
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Studying the effects of Zr-doping in (Bi0.5Na0.5)TiO3via diffraction and spectroscopy
Peter E. R.
Blanchard
*ab,
Samuel
Liu
c,
Brendan J.
Kennedy
c,
Chris D.
Ling
c,
Zhaoming
Zhang
d,
Maxim
Avdeev
d,
Ling-Yun
Jang
e,
Jyh-Fu
Lee
e,
Chih-Wen
Pao
e and
Jeng-Lung
Chen
e
aCanadian Light Source, Saskatoon, SK, S7N 2V3, Canada. E-mail: peter.blanchard@lightsource.ca; Tel: 306-966-8422
bDepartment of Chemistry, University of Saskatchewan, Saskatoon, SK, S7N 5C9, Canada
cSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia
dAustralian Nuclear Science and Technology Organisation, Lucas Heights, NSW 2234, Australia
eExperimental Facility Division, National Synchrotron Radiation Research Center, Hsinchu 30076, Taiwan
First published on 13th October 2014
Abstract
The structural properties of (Bi0.5Na0.5)Ti1−xZrxO3 (where 0 ≤ x ≤ 0.7) have been investigated using powder diffraction and X-ray absorption spectroscopy. Diffraction measurements on (Bi0.5Na0.5)TiO3 confirm that both monoclinic Cc and rhombohedral R3c phases are present at room temperature. Doping small amounts of Zr into the B site of (Bi0.5Na0.5)TiO3 initially stabilizes the rhombohedral phase before the orthorhombic Pnma phase begins to form at x = 0.5. Analysis of the Ti K-edge and Zr L3-edge XANES spectra show that the crystallographic phase change has very little effect on the local structure of Ti4+/Zr4+ cations, suggesting that there is little change in the cation off-center displacement within the BO6 octahedra with each successive phase change.
Introduction
Lead-based ABO3 perovskites, particularly PbTi1−xZrxO3, remain the industry standard for ferroelectric and piezoelectric applications, such as transducers, actuators, and sensors.1 Due to the adverse environmental impact of lead-based materials, considerable efforts have been directed towards developing lead-free ferroelectric and/or piezoelectric materials as alternatives to PbTi1−xZrxO3.2,3 Since the Pb2+ 6s2 lone pair is known to play an important role in influencing the ferroelectric properties of these materials,4 recent attempts to develop lead-free perovskites have focused on replacing Pb2+ with isoelectronic Bi3+. Of these Bi-based perovskites, (Bi0.5Na0.5)TiO3 has been identified as a promising alternative to PbTi1−xZrxO3. (Bi0.5Na0.5)TiO3 was first studied by Smolenski in 1961 and it has subsequently been established that this compound exhibits strong ferroelectricity with unusually high remnant polarization (Pr = 38 μC cm−2) and Curie temperature (593 K).5 However, poling (Bi0.5Na0.5)TiO3 is difficult due to its high coercive field (Ec = 73 kV cm−1), resulting in relatively weak piezoelectric properties (d33 = 73–80 pC N−1).5 The piezoelectric properties of perovskites can be improved by doping the A and/or B site with cations that allow for the formation of a morphotropic phase boundary (MPB), a region in a solid solution where two or more ferroelectric phases co-exist. High piezoelectric behavior is often observed at the MPB as the polarization direction can be easily rotated by external stress or electric field.6 The rhombohedral-tetragonal MPB known to exist in the PbTi1−xZrxO3 solid solution at x = 0.52 has been extensively studied.7 MPB's have also been observed in several (Bi0.5Na0.5)TiO3-based solid solutions, including (Bi0.5Na0.5)1−xBaxTiO3,8 (Bi0.5Na0.5)1−xPbxTiO3,9 (Bi0.5Na0.5−x/2Kx/2)TiO3,10 (Bi0.5−x/2Kx/2Na0.5)Ti1−xNbxO3,11 and (Bi0.5+x/2Na0.5−x/2)Ti1−xCoxO3.12The crystallographic structure of (Bi0.5Na0.5)TiO3 at room temperature was originally described as an A site substituted perovskite that adopts a rhombohedral structure in space group R3c, in which the Bi3+/Na+ and Ti4+ cations are displaced parallel to each other along the [111] direction.13,14 More recent studies have raised doubt as to the accuracy of this description. An infrared and Raman spectroscopic study by Petzelt et al. suggested that localized monoclinic distortions in space group Cm were present.15 Gorfman et al. later suggested from X-ray diffraction (XRD) analysis that Bi0.5Na0.5TiO3 actually forms a monoclinic structure in space group Cc rather than a rhombohedral structure.16 Recently, Aksel et al. demonstrated that the Cc model provided a better fit to the synchrotron X-ray diffraction (S-XRD) pattern of (Bi0.5Na0.5)TiO3 than the R3c model.17 Aksel et al. later confirmed the Cc monoclinic structure in a combined X-ray and neutron total scattering study and noted large displacement parameters of Bi3+/Na+, likely due to the deviations in the Na+ and Bi3+ cations from their ideal positions.18 Evidence of a monoclinic phase in (Bi0.5Na0.5)TiO3 has also been observed in several electron microscopy experiments.19–21 Rao et al. have suggested that both rhombohedral (R3c) and monoclinic (Cc) phases were present at room temperature and that the relative amounts of these are highly dependent on synthesis conditions.22 The existence of two ferroelectric phases (rhombohedral R3c and monoclinic Cm) was observed in PbTi1−xZrxO3.23 (This too has been disputed with a recent X-ray and neutron diffraction study suggesting that PbTi1−xZrxO3 actually forms a single monoclinic structure in space group Cc.24).
