
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-active multinuclear Pd(II) complexes with bis- and tris-mesoionic carbenes†
Ramananda
Maity
,
Margarethe
van der Meer
and
Biprajit
Sarkar
*
Institut für Chemie und Biochemie, Anorganische Chemie, Freie Universität Berlin, Fabeckstraße 34-36, D-14195, Berlin, Germany. E-mail: biprajit.sarkar@fu-berlin.de; Fax: +49-30-838-53310
First published on 22nd October 2014
Abstract
Synthesis of a ligand platform to generate di- and tri-mesoionic carbenes is reported together with their multinuclear Pd(II) complexes. Complete structural characterization and preliminary electrochemical data are presented.
N-heterocyclic carbenes (NHCs) are privileged ligands in organometallic chemistry.1 While homogeneous catalysis has traditionally dominated NHC research,1 these ligands are useful in a variety of other fields, as has been seen in recent years for supramolecular chemistry.2 Their mesoionic carbene (MIC) counterparts are currently gaining immense popularity.3 We have recently used metal complexes of these ligands as catalysts for the “click” reactions,4a–c Suzuki–Miyaura cross-coupling reactions,4d reduction of aromatic nitro compounds4e and for oxidation catalysis.4f,g
Most work on triazolylidene ligands has been on their monodentate forms. Metal complexes of these ligands have been in general used for a variety of homogeneous catalytic processes.3–5
Only a few bis-MIC ligands possessing a metal center at each MIC donor have been reported in the literature.6,7 There have been no reports on tris-MIC ligands and their complexes till now. Bis-carbene ligands can have certain advantages over their mono-counterparts.8 We present here the synthesis of a phenylene based ligand platform for generating MIC ligands of various denticity and report on the mono-, di- and trinuclear Pd(II)-complexes with these ligands. Apart from synthetic and structural aspects, we also present preliminary results on the redox properties of these complexes, a field that has been largely ignored to date for metal complexes of MIC ligands.3b
Reaction of 13a with PdCl2 in the presence of a base resulted in the formation of the mononuclear complex [2], which was characterized by 1H and 13C NMR spectroscopy. The related mononuclear PEPPSI1e type complexes with MIC ligands have been reported earlier in the literature.5a,7c [2] was synthesized here to be able to compare a mononuclear case with the di- and trinuclear complexes where all substituents on ligands are the same. The reaction of the bis-triazolium salt 36 with PdCl2 in pyridine yielded the dinuclear complex [4].
The complex [4] is well soluble in dichloromethane and sparingly soluble in chloroform. The formation of [4] was easily monitored by 1H NMR spectroscopy which showed the absence of the triazolium C5–H signals (δ = 9.25 ppm)6 observed for the original bis-triazolium salt. A singlet was observed for the aryl C–H protons at δ = 8.21 ppm. The resonances for the C–H protons of pyridine rings were detected as multiplets at δ = 8.92–8.93, 7.60–7.63 and 7.21–7.24 ppm. Upon complex formation the resonance for α-hydrogen atoms of the pyridine ring (δ = 8.92–8.93) was more downfield shifted compared to their corresponding resonance in free pyridine (δ = 8.62).9 The 13C{1H} NMR spectrum of [4] showed the characteristic resonance for the Ctrz–Pd carbon atoms at δ = 128.3 ppm, which is not significantly shifted from the C5 resonance of the precursor triazolium salt 3 (δ = 129.6 ppm).6 The ESI mass spectrum (positive ions) of [4] showed a peak for the cationic complex ion [[4] − I + CH3CN]+ which supports the formation of the dipalladium complex [4] depicted in Scheme 1.
