
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of a room temperature molecular spin switch. The light-driven coordination induced spin state switch (LD-CISSS) approach†
M.
Dommaschk
a,
C.
Schütt
a,
S.
Venkataramani
b,
U.
Jana
c,
C.
Näther
d,
F. D.
Sönnichsen
a and
R.
Herges
*a
aOtto-Diels-Institut für Organische Chemie, Christian-Albrechts-Universität, Otto-Hahn-Platz 4, D-24098 Kiel, Germany. E-mail: rherges@oc.uni-kiel.de
bIndian Institute for Science Education and Research (IISER) Mohali, Knowledge City, Sector 81, SAS Nagar, Manauli PO 140306, India
cDepartment of Chemistry, Jadavpur University, Kolkata-700032, India
dInstitut für Anorganische Chemie, Christian-Albrechts-Universität, Max-Eyth-Straße 2, 24098 Kiel, Germany
First published on 13th October 2014
Abstract
Extensive use of quantum chemical calculations has been made to rationally design a molecule whose spin state can be switched reversibly using light of two different wavelengths at room temperature in solution. Spin change is induced by changing the coordination number of a nickel complex. The coordination number in turn is switched using a photochromic ligand that binds in one configuration and dissociates in the other. We demonstrate that successful design relies on a precise geometry fit and delicate electronic tuning. Our designer complex exhibits an extremely high long-term switching stability (more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles) and a high switching efficiency. The high-spin state is extraordinarily stable with a half-life of 400 days at room temperature. Switching between the dia- and paramagnetic state is achieved with visible light (500 and 430 nm). The compound can also be used as a molecular logic gate with light and pH as input and the magnetic state as non-destructive read-out.
000 cycles) and a high switching efficiency. The high-spin state is extraordinarily stable with a half-life of 400 days at room temperature. Switching between the dia- and paramagnetic state is achieved with visible light (500 and 430 nm). The compound can also be used as a molecular logic gate with light and pH as input and the magnetic state as non-destructive read-out.
1. Introduction
Bistability of spin states has long been restricted to solid state systems or very low temperatures. Spin state switching in isolated molecules or in homogenous solution1 at room temperature, however, is still a challenge.2–4 A number of interesting applications are waiting to be exploited for these systems, such as switchable contrast agents for MRI,5,6 light-controlled magnetic levitation, switchable spin labels,7 or spintronics.8,9 Unsurprisingly, a number of groups are aiming at this end using different approaches. Starting point of the LD-LISC (light-driven ligand-induced spin change) approach is the spin crossover of Fe2+ as a function of the ligand field strength. Photochromic compounds are used as light switchable ligands. The two configurations of the photochromic ligands exhibit different ligand field strengths10,11 which eventually give rise to different spin states.12 Redox processes can be used to change the spin state of transition metals.13 Photo switchable antiferromagnetic coupling was demonstrated in nitroxide substituted diarylethenes.9,14 A magnetic switch from paramagnetic to ferromagnetic has been achieved by moving two Co-phthalocyanine molecules (S = 1/2) on top of each other (S = 1) with a STM tip.15 Reversible spin-crossover was also observed by electron injection in single molecules on a surface with an STM tip.16,17Recently we presented the first molecular spin switch that can be operated by visible light at room temperature in homogenous solution with no measurable fatigue over more than 20000 cycles.18 Our approach is based on the well-known fact that a number of transition metal ions, such as Fe2+, Fe3+, Mn2+, Mn3+, Co2+, and Ni2+ change their spin state upon changing the coordination number. Ni2+ was chosen as the transition metal for several reasons: 1. There is a reliable spin state switch between low-spin (S = 0) and high-spin (S = 1) if the coordination is changed from square planar (coordination number CN = 4) to square pyramidal (CN = 5) or square bipyramidal (CN = 6).19–21 (see Fig. 1) 2. Ni2+ high-spin complexes still exhibit reasonably sharp NMR spectra for analysis, and paramagnetic shifts can be used to determine the ratio of high-spin/low-spin Ni ions in solution22–24 3. Ni2+ complexes are easier to calculate than Fe2+ compounds (more reliable convergence of the wavefunction to the lowest electronic state). In the first place we accepted the disadvantage that the high-spin state of Ni2+ (S = 1) has a lower magnetic moment than Fe2+ (S = 2). We chose Ni-porphyrin as a square planar complex (CN = 4) and an azopyridine tethered to the porphyrin as the photoswitch and light-controlled axial ligand.
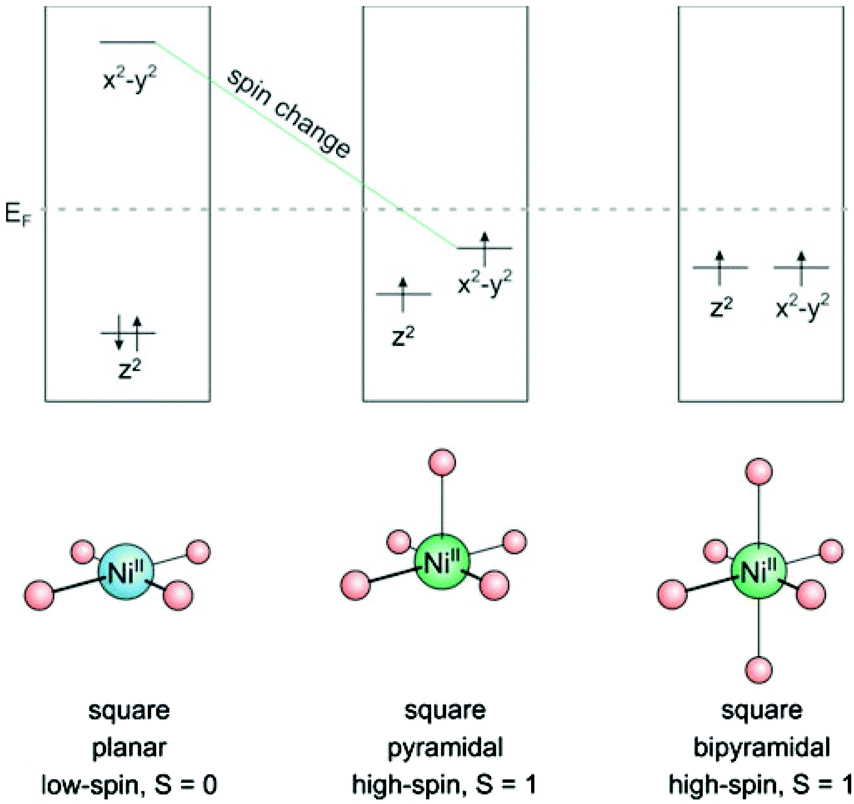 | ||
| Fig. 1 Schematic illustration of coordination induced spin state switch in Ni-porphyrin. | ||
We now present a detailed analysis of the prerequisites of the design and optimization of spin switches based on this Light-Driven Coordination Induced Spin State Switch (LD-CISSS) approach. The optimal system is completely diamagnetic (low-spin) in one state and completely paramagnetic (high-spin) in the other state. However, 100% switching efficiency is difficult to achieve in an ensemble of individual molecules. The overall magnetic switching efficiency depends on the (cis–trans) switching efficiency of the photochromic ligand and on the association constants in both configurations of the ligand. The binding constant should ideally be zero in one state and large in the other. Unfortunately, the situation becomes considerably more complicated under “real” conditions where solvent effects and intermolecular binding interfere. The solvent itself can bind as an axial ligand and convert the nickel complex to the high-spin state, even in the non-binding state of the photochromic ligand. At a first glance, a high binding constant of the photochromic ligand in the binding state should be desirable. However, two problems arise if the association constant is too large. Switching to the non-binding state is thermodynamically less favourable and could be impaired. Moreover, favouring intramolecular binding would also favour intermolecular association. At increasing concentrations an increasing proportion of the complex would form dimers or oligomers and would always be high-spin. Further complications arise from the fact that nickel-porphyrins can add a second axial ligand to form a distorted octahedral complex (Fig. 1). Addition of the first ligand (e.g. photochromic ligand) changes spin state from low- to high-spin and activates the addition of a second axial ligand (K2 usually is larger than K1). Weakly coordinating solvents that do not bind to the square planar Ni porphyrin could still bind to the square pyramidal complex (binding state of the photochromic ligand) and stabilize the high-spin state which would increase the switching efficiency. Another issue is the life time of the thermodynamically less stable state (usually the cis isomer in azobenzenes) which should be large in an ideal system. Many photochromic systems including azobenzenes and spiropyrans undergo thermal back reaction to the more stable state. Coordination could be used to stabilize the binding state of the photochromic ligand if the less stable configuration is binding. A positive feedback from coordination could even improve the switching efficiency of the photochromic ligand. On the other hand the switching of the photochromic ligand often is completely quenched if a chromophore such as a porphyrin is in close proximity or in conjugation. This has to be considered in the design of the tether that connects the porphyrin and the photochromic ligand.
Hence, a number of preconditions have to meet to design molecular spin switches based on the LD-CISSS approach: perfect geometric and electronic design of the switching ligand including the tether, a delicate tuning of the electronic properties of the Ni porphyrin and of the donor strength of the photochromic ligand, the choice of the solvent etc. Detailed information about these parameters is of utmost importance for the design of optimized molecular spin switches for various applications such as contrast agents for MRI or switchable spin labels.
We here report on detailed investigations of the above parameters using NMR and UV-vis spectroscopy and single crystal structure analysis. To elucidate the mechanism of the spin switch we performed quantum chemical calculations and compare experimental and theoretical data of two Ni porphyrins (Ni-TPPF20 and Ni-TPP).
