Concentrated electrolytes: decrypting electrolyte properties and reassessing Al corrosion mechanisms†
Dennis W.
McOwen
a,
Daniel M.
Seo
a,
Oleg
Borodin
b,
Jenel
Vatamanu
c,
Paul D.
Boyle
d and
Wesley A.
Henderson
*a
aIonic Liquids & Electrolytes for Energy Technologies (ILEET) Laboratory, Department of Chemical & Biomolecular Engineering, North Carolina State University, 911 Partners Way, Raleigh, North Carolina 27695, USA. E-mail: whender@ncsu.edu
bElectrochemistry Branch, Sensor & Electron Devices Directorate, U.S. Army Research Laboratory, Adelphi, Maryland 20783, USA
cDepartment of Material Science & Engineering, University of Utah, Salt Lake City, Utah 84112, USA
dX-ray Structural Facility, Department of Chemistry, North Carolina State University, 2620 Yarbrough Drive, Raleigh, North Carolina 27695, USA
First published on 30th October 2013
Abstract
Highly concentrated electrolytes containing carbonate solvents with lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) have been investigated to determine the influence of eliminating bulk solvent (i.e., uncoordinated to a Li+ cation) on electrolyte properties. The phase behavior of ethylene carbonate (EC)–LiTFSI mixtures indicates that two crystalline solvates form—(EC)3:LiTFSI and (EC)1:LiTFSI. Crystal structures for these were determined to obtain insight into the ion and solvent coordination. Between these compositions, however, a crystallinity gap exists. A Raman spectroscopic analysis of the EC solvent bands for the 3–1 and 2–1 EC–LiTFSI liquid electrolytes indicates that ∼86 and 95%, respectively, of the solvent is coordinated to the Li+ cations. This extensive coordination results in significantly improved anodic oxidation and thermal stabilities as compared with more dilute (i.e., 1 M) electrolytes. Further, while dilute EC–LiTFSI electrolytes extensively corrode the Al current collector at high potential, the concentrated electrolytes do not. A new mechanism for electrolyte corrosion of Al in Li-ion batteries is proposed to explain this. Although the ionic conductivity of concentrated EC–LiTFSI electrolytes is somewhat low relative to the current state-of-the-art electrolyte formulations used in commercial Li-ion batteries, using an EC–diethyl carbonate (DEC) mixed solvent instead of pure EC markedly improves the conductivity.
Broader contextMost Li-ion batteries employ a non-aqueous electrolyte that contains the thermally unstable and reactive LiPF6 salt at a relatively dilute (∼1 M) concentration. Similarly dilute electrolytes containing the more thermally and hydrolytically stable LiTFSI (or LiN(SO2CF3)2) salt have largely been excluded from commercial use due to the tendency of such electrolytes to corrode the battery's aluminum current collector at high potentials. Typically, this corrosion has been attributed to the TFSI− anion. In this paper, strikingly different highly concentrated LiTFSI electrolytes are studied and dramatic changes in properties are noted including the drastic reduction in the volatility of the electrolyte solvent. In addition, contrary to expectations, by using a sufficiently high concentration of LiTFSI, the corrosion of aluminum can actually be suppressed. Spectroscopic, computational and electrochemical data are used to argue that TFSI− and similar anions are not directly responsible for the observed corrosion, but instead it is the uncoordinated solvent which is reacting with the aluminum. The molecular-level insights provided for the ion and solvent interactions may result in a paradigm shift in the design of non-aqueous electrolytes for next generation Li-ion batteries with tailored properties for specific applications. |
1 Introduction
Li-ion batteries are ubiquitous in the world of portable electronics and, in recent years, have begun to penetrate the automotive market in the form of plug-in hybrid electric vehicles and electric vehicles. The next generation of Li-ion batteries, however, must have an increased energy density and be less susceptible to degradation and/or catastrophic failure (e.g., venting/explosion hazards) if such batteries are to become an economic and reliable form of energy storage for vehicles. The thermal decomposition of the state-of-the-art electrolyte used in commercial Li-ion batteries (lithium hexafluorophosphate (LiPF6) in mixed carbonate solvents) is well known to occur at only slightly elevated temperatures (e.g., ∼60 °C).1–5 This reaction is primarily a consequence of the choice of lithium salt (LiPF6), and the danger it poses is compounded by the use of volatile and flammable organic solvents. In addition, the state-of-the-art electrolyte has been found to be incompatible with high-voltage cathode materials (>4.5 V), especially at elevated temperature.6–9 Such electrodes, however, are one of the most promising means of dramatically increasing the energy density of Li-ion batteries as the stored energy is equal to the product of the voltage and charge capacity. Increasing the voltage also enables access to a larger fraction of the theoretical capacity of a given cathode material. For example, the NMC (i.e., LiNi1/3Mn1/3Co1/3O2) cathode has a theoretical capacity of 270 mA h g−1. Cycling NMC to 4.3 V vs. Li/Li+ results in an observed capacity of ∼150 mA h g−1. Increasing the voltage to 4.7 V increases this capacity to over 200 mA h g−1.10 The increased capacity, in concert with the higher discharge voltage, results in a tremendous increase in the delivered energy from the battery. Unfortunately, a cell cycled to this high potential is unstable with the state-of-the-art electrolyte and the capacity fades quickly. This may arise from reaction of the electrolyte with the cathode at high voltage resulting in the dissolution of cathode components which then migrate and deposit on the anode leading to cell failure.11 New electrolyte formulations are therefore necessary with improved safety characteristics and stability with high-voltage cathodes, but the identification of such electrolytes has thus far proven elusive.Recently, Yoshida et al. reported a dramatic increase in the anodic stability of triglyme (G3) and tetraglyme (G4) electrolytes with LiTFSI through the formation of [glyme–Li]+TFSI− complexes and the elimination of excess solvent by increasing the salt concentration up to a 1–1 solvent–lithium salt ratio.12,13 This remarkably high anodic stability for the [G4–Li]+TFSI− complex was reported prior to this in a much earlier publication.14 Related electrolytes based upon highly concentrated liquid complexes of LiTFSI with acetamide, 2-oxazolidinone, ethylene urea, etc. have also been reported.15–18 The improved electrochemical stability (relative to more dilute electrolytes) is reported to originate from the coordination bond formation of the glyme molecules to the Li+ cations which effectively stabilizes the solvent's electron lone-pairs, thereby reducing their susceptibility to oxidation. Interestingly, it has also been reported that stable cycling of a graphite anode can be achieved with highly concentrated (>2.7 M) propylene carbonate (PC) or dimethyl sulfoxide (DMSO)-based electrolytes with lithium salts, whereas solvent decomposition and graphite exfoliation occurred for more dilute PC-based electrolytes.19–22 The cycling efficiency of Li metal anodes (i.e., suppression of dendritic lithium formation) has also been greatly improved using concentrated PC-based electrolytes.23 Clearly, radically different electrolyte properties may be obtained with highly concentrated solvent–lithium salt electrolytes.
Here, we extend this approach to the carbonate-based solvents which are commonly used in Li-ion battery electrolytes. In these highly concentrated LiTFSI electrolytes, nearly 95% of the solvent molecules are found to be coordinated to the Li+ cations. Investigation of the anodic stability of electrolytes containing LiTFSI with either EC or conventional solvent mixtures—3/7 (v/v) EC/DEC and EC/PC mixed solvents—results in a marked increase in the onset potential for oxidation on a Pt electrode as the salt concentration is increased. Most notably, however, the oxidative corrosion of Al, which is extensively used in batteries as a cathode current collector, by the LiTFSI-based electrolytes is effectively suppressed. Furthermore, because only about 5% of the solvent is uncoordinated at the highest concentrations, the electrolyte volatility is dramatically decreased, thereby reducing the dangers of cell overpressure and electrolyte flammability. Explanations for these properties are provided based upon the concept of concentrated electrolytes for which there is no significant bulk solvent (uncoordinated to a Li+ cation) present.
