
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedented dinuclear silver(I)-mediated base pair involving the DNA lesion 1,N6-ethenoadenine†
Soham
Mandal
ab,
Alexander
Hepp
a and
Jens
Müller
*ab
aWestfälische Wilhelms-Universität Münster, Institut für Anorganische und Analytische Chemie, Corrensstr. 28/30, 48149 Münster, Germany. E-mail: mueller.j@uni-muenster.de; Fax: +49 251 8336007; Tel: +49 251 8336006
bWestfälische Wilhelms-Universität Münster, NRW Graduate School of Chemistry, Corrensstr. 28/30, 48149 Münster, Germany
First published on 28th November 2014
Abstract
The DNA lesion 1,N6-ethenoadenine (εA) has been investigated with respect to its metal-binding properties. A synthetic DNA duplex comprising an εA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :εA mispair readily forms doubly silver(I)-mediated base pairs εA–Ag(I)2–εA, representing the first example for a dinuclear metal-mediated homo base pair of a purine derivative. It also constitutes the first example for a Hoogsteen-type metal-mediated homo base pair within a B-DNA duplex.
:εA mispair readily forms doubly silver(I)-mediated base pairs εA–Ag(I)2–εA, representing the first example for a dinuclear metal-mediated homo base pair of a purine derivative. It also constitutes the first example for a Hoogsteen-type metal-mediated homo base pair within a B-DNA duplex.
The incorporation of transition metal ions into nucleic acids by means of metal-mediated base pairs represents an elegant and efficient method for the site-specific introduction of metal-based functionality into the self-assembling biomacromolecules.1 In metal-mediated base pairs, the hydrogen bonds between complementary nucleobases are formally replaced by coordinative bonds to a central metal ion. Numerous artificial nucleosides bearing ligands as aglycones have been introduced to broaden the scope of metal-mediated base pairing.2 Experimental structure determinations both by X-ray crystallography and by NMR spectroscopy have clearly proven that the formation of metal-mediated base pairs is compatible with the well-known B-DNA conformation.3 Some prominent possible applications for such DNA-metal conjugates include charge transfer,4 expansion of the genetic code,5 sensors of various types,1c and the use of DNA as a logic gate,6 to name just a few.
While most metal-mediated base pairs contain one metal ion per base pair,7 few examples are known of base pairs which comprise two metal ions. These include homo base pairs of various pyrimidine derivatives8 as well as the hetero base pair 1,3-dideazaadenine–Ag(I)2–thyminate.9 Dinuclear metal-mediated base pairs represent a particular challenge as two positively charged entities need to be positioned at very short distance. On the other hand, they enable a high functionalization of nucleic acids with metal ions and thereby significantly increase the scope of DNA-based nanotechnology.9 We report here the first example of a doubly metal-mediated homo base pair derived from a purine nucleobase. The exocyclic etheno adduct of adenine, 1,N6-ethenoadenine (εA, Scheme 1), is a DNA lesion that is known to form in the presence of e.g. vinyl chloride,10 although endogenous pathways for its formation in vivo have also been suggested.11 Its metal binding properties are superior to those of the parent compound adenine.12
 | ||
| Scheme 1 1,N6-ethenodeoxyadenosine including atom numbering scheme. | ||
To investigate metal-mediated base pair formation, an oligonucleotide duplex with a central εA:εA mispair was synthesized (Scheme 2). This particular sequence was chosen because it had been used in various earlier studies on other metal-mediated base pairs and therefore allows a good comparison of the observations.7a,b,d,13 The melting temperature Tm of the unmetalated duplex comprising the central εA:εA mispair amounts to 35.7 °C. It is significantly more stable than previously investigated other mispairs such as imidazole:imidazole, which showed Tm values in the range of 21.7–30.4 °C.7b Its stability is lower though than that of the corresponding duplexes with regular A:T (42.5 °C) or G:C (45.4 °C) base pairs.13 Hence, the metal-free duplex is destabilized with respect to the purely canonical duplex because of the εA:εA mispair formation, whereas the large π surface of εA is expected to be the main contributor towards its stabilization with respect to mispairs with a smaller π surface.
 | ||
| Scheme 2 Oligonucleotide double helix under investigation (X = εA). | ||
Upon the addition of Ag(I), the melting temperature of the DNA duplex increases significantly until two equivalents of Ag(I) are present per base pair (Fig. 1). From a plot of Tmversus added equivalents of Ag(I) it can clearly be discerned that the first two Ag(I) ions induce the largest stabilization, whereas additional Ag(I) has only very minor effects on the thermal stability of the duplex. This weak additional stabilization by Ag(I) has previously been attributed to an electrostatic interaction with the negatively charged DNA backbone.13 Taken together, this is a first good indication for the formation of a doubly metal-mediated base pair εA–Ag(I)2–εA.