Given the recent controversy over the room temperature structure of (Bi0.5Na0.5)TiO3, it is important to revisit the structural evolution of (Bi0.5Na0.5)TiO3-based solid solutions. One such solid solution that deserves further investigation is (Bi0.5Na0.5)Ti1−xZrxO3. Lily et al. first reported (Bi0.5Na0.5)ZrO3 as forming an orthorhombic structure in space group Pnma.25 Rachakom et al. later reported that the rhombohedral-orthorhombic phase transition in (Bi0.5Na0.5)Ti1−xZrxO3 occurs at x ∼ 0.8 based on analysis of laboratory XRD data.26 More recently, Barick et al. reported that only the rhombohedral structure was observed for compositions with x ≤ 0.6.27 As these analyses were performed using laboratory XRD data, which is well known to be of limited power in studies of oxides containing very heavy cations such as Bi or Zr, we elected to reinvestigate the average structure of (Bi0.5Na0.5)Ti1−xZrxO3 using a combination of S-XRD and neutron powder diffraction (NPD). The local structure of the oxides was investigated using X-ray absorption near-edge spectroscopy (XANES). Levin et al., in a recent study on BaTi1−xZrxO3 perovskites, demonstrated that the Ti K-edge lineshape is sensitive to changes in the local distortions of TiO6 octahedra in perovskite materials.28 We have found the Zr L-edge lineshape to also be extremely sensitive to changes in the Zr4+ coordination environment.29–32
Experimental
Synthesis
All reagents (Bi2O3, BaCO3, Na2CO3, ZrO2, and TiO2) were obtained from Sigma-Aldrich or Aithaca with purities better than 99.9%. ZrO2 and TiO2 were calcined at 1000 °C for 12 h before use, while Bi2O3 and BaCO3 were calcined at 500 °C for 12 h. Samples of (Bi0.5Na0.5)Ti1−xZrxO3, (0 ≤ x ≤ 1 in 0.1 increments) were prepared by finely mixing stoichiometric amounts of each reagent under acetone with a mortar and pestle. The powders were then hydrostatically pressed into rods, heated to 800 °C for 4 h, and quenched. This process was repeated until no further changes were observed in the XRD patterns, obtained using a Panalytical X'Pert Pro X-ray diffractometer equipped with a Cu Kα X-ray source. For the Zr-rich samples (x ≥ 0.8 in (Bi0.5Na0.5)Ti1−xZrxO3), diffraction data contained additional reflections due to ZrO2 and an unknown impurity; therefore, detailed analysis was only performed for samples with x ≤ 0.7.Samples of BaTi1−xZrxO3 (0 ≤ x ≤ 1 in 0.1 increments) were prepared as standards for the XANES analysis. Samples were prepared by finely mixing stoichiometric amounts of each reagent under acetone with a mortar and pestle. The powders were hydrostatically pressed into rods, heated to 900 °C for 24 h. Samples were reground, pressed, and heated to 1200 °C for 24 h. This process was repeated until single phase samples were observed in the XRD patterns.
Structure determination
Structure determination was carried out using both synchrotron X-ray and neutron powder diffraction techniques. S-XRD patterns on the (Bi0.5Na0.5)Ti1−xZrxO3 series (where 0 ≤ x ≤ 0.7) were collected on the Powder Diffraction beamline of the Australian Synchrotron.33 Data were collected at room temperature in an angular range of 5° ≤ 2θ ≤ 85° using X-rays of wavelength 0.7749 Å, calibrated against NIST SRM660a LaB6. Each finely ground sample was placed in a 0.3 mm diameter glass capillary that was rotated during the measurements. Neutron powder diffraction (NPD) patterns of (Bi0.5Na0.5)TiO3 were collected at room temperature and 3 K on the high-resolution powder diffractometer Echidna at ANSTO's OPAL facility in Lucas Heights, Australia.34 Data were obtained over the angular range 10° ≤ 2θ ≤ 165° with the wavelength of the incident neutrons fixed at 1.62 Å and 2.44 Å using (335) and (331) reflections of a germanium monochromator, respectively. The samples were packed into 9 mm diameter vanadium cans.The structures were refined with the full-profile Rietveld method using the program LHPM-Rietica program (S-XRD) and the GSAS program with the EXPGUI front end (NPD).35–37 The peak shape of both the S-XRD and NPD data was modeled using a pseudo-Voigt function, also convoluted with asymmetry for the NPD data (resulting from axial divergence). Background was estimated from the linear interpolation between ∼30 background points. Structural and profile parameters were varied during the refinement. An absorption correction was applied to the S-XRD data. The NPD data were not corrected for absorption because of the moderate neutron absorption cross section of each of the constituent atoms.