1.1 General design
The trivial way to change the coordination number of Ni-porphyrin would be to add a ligand to the solution. This would lead to a continuous increase of the higher coordinated complex but not to a switching process, and it would not be reversible. We chose light as the trigger, because it can be applied in different wavelengths for switching back and forth with temporal and spatial control, and unlike chemical triggers light does not leave a trace if it is switched off. So initially we have to design ligands that can be switched between two states: a binding and non-binding form using light of two different wavelengths. To keep the design as simple as possible, we chose 3-phenyl-azopyridine as the photochromic ligand because it combines the switching properties of azobenzene and the coordination of pyridine in a single small molecule. 2-Phenyl-azopyridine and 4-phenyl-azopyridine are not suitable because the first doesn't coordinate to square planar metal complexes (sterical hindrance) and the latter binds in both configurations. Hence, the building blocks to work with are Ni-porphyrin and 3-phenyl-azopyridine. Both units have to be connected with each other by means of a tether in such a way that the lone pair of the pyridine nitrogen is exactly orthogonal to the porphyrin plane in a distance of about 2.2 Å. There are two points where the tether could be attached to the porphyrin (meso or β). The other end of the tether can be connected to either the pyridine (PDL design) or the phenyl ring (RP design) of the 3-phenyl-azopyridine unit (Fig. 2). The choice of the position of attachment at the phenyl-azopyridine is of particular importance. A number of azobenzene, azoporphyrin or stilbazole substituted porphyrins are known. However, none of the molecules with ortho or para connection to the porphyrin exhibits photochromic behavior.25–29 Conjugation to a chromophore with a low lying S1 state probably quenches the π–π* excitation of the azo group and thus prevents photochemical isomerisation.30 Therefore, a direct conjugation path between the azo group and the porphyrin has to be avoided either by an sp3 atom or a meta connection within the tether. Attachment at the ortho position of the pyridine ring would prevent coordination by steric hindrance. So we are left with several combinations for attachment and a plethora of conceivable tethers with different lengths and geometries (Fig. 2).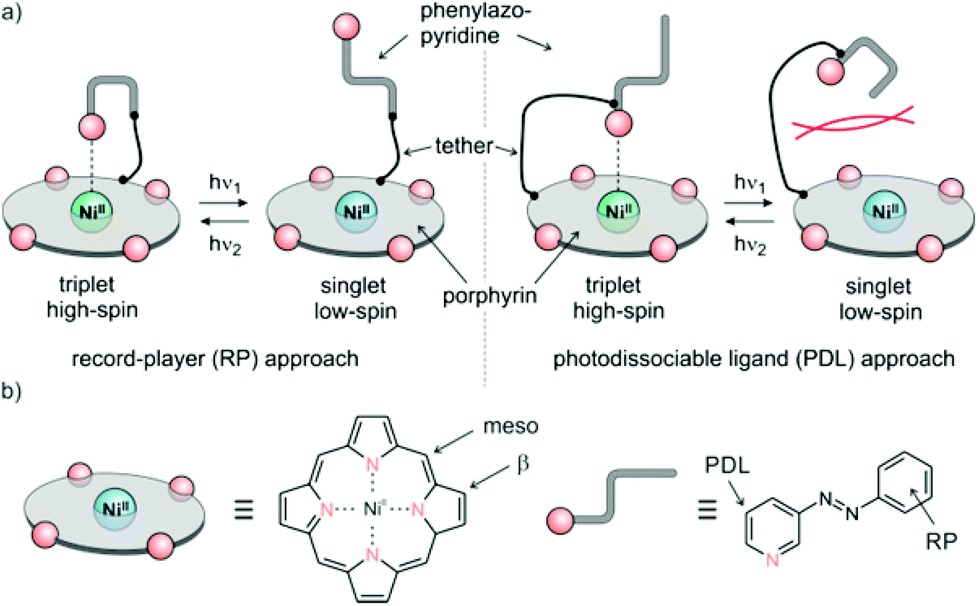 | ||
| Fig. 2 (a) General design approaches (record player (RP), left), photodissociable ligand (PDL) right. (b) Positions of tether attachment. | ||
2. Results
2.1 Computational modeling
At this point we used molecular construction kits to build models and performed density functional theory calculations (B3LYP/6-31G*) on more than 36 different structures that looked promising based on geometry and synthesizability. (a selection of structures is presented in ESI†) For obvious reasons the structures were coined “record players” because the porphyrin resembles a disk, the azopyridine works like a tone arm by placing the pyridine nitrogen lone pair (needle) onto the nickel ion.The structures were ranked according to their theoretically predicted performance. Target parameters are the optimal distance of the pyridine nitrogen and the nickel atom in the binding configuration and a minimal deviation from orthogonal binding to the porphyrin plane. The optimal N–Ni bond length was determined by calculations of phenyl-azopyridine porphyrin complexes without a tether and complete optimisation of all geometry parameters including the N–Ni distance (2.075 Å without and 2.247 Å with pyridine as the sixth ligand, see structure denoted as Ref. in Fig. 3,). Three candidates turned out to comply very well with the geometry constraints above (#1, #2, and #3, Fig. 3).
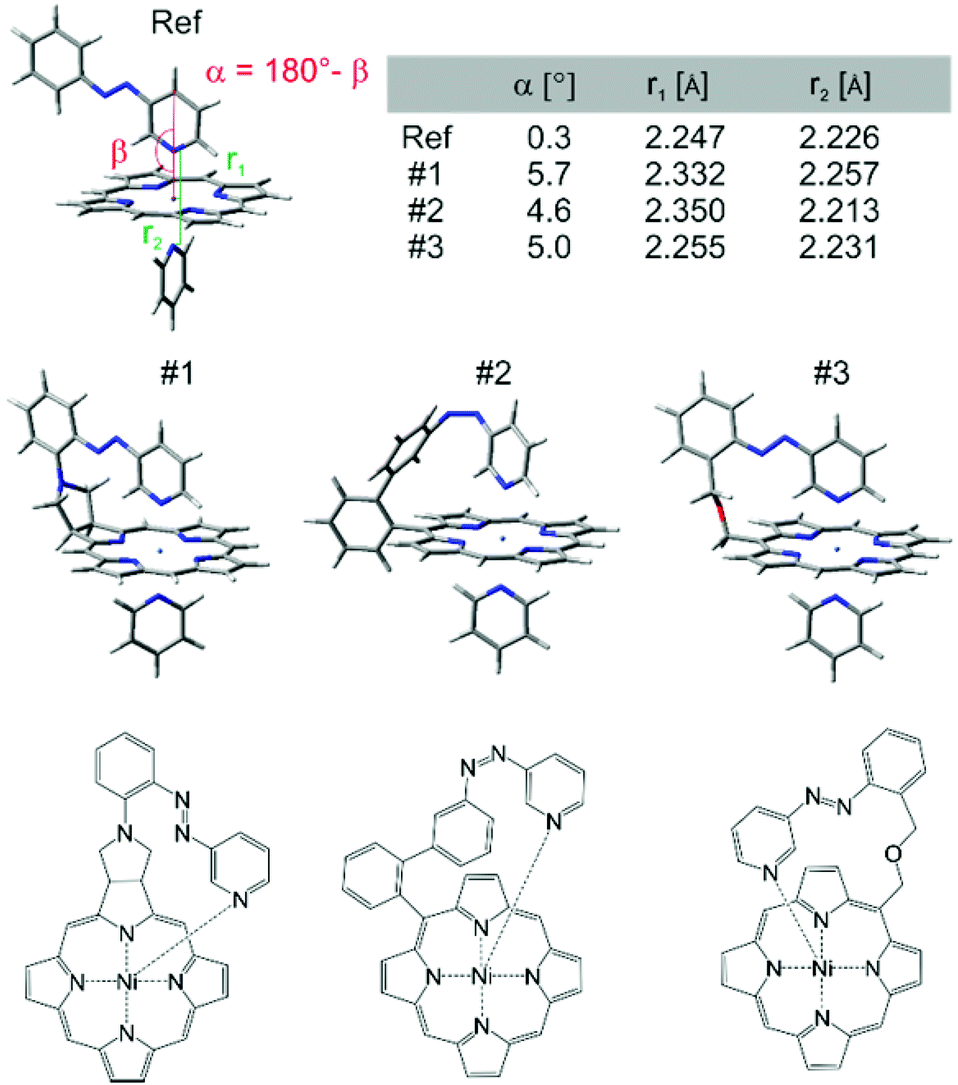 | ||
| Fig. 3 Geometry plots (B3LYP/6-31G*) and structures of Ni-porphyrin with 3-phenyl-azopyridine and pyridine as axial ligands. The reference compound (Ref) without tether is given on top left, and the three highest ranking “record player” structures for LD-CISSS are represented underneath. Note that structure #1 and #3 bind in trans configuration and #2 coordinates in its cis form. Pyridine has been included in the calculations as a second axial ligand because it is known that Ni-porphyrins prefer a 6-coordinate ligand field. It is assumed that upon changing the coordination number from 4 to 5 and concomitant switching from low-spin to high-spin a solvent molecule would coordinate to complete the square bipyramidal ligand field. | ||
“Record player” structure #1 is predicted to be high-spin (binding) in its trans configuration and low-spin in its cis form. A distinct advantage over structures #2 and #3 is the reduced conformational flexibility which should lead to a better switching efficiency. Unfortunately all attempts to synthesize structure #1 failed. The 1,3-dipolar cycloaddition of azomethine ylides to the pyrrole double bonds of porphyrins is known.31–33 Cross coupling of the pyrrolidine nitrogen with 3-(2-iodo-phenylazo)-pyridine, however, resulted in coupling with the pyridine ring. Several other synthetic attempts failed as well. We therefore discontinued the synthesis in favor of record player #2.
