev. Lett., 2009, 058501, 1–4 Search PubMed.
- F. Liu, O. Schaetzle, B. B. Sales, M. Saakes, C. Buisman and H. V. M. Hamelers, Energy Environ. Sci., 2012, 5, 8642–8650 CAS.
- J. Balster, O. Krupenko, I. Pünt, D. F. Stamatialis and M. Wessling, J. Membr. Sci., 2005, 263, 137–145 CrossRef CAS PubMed.
- J. Veerman, M. Saakes, S. J. Metz and G. J. Harmsen, J. Membr. Sci., 2009, 327, 136–144 CrossRef CAS PubMed.
- D. A. Vermaas, M. Saakes and K. Nijmeijer, J. Membr. Sci., 2011, 385–386, 234–242 CrossRef PubMed.
- P. E. Długołęcki, K. Nijmeijer, S. J. Metz and M. Wessling, J. Membr. Sci., 2008, 319, 214–222 CrossRef PubMed.
- A. H. Galama, J. W. Post, M. A. Cohen Stuart and P. M. Biesheuvel, J. Membr. Sci., 2013, 442, 131–139 CrossRef CAS PubMed.
- G. M. Geise, D. R. Paul and B. D. Freeman, Prog. Polym. Sci., 2014, 39, 1–42 CrossRef CAS PubMed.
- D. A. Vermaas, M. Saakes and K. Nijmeijer, J. Membr. Sci., 2014, 453, 312–319 CrossRef CAS PubMed.
- D. A. Vermaas, M. Saakes and K. Nijmeijer, Electrochim. Acta, 2014, 117, 9–17 CrossRef PubMed.
- M. C. Hatzell and B. E. Logan, J. Membr. Sci., 2013, 446, 449–455 CrossRef CAS PubMed.
- F. P. Daly, C. W. Brown and D. R. Kester, J. Phys. Chem., 1972, 76, 3664–3668 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics 2004–2005: A Ready-Reference Book of Chemical and Physical Data, ed., CRC press, 2004 Search PubMed.
- D. A. Vermaas, D. Kunteng, J. Veerman, M. Saakes and K. Nijmeijer, Environ. Sci. Technol., 2014 DOI:10.1021/es4045456.
- V. I. Zabolotsky and V. V. Nikonenko, J. Membr. Sci., 1993, 79, 181–198 CrossRef CAS.
- A. Daniilidis, D. A. Vermaas, R. Herber and K. Nijmeijer, Renewable Energy, 2014, 64, 123–131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ee43501f |
| This journal is © The Royal Society of Chemistry 2014 |