XANES analysis
The Ti K-edge XANES spectra of (Bi0.5Na0.5)Ti−xZrxO3 (0 ≤ x ≤ 0.5) were collected on beamline 17C1 at the National Synchrotron Radiation Research Center (NSRRC) in Hsinchu, Taiwan.38 Powder samples were mixed with the appropriate amount of boron nitride and pressed into pellets. Spectra were collected from ∼200 eV below to ∼300 eV above the Ti K-edge in transmission mode with a step size of 0.25 eV in the near-edge region and a dwell time of 2 s. X-ray energies were scanned using a Si(111) monochromator, which was detuned by 50% to reject higher harmonics. The Ti K-edge was calibrated against metallic Ti foil with the maximum of the first derivative of the Ti K-edge set to 4966.0 eV.The Zr L3-edge XANES spectra were collected on beamline 16A1 at the NSRRC.39 Finely ground samples were dispersed onto Kapton tape and placed in front of the X-ray beam at a 45° angle. Spectra were collected in total fluorescence yield (TFY) mode using a Lytle detector. An energy step-size of 0.2 eV was used near the absorption edge. The Zr L3-edge spectra were calibrated against elemental Zr with the maximum in the first derivative of the L3-edge set to 2222.3 eV.
All XANES spectra were analyzed using the Athena software package.40
Results and discussion
Diffraction analysis
In order to properly determine structural changes in the (Bi0.5Na0.5)Ti1−xZrxO3 series, it was appropriate to first revisit the structure of the end member (Bi0.5Na0.5)TiO3. Fig. 1 shows the refined S-XRD pattern of (Bi0.5Na0.5)TiO3 fitted to various single-phase and two-phase models. R-values and goodness of fits (χ2) from these fits are tabulated in Table 1. Fitting the pattern to the monoclinic Cc phase rather than the rhombohedral R3c model resulted in a decrease in χ2 from 3.228 to 2.885, similar to that originally reported by Aksel et al.17 However, as evident from the insets in Fig. 1, neither of these single-phase models provided a satisfactory fit to all the reflections with the greatest discrepancy being observed for the (123) reflections near 2θ = 54.5° at λ = 0.7749 Å. The fits were noticeably improved when the pattern was fitted to a two-phase model consisting of the rhombohedral R3c phase and either the monoclinic Cm phase (χ2 = 1.842) or monoclinic Cc phase (χ2 = 1.641). These two monoclinic systems are similar in that they both exhibit out-of-phase tilting of the corner sharing BO6 octahedra, with Cm exhibiting a0a0c−—type tilting and Cc exhibiting a−a−a−—type tilting. Alternatively, the monoclinic Cm phase can be viewed as a monoclinically-distorted orthorhombic structure whereas the monoclinic Cc phase can be viewed as a monoclinically-distorted rhombohedral structure. The two-phase models were the only models that could successfully fit the satellite feature in the pseudocubic (123) reflection. Given that the Cc + R3c model results in a slightly better χ2 value than that obtained with the two phase Cm + R3c model, we conclude that the monoclinic phase present in (Bi0.5Na0.5)TiO3 adopts the Cc space group. This is the same monoclinic structure observed in PbTi0.48Zr0.52O3.41 The Rietveld refinements against the S-XRD data showed that this sample of (Bi0.5Na0.5)TiO3 contained 61.5(7)% Cc and 38.5(5)% R3c, which is in good agreement with the phase composition reported by Rao et al.22 (83% Cc and 17% R3c); the small difference between the two studies is likely due to differences in reaction conditions. Aksel et al. previously suggested that reaction conditions play a crucial role in the stability of the monoclinic Cc phase.17 In their study, diffraction patterns for 1100 °C calcined (Bi0.5Na0.5)TiO3 could be fitted equally well to either the monoclinic or the rhombohedral models whereas those from samples annealed at 400 °C could only be fitted to the monoclinic model. | ||
| Fig. 1 S-XRD pattern of (Bi0.5Na0.5)TiO3 fitted to various single-phase and two-phase models. The solid line represents the results of the Rietveld refinement and the vertical markers show the positions of the space group allowed Bragg reflections. The best fit to the pattern was obtained by the two phase Cc + R3c model. The insets highlight the fitting of the pseudocubic (123) reflections. | ||
| Model | R p | R wp | χ 2 |
|---|---|---|---|
| Cc-R3c | 3.380 | 4.482 | 1.641 |
| Cm-R3c | 3.496 | 4.570 | 1.842 |
| Cc | 3.859 | 5.362 | 2.885 |
| Cm | 5.025 | 7.106 | 4.831 |
| R3c | 4.202 | 5.805 | 3.228 |
NPD data were also collected from (Bi0.5Na0.5)TiO3 at 298 and 3 K using both 1.62 and 2.44 Å neutrons. These data were fitted using the same two-phase Cc + R3c model developed in the analysis of the S-XRD data, and the profiles recorded at 298 K are illustrated in Fig. 2. Selected results from the combined refinements are shown in Table 2. Bond distances for the monoclinic phase can be found in Table 3. The refined weight percentage of the monoclinic phase was found to be 64.2(1.2)% at 298 K and this increased slightly to 70.7(5)% on cooling to 3 K. The result at 298 K is similar to that obtained from the analysis of the S-XRD data (61.5(7)%). The refined structure showed significantly larger isotropic atomic displacement parameters of the A-site cations than the B-site at both 298 K and 3 K, consistent with Bi3+/Na+ disorder on the A-site. Refinement of the A-site occupancy showed that it is close to the expected 50% Bi3+/50% Na+ ratio in both the monoclinic and rhombohedral phases.