2 Experimental section
Materials
LiTFSI (battery grade) was purchased from 3M and vacuum dried for 24 h at 120 °C. LiPF6 (battery grade) was purchased from Novolyte and used as-received. EC (Novolyte, >99%, <0.005% H2O), PC (Novolyte, >99%, <0.005% H2O) and DEC (Sigma-Aldrich, >99%, anhydrous) were dried using 3 Å molecular sieves until the H2O content was negligible (<20 ppm), as determined by Karl-Fischer titration. The materials were stored and handled in a Vacuum Atmospheres glovebox with Ar atmosphere (<5 ppm O2 and <1 ppm H2O).Differential scanning calorimetry
DSC experiments were performed using a TA Instruments Q2000 DSC with a liquid N2 cooling unit. The instrument was calibrated with indium (Tm at 156.60 °C) and cyclohexane (solid–solid phase transition at −87.06 °C, Tm at 6.54 °C). Al sample pans were hermetically-sealed in the glovebox. The pans were cooled to −150 °C at 5 °C min−1 and cycled/annealed repeatedly to fully crystallize the samples when possible. The samples were then equilibrated at −150 °C and heated until the samples were fully melted. The final heating traces were used to identify thermal events for the phase diagram.Thermogravimetric analysis
TGA experiments were performed using a TA Instruments Q5000 thermogravimetric analyzer and Pt sample pans. Contact of the sample to ambient air during sample loading was approximately 15 s to minimize moisture absorption. The samples were heated from room temperature to 600 °C at 5 °C min−1 under N2 flow.Crystal structure determination
(EC)3:LiTFSI and (EC)1:LiTFSI single crystals were grown as indicated in the ESI.† Crystals were mounted on a quartz fiber with a small amount of Paratone N oil. X-ray measurements were made on a Bruker-Nonius Kappa Axis X8 Apex2 diffractometer. The unit cell dimensions were determined from symmetry constrained fits of the reflections. The frame integrations were performed using SAINT.24 The resulting raw data were then scaled and absorption corrected using a multi-scan averaging of symmetry equivalent data using SADABS.24 The structures were solved by direct methods using either SIR92 or XS.25,26 All non-hydrogen atoms were obtained from the initial solution. The hydrogen atoms were introduced at idealized positions and were allowed to ride on the parent atom. The structural models were fit to the data using full matrix least-squares based on F2. The calculated structure factors included corrections for anomalous dispersion from the usual tabulation. The structures were refined using the XL program from SHELXTL.26Raman spectroscopic analysis
A Horiba Jobin-Yvon LabRAM HR VIS high-resolution confocal Raman microscope with a 632 nm−1 He–Ne laser excitation source was used to collect Raman spectra. The instrument was calibrated with a monocrystalline Si wafer at 520.7 cm−1. A 50× optical objective was used along with a hermetically-sealed Linkam stage for temperature control. Raman spectra were obtained using a 10–20 s exposure time and five accumulations. Gaussian–Lorentzian functions were used in the Labspec v5.45.09 software to deconvolute the Raman spectra. A correction factor determined from previous QC calculations was used to scale the area of the 895 cm−1 EC solvent band to account for differing Raman scattering activities between the 895 cm−1 band (uncoordinated EC) and the 905 cm−1 band (coordinated EC).27Electrochemical measurements
A ceramic-patterned cell with inert Pt counter and working electrodes from Pine Instruments was used to determine the anodic stability of the electrolytes. The reference electrode was a Ag wire immersed in a solution of 10.0 mM AgCF3SO3 in N-methyl-N-butylpyrrolidinium TFSI (i.e., Ag/Ag+), separated from the electrolyte by a fine glass frit. This reference electrode has been shown to have a potential of 3.4 V vs. Li/Li+ and to be stable within 5 mV for a period of 3 weeks.28 The patterned cell and reference electrode were immersed in the electrolytes in the Ar glovebox and connected to a BioLogic VMP3 potentiostat to conduct the linear sweep voltammetry experiments. The potential was swept from the open circuit voltage (OCV) to 2.45 V vs. Ag/Ag+ at 5.0 mV s−1. After use, the patterned electrode was cycled at 50 mV s−1 in 0.5 M H2SO4 until the current response no longer changed (∼40 cycles) to remove deposits. The Pt patterned cell was dried at 110 °C for 2 h prior to use and the reference electrode was immersed in a vial of reference solution for storage.The ionic conductivity of the electrolytes was determined through electrochemical impedance spectroscopy (EIS) using hermetically-sealed AMEL Instruments two-electrode dip cells with Pt electrodes. The sample temperature was varied using a Binder environmental chamber (−20 °C to 100 °C in 10 °C increments) and measurements were performed with a BioLogic VMP3 potentiostat/impedance analyzer using a 10.0 mV perturbation from 1 MHz to 20 Hz (no DC bias). The conductivity cell constants were determined using aqueous KCl standard solutions at 25 °C. All of the samples were equilibrated at each temperature for at least 45 min before the measurements.
Al corrosion studies were performed using 2032 type coin cells purchased from MTI (Al-clad bottom can, stainless steel top can with polypropylene ring). Coin cells were assembled in the Ar glovebox with an Al foil working electrode and Li metal counter/reference electrode. Both Whatman GF/D glass fiber and Celgard 3501 separators were used with similar results. The cells were crimped with an electric coin cell crimping machine from MTI and conditioned at 50 °C in an oven for 1 h to ensure full separator wetting. The cyclic voltammetry (CV) experiments were performed using a BioLogic VMP3 potentiostat at 5.0 mV s−1 from 2.0 V to 6.0 V for three cycles.
Computational methodology
Molecular dynamics (MD) simulations were performed on EC–LiTFSI electrolytes employing a many-body polarizable force field (APPLE&P) that was previously developed for EC,29 [EC–Li+],29 [TFSI−]30 and [Li+–TFSI−]31 with one modification: the polarizability of the TFSI− anion oxygen atom was reduced from 1.36 Å3 to 1.00 Å3. This modification was performed in order to decrease the [Li+–TFSI−] binding energy from 140.4 to 134.0 kcal mol−1 for the geometry shown in Fig. S8.† DFT calculations performed at M05-2X/6-31+G** and B3LYP/aug-cc-pvTz levels yielded the [Li+–TFSI−] binding energy of −140.0 and −141.3 kcal mol−1, respectively, in good agreement with the prediction from the previous version of the force field. A more accurate G4MP2 calculation, however, resulted in a significantly lower [Li+–TFSI−] binding energy of −135.6 kcal mol−1 indicating a need to modify the force field to reduce the [Li+–TFSI−] binding energy. The revised force field version was denoted as “e47”. QC calculations were performed using Gaussian 09.32 The revised force field predicted the geometry of the [Li+–TFSI−] complex in good agreement with the geometry obtained from G4MP2 calculations, as shown in Fig. S8.†MD simulations were performed for four different EC–LiTFSI concentrations (20–1, 10–1, 5–1, and 2–1) at 60 °C. The MD simulation package Lucretius, which includes many-body polarization, was used for all of the MD simulations. The Ewald method was used for calculating charge–charge and charge–induced dipole interactions with k = 73 vectors used. The Thole screening parameter of 0.2 was used. The interaction between an induced dipole and a partial charge separated by 3 bonds was scaled by 0.8, providing an improved description of the electrostatic potential around the molecules. Multiple timestep integration was used, as well as for the reciprocal part of Ewald, with an inner timestep of 0.5 fs (bonded interactions), a central time step of 1.5 fs for all nonbonded interactions within a truncation of 7.0 Å and an outer timestep of 3.0 fs for all nonbonded between 7.0 Å and the nonbonded truncation distance of 14 Å. A Nose–Hoover thermostat and a barostat were used to control the temperature and pressure with the associated frequencies of 10−2 and 10−5 fs. Equilibration runs were performed in NPT ensemble, while production runs were performed in NVT ensemble. The length of the MD runs and composition of the simulation cells are given in Table S1.† The diffusion coefficient, viscosity, conductivity and enthalpy of vaporization values (Table S1†) were determined as previously described.29,30 The MD simulations predicted density and ionic conductivity in good agreement with experiments, as shown in Table S1,† thus validating to some extent the reliability of the MD predictions.