The synthesis of structure #2 with phenyl groups in the porphyrin meso positions via the mixed aldehyde method was straightforward.33 Unfortunately, irradiation with light of 365 nm which usually leads to efficient trans–cis isomerisation in azobenzenes and azopyridines was incomplete (32% cis). Photo isomerisation of the cis form back to trans with 420 nm was incomplete as well (16% cis left). Back-isomerisation to the pure trans isomer was achieved by heating to 70 °C for several hours. Furthermore the cis isomer does not exhibit paramagnetic behavior as expected. Only 5% of the molecules are in the triplet state. Instead of binding to the Ni2+, the azopyridine “tone arm” rotates away from the porphyrin. This hypothesis was corroborated by UHV STM measurements at 5 K.33 The molecules were deposited on an Au(111) surface and manipulation of the azopyridine unit was achieved by the STM tip. The trans–cis isomerisation is clearly visible in the STM image. The azopyridine arm points straight away from the porphyrin core and it is bent in cis configuration (Fig. 4).
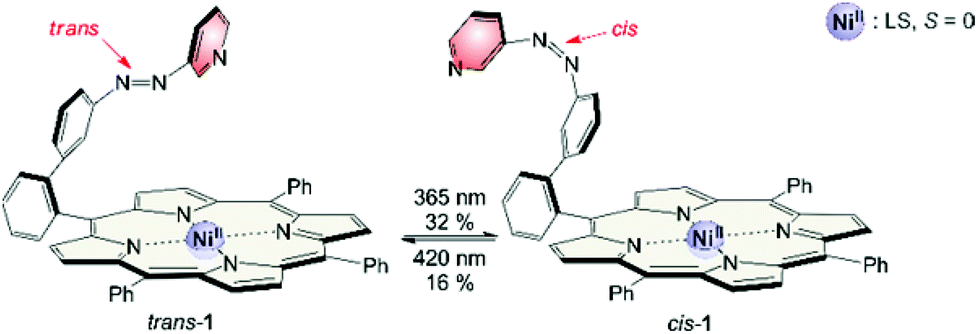 | ||
| Fig. 4 Record player #2 with phenyl groups in meso position (1) does not switch efficiently upon irradiation. The photostationary state (PSS) upon irradiation at 365 nm is 32% cis and at 420 nm 16% cis. Even though the geometry of the cis isomer is optimal for coordination, the “tone arm” doesn't bind to the Ni2+. | ||
To elucidate the reason for this almost complete failure, we performed further DFT calculations with emphasis on the binding energy of pyridines as axial ligands, and on the singlet–triplet gap of Ni-porphyrins with different substituents in meso position. Among 10 different functionals in combination with 5 different basis sets B3LYP/def2TZVP performed best34 in predicting our experimentally determined association energies including the formation of 1:1 and 2:1 complexes of porphyrins with pyridines. Simple PBE/DZP seems to be sufficient for geometry optimisation as was confirmed by comparison with our X-ray structures, and by comparing B3LYP/def2TZVP single point association energies at geometries obtained by optimisation with different functionals and basis sets (for computational details see ESI†). In Table 1 and Fig. 5 the corresponding data for Ni-tetraphenyl-porphyrin (Ni-TPP) and Ni-tetrakis(pentafluoro-phenyl)-porphyrin (Ni-TPPF20) (the latter as an example for an electron poor porphyrin) are presented. The calculated values are compared with experimental association energies that were obtained by NMR titration at different temperatures.22 (Table 1, Fig. 5). It is important to note that neither of the Ni-porphyrins binds pyridine as an axial ligand in its singlet spin state.35 Our calculations reveal a strong repulsion between the Ni ion and the pyridine nitrogen lone pair. Upon including dispersion energy (D3)36 sandwich type structures were found, however, with no coordination between nickel and the pyridine nitrogen. One can assume that these van der Waals complexes do not exist in homogenous solution due to competition of the pyridine with the solvent. Therefore we conclude that there is no axial coordination in the singlet state at all.
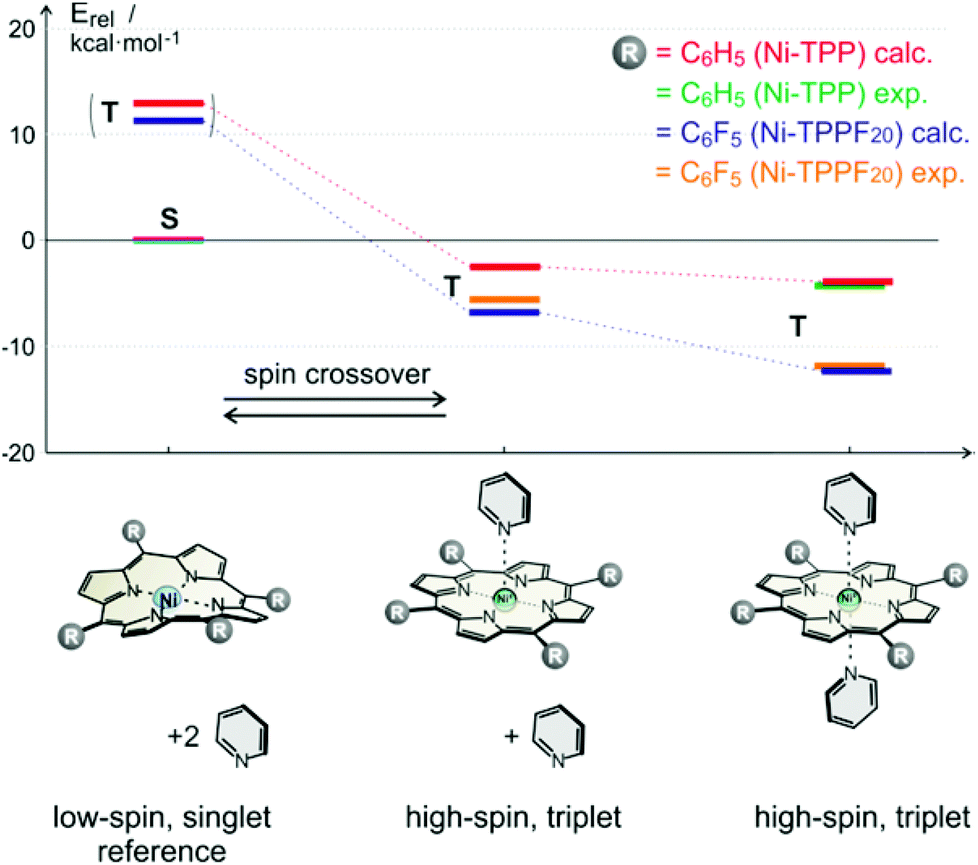 | ||
| Fig. 5 Calculated (B3LYP/def2TZVP//PBE/DZP) and experimental relative association energies (kcal mol−1) of pyridine to Ni-TPP (calc. red, exp. green) and Ni-TPPF20 (calc. blue, exp. yellow) for the respective triplet (high-spin) complexes. The energies are relative to the corresponding porphyrin in its low-spin singlet state (including two unbound pyridine molecules). Note that the experimental22 and calculated association energies agree quite well. The (very small) experimental binding enthalpy for the 1:1 complex of the Ni-tetraphenylporphyrin with pyridine could not be experimentally determined. | ||
| Porphyrin (triplet state) | 0 Pya | 1 Pyb | 2 Py |
|---|---|---|---|
| a Relative energy includes two uncoordinated pyridine molecules. b Relative energy includes one uncoordinated pyridine. c Methods for the determination of the association constant see ESI. | |||
| Ni-TPP calc. | 12.04 | −1.96 | −4.17 |
| Ni-TPP exp.c | — | — | −4.6 (±0.2) |
| Ni-TPPF20 calc. | 10.59 | −6.96 | −11.67 |
| Ni-TPPF20 exp.22 | — | −5.3 (±0.1) | −11.2 (±0.5) |