 | ||
| Fig. 2 Neutron diffraction patterns of (Bi0.5Na0.5)TiO3 collected at 298 K. The Rietveld refinements were performed on patterns collected at wavelengths of 1.62 Å (top) and 2.44 Å (bottom). The solid line represents the results of the Rietveld refinement and the vertical markers show the positions of the space group allowed Bragg reflections for the monoclinic Cc (upper markers) and rhombohedral R3c (lower markers) phases. | ||
| 298 Ka | 3 Kb | ||||
|---|---|---|---|---|---|
| Space group | Cc | R3c | Cc | R3c | |
| a χ 2 = 2.65. At 1.62 Å, Rp = 4.86%, Rwp = 5.70%. At 2.44 Å, Rp = 6.58%, Rwp = 7.84%. b χ 2 = 3.32. At 1.62 Å, Rp = 5.41%, Rwp = 6.72%. At 2.44 Å, Rp = 7.89%, Rwp = 9.44%. | |||||
| a (Å) | 9.5087(8) | 5.4916(6) | 9.4857(11) | 5.4778(8) | |
| b (Å) | 5.4829(5) | 5.4916(6) | 5.4730(4) | 5.4778(8) | |
| c (Å) | 5.5176(5) | 13.4921(16) | 5.5142(7) | 13.4850(14) | |
| β (°) | 125.101(5) | 125.050(6) | |||
| V (Å3) | 235.35(4) | 352.37(7) | 234.36(5) | 350.42(10) | |
| X (%) | 64.2(1.2) | 35.8(2.0) | 70.7(5) | 29.3(15) | |
| Bi/Na | X | 0 | 0 | 0 | 0 |
| Y | 1/4 | 0 | 1/4 | 0 | |
| Z | 0 | 0.253(2) | 0 | 0.242(6) | |
| U iso | 2.25(14) | 6.3(5) | 1.24(12) | 4.3(5) | |
| Ti | X | 0.2474(17) | 0 | 0.2362(19) | 0 |
| y | 0.2442(23) | 0 | 0.246(4) | 0 | |
| z | 0.7131(13) | −0.013(3) | 0.6998(12) | −0.01302 | |
| U iso | 0.53(9) | 9.3(9) | 0.24(17) | 4.0(1.0) | |
| O1 | x | −0.0056(20) | 0.123(2) | −0.0186(15) | 0.120(2) |
| y | 0.2004(16) | 0.305(3) | 0.2013(17) | 0.334(4) | |
| z | 0.4762(25) | 0.083(3) | 0.3965(17) | 0.072(5) | |
| U iso | 1.21(15) | 6.6(3) | 0.12(16) | 3.4(3) | |
| O2 | x | 0.2081(19) | 0.1980(23) | ||
| y | 0.4849(22) | 0.4790(24) | |||
| z | −0.0736(16) | −0.0704(33) | |||
| U iso | −0.01(13) | 1.06(16) | |||
| O3 | x | 0.2593(17) | 0.2618(22) | ||
| y | 0.9667(24) | 0.9804(25) | |||
| z | −0.0683(18) | −0.0703(33) | |||
| U iso | 1.81(24) | 0.91(24) | |||
| 298 K | 3 K | |
|---|---|---|
| Bi/Na–O1 | 2.47(14) | 2.42(15) |
| 2.60(8) | 2.58(17) | |
| 2.83(8) | 2.86(18) | |
| 2.93(14) | 2.97(15) | |
| Bi/Na–O2 | 2.41(3) | 2.39(6) |
| 2.45(3) | 2.45(7) | |
| 2.99(4) | 2.98(6) | |
| 3.04(4) | 3.06(6) | |
| Bi/Na–O3 | 2.44(3) | 2.38(5) |
| 2.52(3) | 2.48(5) | |
| 2.93(3) | 2.99(7) | |
| 2.97(3) | 3.04(7) | |
| Ti–O1 | 1.87(12) | 1.88(12) |
| 1.98(12) | 1.97(13) | |
| Ti–O2 | 1.90(1) | 1.89(2) |
| 2.02(1) | 1.99(2) | |
| Ti–O3 | 1.88(2) | 1.86(3) |
| 1.96(1) | 2.01(3) |
Fitted S-XRD patterns for various members in the (Bi0.5Na0.5)Ti1−xZrxO3 series are shown in Fig. 3. There is a general shift in the peaks to lower 2θ with increasing x consistent with an increase in the unit cell volume due to the progressive replacement of Ti4+ (ionic radius: 0.605 Å) by the larger Zr4+ (ionic radius: 0.72 Å).42 This increase is quantified through the Rietveld refinements and these results are summarized in Table 4 and Fig. 4a. Most compositions studied consisted of two phases (the exceptions being x = 0.3, 0.4, and 0.7). As shown by Fig. 4b, doping Zr4+ into the B site initially stabilizes the rhombohedral phase. This is confirmed by the disappearance of superlattice reflections characteristic of the monoclinic Cc phase (Fig. 3b). Surprisingly, there are quite small increases in the unit cell volume of the monoclinic phase (0.7%) at x = 0.1 compared to the rhombohedral phase (1.6%), suggesting that Zr4+ cations initially preferentially substitute into the rhombohedral phase. Refining the B site occupancy of (Bi0.5Na0.5)Ti0.9Zr0.1O3 suggested the rhombohedral phase was slightly Zr4+-rich (82.5(1.2)% Ti4+; 17.5(1.2)% Zr4+) and the monoclinic phase was Ti-rich (93.6(1.4)% Ti4+; 6.3(1.4)% Zr4+). Rao et al. has previously reported that the rhombohedral phase can be electrically poled in (Bi0.5Na0.5)TiO3.22 Here, the rhombohedral phase can be preferentially stabilized by doping the B site with a larger cation, but a correlation between these two observations is yet to be established. At x = 0.5, superlattice reflections consistent with an orthorhombic Pnma phase begin to form, and by x = 0.7 only the orthorhombic phase is present, which is within the range of the rhombohedral-orthorhombic phase boundaries reported by Rachakom et al. (x = 0.8) and Barick et al. (x = 0.6).26,27