3 Results and discussion
Phase behavior
The formation of crystalline solvate phases is a key consideration for concentrated electrolytes. In most cases, when the lithium salt concentration is increased in organic solvents, solvates crystallize from solution, severely reducing the ionic conductivity. For example, most concentrated mixtures of aprotic solvents with LiPF6 readily crystallize, with the solvates formed often melting well above ambient temperature (ESI Fig. S1†). For other salts, however, low melting crystalline solvates or crystallinity gaps (specific concentration ranges over which it is difficult/impossible for crystalline solvates to crystallize) can exist. A knowledge of the phase behavior of solvent–salt mixtures is therefore critical for the determination of viable concentrated electrolyte formulations.The phase diagram for EC–LiTFSI mixtures is depicted in Fig. 1. Two crystalline solvates form—(EC)3:LiTFSI and (EC)1:LiTFSI—which melt at 26 °C and 50 °C, respectively (note: a 3–1 mixture of EC–LiTFSI remains liquid at room temperature unless subcooled). A crystallinity gap exists between these two compositions from 2.5–1 to 1.7–1 EC–LiTFSI, indicating that electrolytes within this composition range are difficult to crystallize, especially for smaller samples due to slow nucleation kinetics. It was possible to crystallize a larger sample (∼5 mL) of a 2–1 EC–LiTFSI electrolyte after holding the sample at −20 °C for 24 h, but this then melted near 0 °C.
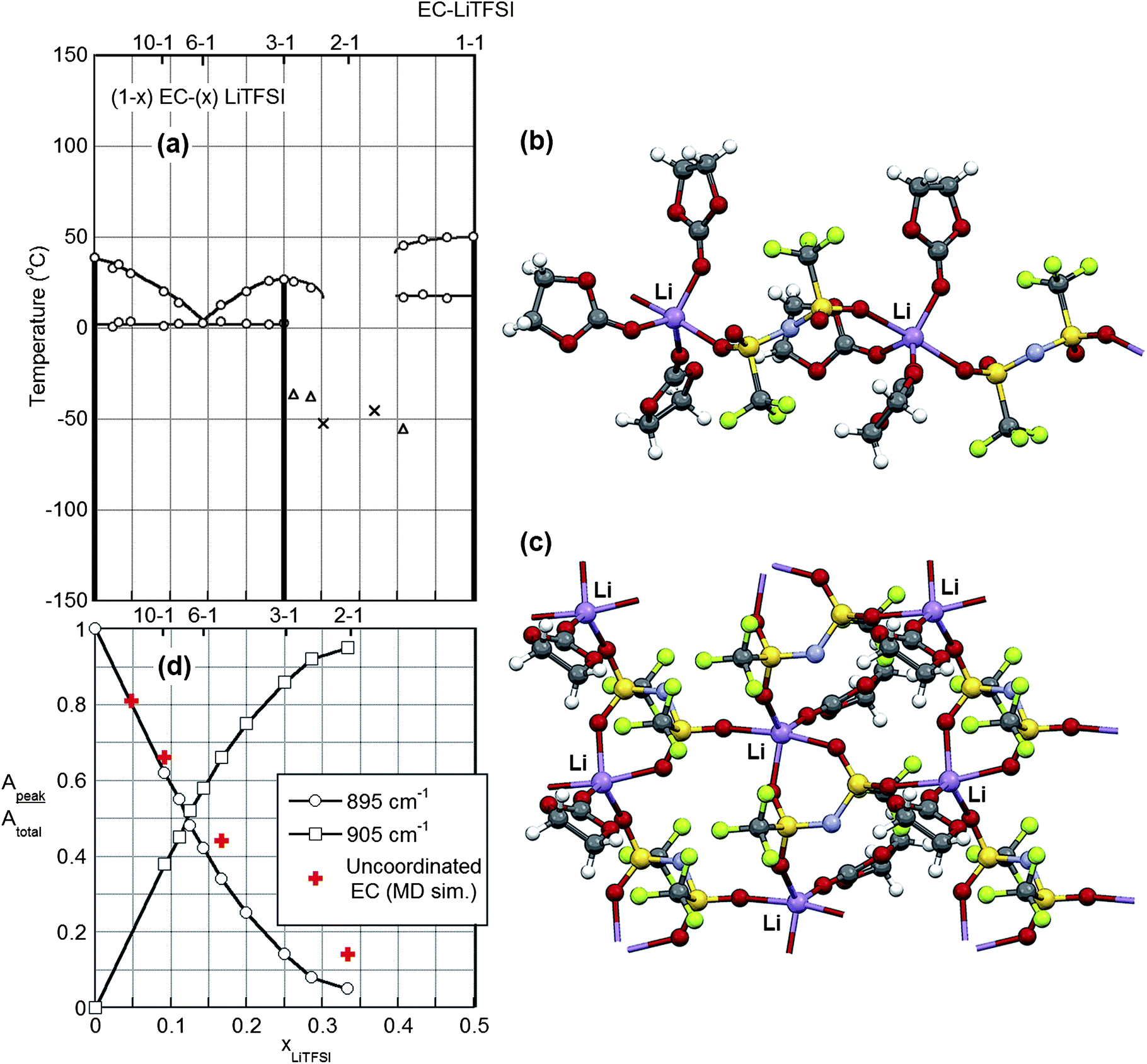 | ||
| Fig. 1 (a) Phase diagram for EC–LiTFSI mixtures. Vertical lines indicate the phases present (at x = 0, 0.25, and 0.50) corresponding to pure EC and the (EC)3:LiTFSI and (EC)1:LiTFSI solvates, respectively. Triangles and crosses indicate the glass transition temperature (Tg) for partially crystalline and fully amorphous samples, respectively. Ion and solvent coordination within the (b) (EC)3:LiTFSI and (c) (EC)1:LiTFSI crystalline solvate structures (Li – purple, O – red, N – blue, S – yellow, F – light green). (d) Summary of Raman spectroscopic analysis of liquid EC–LiTFSI electrolytes at 20 °C indicating the fraction of coordinated (to a Li+ cation) (905 cm−1, squares) and uncoordinated (895 cm−1, circles) EC (uncoordinated solvent from MD simulations performed at 60 °C). | ||
The (EC)3:LiTFSI crystalline solvate structure† (Fig. 1) consists of one symmetry independent (unique) Li+ cation, one TFSI− anion and three EC molecules. The Li+ cation has five-fold coordination by two oxygen atoms from two different TFSI− anions and three carbonyl oxygen atoms from three EC molecules. The anion fluorine atoms do not participate in the Li+ cation coordination. Two oxygen atoms from the TFSI− anion are coordinated to two different Li+ cations. This structure displays positional disorder for the TFSI− anion and one of the EC molecules (not shown in Fig. 1). The TFSI− anion has the C2 conformation.33 The ions and solvent together form one-dimensional (linear) polymeric chains.