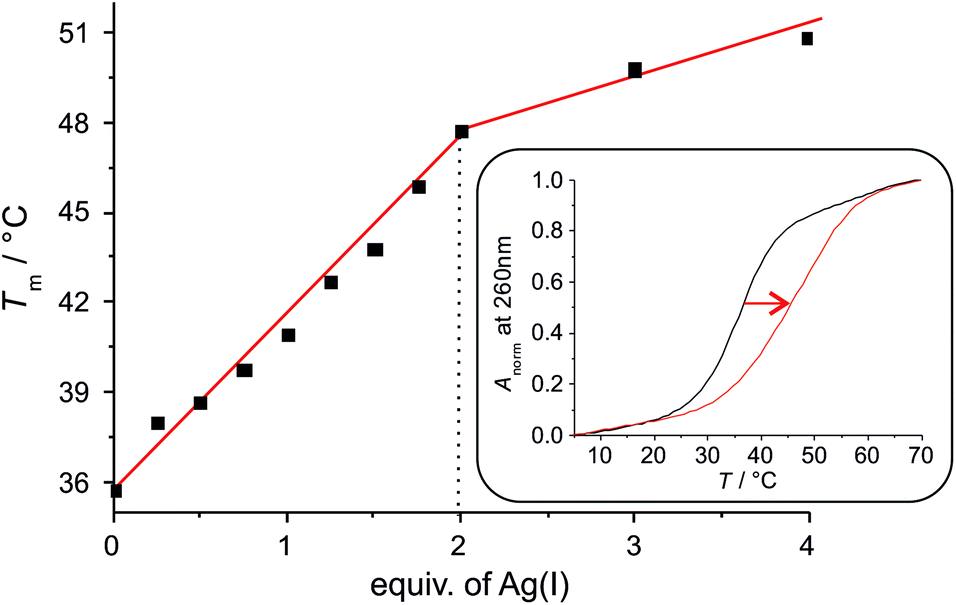 | ||
| Fig. 1 Increase of the melting temperature Tm upon the addition of Ag(I). Inset: UV melting profile of the duplex with one central εA:εA mispair in the absence (black) and presence (red) of two equivalents of Ag(I). Anorm = (A − Amin)/(Amax − Amin). | ||
Additional spectroscopic experiments have been performed to validate in an independent manner the 2:1 stoichiometry of the base pair. Towards this end, CD and UV spectra of the DNA duplex have been recorded in the presence of various amounts of Ag(I). Fig. 2a shows a plot of the CD spectra, indicating a constant positive Cotton effect around 275 nm. In addition, two negative Cotton effects at 232 nm and 244 nm are observed in the absence of Ag(I), merging into a single and more intense negative Cotton effect at 236 nm upon the addition of Ag(I). The CD spectrum in the presence of two equivalents of Ag(I) is reminiscent of that of B-DNA.14 The somewhat unusual CD spectrum in the absence of Ag(I) is likely to be due to the presence of the εA:εA mispair in which the nucleosides probably adopt a conformation not found in a regular B-DNA duplex (e.g. by stacking on top of one another or by extruding one base out of the base stack). A plot of the molar ellipticity at 236 nm versus added equivalents of Ag(I) (Fig. 2b) confirms that the distinct change of the ellipticity occurs only until two equivalents of Ag(I) are present. Excess Ag(I) only leads to minor additional changes in the CD spectrum. The same behaviour is found for the UV spectra of the duplex (Fig. 2c): upon adding the first two Ag(I) ions per εA:εA mispair a significant change of the UV spectrum is observed, whereas excess Ag(I) does not significantly change the spectrum. As the changes in absorbance are most prominent in the wavelength region where the aromatic nucleobases absorb, a binding of the Ag(I) ions to the nucleobases can be concluded. Previous experiments with same sequence comprising entirely canonical base pairs indicate no significant changes of the spectral properties upon the addition of Ag(I).13 Hence, as all experiments nicely show that the binding site with the highest Ag(I) binding affinity binds exactly two Ag(I) ions, the εA:εA mispair can be unambiguously identified as the metal binding site. Scheme 3 shows the proposed structure of this doubly metal-mediated base pair.