 | ||
| Fig. 3 (a) The fitted S-XRD patterns of (Bi0.5Na0.5)Ti1−xZrxO3 (0 ≤ x ≤ 0.7). (b) An enlargement of the S-XRD patterns showing the change in the pseudo-cubic (013) reflection Δ and * represent superlattice reflections corresponding to the monoclinic Cc and orthorhombic Pnma phase, respectively. | ||
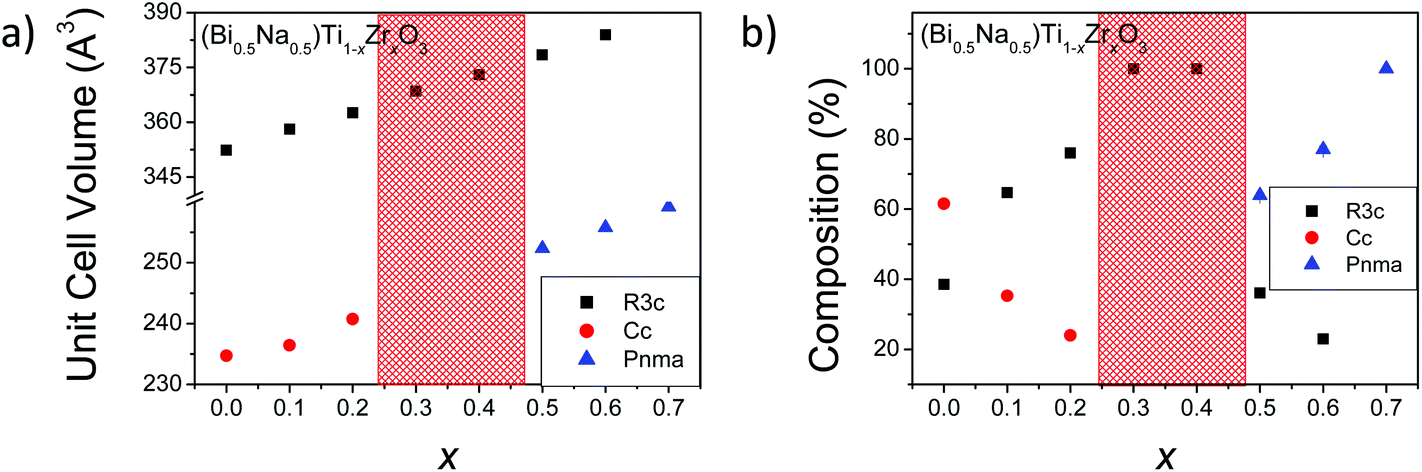 | ||
| Fig. 4 Plots of (a) unit cell volume and (b) phase composition as functions of x in the (Bi0.5Na0.5)Ti1−xZrxO3 solid solution. The shading illustrates the single-phase (rhombohedral) region in the solid solution. | ||
| x | Cc | R3c | Pnma | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a (Å) | b (Å) | c (Å) | β | X (%) | a (Å) | c (Å) | X (%) | a (Å) | b (Å) | c (Å) | X (%) | |
| 0 | 9.5156(2) | 5.4863(2) | 5.5049(2) | 125.243(3) | 61.5(7) | 5.48194(7) | 13.5421(3) | 38.5(5) | ||||
| 0.1 | 9.5872(5) | 5.4961(3) | 5.5078(3) | 125.439(3) | 35.3(5) | 5.5206(2) | 13.5674(5) | 64.7(7) | ||||
| 0.2 | 9.6770(4) | 5.5165(3) | 5.5420(4) | 125.538(3) | 24.0(5) | 5.5456(2) | 13.6126(5) | 76.0(8) | ||||
| 0.3 | 5.5787(2) | 13.6630(2) | 100 | |||||||||
| 0.4 | 5.6042(2) | 13.7156(3) | 100 | |||||||||
| 0.5 | 5.6316(1) | 13.7794(7) | 32.3(8) | 5.6455(1) | 7.9793(1) | 5.6012(1) | 63.9(9) | |||||
| 0.6 | 5.6580(4) | 13.849(2) | 23(1) | 5.6746(1) | 8.0189(1) | 5.6216(1) | 77(1) | |||||
| 0.7 | 5.7036(1) | 8.0535(1) | 5.6422(1) | 100 | ||||||||
XANES analysis
The Ti K-edge XANES spectra of representative members of the (Bi0.5Na0.5)Ti1−xZrxO3 solid solution (x = 0–0.5) are shown in Fig. 5a. For comparison, spectra of the corresponding members of the BaTi1−xZrxO3 solid solution were also collected (Fig. 5b). To remove effects from sample preparation and absorber concentration, all spectra are normalized so that the edge jump is equal to one. This ensures that changes in spectra features are due to electronic effects. Changes in the lineshape of the Ti K-edge of BaTi1−xZrxO3 solid solution are identical to those reported by Levin et al.28 There are two major features to the Ti K-edge, a main edge feature (labeled A) and a pre-edge feature (labeled B), which are highly sensitive to the oxidation state and coordination environment of Ti4+ cations.43 The main-edge feature corresponds to a dipole-allowed transition of a 1s electron into the unoccupied 4p states. There exist obvious differences in the lineshape of the main-edge between the two solid solutions, which are likely due to differences in the hybridization of Ti 4p states with the bonding states of nearest and next-nearest neighboring atoms (i.e. Bi 6p/Na 3p vs. Ba 6p states). Regardless of these changes it is evident that doping Zr4+ into both (Bi0.5Na0.5)TiO3 and BaTiO3 does not cause a shift in the Ti K-edge absorption edge energy, confirming that Ti remains tetravalent in both series. | ||
| Fig. 5 The Ti K-edge XANES spectra of representative members of the (a) (Bi0.5Na0.5)Ti1−xZrxO3 and (b) BaTi1−xZrxO3 solid solutions. Major features of the Ti K-edge pre-edge region of the (Bi0.5Na0.5)Ti1−xZrxO3 and BaTi1−xZrxO3 solid solutions are highlighted in (c) and (d). All Ti K-edge XANES spectra were collected in transmission mode. | ||