The (EC)1:LiTFSI crystalline solvate structure† (Fig. 1), in contrast, consists of one symmetry independent (unique) Li+ cation, one TFSI− anion and one EC molecule. The Li+ cation has five-fold coordination by four oxygen atoms from three different TFSI− anions and the carbonyl oxygen atom from one EC molecule. Again, the anion fluorine atoms do not participate in the Li+ cation coordination. The four oxygen atoms from the TFSI− anion are coordinated to three different Li+ cations. The TFSI− anion has the C2 conformation.33 The ions and solvent together form two-dimensional (planar) polymeric sheets.
Anion and solvent⋯Li+ cation coordination
The two crystalline solvate structures indicate that, in the solid-state, all of the EC solvent molecules are coordinated to the Li+ cations. In addition, the anions are each coordinated to two or more Li+ cations resulting in polymeric aggregates. Similar solvate aggregate clusters are expected to persist in the melt state for concentrated salt mixtures. This is confirmed by MD simulations of a concentrated 2–1 EC–LiTFSI mixture (Fig. 2 and ESI Fig. S11†) in which aggregate clusters predominate with Li+ cations coordinated by four, three, two, one and even zero EC molecules. One of the anions in Fig. 2 is coordinated to a Li+ cation via the nitrogen atom, but this form of coordination is exceptionally rare. Instead, nearly all of the anions are coordinated to the Li+ cations through the anion oxygen atoms. Li+ cations in the center of the aggregated solvates tend to be less solvated (i.e., are more extensively coordinated to anions), whereas those at the ends have greater solvation (i.e., less coordination to anions). In addition to most of the EC solvent molecules being coordinated, the anions also have extensive coordination to the Li+ cations as most of the Li+ cations have either four- or five-fold coordination. | ||
| Fig. 2 Examples of ion and solvent coordination in several aggregate solvates extracted from the MD simulation at 60 °C for a 2–1 EC–LiTFSI mixture (Li – purple, O – red, N – blue, S – yellow, F – light green). | ||
Raman spectroscopy is a powerful tool for characterizing both the solvent and anion coordination to Li+ cations. The TFSI− anion bands in the 740–750 cm−1 region were analyzed as a function of concentration to investigate the relative distribution of uncoordinated and coordinated anions in the electrolytes (Fig. 3). The bands from 739 to 742 cm−1 have previously been assigned to solvent-separated ion pair (SSIP, uncoordinated TFSI−) coordination, while bands from 745 to 755 cm−1 have been assigned to contact ion pair (CIP, TFSI− coordinated to one Li+ cation) and aggregate (AGG, TFSI− coordinated to two or more Li+ cations) coordination.34 The data in Fig. 3 show that, as the concentration of LiTFSI increases, the anion bands shift to higher wavenumber with bands evident at 740, 744–745, 747 and 751 cm−1. In reality, there are numerous forms of TFSI−⋯Li+ cation coordination which may exist for the aggregate species which result in closely positioned, overlapping bands and, in addition, the band at 740 cm−1 may also include CIP anion coordination.31,34 The results do clearly indicate, however, that a change occurs from numerous SSIP (uncoordinated) anions to CIP and then AGG anion coordination with increasing salt concentration, as expected. For the 2–1 EC–LiTFSI concentration, the anion band is centered at 747 cm−1 with shoulders evident at 742 and 751 cm−1. While the 751 cm−1 shoulder signifies AGG anion coordination, the 742 cm−1 shoulder could represent a small amount of SSIP anion coordination or more likely CIP anion coordination from anions at the end of aggregate clusters (e.g., the top right cluster in Fig. 2).31 The Raman spectra demonstrate that the aggregate clusters do indeed persist in the melt state for the concentrated electrolytes with few, if any, uncoordinated anions present. The MD simulations also support this conclusion as they indicate that for the 2–1 concentration 99% of the anions are coordinated to one or more Li+ cations (ESI Table S1 and ESI Fig. S11†).
 | ||
| Fig. 3 Raman spectra of the TFSI− anion expansion–contraction band (740–750 cm−1) at 20 °C (liquid electrolytes), as well as the coordinated EC solvent band at 729 cm−1 (data normalized to the TFSI− anion bands: 735–765 cm−1). | ||
For liquid mixtures, even for the concentrated electrolytes, some of the solvent molecules may be uncoordinated, thus affecting the electrolyte's anodic stability and volatility. Upon Li+ cation coordination, some of the EC solvent Raman bands shift (ESI Fig. S2†). Specifically, the 717 and 895 cm−1 Raman bands of neat EC, which correspond to two different ring breathing vibrational modes, shift to 729 and 905 cm−1, respectively.27,35,36 The fraction of uncoordinated solvent may be determined by dividing the peak area of the uncoordinated band by the total peak area of the coordinated and uncoordinated bands for each vibrational mode. Changes in the Raman scattering activity between the corresponding coordinated/uncoordinated Raman bands must be taken into account, however, using scaling factors obtained from quantum chemical calculations (0.88 for the 717 cm−1 band, and 1.06 for the 895 cm−1 band, see ESI†) for such calculations to be valid.27 The fractions of coordinated and uncoordinated EC molecules as a function of LiTFSI salt concentration obtained from this Raman analysis are presented in Fig. 1. The EC solvent Raman bands at 717 and 729 cm−1 were not used for this evaluation due to the overlap of the anion band with the coordinated EC band (Fig. 3 and ESI Fig. S2†), which introduces error into the deconvolution of the spectra. A comparison of the fractions of coordinated and uncoordinated EC molecules determined from the 717–729 and 895–905 cm−1 regions, however, shows a qualitative agreement despite the error associated with the 717–729 cm−1 region (ESI Fig. S3†).
Evaluation of the 895 and 905 cm−1 EC solvent Raman bands at 20 °C indicates that, for the 2–1 EC–LiTFSI concentration, approximately 95% of the EC is coordinated to the Li+ cations. Overall, the experimental solvation data is in very good agreement with the MD simulation results (Fig. 1), although the results for the more concentrated 5–1 and 2–1 mixtures indicate a somewhat higher fraction of uncoordinated EC than is obtained from the experimental Raman evaluation. There are two explanations for this. Raman data for the mixtures at higher temperature (not shown) reveal that the solvation decreases (and the ionic association correspondingly increases) upon increasing the temperature from 20 °C to 60 °C, but this occurs only to a limited extent for a given sample concentration. In addition, a small fraction of the EC solvent molecules in the most concentrated simulation mixtures are coordinated to the Li+ cations by the ring oxygen atoms instead of the carbonyl oxygen atoms (ESI Table S1†). This is likely not accounted for in the fraction of coordinated solvent calculated from the simulations. It is unclear, however, if this coordination would be accounted for by the experimental methodology as the perturbation effect of such coordination on the solvent band positions/intensities is unknown.27
The experimental/computational work indicate that the solution structure differs markedly for the relatively dilute (i.e., 10–1) and concentrated (i.e., 2–1) EC–LiTFSI electrolytes. For the more dilute electrolytes, the MD simulation (ESI Fig. S10†) consists of a relatively large fraction of uncoordinated TFSI− anions and fully solvated Li+ cations. A significant fraction of the ions are also present as CIP and AGG solvate clusters. This differs somewhat from the model of complete solvation of the Li+ cations which is often used for carbonate solvent mixtures with LiPF6, but then LiPF6 is more dissociated than LiTFSI31 and the typical electrolyte concentration of 1 M corresponds to an EC–LiTFSI mole ratio of about 14–1 (and thus is more dilute than the 10–1 concentration). In contrast, the MD simulations indicate that for the concentrated electrolyte, most of the Li+ cations are coordinated to one or more TFSI− anions resulting in much more extensive ionic association as CIP and AGG solvates (ESI Fig. S11†). This difference in solution structure is expected to strongly influence the kinetics of the Li+ cation transfer at the electrolyte–electrode interfaces.37–48 Notably, the anions may have a much longer residence time in the Li+ cation's coordination shell than the solvent molecules (thus shedding anions may be more difficult than desolvation),49 but the much higher concentration of Li+ cations at the interphases (higher availability for transfer) should not be overlooked.