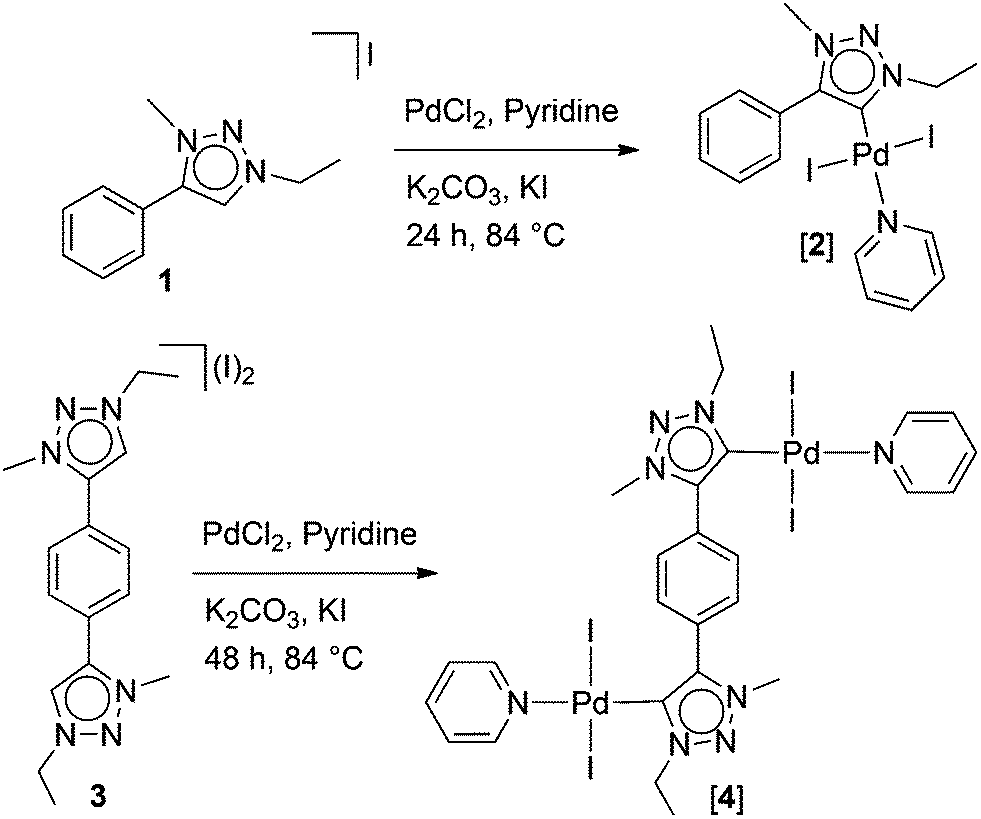 | ||
| Scheme 1 Synthesis of complexes [2] and [4]. | ||
Single crystals suitable for X-ray diffraction studies were obtained by slow evaporation of the saturated dichloromethane–chloroform solution of [4] at ambient temperature. The molecular structure analysis of these crystals confirmed the formation of the dinuclear palladium complex [4] where each palladium atom is coordinated by a carbene C-donor and one pyridine donor in a trans-fashion. The remaining coordination sites at the PdII center are occupied by two iodido donors (Fig. 1).
 | ||
| Fig. 1 ORTEP plot of [4]. Ellipsoids are drawn at 50% probability. Hydrogen atoms and solvent molecules have been omitted for clarity. | ||
The planes of the NHC donors are rotated out of the central aryl ring plane to incorporate the palladium–pyridine moieties. The dihedral angle between the NHC planes and the central aryl ring plane is measured to be 41.97°. Two Pd–Py moieties are oriented in an opposite direction, thus featuring an anti-geometry of the complex. The asymmetric unit contains a half formula unit of the complex [4] and a chloroform molecule. The half unit is related to the other half by a crystallographic inversion center. The coordination geometry around the Pd atom is distorted square planar. The C1–Pd1–N4 bond angle is measured to be 178.8(3)°. This value, like the Pd1–C1 bond length (1.967(10) Å), falls in the range previously described for palladium MIC complexes.5a,7c
The tris-triazole 5 was prepared by an in situ Cu(I) catalyzed click reaction. Subsequent trimethylation of 5 delivered the new tris-triazolium salt 6 (see ESI†). For comparison purposes we also used the tris-imidazolium salt 82d to prepare the corresponding trinuclear Pd(II) complex. The reaction of 1 equiv. of the azolium salts 6 or 8 with 3 equiv. of PdCl2 in the presence of K2CO3 in pyridine led to the formation of trinuclear complexes [7] and [9], respectively (Scheme 2). Complexes [7] and [9] were obtained in good yields of 60.5 and 72%, respectively. Both complexes are stable towards air and moisture in the solid state. These trinuclear complexes are well soluble in dichloromethane. Changing the amount of the base did not result in the formation of orthometalated complexes.6
 | ||
| Scheme 2 Preparation of complexes [7] and [9]. | ||
The formation of complexes [7] and [9] was confirmed by 1H, 13C, 2D correlation NMR spectroscopy and mass spectrometry (ESI†). The 1H NMR spectra of both complexes show the absence of azolium C–H protons which were observed at δ = 9.27 (6) and 10.07 (8) ppm for their parent azolium salts. The resonance for the aryl C–H protons (δ = 9.68 ppm) in complex [9] is significantly downfield shifted compared to their corresponding resonance (δ = 8.95 ppm) in complex [7]. The resonances for the characteristic carbene carbon atoms were observed at δ = 133.4 and 147.9 ppm in the 13C{1H} NMR spectra of complexes [7] and [9], respectively. The resonance for the imidazol-2-ylidene carbon atoms (147.9 ppm for [9]) appeared more downfield shifted compared to 1,2,3-triazol-5-ylidene carbon atoms (δ = 133.4 ppm for [7]). These resonances fall in the range reported for mono- and dinuclear palladium(II) complexes of similar type.5a,10 The resonance for the α-carbon atoms of the pyridine rings appeared downfield shifted ([7]: δ = 154.5 ppm; [9]: 154.4 ppm) compared to their corresponding resonances in free pyridine.9