Although Zr4+ doping does not result in an appreciable shift in the energy of the Ti K-edge in either (Bi0.5Na0.5)Ti1−xZrxO3 or BaTi1−xZrxO3, there are noticeable changes in the shape of the pre-edge feature for both solid solutions. The pre-edge corresponds to a dipole-forbidden transition of a 1s electron into the unoccupied 3d states. There are four significant features (labeled B1–B4) observed in the pre-edge. Feature B1 is believed to correspond to a dipole-forbidden transition of a 1s electron into the t2g states for octahedrally coordinated Ti.44,45 In perovskite systems, feature B2 corresponds to a dipole-allowed transition of a 1s electron into hybridized 3d–4p states with eg symmetry.45 3d–4p mixing occurs in the presence of static or dynamic violation of the inversion symmetry of the absorbing Ti atom (i.e., Ti off-centering). As such, the intensity of B2 is sensitive to the displacement of Ti4+ cations from the center of the TiO6 octahedron. For BaTi1−xZrxO3, B2 shows a small decrease in intensity with increasing x, suggesting a decrease in Ti 3d/4p hybridization. As previously reported by Levin et al., this is consistent with a decrease in Ti off-centering with increasing x.28 In comparison, there is a small increase in the intensity of B2 in the (Bi0.5Na0.5)Ti1−xZrxO3 solid solution with increasing x, suggesting a small increase in Ti4+ cation displacement. However, the intensity change is smaller than that observed in BaTi1−xZrxO3, suggesting that Zr-doping has a much smaller affect on Ti4+ cation displacement in (Bi0.5Na0.5)Ti1−xZrxO3.
Information on the local ordering of Ti4+/Zr4+ cations can be inferred from features B3 and B4. Features B3 corresponds to the transition of a 1s electron into the unoccupied 3d states of neighboring Ti4+ cations.45 In both solid solutions, the intensity of B3 decreases as Zr4+ content increases, consistent with a decrease in the number of neighboring Ti 3d states. Coincidently, feature B4 increases in intensity with increasing x. This feature has been previously assigned as the transition of a 1s electron into the 4d states of neighboring Zr atoms.45 Overall, the intensity changes in B3 and B4 suggest that Zr4+ and Ti4+ cations are randomly distributed in (Bi0.5Na0.5)Ti1−xZrxO3 and that there is little or no local clustering of TiO6 and ZrO6 octahedra.
Information on the Zr4+ cation displacement within BO6 octahedra can, in principle, be obtained from the Zr L3-edge XANES spectra (Fig. 6), which corresponds to a dipole-allowed transition of a 2p3/2 electron into the unoccupied 3d states. The lineshape of the Zr L3-edge has recently been shown to be extremely sensitive to coordination environment of Zr4+ cations.29–32 There are two major features observed in the Zr L3-edge XANES spectra of (Bi0.5Na0.5)Ti1−xZrxO3 (Fig. 6a) and BaTi1−xZrxO3 (Fig. 6b). In octahedral systems, these features correspond to transitions to the t2g (lower energy) and eg (higher energy) states.46,47 Whilst the t2g peak appears as a single peak, splitting of the eg peak is apparent in both series. Similar peak splitting has been observed in the Zr L3-edge XANES spectra of PbTi1−xZrxO3.48 Ikeno et al. have recently suggested that splitting of the t2g and eg peaks in 6-coordinate zirconates can occur as a consequence of the displacement of Zr4+ from the center of the ZrO6 octahedra.47 This suggestion is consistent with the changes in the lineshape of the eg peak observed across the BaTi1−xZrxO3 series, as a consequence of changes in Zr off-centering. The eg peak becomes more symmetrical with increasing x in BaTi1−xZrxO3, indicating a reduction in eg peak splitting due to a decrease in Zr off-centering. No significant changes were observed in the lineshape of the Zr L3-edge spectra in (Bi0.5Na0.5)Ti1−xZrxO3 indicating that the off-center displacement of Zr4+ within the BO6 octahedra is essentially independent of the Ti/Zr ratio.