Solvent volatility/thermal stability
The extensive solvent coordination for the concentrated electrolytes has a dramatic influence on the volatility. Fig. 4 shows the TGA results for each of the EC–LiTFSI electrolytes, as compared to a 1.0 M LiPF6 in EC/DEC 3/7 (v/v) electrolyte. The state-of-the-art electrolyte begins to lose mass immediately after the heating begins at 25 °C, attesting to its high volatility. Nearly half of this electrolyte has volatilized upon heating to 50 °C and only nonvolatile decomposition products remain at 150 °C due to solvent vaporization and LiPF6 conversion to PF5(g) and LiF(s).1,2 By contrast, all of the EC–LiTFSI electrolytes are significantly more thermally robust due, in part, to the absence of the volatile DEC, but also due to the increased thermal stability of LiTFSI as compared to LiPF6. Perhaps the most intriguing aspect of these measurements, however, is the trend with respect to LiTFSI concentration—as the concentration of LiTFSI increases, the volatility of the EC significantly decreases. For example, the 10–1 electrolyte loses approximately 5 wt% upon heating to 100 °C, whereas the 2–1 EC–LiTFSI electrolyte loses negligible mass at this temperature. This is attributed to the coordination of the solvent molecules to the Li+ cations making them less susceptible to volatilization, as well as the lower concentration of the solvent at the liquid–gas interface. The MD simulations for the EC–LiTFSI mixtures support this as the computed enthalpy of vaporization rises precipitously as the salt concentration increases (ESI Table S1†). | ||
| Fig. 4 TGA heating thermograms (5 °C min−1) of 1.0 M LiPF6 in EC/DEC 3/7 (v/v), pure EC and EC–LiTFSI mixtures. | ||
Anodic stability
The extensive coordination found in the concentrated electrolytes takes place through the electron lone-pairs on the solvent molecules and anions which form coordination bonds to Li+ cations. This bonding stabilizes the electron lone-pairs, making both the solvent and anions less prone to oxidation. To demonstrate this effect, the anodic stability of the EC–LiTFSI electrolytes was investigated on both Pt and Al electrodes (Fig. 5). When an inert Pt electrode was used, the onset potential for oxidation of the electrolyte increases with increasing LiTFSI concentration, while the oxidation current is reduced. It must be noted, however, that the oxidation of the electrolytes is occurring near 5.4 V vs. Li/Li+ where the TFSI− anion itself is known to oxidize.50 A possible oxidation mechanism for the EC solvent molecules reacting with the TFSI− anions is given in the ESI (Fig. S12†) and the calculated oxidation potential is in good agreement with the results in Fig. 5. The effect of solvent coordination may therefore not be playing a prominent role in the results due to the inherent high oxidative stability of EC (with the Pt electrode), but this effect is much more dramatic on an active electrode such as Al.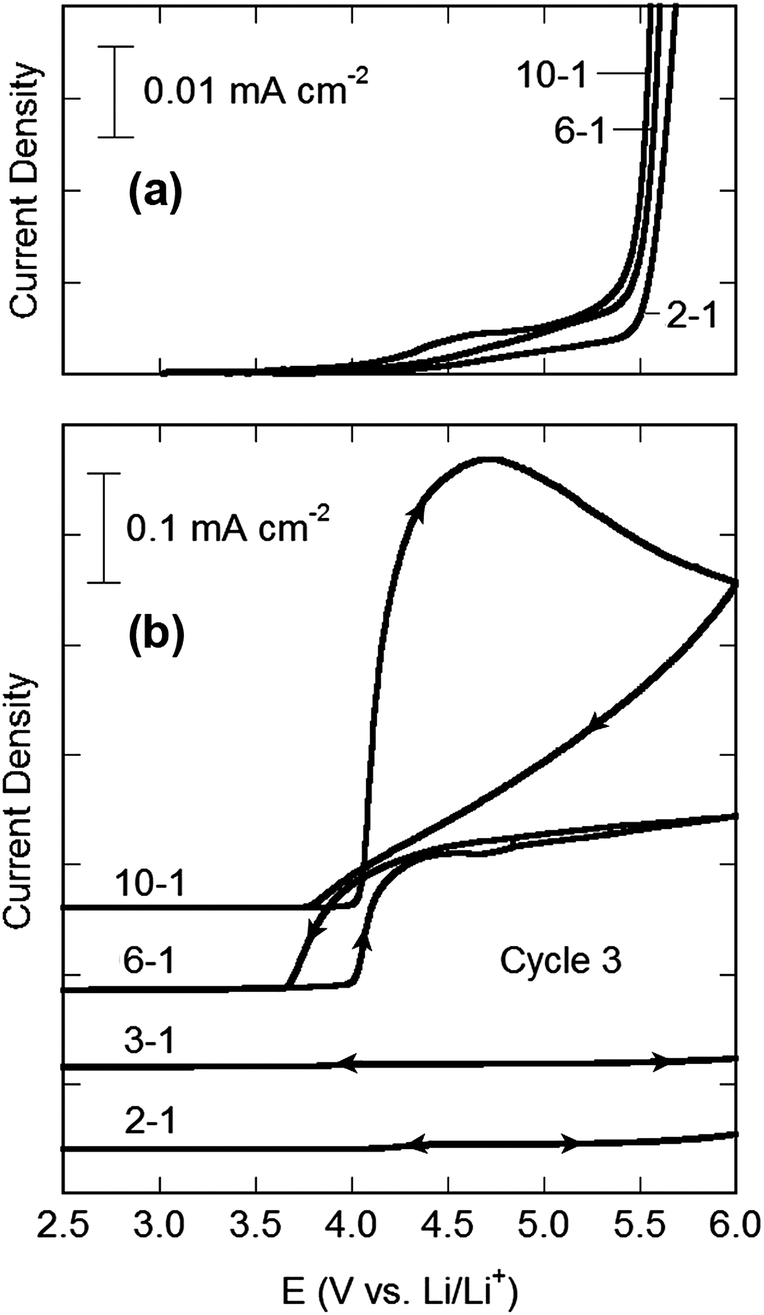 | ||
| Fig. 5 Anodic stability (25 °C, 5.0 mV s−1) of EC–LiTFSI electrolytes: (a) Pt counter and working electrodes with a Ag/Ag+ reference electrode (3.4 V vs. Li/Li+) and (b) 3rd cycle from CVs of coin cells with Al foil working electrodes and Li metal counter/reference electrodes. | ||
It has been shown in numerous studies that LiTFSI, when mixed with organic aprotic solvents such as EC, extensively corrodes Al electrodes near 4.0 V (vs. Li/Li+).51–61 This is of particular importance because, as noted above, Al metal is used as the current collector for most Li-ion battery cathode materials and is thus a nearly indispensable component of commercial Li-ion batteries. In fact, the corrosive behavior of LiTFSI electrolytes towards Al has been one key factor in largely excluding its use from such batteries. To explore the corrosion characteristics of the concentrated electrolytes, half-cells containing Al working electrodes were cycled (5 mV s−1) between 2.0 and 6.0 V vs. Li/Li+ for three cycles at 25 °C. The CVs are presented in Fig. 5. The sudden increase in current density near 4.0 V for the half cells containing the 10–1 and 6–1 EC–LiTFSI electrolytes results from the corrosive reaction of the electrolyte with the Al electrode. Remarkably, for the highest concentrations (3–1 and 2–1), only a negligible current density is observed beyond 4.0 V for all three cycles, indicating that the corrosion of Al has effectively been suppressed, despite the exceptionally high concentration of LiTFSI present.