Electron withdrawing substituents in meso position of Ni-porphyrins increase the binding energy of axial ligands by lowering the dz2 orbital. It has been shown that the association constants of piperidine as axial ligands to Ni-porphyrins with a number of different para substituted phenyl groups at meso position follow a Hammett relationship. The binding constant (2:1 complex) of the meso-tetrakis-(4-nitrophenyl) derivative is more than 10 times higher than the association constant of the parent Ni-tetraphenyl-porphyrin.37 A reverse electronic effect is observed for axial ligands. Electron donor substituents in 4-position of pyridine increase the binding energies to Ni-porphyrin also following an approximate Hammett (or basicity) relationship.22,38 Our calculations confirm the increase in binding strength of pyridine as a function of electron withdrawing substituents in meso-position of the porphyrin. The calculated association energy of pyridine to Ni-TPPF20 (−6.96 kcal mol−1) is 3.6 times higher than to the Ni-TPP (−1.96 kcal mol−1). This effect can be easily rationalized using qualitative MO theory. Triplet (high-spin) Ni-porphyrin has an unpaired electron in the dz2 as well as in the dx2−y2 orbital. The dz2 orbital is mainly responsible for the binding strength of axial ligands. Electron withdrawing substituents at the porphyrin meso-position lower the energy of the dz2 orbital and increase binding. Concomitant with a higher binding energy is a larger singlet–triplet gap in the triplet state. Hence the propensity to undergo spin state change from singlet (low-spin) to triplet (high-spin) should increase with increasing electron withdrawing power of substituents in meso position of the porphyrin ring. An analogous effect should also apply for the record player design. This is why we set out to synthesize record player #2 with electron withdrawing pentafluorophenyl substituents (2) in the three available meso positions. The syntheses and a rough characterisation were presented previously.18
2.2 Photochromism
Record player 2 shows an unexpected photochromic behavior. We found that the highest conversion to the cis isomer (75%) is achieved by irradiation into the porphyrin Q band at around 495–530 nm. By irradiation into the Soret band of the cis isomer at 420–435 nm the molecule can be converted into trans configuration almost quantitatively (<3% cis) (Fig. 6). The photostationary states (PSS) strongly depend on the solvent (Table 2). Dichloromethane and chloroform decrease switching efficiency which could be due to traces of hydrochloric acid. Coordinating solvents improve trans–cis isomerisation. It seems that a solvent molecule stabilizes the paramagnetic cis isomer by formation of the six fold coordinated complex.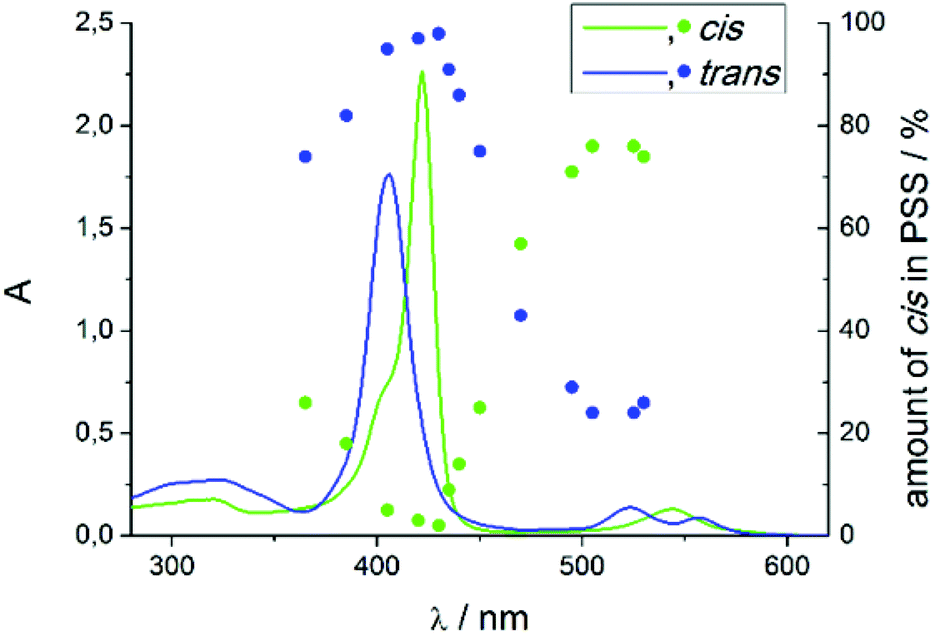 | ||
| Fig. 6 UV/Vis spectra of trans and cis record player 2 (blue and green line) and the photo steady states (blue and green points) after irradiation with the corresponding wavelength in dimethylsulfoxide (DMSO). The isomers were separated by HPLC. | ||
| Solvent | % cis PSS 420 nm | % cis PSS 495 nm |
|---|---|---|
| Acetone-d6 | <5 | 61 |
| Acetonitrile-d3 | <5 | 65 |
| Benzene-d6 | <5 | 63 |
| Chloroform-d | <5 | 18 |
| Cyclohexane-d12 | <5 | 59 |
| Dichloromethane-d2 | <5 | 45 |
| DMSO-d6 | <5 | 75 |
| Methanol-d4 | <5 | 69 |
| Pentafluoro benzonitrile | <5 | 67 |
| Tetrachloromethane | <5 | 57 |
| Tetrahydrofuran-d8 | <5 | 75 |
| Toluene-d8 | <5 | 65 |
This stabilisation also affects the thermal half-life of the cis form. In coordinating solvents the cis isomer is highly stable. The half-life in DMSO is about 400 days (20 °C), which, to our knowledge, is the highest value ever observed for an azo compound. In non-coordinating solvents it is much shorter (dichloromethane: 63 d (20 °C), chloroform: 29 d (20 °C)), but still longer than for usual azo compounds.39,40 The photo physics of the isomerisation is very unusual and not yet understood. Particularly the energy transfer from the porphyrin to the azopyridine unit is unclear. Mechanistic investigations on metal free and Zn containing record player molecules are under way. The fact that the meso phenyl substituted nickel record player (1) has poor switching properties (Fig. 7) leads to the conclusion that the isomerisation is strongly coupled to the spin state switch.
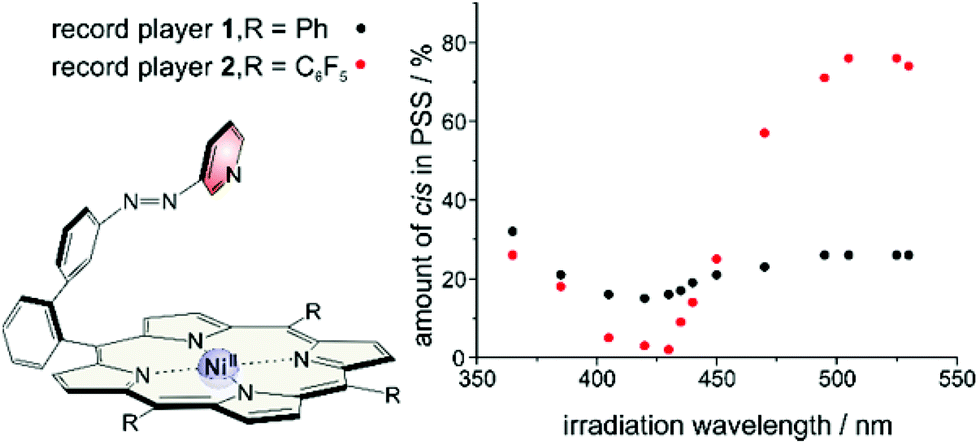 | ||
| Fig. 7 Switching properties of meso phenyl (1) and meso pentafluorophenyl (2) substituted record players. | ||
This pertains to both directions. Irradiation into the Q band (495–530 nm) will only enrich the cis isomer if coordination takes place, and irradiation into the Soret band (405–435 nm) will only convert the molecule to its trans configuration if a decoordination occurs. Upon irradiation with a shorter wavelength (365 nm) the photochromic behaviors of 1 and 2 become quite similar and independent of the spin state switch. In this region the ππ* band of the azopyridine is located. So the spin state switch seems only to be crucial for the isomerisation if it is indirectly induced by irradiation into a porphyrin absorption band (Soret or Q band) (Fig. 8).
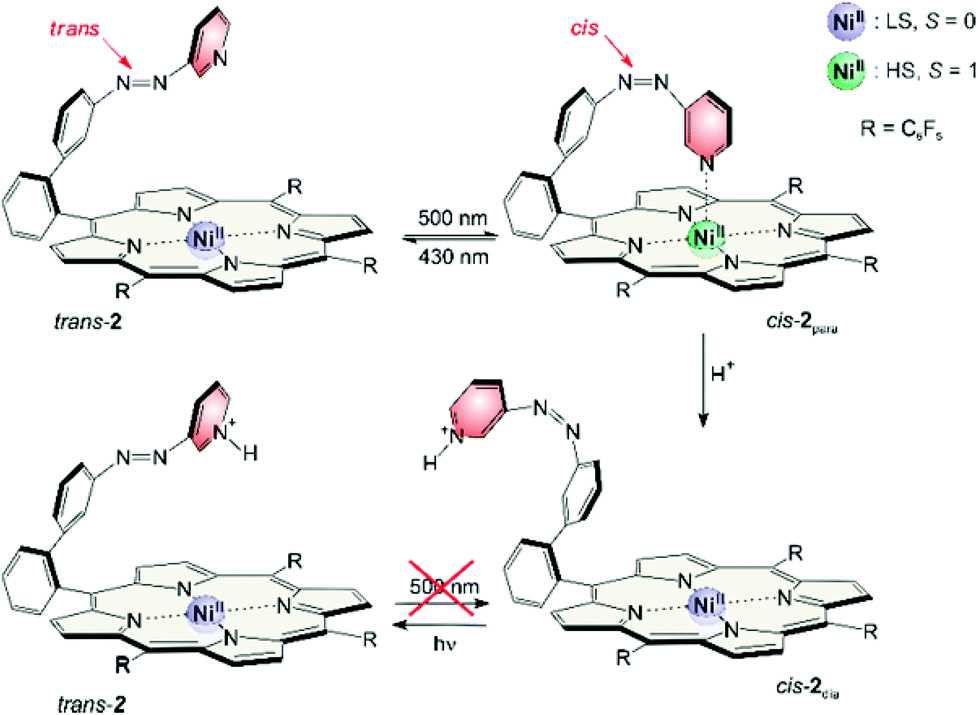 | ||
| Fig. 8 Photo switching between trans-2 and cis-2para was achieved with 500 and 430 nm (top). After conversion into the respective protonated forms, photo conversion is only possible from the cis to the trans configuration (bottom). | ||
Magnetic states and photo switching can be manipulated by changing the pH value. Addition of acids (<1% TFA) converts cis-2 to a protonated form which is completely diamagnetic. Photo induced back-isomerisation is possible with any wavelength. Protonated trans-2 does not isomerise to the cis configuration (e.g. by irradiation into the Q band).