 | ||
| Fig. 6 The Zr L3-edge XANES spectra of representative members of the (a) (Bi0.5Na0.5)Ti1−xZrxO3 and (b) BaTi1−xZrxO3 solid solution. All spectra were collected in fluorescence mode. Due to absorption effects, all spectra were normalized to the height of the t2g peak to highlight changes in the shape of the eg peak. | ||
Conclusion
Our results from diffraction analysis of (Bi0.5Na0.5)TiO3 are in good agreement with those reported by Rao et al.22 S-XRD analysis confirms that (Bi0.5Na0.5)TiO3 forms both monoclinic (space group Cc) and rhombohedral (space group R3c) phases at room temperature. We have identified the evolutionary nature of the phase change in (Bi0.5Na0.5)Ti1−xZrxO3, as distinct from the discrete rhombohedral to orthorhombic phase transition suggested previously.26,27 The sequence of the phase changes is Cc-R3c (x = 0–0.2) → R3c (x = 0.3–0.4) → R3c-Pnma (x = 0.5–0.6) → Pnma (x = 0.7), i.e., Zr doping initially stabilizes the rhombohedral phase before the orthorhombic (space group Pnma) phase forms at x = 0.5. Although the phase changes were obvious from the diffraction analysis, XANES analysis of (Bi0.5Na0.5)Ti1−xZrxO3 showed that Zr doping has little effect on the off-center displacement of Ti4+/Zr4+ cations within the BO6 octahedra. In contrast to our previous studies on zirconate pyrchores,29–32 diffraction analysis proved to be superior in analyzing the structural changes in (Bi0.5Na0.5)Ti1−xZrxO3. This is likely due to similar local distortions within the BO6 octahedra in all three phases, resulting in similar degrees of p–d hybridization. Regardless, the important implication of our work is that the rhombohedral phase can be preferentially stabilized in (Bi0.5Na0.5)TiO3 by doping the B site with a larger cation, which has not been observed until now. A similar observation was made in our neutron analysis of (Bi0.5Na0.5)TiO3, where cooling resulted in increased stabilization of the monoclinic phase. Control over the phase composition of these materials is an important step in optimizing their electronic properties (i.e., maximizing the number of phases at the MPB). Further investigation is required to determine if it is possible to stabilize the monoclinic phase by chemical substitution (i.e., A site substitution).Acknowledgements
This work was, in part, performed at the Powder Diffraction beamline at the Australian Synchrotron with the assistance of Dr Helen Brand. We acknowledge the Australian Research Council and the Australian Institute of Nuclear Science and Engineering for support of this work. The work performed at the NSRRC was supported by the Australian Synchrotron International Access Program.References
- G. H. Haertling, J. Am. Ceram. Soc., 1999, 82, 797–818 CrossRef CAS PubMed.
- E. Ringgaard and T. Wurlitzer, J. Eur. Ceram. Soc., 2005, 25, 2701–2706 CrossRef CAS PubMed.
- P. K. Panda, J. Mater. Sci., 2009, 44, 5049–5062 CrossRef CAS.
- R. E. Cohen, Nature, 1992, 358, 136–138 CrossRef CAS.
- G. A. Smolenskii, V. A. Isupov, A. I. Agranovskaya and N. N. Krainik, J. Sov. Phys. Solid State, 1961, 2, 2651–2654 Search PubMed.
- M. Ahart, M. Somayazulu, R. E. Cohen, P. Ganesh, P. Dera, H. Mao, R. J. Hemley, Y. Ren, P. Liermann and Z. Wu, Nature, 2008, 451, 545–548 CrossRef CAS PubMed.
- B. Jaffe, W. R. Cook and H. Jaffe, Piezoelectric Ceramics, Academic, London, 1971 Search PubMed.
- L. Gao, Y. Huang, Y. Hu and H. Du, Ceram. Int., 2007, 33, 1041–1046 CrossRef CAS PubMed.
- K. Sakata, T. Takenaka and Y. Naitou, Ferroelectrics, 1992, 131, 219–226 CrossRef CAS.
- A. Sasaki, T. Chiba, Y. Mamiya and E. Otsuki, Jpn. J. Appl. Phys., 1999, 38, 5564–5567 CrossRef CAS.
- A. B. Kounga, S. T. Zhang, W. Jo and T. Granzow, Appl. Phys. Lett., 2008, 92, 222902 CrossRef PubMed.
- F. F. Guo, B. Yang, S. T. Zhang, X. Liu, L. M. Zheng, Z. Wang, F. M. Wu, D. L. Wang and W. W. Cao, J. Appl. Phys., 2012, 111, 124113 CrossRef PubMed.
- J. A. Zvirgzds, P. P. Kapostins, J. V. Zvirgzde and T. V. Kruzina, Ferroelectrics, 1982, 40, 75–77 CrossRef CAS.
- G. O. Jones and P. A. Thomas, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 168–178 CAS.
- J. Petzelt, S. Kamba, J. Fábry, D. Noujni, V. Porokhonsky, A. Pashkin, I. Franke, K. Roleder, J. Suchanicz, R. Klein and G. E. Kugel, J. Phys.: Condens. Matter, 2004, 16, 2719–2731 CrossRef CAS.
- S. Gorfman and P. A. Thomas, J. Appl. Crystallogr., 2010, 43, 1409–1414 CrossRef CAS.
- E. Aksel, J. S. Forrester, J. L. Jones, P. A. Thomas, K. Page and M. R. Suchomel, Appl. Phys. Lett., 2011, 98, 152901 CrossRef PubMed.
- E. Aksel, J. S. Forrester, J. C. Nino, K. Page, D. P. Shoemaker and J. L. Jones, Phys. Rev. B: Condens. Matter, 2013, 87, 104113 CrossRef.