This suppression of Al corrosion for a concentrated EC/DEC–LiTFSI electrolyte was also noted in a recent report.62 The notion that Al corrosion by LiTFSI electrolytes can be inhibited by increasing the concentration of LiTFSI may, at first, seem counterintuitive. The authors of this work found that significant pitting corrosion occurred when an EC/DEC 3/7 (v/v)–LiTFSI electrolyte with a 1.0 M concentration (approximately 9–1 EC/DEC–LiTFSI, ESI Fig. S4†) was cycled to high potential with an Al electrode, but much less pitting was found for a 1.8 M (4.5–1 EC/DEC–LiTFSI) electrolyte,62 in agreement with the results presented here. X-ray photoelectron spectroscopy (XPS) analysis afterwards found that the Al surface in contact with the 1.0 M electrolyte (polarized at 4.3 V vs. Li/Li+ for 3 h) consisted principally of Al and O, along with small amounts of Li and F.62 In sharp contrast, the surface layer which resulted from contact with the 1.8 M electrolyte had a relatively large amount of Li and F, with lesser amounts of Al and O.62 It was suggested that, for the more dilute concentration, the thin surface film was principally composed of AlF3 and Al2O3, whereas for the 1.8 M electrolyte, the surface instead consisted of a large amount of LiF, in addition to AlF3 and Al2O3.62 The proposed explanation for this was linked to the dissociation tendency (ionic association) of the LiTFSI salt. For the lower concentration, the salt was postulated to be separated into solvated Li+ cations and uncoordinated anions. Thus, at high potential, the TFSI− anions decompose (oxidize) on the Al surface producing fluoride (F−) anions which reacted with the Al to form AlF3. For the more concentrated mixture, however, it was suggested that the Li+ cations and TFSI− anions were associated with one another and this ion proximity resulted in the formation of the LiF which is what inhibits the Al corrosion. This explanation is problematic, however, as very little F was actually noted on the Al surface for the 1.0 M LiTFSI electrolyte.
An alternative explanation is provided below for the variability in the Al corrosion using the concentrated electrolyte data from the present work and a discussion about uncoordinated solvent and anion stability. Fig. 6 shows the interfacial interactions obtained from previous computational MD simulations of a graphite surface in contact with an EC/DMC–LiPF6 electrolyte (with approximately a 3/7 EC/DMC solvent mole ratio and 12–1 total solvent–LiPF6 concentration) upon increasing polarization.63 As no chemistry is present in these simulations, these interactions should mirror those of other charged interfaces (i.e., with Al and Pt). At the unpolarized (0 V) graphite surface, a mixture of both EC and DMC molecules is found, with no significant preferential orientation, and a low anion concentration which is representative of the overall bulk electrolyte concentration. As the graphite surface is positively polarized, however, the PF6− anion concentration at the surface progressively increases. In addition, the EC molecules displace the DMC molecules, despite the larger concentration of the latter in the bulk electrolyte, and the EC molecules tend to orient themselves so that the electron lone-pairs from both the carbonyl and one ring oxygen atom are directed at the surface. This occurs because the EC molecules are more readily polarizable than the DMC molecules, as reflected in the disparity in the dielectric constants of the solvents (ε: 90.4 for EC, 3.1 for DMC). Thus, both the anions and EC molecules stabilize the positive charge dispersed on the carbon surface atoms at high potentials.
 | ||
| Fig. 6 (a) MD simulation snapshots (spaceview) of a graphite–electrolyte (EC/DMC–LiPF6) interface as the electrode potential (between two graphite electrodes—with only the positively polarized electrode shown) is progressively increased (note that the boundaries are periodic) (Li – purple, C – gray, O – red, F – green with two views—rotated 90°—shown for each potential)63 and (b) size comparison for the PF6−, N(SO2CF3)2− (i.e., TFSI−), N(SO2C2F5)2− and N(SO2CF3)(SO2C4F9)− anions. | ||
It is proposed here that the reason that electrolytes with LiTFSI and LiCF3SO3 strongly corrode Al, in sharp contrast to those with LiPF6 and LiBF4, is because these anions with C–F bonds are essentially too stable (rather than being overly reactive). All four anions display a similar oxidative stability on inert glassy carbon or Pt electrodes, whereas TFSI− is found to be more stable than PF6− on a tungsten electrode.64,65 But an Al surface is much more active than Pt (once the native, dense Al2O3 layer formed from the reaction with O2 is removed). Thus, it is proposed that as the potential increases, the solvent molecules at the surface react to form Al2O3, while the PF6− (or BF4−) anions react either with the Al to form AlF3, or with the neighboring solvent molecules to form HF.66 The HF then reacts with the Al2O3 to form AlF3 and AlOF,55 or with the Li+ cations to form LiF, all of which serve to passivate the surface. These reactions occur at a much lower potential than is noted for the PF6− (or BF4−) anion or uncoordinated solvent oxidation on a Pt surface. In contrast, the TFSI− anions do not degrade significantly until a much higher potential. Thus, the surface reactions involve principally the oxidation of the uncoordinated solvent with the Al to form Al2O3 and this does not effectively passivate the surface from further reactions (perhaps including the formation of Al–TFSI complexes which disperse in the electrolyte solution as part of the surface pitting). In contrast to the extensive corrosion noted for LiTFSI and LiCF3SO3 electrolytes with aprotic solvents, a reduction in Al corrosion has been noted for lithium salts with bulkier imide anions, such as N(SO2C2F5)2− or N(SO2CF3)(SO2C4F9)−,51,55,56 despite the fact that the Al surfaces after oxidation at high potentials for all of these electrolytes have nearly the same composition.55 This can be explained by a solvent exclusion effect. If the PF6− anions in Fig. 6a are replaced with these bulky anions (Fig. 6b), then the surface would be effectively covered almost exclusively with the anions, thus effectively suppressing the continuous oxidation of the solvent. XPS data does indicate that anions are strongly absorbed on the oxidized Al surface, even after washing the electrodes.55 The solvent used has also been found to influence the Al corrosion, with LiTFSI-based electrolytes with ethers or fluorinated solvents having less corrosion than those with cyclic carbonate (EC and PC) solvents,54,58 even though carbonate solvents have been found to be more stable than ether solvents when using a Pt electrode.67 The lower dielectric constants of the ethers and fluorinated solvents, relative to the cyclic carbonates, will result in a lower concentration of these solvent molecules at the polarized surface (replaced by a greater number of anions), thus significantly reducing the solvent oxidation.