Single crystals of both complexes [7] and [9] were obtained by slow evaporation of the solvents from a concentrated dichloromethane–chloroform solution at room temperature. X-ray diffraction analysis of these crystals confirmed the formation of trinuclear palladium complexes [7] (Fig. 2) and [9] (Fig. 3).
 | ||
| Fig. 2 ORTEP plot of [7]. Ellipsoids are drawn at 50% probability. Hydrogen atoms and solvent molecules have been omitted for clarity. | ||
 | ||
| Fig. 3 ORTEP plot of [9]. Ellipsoids are drawn at 50% probability. Hydrogen atoms and solvent molecules have been omitted for clarity. One of the iodine atoms is disordered over two positions and only one position is shown. | ||
The metric parameters for complex [7] are almost identical to the previously described dinuclear complex [4]. To coordinate with palladium atoms the NHC donor planes are rotated out of the central aryl ring plane by 34.21 to 44.04° in complex [7] and by 35.56 to 48.26° in complex [9]. The Pd–CMIC bond distances (range: 1.950(5)–1.952(5) Å) are slightly shorter in complex [7] compared to their corresponding bond distances in complex [9] (range: 1.962(3)–1.970(3) Å). The Pd–CNHC in [9] bond distances are in similar range described for previously reported palladium NHC complexes.11 The N–CNHC–N bond angles (range: 105.2(3)–105.5(5)°) in [9] are slightly larger than C–CMIC–N bond angles (range: 102.6(5)–102.9(4)°) in [7].
Mononuclear Pd(II)-MIC complexes of the PEPPSI type have been used as pre-catalysts for cross-coupling reactions.5a,7c The initial step in these catalytic cycles is believed to be reductive elimination which results in the in situ generation of the catalytically active Pd(0) species. In general, electrochemistry can help in addressing questions related to such elementary steps, as has been shown for studies with Pd-NHC and Pd-phosphine complexes.12 To the best of our knowledge no electrochemical investigation has been carried out on Pd-MIC complexes so far.3b–d,5a,7c We were interested in comparing the electrochemical responses of the complexes as a function of their nuclearity and we wanted to compare the results of the Pd-MIC complexes with that of the tris-palladium NHC counterpart [9]. The mononuclear complex [2] displays an irreversible reduction peak at −1.91 V (Fig. 4). The corresponding re-oxidation peak is observed at −0.11 V. This oxidation peak shows up only after the reductive side has been scanned proving this to be a follow-up of the reduction peak at −1.91 V. This process is assigned to the reduction of Pd(II) to Pd(0) combined with the ligand loss, making this step irreversible.12 The complexes [4] and [7] also display an irreversible reduction step at potentials comparable to that of [2] (Fig. 4 and Table S4†). These processes are also associated with re-oxidation steps at more positive potentials just like for [2]. However, for the dinuclear complex [4] one further reduction step is observed and for the trinuclear complex [7] two further reduction steps are observed. These data thus likely point to a stepwise reduction of the Pd(II) centers in these multinuclear complexes suggesting strong electrochemical coupling among them. For the trinuclear complex [9] containing a tris-imidazol-2-ylidene ligand, only one irreversible reduction step is observed at −1.80 V indicating a different electronic situation in [9] as compared to [4] and [7].