 | ||
| Fig. 2 (a) CD spectra of the duplex with one central εA:εA mispair upon the addition of Ag(I). (b) Change of molar ellipticity at 236 nm upon the addition of Ag(I). (c) Change of absorbance at 257 nm upon the addition of Ag(I). | ||
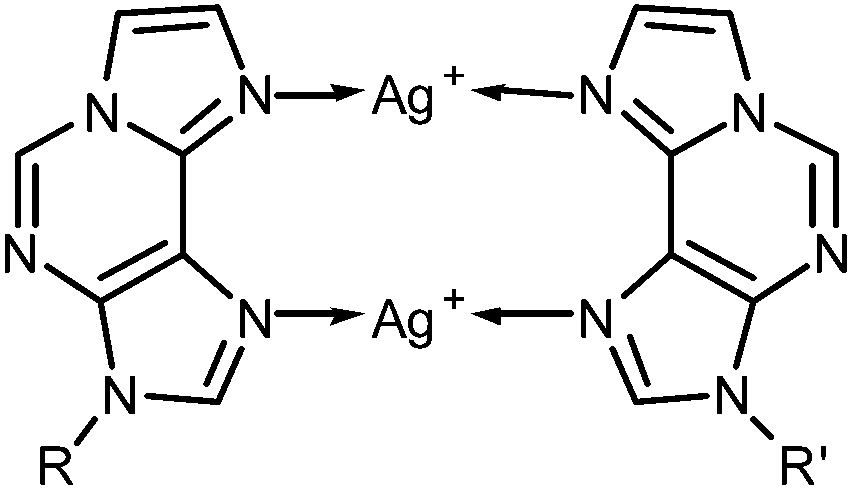 | ||
| Scheme 3 Structure of the εA–Ag(I)2–εA base pair (R, R′ = DNA backbone). | ||
The model nucleobase 9-ethyl-1,N6-ethenoadenine was applied for another independent proof of the metal-binding stoichiometry by performing 1H NMR- and 15N NMR-spectroscopy-based titrations of the ligand with Ag(I) ions (Fig. 3 and ESI†). As can clearly be discerned from Fig. 3a, the chemical shifts of the aromatic protons change until one Ag(I) ion is present per ligand, corresponding to two Ag(I) ions per pair of ligands. 15N NMR spectroscopy unambiguously confirms N6 and N7 as the sites of metalation, as both nitrogen atoms experience an upfield shift of >40 ppm upon binding of Ag(I) whereas the chemical shifts of the other three nitrogen atoms change by only 1–6 ppm (Fig. 3b). Taken together, these NMR data confirm the formation of a dinuclear complex also in solution. In contrast to the situation inside the DNA duplex, the dinuclear complex can adopt two distinct conformations in solution. In the case of transoid ethyl substituents, two identical Ag(I) binding sites are present (N6–Ag(I)–N7), whereas a cisoid orientation of the ethyl substituents (Scheme 3) leads to two distinct Ag(I) environments (N6–Ag(I)–N6 and N7–Ag(I)–N7). Unfortunately, it is not possible to differentiate between these conformations based on the NMR data, as the Ag(I) ions bind in a labile fashion, leading to a fast exchange on the NMR time scale (as indicated by the smooth change of the 1H and 15N NMR chemical shifts upon metalation). As the B-DNA geometry enforces a cisoid orientation of the glycosidic bonds, and the 15N NMR data show that both N6 and N7 are metalated, it can nonetheless be concluded that inside the DNA duplex the metal-mediated base pair adopts the structure shown in Scheme 3.