Assuming light (430/500 nm) and pH (high/low) as input and the magnetic state (dia-/paramagnetic) as output, record player 2 can viewed as a molecular logic gate (truth table see Table 3). The molecule is extremely robust (several thousand switching cycles), and the spin state allows a nondestructive readout. Assuming 500 nm light, pH high and paramagnetic spin state as digital 1 the molecule corresponds to an AND gate.
| Input hν | Input pH | Output hν & pH |
|---|---|---|
| 500 nm | OH− | para |
| 500 nm | H+ | dia |
| 430 nm | OH− | dia |
| 430 nm | H+ | dia |
2.3 Methods for magnetic measurements
Beside the photochromism the magnetic properties are the second important (and probably the most interesting) features of the record player 2. The LD-CISSS approach is based on the assumption that the nickel complex is completely diamagnetic in trans and completely paramagnetic in cis configuration. However, one can essentially assume that the trans isomer should exhibit at least some residual paramagnetism because of intermolecular coordination, and the cis form might not be 100% paramagnetic because of incomplete binding of the azopyridine arm. To quantify the paramagnetism of a Ni-porphyrin the chemical shifts of the pyrrole protons in 1H NMR spectra were investigated. Diamagnetic Ni-TPPF20 gives rise to a shift of about 8.9 (+/−0.4) ppm depending on the solvent while the proton signals of a paramagnetic complex appear at 53 (+/−0.4) ppm. This fact is shown in Fig. 9. By addition of pyridine a paramagnetic complex is formed which leads to a downfield shift of 44 ppm (from 8.9 to 53 ppm). The maximum shift only depends on the solvent (+/−0.4 ppm) but not on the nature of the axial ligand (see ESI†). The magnetic moment of the high-spin species (2.9 B.M.) was confirmed by Evans measurements.33,41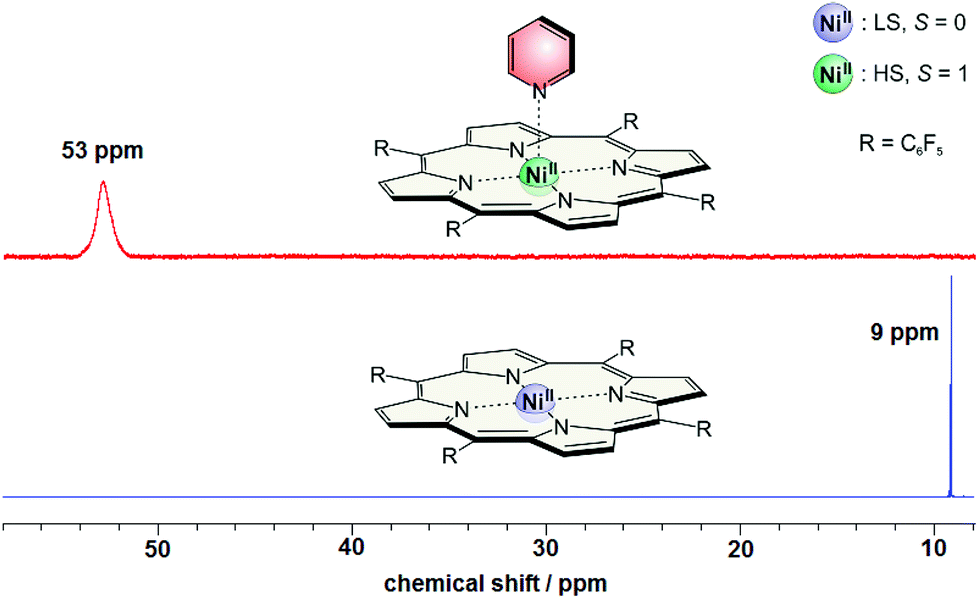 | ||
| Fig. 9 1H NMR spectra of Ni-TPPF20 with (red) and without (blue) pyridine (500 MHz, acetonitrile, 300 K). | ||
Because of the fast ligand exchange a time average of the pyrrole signals of dia- and paramagnetic Ni-porphyrin molecules is observed. The chemical shift δ is a linear function of the mole fraction of paramagnetic nickel (amount of paramagnetic nickel divided by the total amount of nickel):
 | (1) |
There is formation of square pyramidal complexes as well. However, square pyramidal and square bipyramidal complexes exhibit approximately the same paramagnetic shifts which can be shown by titration experiments (see ESI†).
2.4 Intermolecular coordination
Dilution series of trans-2 show a concentration dependence of the chemical shifts of the pyrrole signals (Fig. 10). A concentrated solution gives rise to a slightly downfield shift indicating paramagnetism due to intermolecular coordination. The magnitude of this effect strongly depends on the solvent. Polar (coordinating) solvents like acetonitrile and DMSO suppress intermolecular coordination while nonpolar solvents like cyclohexane have no effect. There is a linear correlation between the average paramagnetic shift (δpara) and the concentration (Fig. 11). The slope (kint) is a solvent specific parameter representing the tendency for the formation of paramagnetic aggregates.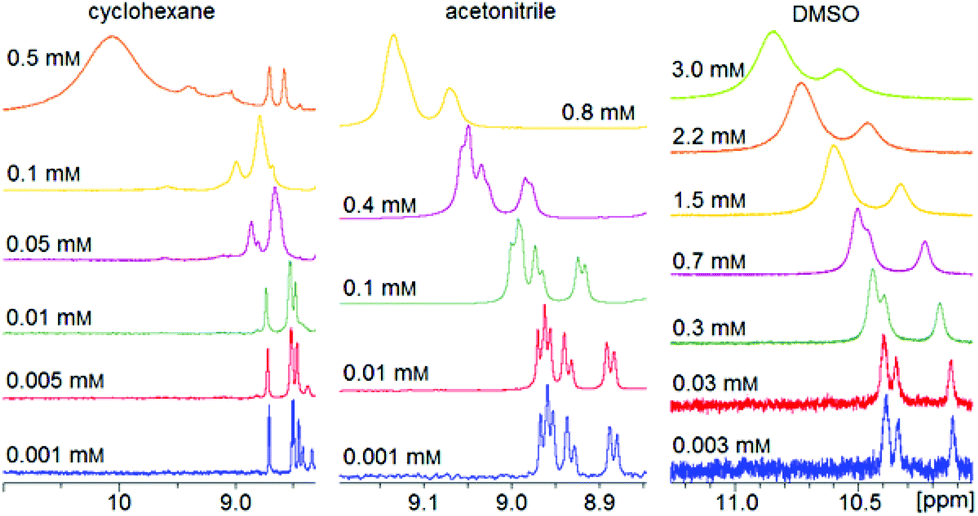 | ||
| Fig. 10 1H NMR spectra of record player 2 in trans configuration in different solvents (500 MHz, 300 K). The downfield shift and signal broadening indicates intermolecular coordination. | ||
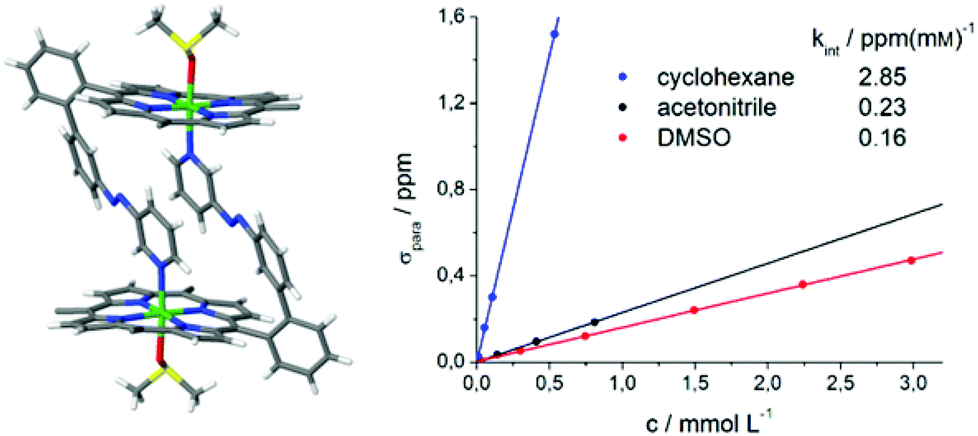 | ||
| Fig. 11 Crystal structure of trans-2 shows formation of dimers and a sixfold coordination sphere of Ni2+ complemented by one axial DMSO molecule (left). The pentafluorophenyl groups at the porphyrin rings are omitted for clarity. For details see ESI Fig. S4.† The linear correlation between paramagnetic shift of pyrrole signals (δpara) and concentration of trans-2 (right) confirms that there is also intermolecular coordination in solution. | ||
The crystal structure of trans-2 (crystallized from DMSO) provides information for the mode of interaction, and the strong solvent dependence of intermolecular coordination. trans-2 forms head to tail dimers (Fig. 11). The remaining axial coordination sites are complemented with DMSO. We interpret the reduced intermolecular coordination in DMSO, acetonitrile, methanol, and THF by competition of dimer (or oligomer) formation and coordination by the solvent. For most applications a low degree of intermolecular aggregation (small kint, Fig. 13) is advantegeous. For a efficient LD-CISSS the trans configuration should be as diamagnetic as possible in solution. From the kint value for cyclohexane (2.85 ppm mM−1) a mole fraction of 6.5% paramagnetic species in a 1 mM solution due to intermolecular coordination can be derived. In DMSO (0.4%) this effect is negligible.