 | ||
| Fig. 4 Cyclic voltammograms of the complexes in DMF–0.1 M Bu4NPF6 at 298 K. Scan rate: 250 mV s−1. Fc/Fc+ was used as an internal standard. | ||
In conclusion, we have presented here a synthetic strategy for building multinuclear complexes with MIC ligands based on a phenylene platform. [7] is the first example of a trinuclear complex containing MIC donors. Electrochemical data clearly show that the mononuclear complex [2] undergoes a reductive elimination step, and that electrochemical communication exists between the palladium centers in the multinuclear complexes [4] and [7]. These results differ from the electrochemical response of the imidazole-2-ylidene containing complex [9] where no such communication is observed. In view of the modular synthesis for introducing additional triazolylidene units into the phenylene rings, and the lability of the pyridine ligands in these PEPPSI complexes, the systems reported here are likely to be appropriate synthons for generating supramolecular assemblies. Additionally, the electrochemical data presented here should be helpful in deciphering mechanisms (including cooperativity) of cross-coupling reactions with these and other Pd-MIC complexes. Current research in our laboratory is focused in these directions.
The authors thank the Fonds der Chemischen Industrie (FCI) and the Freie Universität Berlin for financial support.
Notes and references
- For selected reviews see: (a) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862 CrossRef PubMed; (b) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810 CrossRef CAS PubMed; (c) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed; (d) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (e) E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768 CrossRef CAS PubMed; (f) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (g) C. C. Loh and D. Enders, Chem. – Eur. J., 2012, 18, 10212 CrossRef CAS PubMed; (h) J. D. Egbert, C. S. J. Cazin and S. P. Nolan, Catal. Sci. Technol., 2013, 3, 912 RSC.
- For selected examples see: (a) C. Radloff, F. E. Hahn, T. Pape and R. Fröhlich, Dalton Trans., 2009, 7215 RSC; (b) C. Radloff, J. J. Weigand and F. E. Hahn, Dalton Trans., 2009, 9392 RSC; (c) Y. Han, L. J. Lee and H. V. Huynh, Chem. – Eur. J., 2010, 16, 771 CrossRef CAS PubMed; (d) A. Rit, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2010, 132, 4572 CrossRef CAS PubMed; (e) R. Maity, A. Rit, C. Schulte to Brinke, C. G. Daniliuc and F. E. Hahn, Chem. Commun., 2013, 49, 1011 RSC; (f) C. Segarra, G. Guisado-Barrios, F. E. Hahn and E. Peris, Organometallics, 2014, 30, 5077 CrossRef.
- (a) P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534 CrossRef CAS PubMed; (b) K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145 RSC; (c) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755 CrossRef CAS PubMed; (d) J. M. Aizpurua, R. M. Fratila, Z. Monasterio, N. Perez-Esnaola, E. Andreieff, A. Irastorza and M. Sagartzazu-Aizpurua, New J. Chem., 2014, 38, 474 RSC; (e) D. Schweinfurth, N. Deibel, F. Weisser and B. Sarkar, Nat. Chem., 2011, 59, 937 CAS; (f) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759 CrossRef CAS PubMed; (g) J. D. Crowley, A. Lee and K. J. Kilpin, Aust. J. Chem., 2011, 64, 1118 CrossRef CAS.
- (a) S. Hohloch, C.-Y. Su and B. Sarkar, Eur. J. Inorg. Chem., 2011, 3067 CrossRef CAS; (b) S. Hohloch, D. Scheiffele and B. Sarkar, Eur. J. Inorg. Chem., 2013, 3956 CrossRef CAS; (c) S. Hohloch, B. Sarkar, L. Nauton, F. Cisnetti and A. Gautier, Tetrahedron Lett., 2013, 54, 1808 CrossRef CAS PubMed; (d) S. Hohloch, W. Frey, C.-Y. Su and B. Sarkar, Dalton Trans., 2013, 42, 11355 RSC; (e) S. Hohloch, L. Suntrup and B. Sarkar, Organometallics, 2013, 32, 7376 CrossRef CAS; (f) S. Hohloch, L. Hettmanczyk and B. Sarkar, Eur. J. Inorg. Chem., 2014, 3164 CAS; (g) A. Bolje, S. Hohloch, D. Urankar, A. Pevec, M. Gazvoda, B. Sarkar and J. Kosmrlj, Organometallics, 2014, 33, 2588 CrossRef CAS.
- For selected examples see: (a) D. Canseco-Gonzalez, A. Gniewek, M. Szulmanowicz, H. Müller-Bunz, A. M. Trzeciak and M. Albrecht, Chem. – Eur. J., 2012, 18, 6055–6062 CrossRef CAS PubMed; (b) T. Nakamura, T. Terashima, K. Ogata and S. Fukuzawa, Org. Lett., 2011, 13, 620 CrossRef CAS PubMed; (c) J. R. Wright, P. C. Young, N. T. Lucas, A.-L. Lee and J. D. Crowley, Organometallics, 2013, 32, 7065 CrossRef CAS PubMed; (d) J. Huang, J.-T. Hong and S. H. Hong, Eur. J. Org. Chem., 2012, 6630 CAS; (e) E. C. Keske, O. V. Zenkina, R. Wang and C. M. Crudden, Organometallics, 2012, 31, 6215 CrossRef CAS.
- R. Maity, S. Hohloch, C.-Y. Su, M. van der Meer and B. Sarkar, Chem. – Eur. J., 2014, 20, 9952–9961 CrossRef CAS PubMed.
- (a) M. T. Zamora, M. J. Farguson and M. Cowie, Organometallics, 2012, 31, 5384 CrossRef CAS; (b) J. Cai, X. Yang, K. Arumugam, C. W. Bielawski and J. L. Sessler, Organometallics, 2011, 30, 5033 CrossRef CAS PubMed; (c) E. C. Keske, O. V. Zenkina, R. Wang and C. M. Crudden, Organometallics, 2012, 31, 456 CrossRef CAS; (d) K. J. Kilpin, U. S. D. Paul, A.-L. Lee and J. D. Crowley, Chem. Commun., 2011, 47, 328 RSC.
- (a) G. Guisado-Barrios, J. Hiler and E. Peris, Chem. – Eur. J., 2013, 19, 10405 CrossRef CAS PubMed; (b) S. Sabater, J. A. Mata and E. Peris, Eur. J. Inorg. Chem., 2013, 4764 CrossRef CAS; (c) S. Gonell, M. Poyatos and E. Peris, Angew. Chem., Int. Ed., 2013, 125, 7147 CrossRef.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176 CrossRef CAS.
- (a) G. Wang, G. Liu, Y. Du, W. Li, S. Yin, S. Wang, Y. Shi and C. Cao, Transition Met. Chem., 2014, 39, 691 CrossRef CAS; (b) S. Ruiz-Botella and E. Peris, Organometallics, 2014, 33, 5509 CrossRef CAS.
- (a) R. Maity, H. Koppetz, A. Hepp and F. E. Hahn, J. Am. Chem. Soc., 2013, 135, 4966 CrossRef CAS PubMed; (b) R. Maity, A. Rit, C. Schulte to Brinke, J. Kösters and F. E. Hahn, Organometallics, 2013, 32, 6174 CrossRef CAS.
- For a detailed review see: A. Jutand, Chem. Rev., 2008, 108, 2300 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation and spectroscopic properties of complexes. CV table. CCDC 1019285 ([4]·2CHCl3), 1019286 ([7]·CHCl3) and 1021276 ([9]). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03239j |
| This journal is © The Royal Society of Chemistry 2015 |