The high oxidative stability of the TFSI− anion with Al is further confirmed by the lack of Al oxidation until very high potentials for ionic liquids with TFSI− anions, even when mixed with LiTFSI, despite the very high concentration of TFSI− anions present.68–70 In fact, MD simulations suggest that upon positively polarizing an electrode surface, the electrode is effectively covered solely by the anions for such electrolytes.71 For the highly concentrated solvent–LiTFSI electrolytes, it is expected that the solubility of Al–TFSI complexes will be much lower (than for dilute electrolytes) as the solvent molecules and anions are already extensively coordinated. The high concentration of TFSI− anions at the electrode–electrolyte interface (similar to the interface with ionic liquids) will also serve as a barrier which hinders the access of solvent molecules to the electrode and loss of material (dissolution) from the electrode surface. In addition, for the concentrated electrolytes, the absence of uncoordinated solvent, relatively low amount of solvent present in general and extensive coordination of both the solvent molecules and anions to Li+ cations (which increases their oxidative stability) further combine to inhibit the oxidation reactions at the Al electrode surface until much higher potentials than is found for dilute electrolytes (Fig. 5).
Ionic conductivity
One particular limitation for the concentrated EC–LiTFSI electrolytes is the effect of the high level of coordination on the ionic conductivity (Fig. 7). Concentrations of 3–1 and 2–1 EC–LiTFSI have conductivity values at 20 °C of 0.7 and 0.1 mS cm−1, respectively, which will negatively affect the rate capability of a Li-ion battery. State-of-the-art electrolyte formulations, however, use a mixed solvent blend, such as EC/DEC, to both inhibit solvate formation and significantly increase the conductivity. For the concentrated electrolytes, if the EC is substituted with an EC/PC 3/7 (v/v) mixture (i.e., a 2–1 EC/PC–LiTFSI mixture represents a 2–1 total solvent–LiTFSI (molar basis) composition in which the solvent ratio is 3/7 (v/v)), little effect is noted for the electrolyte conductivity (ESI Fig. S5†). In contrast, a similar ratio of EC/DEC increased the conductivity of the most concentrated electrolytes, especially at low temperature. For example, at 20 °C, the conductivity of the 3–1 and 2–1 EC/DEC–LiTFSI electrolytes is 1.5 and 0.5 mS cm−1, respectively. While the inclusion of DEC increases the volatility of the concentrated electrolytes (ESI Fig. S6†), the most concentrated EC/DEC electrolytes are still much less volatile than the dilute mixtures. Furthermore, the corrosion of Al continues to be suppressed in concentrated electrolytes using either EC/PC or EC/DEC solvent mixtures (ESI Fig. S7†). A mixed solvent approach is therefore a promising strategy for further tuning the properties of concentrated electrolytes. | ||
| Fig. 7 Variable-temperature ionic conductivity of (a) EC–LiTFSI and (b) EC/DEC 3/7 (v/v)–LiTFSI electrolytes (conductivity measurements were performed at 10 °C intervals). | ||
4 Conclusions
Concentrated EC–LiTFSI electrolytes have a dramatically improved thermal and anodic stability relative to more dilute mixtures. In particular, the electrolytes with a high concentration of LiTFSI effectively suppress the corrosion of Al. An explanation for the stabilization behavior of concentrated electrolytes is obtained from crystalline solvate structures, MD simulation/QC calculation results and the Raman spectroscopic analysis of the mixtures. In the liquid state, nearly all of the EC molecules are coordinated to the Li+ cations through their carbonyl oxygen atoms and the anions are coordinated to multiple Li+ cations. The anodic stability arises from this extensive coordination—electron lone-pairs on both the solvent and anions are stabilized by extensive coordination to the positively-charged Li+ cations. A dramatic reduction in the volatility of the coordinated solvent, as compared to the uncoordinated solvent, is also found. The main drawback of the concentrated EC–LiTFSI electrolytes is the relatively low ionic conductivity (<1.0 mS cm−1) at ambient temperature. A mixture of EC/DEC with LiTFSI, however, did significantly increase the conductivity of the concentrated electrolytes, especially at low temperatures. Concentrated electrolytes are thus a promising approach for tailoring electrolyte properties for demanding electrochemical applications.Acknowledgements
The experimental work was fully supported by the DOE BATT program (contract number DE-AC02-05-CH11231). The computational work was supported via an Interagency Agreement between the U.S. Department of Energy and the U.S. Army Research Laboratory under DE-IA01-11EE003413 for the Office of Vehicle Technologies Programs including the ARB Program. The Army Research Lab is gratefully acknowledged for providing the Li metal used in this study. PDB would like to thank the Department of Chemistry of North Carolina State University and the State of North Carolina for funding the purchase of the APEXII diffractometer.Notes and references
- C. L. Campion, W. Li and B. L. Lucht, J. Electrochem. Soc., 2005, 152, A2327 CrossRef CAS PubMed.
- S. E. Sloop, J. K. Pugh, S. Wang, J. B. Kerr and K. Kinoshita, Electrochem. Solid-State Lett., 2001, 4, A42 CrossRef CAS PubMed.
- D. Chalasani, J. Li, N. M. Jackson, M. Payne and B. L. Lucht, J. Power Sources, 2012, 208, 67 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS.
- X. Sun, H. S. Lee, X.-Q. Yang and J. McBreen, Electrochem. Solid-State Lett., 2002, 5, A248 CrossRef CAS PubMed.
- L. Yang, B. Ravdel and B. L. Lucht, Electrochem. Solid-State Lett., 2010, 13, A95 CrossRef CAS PubMed.
- S. Dalavi, M. Xu, B. Knight and B. L. Lucht, Electrochem. Solid-State Lett., 2012, 15, A28 CrossRef CAS PubMed.
- D. D. MacNeil, Z. Lu, Z. Chen and J. R. Dahn, J. Power Sources, 2002, 108, 8 CrossRef CAS.
- L. Hu, Z. Zhang and K. Amine, J. Power Sources, 2013, 236, 175 CrossRef CAS PubMed.
- J. Choi and A. Manthiram, J. Electrochem. Soc., 2005, 152, A1714 CrossRef CAS PubMed.
- H. Zheng, Q. Sun, G. Liu, X. Song and V. S. Battaglia, J. Power Sources, 2012, 207, 134 CrossRef CAS PubMed.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121 CrossRef CAS PubMed.
- T. Tamura, K. Yoshida, T. Hachida, M. Tsuchiya, M. Nakamura, Y. Kazue, N. Tachikawa, K. Dokko and M. Watanabe, Chem. Lett., 2010, 39, 753 CrossRef CAS.
- T. M. Pappenfus, W. A. Henderson, B. B. Owens, K. R. Mann and W. H. Smyrl, J. Electrochem. Soc., 2004, 151, A209 CrossRef CAS PubMed.
- Y. Hu, Z. Wang, H. Li, X. Huang and L. Chen, J. Electrochem. Soc., 2004, 151, A1424 CrossRef CAS PubMed.
- H. Liang, H. Li, Z. Wang, F. Wu, L. Chen and X. Huang, J. Phys. Chem. B, 2001, 105, 9966 CrossRef CAS.
- R. Chen, F. Wu, L. Li, B. Xu, X. Qiu and S. Chen, J. Phys. Chem. C, 2007, 111, 5184 CAS.
- R. Chen, F. Wu, L. Li, X. Qiu, L. Chen and S. Chen, Vib. Spectrosc., 2007, 44, 297 CrossRef CAS PubMed.
- S.-K. Jeong, M. Inaba, Y. Iriyama, T. Abe and Z. Ogumi, Electrochem. Solid-State Lett., 2003, 6, A13 CrossRef CAS PubMed.
- S.-K. Jeong, M. Inaba, Y. Iriyama, T. Abe and Z. Ogumi, J. Power Sources, 2008, 175, 540 CrossRef CAS PubMed.
- S. Takeuchi, K. Miyazaki, F. Sagane, T. Fukutsuka, S.-K. Jeong and T. Abe, Electrochim. Acta, 2011, 56, 10450 CrossRef CAS PubMed.