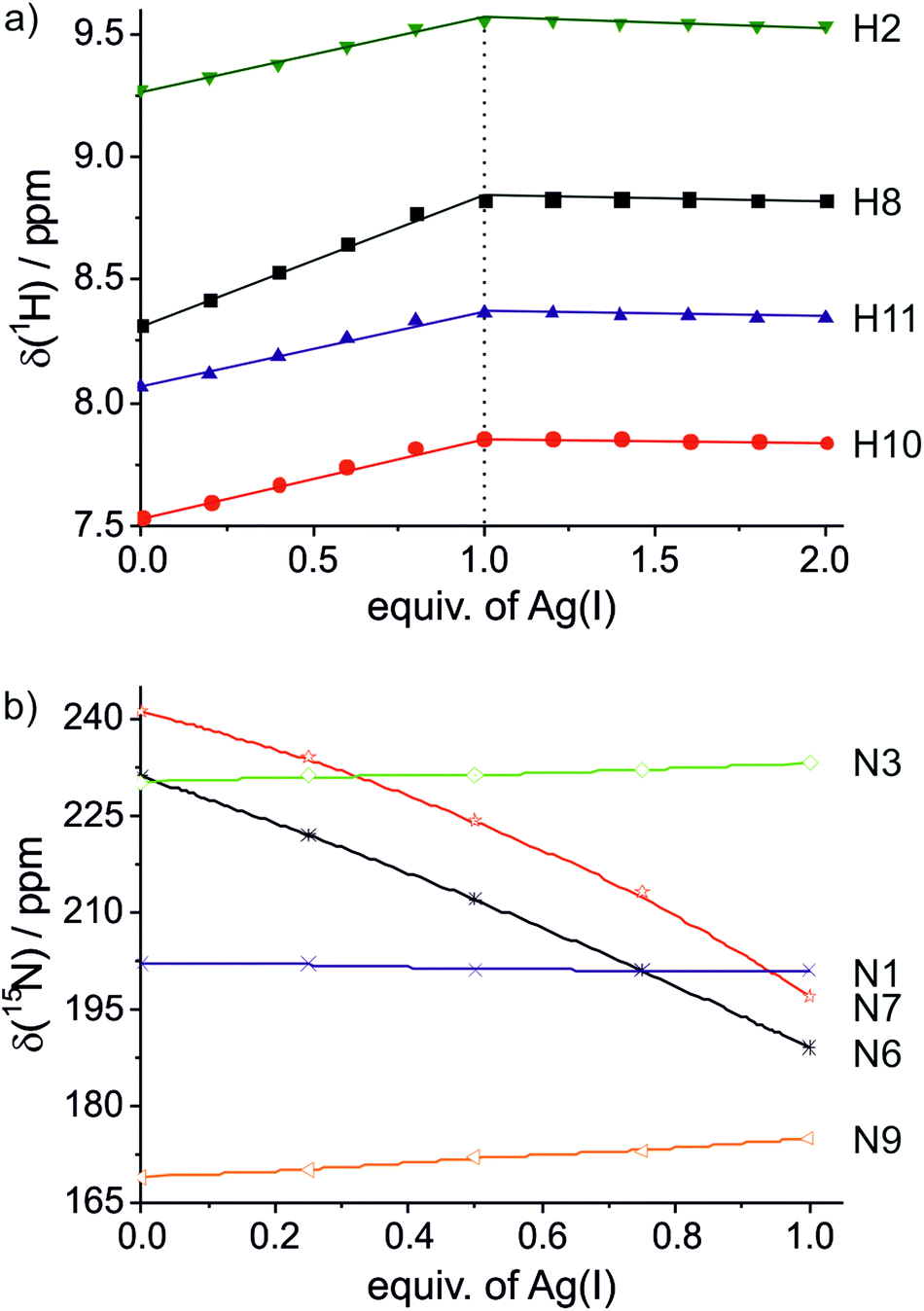 | ||
| Fig. 3 Chemical shifts of the model nucleobase 9-ethyl-1,N6-ethenoadenine in the presence of varying amounts of Ag(I). (a) 1H NMR; (b) 15N NMR. | ||
Conclusions
The exocyclic etheno adduct of adenine, 1,N6-ethenoadenine (εA), is capable of forming doubly Ag(I)-mediated homo base pairs. This is the first time that doubly metal-mediated homo base pairs are being reported for a purine derivative, and the first example for a Hoogsteen-type metal-mediated base pair within a regular B-DNA context. Particularly in view of the commercial availability of εA, this finding represents a significant step forward towards easily prepared, highly metal-functionalized nucleic acids.Financial support by the NRW Graduate School of Chemistry (GSC-MS) and the DFG (SFB 858) is greatly acknowledged. This work was performed in the context of COST Action CM1105.
Notes and references
- (a) J. Müller, Eur. J. Inorg. Chem., 2008, 3749–3763 CrossRef; (b) D. A. Megger, N. Megger and J. Müller, Met. Ions Life Sci., 2012, 10, 295–317 CAS; (c) P. Scharf and J. Müller, ChemPlusChem, 2013, 78, 20–34 CrossRef CAS.
- (a) G. H. Clever and M. Shionoya, Met. Ions Life Sci., 2012, 10, 269–294 CAS; (b) Y. Takezawa and M. Shionoya, Acc. Chem. Res., 2012, 45, 2066–2076 CrossRef CAS PubMed; (c) G. H. Clever and M. Shionoya, Coord. Chem. Rev., 2010, 254, 2391–2402 CrossRef CAS PubMed; (d) G. H. Clever, C. Kaul and T. Carell, Angew. Chem., Int. Ed., 2007, 46, 6226–6236 CrossRef CAS PubMed; (e) A. Ono, H. Torigoe, Y. Tanaka and I. Okamoto, Chem. Soc. Rev., 2011, 40, 5855–5866 RSC.
- (a) S. Johannsen, N. Megger, D. Böhme, R. K. O. Sigel and J. Müller, Nat. Chem., 2010, 2, 229–234 CrossRef CAS PubMed; (b) S. Kumbhar, S. Johannsen, R. K. O. Sigel, M. P. Waller and J. Müller, J. Inorg. Biochem., 2013, 127, 203–210 CrossRef CAS PubMed; (c) H. Yamaguchi, J. Šebera, J. Kondo, S. Oda, T. Komuro, T. Kawamura, T. Dairaku, Y. Kondo, I. Okamoto, A. Ono, J. V. Burda, C. Kojima, V. Sychrovský and Y. Tanaka, Nucleic Acids Res., 2014, 42, 4094–4099 CrossRef CAS PubMed; (d) Y. Kondo, T. Yamada, C. Hirose, I. Okamoto, Y. Tanaka and A. Ono, Angew. Chem., Int. Ed., 2014, 53, 2385–2388 CrossRef PubMed.
- (a) S. Liu, G. H. Clever, Y. Takezawa, M. Kaneko, K. Tanaka, X. Guo and M. Shionoya, Angew. Chem., Int. Ed., 2011, 50, 8886–8890 CrossRef CAS PubMed; (b) T. Ehrenschwender, W. Schmucker, C. Wellner, T. Augenstein, P. Carl, J. Harmer, F. Breher and H.-A. Wagenknecht, Chem. – Eur. J., 2013, 19, 12547–12552 CrossRef CAS PubMed.
- C. Kaul, M. Müller, M. Wagner, S. Schneider and T. Carell, Nat. Chem., 2011, 3, 794–800 CrossRef CAS PubMed.
- D.-L. Ma, H.-Z. He, D. S.-H. Chan and C.-H. Leung, Chem. Sci., 2013, 4, 3366–3380 RSC.
- (a) T. Richters and J. Müller, Eur. J. Inorg. Chem., 2014, 437–441 CrossRef CAS; (b) T. Richters, O. Krug, J. Kösters, A. Hepp and J. Müller, Chem. – Eur. J., 2014, 20, 7811–7818 CrossRef CAS PubMed; (c) K. Petrovec, B. J. Ravoo and J. Müller, Chem. Commun., 2012, 48, 11844–11846 RSC; (d) K. Seubert, C. Fonseca Guerra, F. M. Bickelhaupt and J. Müller, Chem. Commun., 2011, 47, 11041–11043 RSC; (e) F.-A. Polonius and J. Müller, Angew. Chem., Int. Ed., 2007, 46, 5602–5604 CrossRef CAS PubMed; (f) D. Böhme, N. Düpre, D. A. Megger and J. Müller, Inorg. Chem., 2007, 46, 10114–10119 CrossRef PubMed.
- (a) I. Okamoto, K. Iwamoto, Y. Watanabe, Y. Miyake and A. Ono, Angew. Chem., Int. Ed., 2009, 48, 1648–1651 CrossRef CAS PubMed; (b) I. Okamoto, T. Ono, R. Sameshima and A. Ono, Chem. Commun., 2012, 48, 4347–4349 RSC; (c) H. Mei, I. Röhl and F. Seela, J. Org. Chem., 2013, 78, 9457–9463 CrossRef CAS PubMed.
- D. A. Megger, C. Fonseca Guerra, J. Hoffmann, B. Brutschy, F. M. Bickelhaupt and J. Müller, Chem. – Eur. J., 2011, 17, 6533–6544 CrossRef CAS PubMed.
- J. A. Hall, R. Saffhill, T. Green and D. E. Hathway, Carcinogenesis, 1981, 2, 141–146 CrossRef CAS.
- F.-L. Chung, H.-J. C. Chen and R. G. Nath, Carcinogenesis, 1996, 17, 2105–2111 CrossRef CAS PubMed.
- K. H. Scheller and H. Sigel, J. Am. Chem. Soc., 1983, 105, 3005–3014 CrossRef CAS.
- I. Sinha, J. Kösters, A. Hepp and J. Müller, Dalton Trans., 2013, 42, 16080–16089 RSC.
- M. Vorlíčková, I. Kejnovská, K. Bednářová, D. Renčiuk and J. Kypr, Chirality, 2012, 24, 691–698 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic characterization, stack plot of 1H NMR spectra. See DOI: 10.1039/c4dt02663b |
| This journal is © The Royal Society of Chemistry 2015 |