2.5 Intramolecular coordination
NMR signals of cis record player 2 do not show any concentration dependence. But we found that pyrrole signals have different chemical shifts depending on the solvent (Table 4). The maximum shift δmax (eqn (1)) of 53 ppm was confirmed by addition of pyridine-d5. However, in non-coordinating solvents such as cyclohexane or benzene the chemical shift of the pyrrole protons is lower (46 ppm) which indicates that the cis isomer is not completely paramagnetic. Obviously there is a fast equilibrium between the intramolecularly coordinated paramagnetic species (cis-2para) and a non-coordinated diamagnetic form (cis-2dia). The two species are magnetic conformers (Fig. 12).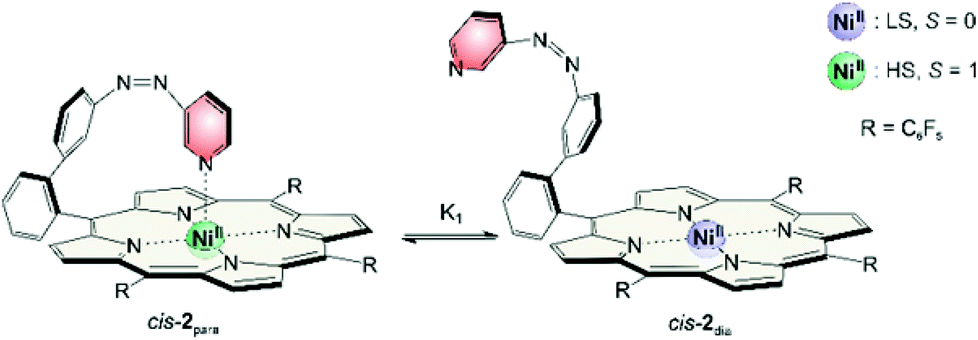 | ||
| Fig. 12 Equilibrium of the magnetic conformers cis-2para (left) and cis-2dia (right). | ||
| Solvent | Shift of pyrrole proton/ppm | % cis-2para |
|---|---|---|
| Acetone | 42 | 75 |
| Acetonitrile | 44 | 80 |
| Benzene | 46 | 84 |
| Cyclohexane | 46 | 84 |
| Dichloromethane | 37 | 64 |
| DMSO | 49 | 91 |
| Methanol | 47 | 86 |
| Pentafluoro benzonitrile | 45 | 82 |
| Tetrachloromethane | 43 | 77 |
| Tetrahydrofuran | 50 | 93 |
| Toluene | 46 | 84 |
In DMSO and tetrahydrofuran the proportions of cis-2para (91 and 93%) are quite close to the maximum value. The properties of these polar solvents obviously favor the paramagnetic conformer. Previous studies on the association constants of axial ligands to Ni-porphyrins could give a hint why this is the case. Binding of the first axial ligand (K1) is less exergonic than association of the second (K2) (except for very strong ligands). DMSO and THF are not sufficiently strong ligands to efficiently bind to the square planar Ni-porphyrin (K1 is small). However, intramolecular coordination of the azopyridine in the cis-isomer activates the sixth binding site sufficiently to bind a weak ligand such as DMSO.33 Hence, upon addition of a solvent molecule the square bipyramidal complex is formed (K2) which is now stabilized and which cannot directly convert into its diamagnetic form. Note that DMSO also complements the sixth binding site in the X-ray structure of trans-2. It is striking that the same solvents giving rise to a large downfield pyrrole shift in cis-2 also favor the conversion of trans-2 to cis-2 upon irradiation at 495 nm (see Table 2).
2.6 Temperature dependence of NMR shifts
The high upfield shift of NMR signals of the paramagnetic complex is mainly due to the hyperfine contact shift.42–44 Isotropic shifts (δpara = δobs − δdia) can be determined accurately comparing cis-2 with the corresponding diamagnetic zinc derivative. Temperature dependence of chemical shifts show Curie behavior with a reasonable good fit. The slopes correlate with the magnitude of the hyperfine coupling. For the ortho pyridine protons the interaction is strongest. At 300 K the signals are already at about 100 ppm. Signals vanish at lower temperatures because they become too broad, and relaxation is too fast. All other shifts are shown in Fig. 13. Some intercepts differ significantly from Curie law which can be explained by the presence of a second spin species namely the diamagnetic cis-2dia. This seems reasonable taking into account that measurements are performed in dichloromethane in which the amount of diamagnetic cis isomer is high (36%).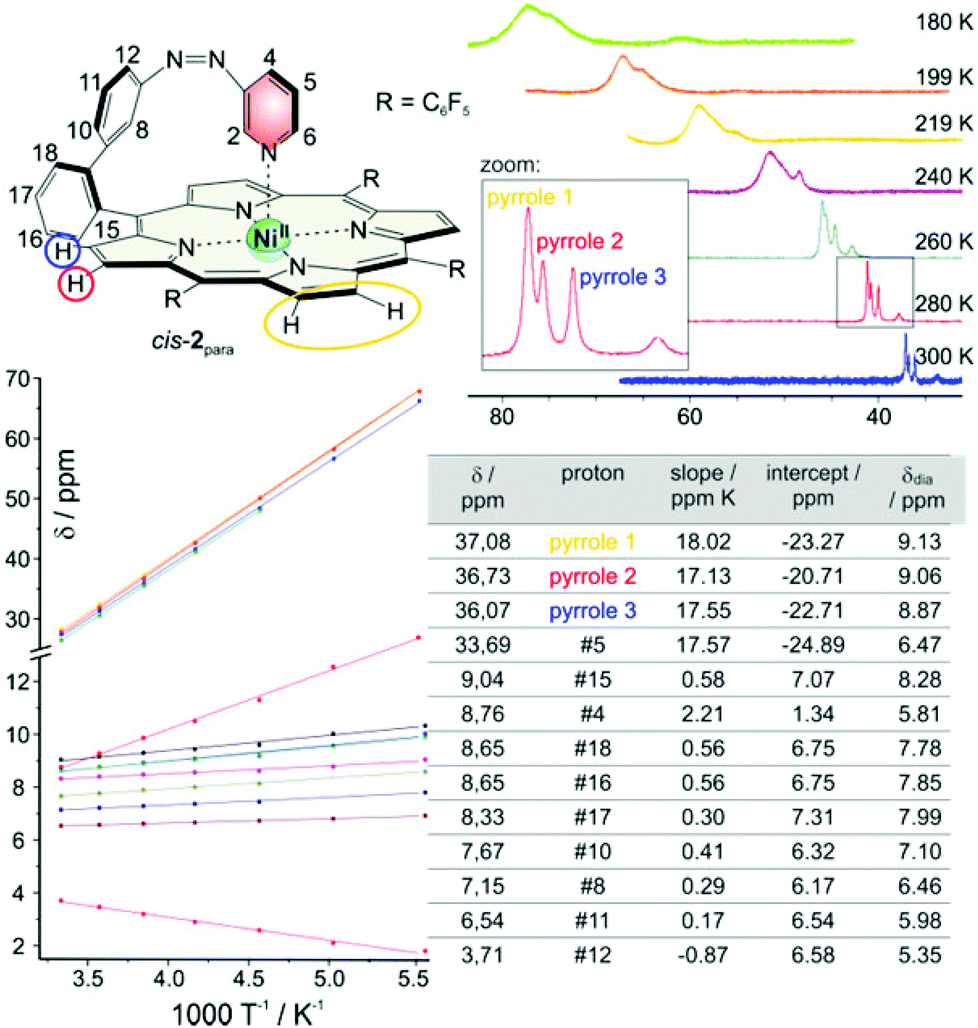 | ||
| Fig. 13 Temperature dependence of chemical shifts of cis-2 in 1H NMR spectra (500 MHz, dichloromethane-d2). Shifts of the pyrrole protons as a function of temperature are shown top right. The table bottom right shows the chemical shifts of all protons (except those of ortho pyridine #2 and #6) at 300 K (δ), the slopes and intercepts of the corresponding Curie plots (bottom left) and the diamagnetic shifts of the corresponding zinc derivative (δdia).18 | ||
From temperature dependent NMR measurements thermodynamic parameters ΔH and ΔS can be determined as well. The intramolecular association constant K1 (see Fig. 12) can be calculated directly from the observed pyrrole proton shifts in 1H NMR spectra (ESI†). Temperature dependence of K1 gives ΔH and ΔS by the Gibbs free enthalpy relation. The experimental values are in good agreement with the calculated (B3LYP/def2TZVP//PBE/SVP) energies which again confirms that the used level of theory is suitable for the investigated complexes (Table 5).
| cis-1 | cis-2 | |
|---|---|---|
| K 1 (300 K) | 0.0616 | 7.4674 |
| ΔH (exp.) | −1.19 (±0.09) | −3.95 (±0.13) |
| ΔH (calc.) | −0.25 | −3.26 |
| ΔS (exp.) | −9.52 (±0.28) | −9.18 (±0.43) |
| ΔG300 K (exp.) | 1.67 (±0.17) | −1.20 (±0.26) |
The values for the association constant (7.47 at 300 K), enthalpy (−3.95 kcal mol−1) and entropy (−9.18 cal mol−1 T−1) of cis-2 are in good agreement with values obtained for free pyridine and azopyridine ligands.22–24 Binding enthalpy is smaller compared to pyridines which is compensated by a smaller entropy. As expected the binding enthalpy for the meso phenyl substituted record player 1 is much lower (−1.19 kcal mol−1) whereas entropy is almost equal (−9.52 cal mol−1 K−1). The thermodynamic data above explain why the phenyl substituted record player cis-1 is not paramagnetic. Even though the geometry is suitable for coordination the binding is endergonic (ΔG = 1.67 kcal mol−1). Coordination of the azopyridine “tonearm” in electron deficient cis-2, however, is clearly exergonic (ΔG = −1.20 kcal mol−1).
2.7 Structure and molecular dynamics
Record player cis-2para does not exhibit symmetry (C1). However, the NMR spectra are rather in agreement with a time averaged Cs symmetry. The fact that there are only three pyrrole proton signals in cis-2 (Fig. 10 and 13) can be explained by the fast conversion of the two respective enantiomers. The 19F NMR spectra add more evidence for this process. cis-2 exhibits four signals for ortho fluorine (1:2:2:1), two signals for para fluorine (1:2) and three signals for meta fluorine (1:2:3) atoms (Fig. 14).
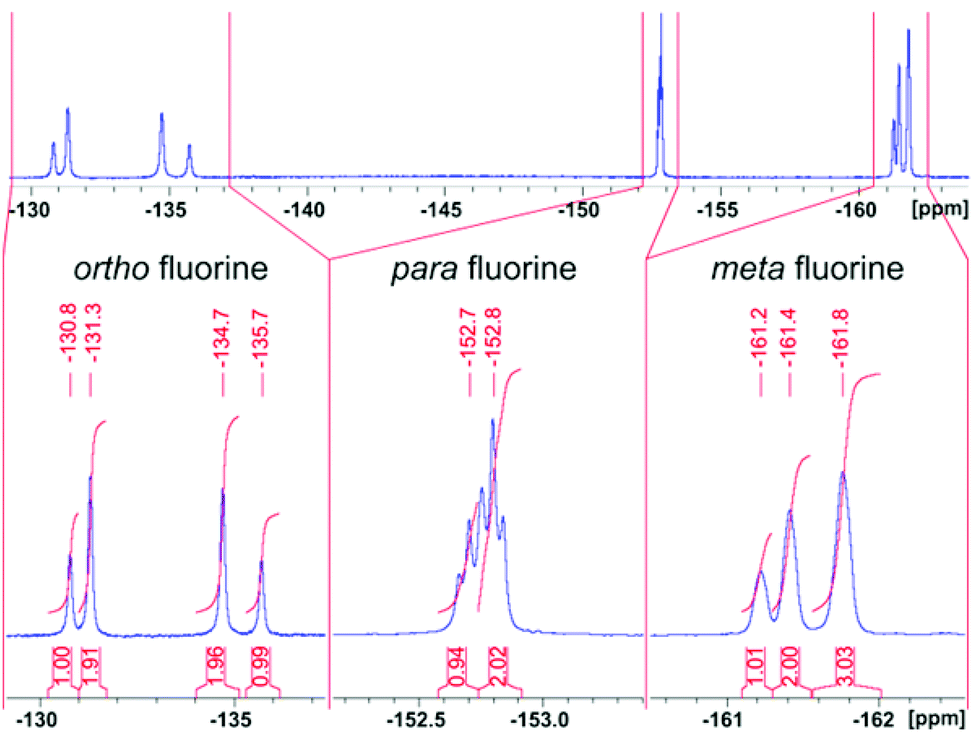 | ||
| Fig. 14 19F-NMR spectra of cis-2 (500 MHz, dichloromethane, 300 K). | ||
Information about the dynamic behavior of cis-2 can be derived from the signal pattern. Since more than three ortho fluorine signals are observed the rotation of the pentafluorophenyl groups in meso position of the porphyrin must be hindered. Hence the molecule has two sides (σ and σ′) with chemical non-equivalent fluorine atoms. cis-2 as shown in Fig. 12 has C1 symmetry which should exhibit six ortho fluorine signals. The fact that there are only four signals and that the ratio given by the integrals is 1:2:2:1 indicates that the two meso substituents A must be equal. (Fig. 15) This is the case if cis-1 is in equilibrium with its enantiomer and conversion is faster than NMR time scale. The conversion is only a reorientation to a different conformation and therefore should not need much activation energy (Fig. 15). However, racemisation can only proceed via a (short-lived) decoordinated (diamagnetic) state. 19F signals of meta and para fluorine are in agreement with this hypothesis.
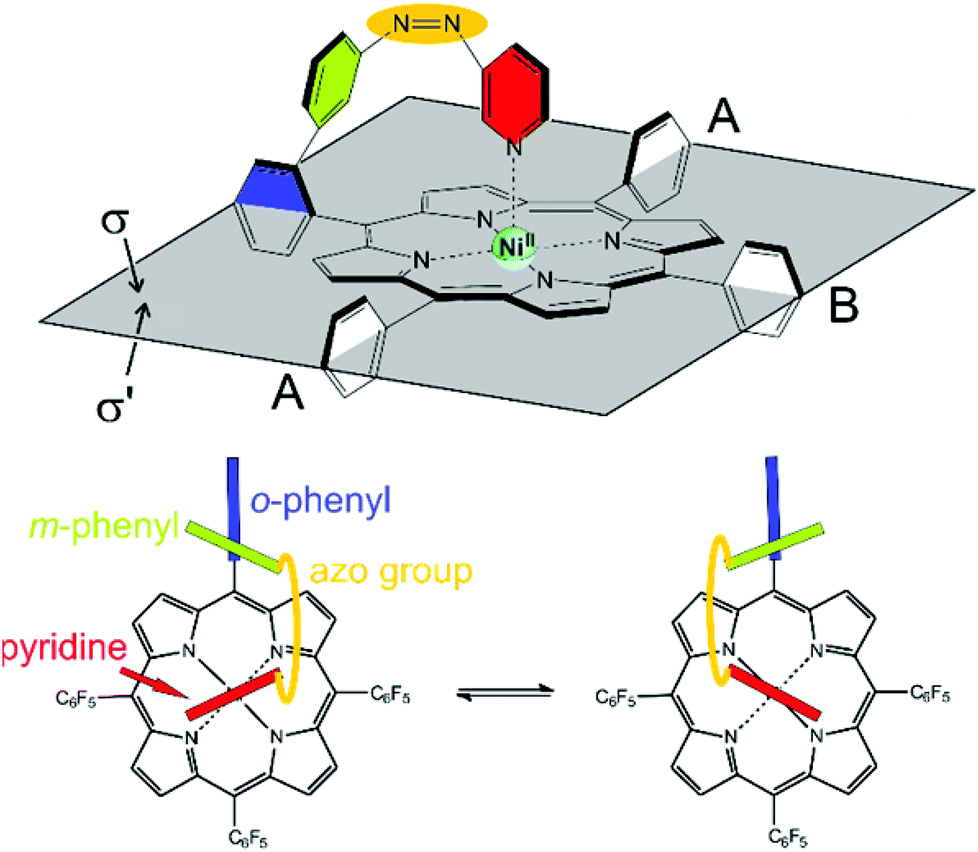 | ||
| Fig. 15 Top: Record player cis-2para has two symmetry non-equivalent sides σ and σ′. Neither of the meso substituents (pentafluorophenyl and “tone arm”) rotate around the porphyrin plane on the NMR time scale at 300 K. For the sake of clarity the fluorine atoms at the meso positions A and B are omitted. Bottom: top view on the conversion between the two enantiomers of cis-2. The conformational equilibrium is fast on the NMR time scale at 300 K. | ||
3. Conclusions
We present the rational design of a molecule whose spin state can be switched with visible light at room temperature in homogenous solution. Our approach is based on a three-part system: a photoswitchable azopyridine unit, a Ni-porphyrin, and a tether that connects the latter two building blocks (“record player” or “photodissociable ligand” design). Upon irradiation the azopyridine reversibly changes configuration which leads to coordination/decoordination of the pyridine to the Ni ion. The change of coordination number in turn switches the spin state of the Ni2+ between low-spin (diamagnetic) and high-spin (paramagnetic). We coined our approach light-driven coordination-induced spin state switching (LD-CISSS). To find a system with high switching efficiency, a number of systems differing in tether structure, and position of attachment were evaluated in silico by DFT calculations. Among 36 candidates three structures were identified as potentially operative, and worthwhile being synthesized. One of the three structures was actually prepared, and thoroughly investigated. While the basic properties have been published recently,18 we here present a detailed experimental and computational study of the system. DFT calculations and experiments reveal that both, geometry and electronic requirements have to be accurately met to obtain an efficient system. The tether has to be constructed in such a way that the pyridine nitrogen lone pair is placed exactly perpendicular to the porphyrin plane, and at a distance of 2.25 to 2.35 Å. Moreover, the Ni-porphyrin must be electron poor. The “record player” compound with phenyl substituents at the meso positions exhibits no spin state switching, whereas the corresponding system with three pentafluorophenyl substituents is highly efficient. According to DFT calculations, and corresponding NMR experiments, electron withdrawing substituents at the porphyrin meso positions strongly improve the coordination of the pyridine to the Ni ion, which is a prerequisite for the spin change. Switching efficiency also strongly depends on the solvent. Weakly coordinating solvents prevent intermolecular coordination, and concomitantly improve intramolecular binding. The mole fraction of paramagnetic cis-2 in DMSO is 91% and 93% in THF. Thermal isomerisation of the cis isomer back to the thermodynamically more stable trans form is slowed down considerably as well in weakly coordinating solvents. Thermal half-life of the high-spin cis isomer in DMSO is extraordinarily long (400 days at 20 °C). So DMSO seems to be the solvent of choice. Considering the extreme long-term switching stability (more than 20000 cycles with no detectable side products) and the fact that it can be switched with visible light, combined with the above mentioned properties, “record player” 2 is superior to conventional azobenzenes in most aspects. Spin state switching is inhibited at low pH. If light (500/430 nm) and pH (low/high) is used as input, and the magnetic state (dia/paramagnetic) as output the molecule can be used as a logic gate (AND gate). The fact that the magnetic state can be used as output has the advantage that it provides means for a non-destructive readout (orthogonal to light and pH).
Room temperature switchable molecular magnets provide the potential for a number of interesting applications such as information storage45,46 and processing,47 sensor applications,48 switchable contrast agents for MRI49–51 or light induced magnetic levitation.41 Further improvement based on the detailed information provided above should be possible. However, the mechanism of the unusual energy transfer from the porphyrin unit to the azopyridine ligand has yet to be elucidated.
Acknowledgements
This work has been supported by the Deutsche Forschungsgemeinschaft within the Sonderforschungsbereich 677 “Function by Switching”.Notes and references
- M. P. Shores, C. M. Klug and S. R. Fiedler, Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Ltd., 1st edn, 2013, ch. 10, pp. 281–301 Search PubMed.
- C. Gandolfi, G. G. Morgan and M. Albrecht, Dalton Trans., 2012, 41, 3726–3730 RSC.
- P. N. Martinho, Y. Ortin, B. Gildea, C. Gandolfi, G. McKerr, B. O'Hagan, M. Albrecht and G. G. Morgan, Dalton Trans., 2012, 41, 7461–7463 RSC.
- M. P. Shores, C. M. Klug and S. R. Fiedler, Spin-Crossover Materials, ed. M. A. Halcrow, 2013, pp. 281–301 Search PubMed.
- R. N. Muller, L. Vander Elst and S. Laurent, J. Am. Chem. Soc., 2003, 125, 8405–8407 CrossRef CAS PubMed.
- C. Rajadurai, M. Ruben and D. Kruk, EP2 072 062 A1, 2009 Search PubMed.
- G. M. Clore and J. Iwahara, Chem. Rev., 2009, 109, 4108–4139 CrossRef CAS PubMed.
- A. R. Rocha, V. M. Garcia-suarez, S. W. Bailey, C. J. Lambert, J. Ferrer and S. Sanvito, Nat. Mater., 2005, 4, 335–339 CrossRef CAS PubMed.
- K. Hamachi, K. Matsuda, T. Itoh and H. Iwamura, Bull. Chem. Soc. Jpn., 1998, 71, 2937 CrossRef CAS.
- M. M. Paquette, B. O. Patrick and N. L. Frank, J. Am. Chem. Soc., 2011, 133, 10081–10093 CrossRef CAS PubMed.
- A. Bannwarth, S. O. Schmidt, G. Peters, F. D. Sönnichsen, W. Thimm, R. Herges and F. Tuczek, Eur. J. Inorg. Chem., 2012, 2012, 2776–2783 CrossRef CAS.
- Y. Hasegawa, S. Kume and H. Nishihara, Dalton Trans., 2009, 280–284 RSC.
- R. K. Wilson and S. Brooker, Dalton Trans., 2013, 42, 12075–12078 RSC.
- N. Tanifuji, M. Irie and K. Matsuda, J. Am. Chem. Soc., 2005, 127, 13344–13353 CrossRef CAS PubMed.
- X. Ge, C. Manzano, R. Berndt, L. T. Anger, F. Köhler and R. Herges, J. Am. Chem. Soc., 2009, 131, 6096–6098 CrossRef CAS PubMed.
- T. G. Gopakumar, F. Matino, H. Naggert, A. Bannwarth, F. Tuczek and R. Berndt, Angew. Chem., Int. Ed., 2012, 51, 6262–6266 CrossRef CAS PubMed.
- T. G. Gopakumar, M. Bernien, H. Naggert, F. Matino, C. F. Hermanns, A. Bannwarth, S. Mühlenberend, A. Krüger, D. Krüger, F. Nickel, W. Walter, R. Berndt, W. Kuch and F. Tuczek, Chem. – Eur. J., 2013, 19, 15702–15709 CrossRef CAS PubMed.
- S. Venkataramani, U. Jana, M. Dommaschk, F. D. Sönnichsen, F. Tuczek and R. Herges, Science, 2011, 331, 445–448 CrossRef CAS PubMed.
- D. Achey and G. J. Meyer, Inorg. Chem., 2013, 52, 9574–9582 CrossRef CAS PubMed.
- T. J. Lotz and T. A. Kaden, J. Chem. Soc., Chem. Commun., 1977, 15–16 RSC.
- Y. Song, R. E. Haddad, S.-L. Jia, S. Hok, M. M. Olmstead, D. J. Nurco, N. E. Schore, J. Zhang, J.-G. Ma, K. M. Smith, S. Gazeau, J. Pécaut, J.-C. Marchon, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2005, 127, 1179–1192 CrossRef CAS PubMed.
- S. Thies, C. Bornholdt, F. Köhler, F. D. Sönnichsen, C. Näther, F. Tuczek and R. Herges, Chem. – Eur. J., 2010, 16, 10074–10083 CrossRef CAS PubMed.
- S. Thies, H. Sell, C. Bornholdt, C. Schütt, F. Köhler, F. Tuczek and R. Herges, Chem. – Eur. J., 2012, 18, 16358–16368 CrossRef CAS PubMed.
- S. Thies, H. Sell, C. Schütt, C. Bornholdt, C. Näther, F. Tuczek and R. Herges, J. Am. Chem. Soc., 2011, 133, 16243–16250 CrossRef CAS PubMed.
- H. K. Hombrecher and K. Lüdtke, Tetrahedron, 1993, 49, 9489–9494 CrossRef CAS.
- K. H. Neumann and F. Vogtle, J. Chem. Soc., Chem. Commun., 1988, 520–522 RSC.
- C. A. Hunter and L. D. Sarson, Tetrahedron Lett., 1996, 37, 699–702 CrossRef CAS.
- H. Sugimoto, K. Kuramoto and S. Inoue, J. Chem. Soc., Perkin Trans. 1, 2002, 1826–1830 RSC.
- L. J. Esdaile, P. Jensen, J. C. McMurtrie and D. P. Arnold, Angew. Chem., Int. Ed., 2007, 46, 2090–2093 CrossRef CAS PubMed.
- M. V. Peters, R. Goddard and S. Hecht, J. Org. Chem., 2006, 71, 7846–7849 CrossRef CAS PubMed.
- A. M. G. Silva, A. C. Tom'e, M. G. P. M. S. Neves, A. M. S. Silva and J. A. S. Cavaleiro, J. Org. Chem., 2005, 70, 2306–2314 CrossRef CAS PubMed.
- A. M. G. Silva, P. S. S. Lacerda, A. C. Tom'e, M. G. P. M. S. Neves, A. M. S. Silva, J. A. S. Cavaleiro, E. A. Makarova and E. A. Lukyanets, J. Org. Chem., 2006, 71, 8352–8356 CrossRef CAS PubMed.
- F. Matino, G. Schull, U. Jana, F. Köhler, R. Berndt and R. Herges, Chem. Commun., 2010, 46, 6780–6782 RSC.
- A. A. Starikova, R. M. Minyaev, A. G. Starikov and V. I. Minkin, Eur. J. Inorg. Chem., 2013, 2013, 4203–4219 CrossRef CAS.
- D. Kim, Y. O. Su and T. G. Spiro, Inorg. Chem., 1986, 25, 3988–3993 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. A. Walker, E. Hui and J. M. Walker, J. Am. Chem. Soc., 1975, 97, 2390–2397 CrossRef CAS.
- S. J. Cole, G. C. Curthoys, E. A. Magnusson and J. N. Phillips, Inorg. Chem., 1972, 11, 1024–1028 CrossRef CAS.
- R. J. W. Le Fevre and J. Northcott, J. Chem. Soc., 1953, 867–870 RSC.
- E. R. Talaty and J. C. Fargo, Chem. Commun., 1967, 65–66 RSC.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- J. A. Happe and R. L. Ward, J. Chem. Phys., 1963, 39, 1211–1218 CrossRef CAS PubMed.
- C. Belle, C. Bougault, M.-T. Averbuch, A. Durif, J.-L. Pierre, J.-M. Latour and L. Le Pape, J. Am. Chem. Soc., 2001, 123, 8053–8066 CrossRef CAS PubMed.
- I. Bertini, P. Turano and A. J. Vila, Chem. Rev., 1993, 93, 2833–2932 CrossRef CAS.
- L. Wei, K. Padmaja, W. J. Youngblood, A. B. Lysenko, J. S. Lindsey and D. F. Bocian, J. Org. Chem., 2003, 69, 1461–1469 CrossRef PubMed.
- K. L. Kompa and R. D. Levine, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 410–414 CrossRef CAS PubMed.
- K. Szacilowski, Chem. Rev., 2008, 108, 3481–3548 CrossRef CAS PubMed.
- J. M. Perez, L. Josephson, T. O'Loughlin, D. Hogemann and R. Weissleder, Nat. Biotechnol., 2002, 20, 816–820 CrossRef CAS PubMed.
- Christian-Albrechts-Universität and Universitätsklinikum S.-H., WO2012/022299, patent pending Search PubMed.
- R. Herges, Nachr. Chem., 2011, 59, 817–821 CrossRef CAS.
- R. B. Lauffer, Chem. Rev., 1987, 87, 901–927 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1023485. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03048f |
| This journal is © The Royal Society of Chemistry 2014 |