- Y. Yamada, Y. Takazawa, K. Miyazaki and T. Abe, J. Phys. Chem. C, 2010, 114, 11680 CAS.
- S.-K. Jeong, H.-Y. Seo, D.-H. Kim, H.-K. Han, J.-G. Kim, Y. B. Lee, Y. Iriyama, T. Abe and Z. Ogumi, Electrochem. Commun., 2008, 10, 635 CrossRef CAS PubMed.
- Bruker, APEX2, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2007, 64, 112 CrossRef PubMed.
- J. L. Allen, O. Borodin, D. M. Seo and W. A. Henderson, J. Phys. Chem. C, 2013 Search PubMed , under review.
- G. A. Snook, A. S. Best, A. G. Pandolfo and A. F. Hollenkamp, Electrochem. Commun., 2006, 8, 1405 CrossRef CAS PubMed.
- O. Borodin and G. D. Smith, J. Phys. Chem. B, 2009, 113, 1763 CrossRef CAS PubMed.
- O. Borodin, J. Phys. Chem. B, 2009, 113, 11463 CrossRef CAS PubMed.
- D. M. Seo, O. Borodin, S.-D. Han, P. D. Boyle and W. A. Henderson, J. Electrochem. Soc., 2012, 159, A1489 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman and G. Scalmani, Gaussian 09, Revision B, 2009 Search PubMed.
- M. Herstedt, M. Smirnov, P. Johansson, M. Chami, J. Grondin, L. Servant and J. C. Lasségues, J. Raman Spectrosc., 2005, 36, 762 CrossRef CAS.
- D. M. Seo, P. D. Boyle, R. D. Sommer, J. S. Daubert, O. Borodin and W. A. Henderson, J. Phys. Chem. C, 2013 Search PubMed , under review.
- J. R. Durig, G. L. Coulter and D. W. Wertz, J. Mol. Spectrosc., 1968, 27, 285 CrossRef CAS.
- B. Fortunato, P. Mirone and G. Fini, Spectrochim. Acta, Part A, 1971, 27, 1917 CrossRef CAS.
- T. Abe, M. Ohtsuka, F. Sagan, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2004, 151, A1950 CrossRef CAS PubMed.
- T. Abe, H. Fukuda, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2004, 151, A1120 CrossRef CAS PubMed.
- T. Abe, F. Sagane, M. Ohtsuka, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2005, 152, A2151 CrossRef PubMed.
- K. Xu, Y. Lam, S. S. Zhang, T. R. Jow and T. B. Curtis, J. Phys. Chem. C, 2007, 111, 7411 CAS.
- K. Xu, J. Electrochem. Soc., 2007, 154, A162 CrossRef CAS PubMed.
- K. Xu, J. Electrochem. Soc., 2009, 156, A751 CrossRef CAS PubMed.
- K. Xu, A. V. Cresce and U. Lee, Langmuir, 2010, 26, 11538 CrossRef CAS PubMed.
- K. Xu and A. V. Cresce, J. Mater. Chem., 2011, 21, 9849 RSC.
- A. V. Cresce and K. Xu, Electrochem. Solid-State Lett., 2011, 14, A154 CrossRef PubMed.
- K. Xu and A. V. Cresce, J. Mater. Res., 2012, 27, 2327 CrossRef CAS.
- A. V. Cresce, O. Borodin and K. Xu, J. Phys. Chem. C, 2012, 116, 26111 Search PubMed.
- X. Bogle, R. Vazquez, S. Greenbaum, A. V. Cresce and K. Xu, J. Phys. Chem. Lett., 2013, 4, 1664 CrossRef CAS.
- D. M. Seo, O. Borodin, D. Balogh, M. O'Connell, Q. Ly, S.-D. Han, S. Passerini and W. A. Henderson, J. Electrochem. Soc., 2013, 160, A1061 CrossRef CAS PubMed.
- J. Jin, H. H. Li, J. P. Wei, X. K. Bian, Z. Zhou and J. Yan, Electrochem. Commun., 2009, 11, 1500 CrossRef CAS PubMed.
- L. J. Krause, W. Lamanna, J. Summerfield, M. Engle, G. Korba, R. Loch and R. Atanasoski, J. Power Sources, 1997, 68, 320 CrossRef CAS.
- L. Péter and J. Arai, J. Appl. Electrochem., 1999, 29, 1053 CrossRef.
- H. Yang, K. Kwon, T. M. Devine and J. W. Evans, J. Electrochem. Soc., 2000, 147, 4399 CrossRef CAS PubMed.
- X. Wang, E. Yasukawa and S. Mori, Electrochim. Acta, 2000, 45, 2677 CrossRef CAS.
- K. Kanamura, T. Umegaki, S. Shiraishi, M. Ohashi and Z. Takehara, J. Electrochem. Soc., 2002, 149, A185 CrossRef CAS PubMed.
- M. Morita, T. Shibata, N. Yoshimoto and M. Ishikawa, Electrochim. Acta, 2002, 47, 2787 CrossRef CAS.
- D. Di Censo, I. Exnar and M. Graetzel, Electrochem. Commun., 2005, 7, 1000 CrossRef CAS PubMed.
- T. Kawamura, T. Tanaka, M. Egashira, I. Watanabe, S. Okada and J. Yamaki, Electrochem. Solid-State Lett., 2005, 8, A459 CrossRef CAS PubMed.
- B. Markovsky, F. Amalraj, H. E. Gottlieb, Y. Gofer, S. K. Martha and D. Aurbach, J. Electrochem. Soc., 2010, 157, A423 CrossRef CAS PubMed.
- S.-T. Myung, Y. Hitoshi and Y.-K. Sun, J. Mater. Chem., 2011, 21, 9891 RSC.
- J. L. Allen, D. W. McOwen, S. A. Delp, E. T. Fox, J. S. Dickmann, S.-D. Han, Z.-B. Zhou, T. R. Jow and W. A. Henderson, J. Power Sources, 2013, 237, 104 CrossRef CAS PubMed.
- K. Matsumoto, K. Inoue, K. Nakahara, R. Yuge, T. Noguchi and K. Utsugi, J. Power Sources, 2013, 231, 234 CrossRef CAS PubMed.
- J. Vatamanu, O. Borodin and G. D. Smith, J. Phys. Chem. C, 2012, 116, 1114 CAS.
- V. R. Koch, L. A. Dominey, C. Nanjundiah and M. J. Ondrechen, J. Electrochem. Soc., 1996, 143, 798 CrossRef CAS PubMed.
- M. Ue, M. Takeda, M. Takehara and S. Mori, J. Electrochem. Soc., 1997, 144, 2684 CrossRef CAS PubMed.
- O. Borodin, W. Behl and T. R. Jow, J. Phys. Chem. C, 2013, 117, 8661 CAS.
- F. Ossola, G. Pistoia, R. Seeber and P. Ugo, Electrochim. Acta, 1988, 33, 47 CrossRef CAS.
- B. Garcia and M. Armand, J. Power Sources, 2004, 132, 206 CrossRef CAS PubMed.
- R.-S. Kühnel, M. Lübke, M. Winter, S. Passerini and A. Balducci, J. Power Sources, 2012, 214, 178 CrossRef PubMed.
- C. Peng, L. Yang, Z. Zhang, K. Tachibana and Y. Yang, J. Power Sources, 2007, 173, 510 CrossRef CAS PubMed.
- J. Vatamanu, O. Borodin and G. D. Smith, J. Am. Chem. Soc., 2010, 132, 14825 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 875125 and 875816. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ee42351d |
| This journal is © The Royal Society of Chemistry 2014 |