- V. Dorcet and G. Trolliard, Acta Mater., 2008, 56, 1753–1761 CrossRef CAS PubMed.
- G. Trolliard and V. Dorcet, Chem. Mater., 2008, 20, 5074–5082 CrossRef CAS.
- S. Gorfman, A. M. Glazer, Y. Noguchi, M. Miyayama, H. Luo and P. A. Thomas, J. Appl. Crystallogr., 2012, 45, 444–452 CAS.
- B. N. Rao, A. N. Fitch and R. Ranjan, Phys. Rev. B: Condens. Matter, 2013, 87, 060102(R) CrossRef.
- H. Yokota, N. Zhang, A. E. Taylor, P. A. Thomas and A. M. Glazer, Phys. Rev. B: Condens. Matter, 2009, 80, 104109 CrossRef.
- R. S. Solanki, S. K. Mishra, A. Senyshyn, S. Yoon, S. Baik, N. Shin and D. Pandey, Appl. Phys. Lett., 2013, 102, 052903 CrossRef PubMed.
- K. Lily, K. Kumari, K. Prasad and K. L. Yadar, J. Mater. Sci., 2007, 42, 6252–6259 CrossRef CAS.
- A. Rachakom, P. Jaiban, S. Jiansirisomboon and A. Watcharapasorn, Nanoscale Res. Lett., 2012, 7, 57 CrossRef PubMed.
- B. K. Barick, R. N. P. Choudhary and D. K. Pradhan, Ceram. Int., 2013, 39, 5695–5704 CrossRef CAS PubMed.
- I. Levin, E. Cockayne, V. Krayzman, J. C. Woicik, S. Lee and C. A. Randall, Phys. Rev. B: Condens. Matter, 2011, 83, 094122 CrossRef.
- P. E. R. Blanchard, R. Clements, B. J. Kennedy, C. D. Ling, E. Reynolds, M. Avdeev, A. P. J. Stampfl, Z. Zhang and L. Y. Jang, Inorg. Chem., 2012, 51, 13237–13244 CrossRef CAS PubMed.
- E. Reynolds, P. E. R. Blanchard, B. J. Kennedy, C. D. Ling, S. Liu, M. Avdeev, Z. Zhang, G. J. Cuello, A. Tadich and L. Y. Jang, Inorg. Chem., 2013, 52, 8409–8415 CrossRef CAS PubMed.
- P. E. R. Blanchard, S. Liu, B. J. Kennedy, C. D. Ling, Z. Zhang, M. Avdeev, B. C. C. Cowie, L. Thomsen and L. Y. Jang, Dalton Trans., 2013, 42, 14875–14882 RSC.
- Z. Zhang, S. C. Middleburgh, M. de los Reyes, G. R. Lumpkin, B. J. Kennedy, P. E. R. Blanchard, E. Reynolds and L. Y. Jang, J. Phys. Chem. C, 2013, 117, 26740–26749 CAS.
- K. S. Wallwork, B. J. Kennedy and D. Wang, AIP Conf. Proc., 2007, 879, 879–882 CrossRef CAS PubMed.
- K. D. Liss, B. Hunter, M. Hagen, T. Noakes and S. Kennedy, Physica B, 2006, 385–86, 1010–1012 CrossRef PubMed.
- B. A. Hunter and C. J. Howard, RIETICA A Computer Program for Rietveld Analysis of X-Ray and Neutron Powder Diffraction Patterns, 1998 Search PubMed.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR 86-748, 1994.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- K. L. Tsang, C. H. Lee, Y. C. Jean, T. E. Dann, J. R. Chen, K. L. D'Amico and T. Oversluizen, Rev. Sci. Instrum., 1995, 66, 1812–1814 CrossRef CAS PubMed.
- T. E. Dann, S. C. Chung, L. J. Huang, J. M. Juang, C. I. Chen and K. L. Tsang, J. Synchrotron Radiat., 1998, 5, 664–666 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- D. M. Hatch, H. T. Stokes, R. Ranjan, R. Ragini, S. K. Mishra, D. Pandey and B. J. Kennedy, Phys. Rev. B: Condens. Matter, 2002, 65, 212101 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- T. Yamamoto, X-Ray Spectrom., 2008, 37, 572–584 CrossRef CAS.
- D. Cabaret, A. Bordage, A. Juhin, M. Arfaoui and E. Gaudry, Phys. Chem. Chem. Phys., 2010, 12, 5619–5633 RSC.
- R. V. Vedrinskii, V. L. Kraizman, A. A. Novakovich, P. V. Demekhin and S. V. Urazhdin, J. Phys.: Condens. Matter, 1998, 10, 9561–9580 CrossRef CAS.
- L. Galoisy, E. Pelegrin, M. A. Arrio, P. Ildefonse and G. Calas, J. Am. Ceram. Soc., 1999, 82, 2219–2224 CrossRef CAS PubMed.
- H. Ikeno, M. Krause, T. Höche, C. Patzig, Y. Hu, A. Gawronski, I. Tanaka and C. Rüssel, J. Phys.: Condens. Matter, 2013, 25, 165505 CrossRef PubMed.
- S. C. Ray, H. C. Hsueh, C. H. Wu, C. W. Pao, K. Asokan, M. T. Liu, H. M. Tsai, C. H. Chuang, W. F. Pong, J. W. Chiou, M. H. Tsai, J. M. Lee, L. Y. Jang, J. M. Chen and J. F. Lee, Appl. Phys. Lett., 2011, 99, 042909 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |