
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible CO exchange at platinum(0). An example of similar complex properties produced by ligands with very different stereoelectronic characteristics†
Sebastian J. K.
Forrest
,
Paul G.
Pringle
*,
Hazel A.
Sparkes
and
Duncan F.
Wass
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: paul.pringle@bristol.ac.uk; duncan.wass@bristol.ac.uk
First published on 10th September 2014
Abstract
The ligands 1,2-C6H4(CH2PtBu2)2 (La) and 1,2-C6H4(PtBu2)(CH2PtBu2) (Lb) displace norbornene (nbe) from [Pt(η2-nbe)3] to give [PtL(η2-nbe)] where L = La (1a) or Lb (1b); 1a is fluxional on the NMR timescale. Reaction of 1a,b with CO gives the corresponding monocarbonyls [PtL(CO)] where L = La (2a) or Lb (2b) which then react further, and reversibly, to give the dicarbonyls [PtL(CO)2] where L = La (3a) or Lb (3b). The CO interchange between 2a,b and 3a,b is compared with the only other such system (2f and 3f), which are complexes of (C2F5)2PCH2CH2P(C2F5)2 (Lf). Ethene reacts smoothly with 2a to give (4a) and H2 with 2a generates some [PtH2(La)]. Protonation of 2a gives [Pt(La)(H)(CO)][B(C6F5)4] (5a) whose crystal structure has been determined. Similarly protonation of 2b gives [Pt(Lb)(H)(CO)][B(C6F5)4] as a mixture of geometric isomers 5b–6b.
Introduction
The bulky o-xylenyl diphosphine La (Chart 1) was first prepared in 1976 by Shaw et al. who also reported cis-[PtH2(La)], the first example of a cis-dihydridoplatinum(II) complex.1 Spencer et al. later reported that treatment of complexes of the type [Pt(La)(η2-alkene)] with a Brønsted acid produced the first examples of β-agostic C–H–Pt complexes.2,3 More recently, La has been used in the commercialised Pd-catalysed ethene hydromethoxycarbonylation (Lucite Process) for the production of methyl methacrylate, shown in eqn (1).4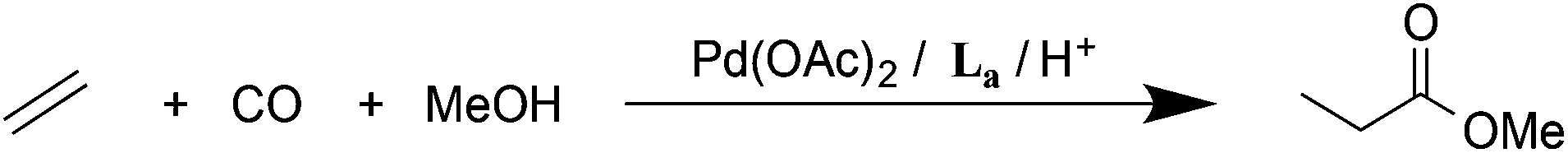 | (1) |
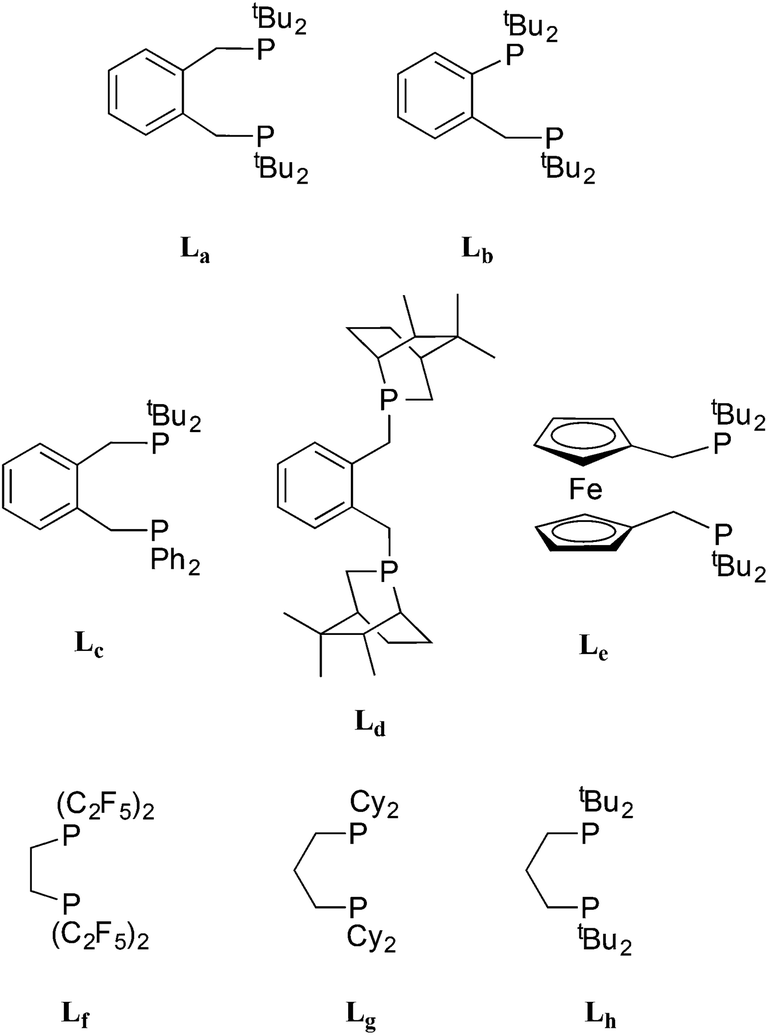 | ||
| Chart 1 Structures of ligands discussed. | ||
This application has galvanised academic interest in the Pd and Pt chemistry of La and related ligands (e.g. ligands Lb–Le shown in Chart 1)5–10 with the aim of understanding the special qualities of the La chelate. For example, using isotopically labelled MeOD and 13CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, Iggo et al.6,7 identified by NMR the key intermediates in the carbonylation cycle with Pd–La and Pt–La catalysts; they determined that the Pt-catalysis (Scheme 1) is inhibited by the reversible binding of CO to the Pt(II) intermediates while the corresponding Pd–La cycle is not similarly inhibited because of the lower affinity of Pd(II) for CO.
CH2, Iggo et al.6,7 identified by NMR the key intermediates in the carbonylation cycle with Pd–La and Pt–La catalysts; they determined that the Pt-catalysis (Scheme 1) is inhibited by the reversible binding of CO to the Pt(II) intermediates while the corresponding Pd–La cycle is not similarly inhibited because of the lower affinity of Pd(II) for CO.
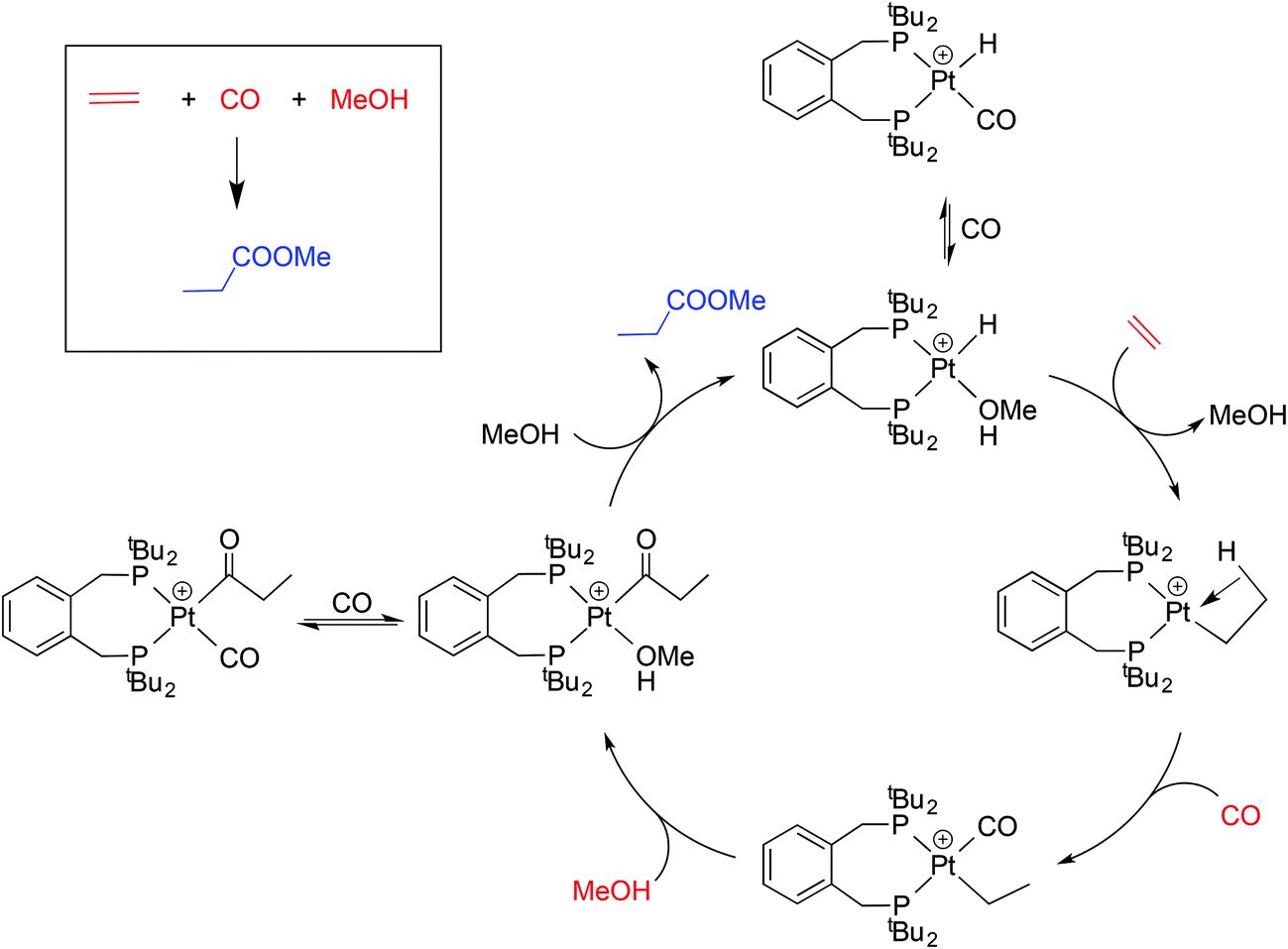 | ||
| Scheme 1 Outline of the mechanism of Pt-catalysed hydromethoxycarbonylation of ethene7 including two exocyclic equilibria. | ||
It has been established that the activity and longevity of the Pd-catalyst for carbonylation can be improved by ligand modification.6 For example, we have shown that changing the backbone from xylenediyl in La to toluenediyl in Lb led to a more active Pd-catalyst.8 This has prompted us to investigate and compare the coordination chemistry of La and Lb, particularly that which may be relevant to the ethene hydromethoxycarbonylation catalysis (eqn (1)).9
The dichloroplatinum(II) complexes of La and Lb have been shown to be fluxional on the NMR timescale.11 The conformations of the chelate rings in [PtCl2(L)], as determined by X-ray crystallography, are depicted as A (L = La) and B (L = Lb) in Chart 2 and the observed fluxionality is associated with ring inversion.
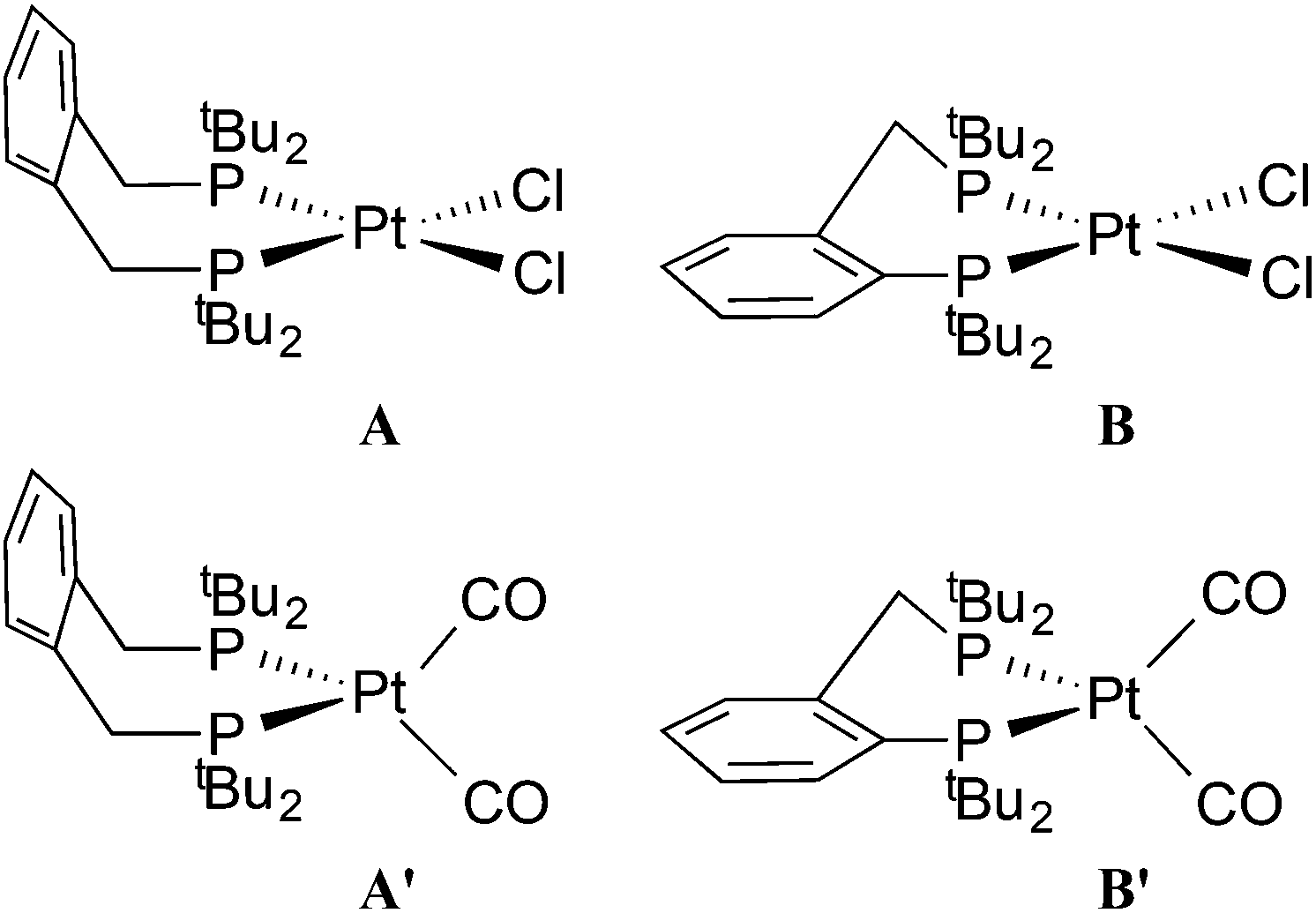 | ||
| Chart 2 Conformations of the chelate rings in square planar (A and B) and tetrahedral (A′ and B′) complexes. | ||
Here we present the characterisation of Pt(0)–carbonyl complexes of the electron-rich, bulky ligands La and Lb, that have the property of reversible CO-interchange between three- and four-coordinate complexes, only previously observed with Pt(0) complexes of the electron-poor Lf. The reactions of the carbonyl complexes of La and Lb with ethene, H2 and H+ are also described and the relevance of this chemistry to the carbonylation catalysis shown in Scheme 1 is discussed.
Results and discussion
The platinum(0) chemistry of La and Lb is summarised in Scheme 2. Addition of 1 equiv. of La or Lb to [Pt(η2-nbe)3] (nbe = norbornene) gave the corresponding [Pt(L)(η2-nbe)] (1a, L = La; 1b, L = Lb). The 31P NMR spectrum of the previously reported21a at −60 °C in toluene, showed 2 singlets in the ratio of ca. 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (δ 49.2 ppm JPtP = 3325 Hz and δ 47.3 ppm JPtP = 3308 Hz) which coalesce at 0 °C, consistent with the interchange of the diastereoisomeric rotamers 1a and 1a′ shown in eqn (2). The 1H NMR spectrum of 1a at −40 °C shows two multiplets for the diphos CH2 which coalesce at −20 °C and appear as a broad singlet (w1/2 = 23 Hz) at room temperature; only one A9XX′ signal for the tBu groups was discerned across the temperature range −40 to +20 °C indicating the insensitivity of this signal to the inequivalence of these groups. Two mechanisms can be postulated for the interchange shown in eqn (2): chelate ring inversion and Pt–nbe bond rotation. Both are plausible since there is precedent for Pt–La chelate ring inversion on the NMR timescale8–11 and slow rotation about the Pt–nbe bonds in [Pt(Ld)(η2-nbe)], a complex of the xylenediyl ligand Ld (Chart 1), has been observed.12
:3 (δ 49.2 ppm JPtP = 3325 Hz and δ 47.3 ppm JPtP = 3308 Hz) which coalesce at 0 °C, consistent with the interchange of the diastereoisomeric rotamers 1a and 1a′ shown in eqn (2). The 1H NMR spectrum of 1a at −40 °C shows two multiplets for the diphos CH2 which coalesce at −20 °C and appear as a broad singlet (w1/2 = 23 Hz) at room temperature; only one A9XX′ signal for the tBu groups was discerned across the temperature range −40 to +20 °C indicating the insensitivity of this signal to the inequivalence of these groups. Two mechanisms can be postulated for the interchange shown in eqn (2): chelate ring inversion and Pt–nbe bond rotation. Both are plausible since there is precedent for Pt–La chelate ring inversion on the NMR timescale8–11 and slow rotation about the Pt–nbe bonds in [Pt(Ld)(η2-nbe)], a complex of the xylenediyl ligand Ld (Chart 1), has been observed.12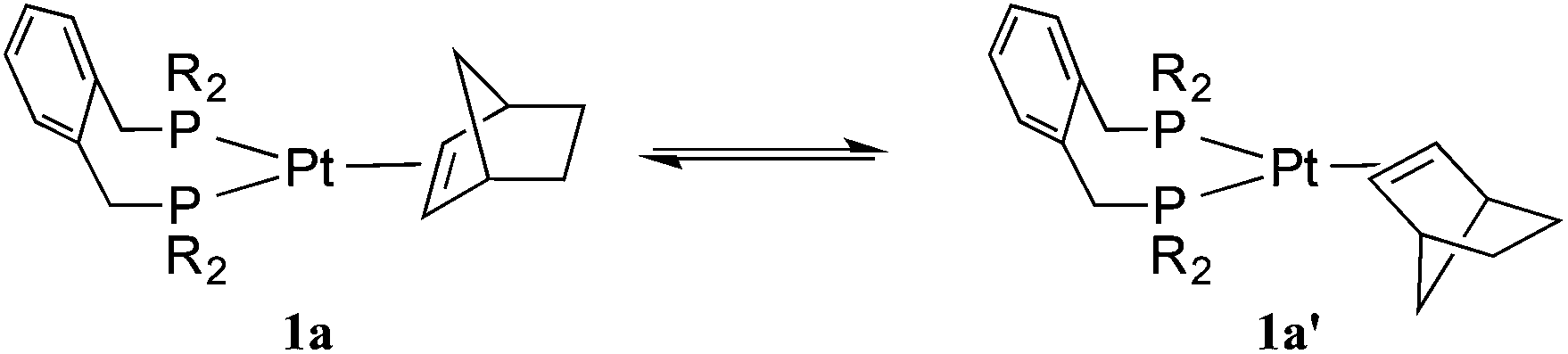 | (2) |
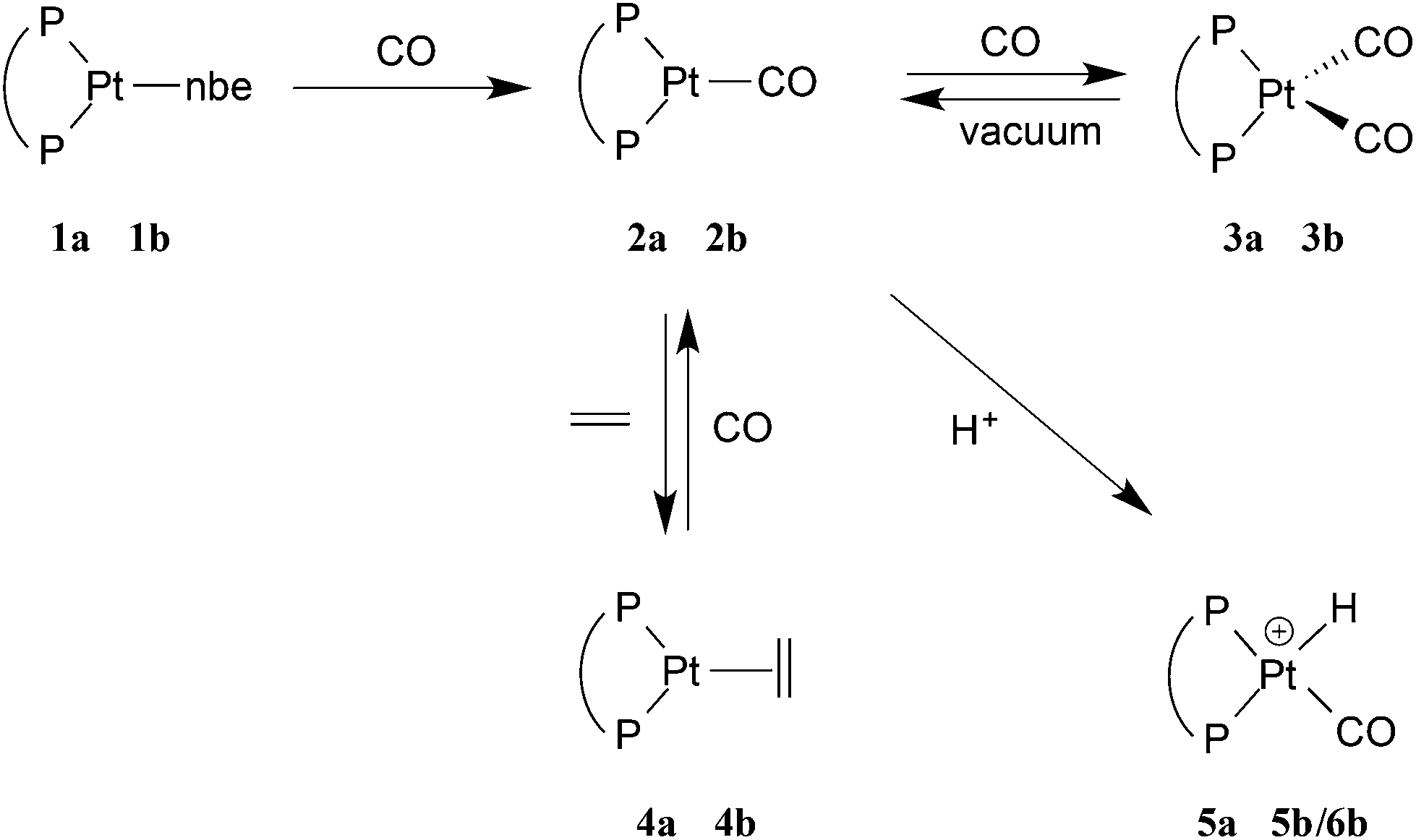 | ||
| Scheme 2 When diphos = La, the complex is denoted na, and when diphos = Lb, the complex is denoted nb. | ||
In the case of 1b, its 31P NMR spectrum shows sharp doublets over the whole temperature range of +23 to −90 °C which we interpret as due to there being a thermodynamic preference for one of the rotamers analogous to those shown in eqn (2). The 1H NMR spectrum of 1b showed 2 multiplets for the diphos CH2 and 4 sharp A9XX′ multiplets for the tBu groups, consistent with the presence of a predominant isomer.
The displacement of the nbe from 1a and 1b to give the monocarbonyls 2a and 2b was achieved by bubbling CO through their toluene solutions and removing the solvent with the displaced nbe by evaporation to dryness. The colourlessness of 1a/1b contrasts with the vivid orange-red of 2a/2b.
Complexes 2a and 2b exhibit one ν(CO) band at 1907 and 1898 cm−1 respectively, (cf. for [Pt(PCy3)2(CO)], ν(CO) = 1916 cm−1).13 The 31P NMR spectrum of 2a shows the expected singlet at 70.0 ppm with a 1J(PPt) of 3647 Hz and its 1H NMR spectrum shows one broad signal for the diphos CH2 group and one A9XX′ multiplet for the tBu groups, as expected for a conformationally labile chelate.8–10 Unexpectedly, the 31P NMR spectrum of 2b showed a single broad signal (w1/2 = 40 Hz) at 78.5 ppm with 1J(PPt) of 3450 Hz which below −20 °C is resolved into an AB pattern with δ 77.9 and 76.4, 2J(PP) = 93 Hz and 1J(PPt) = 3556 and 3250 Hz respectively (see Fig. 1).
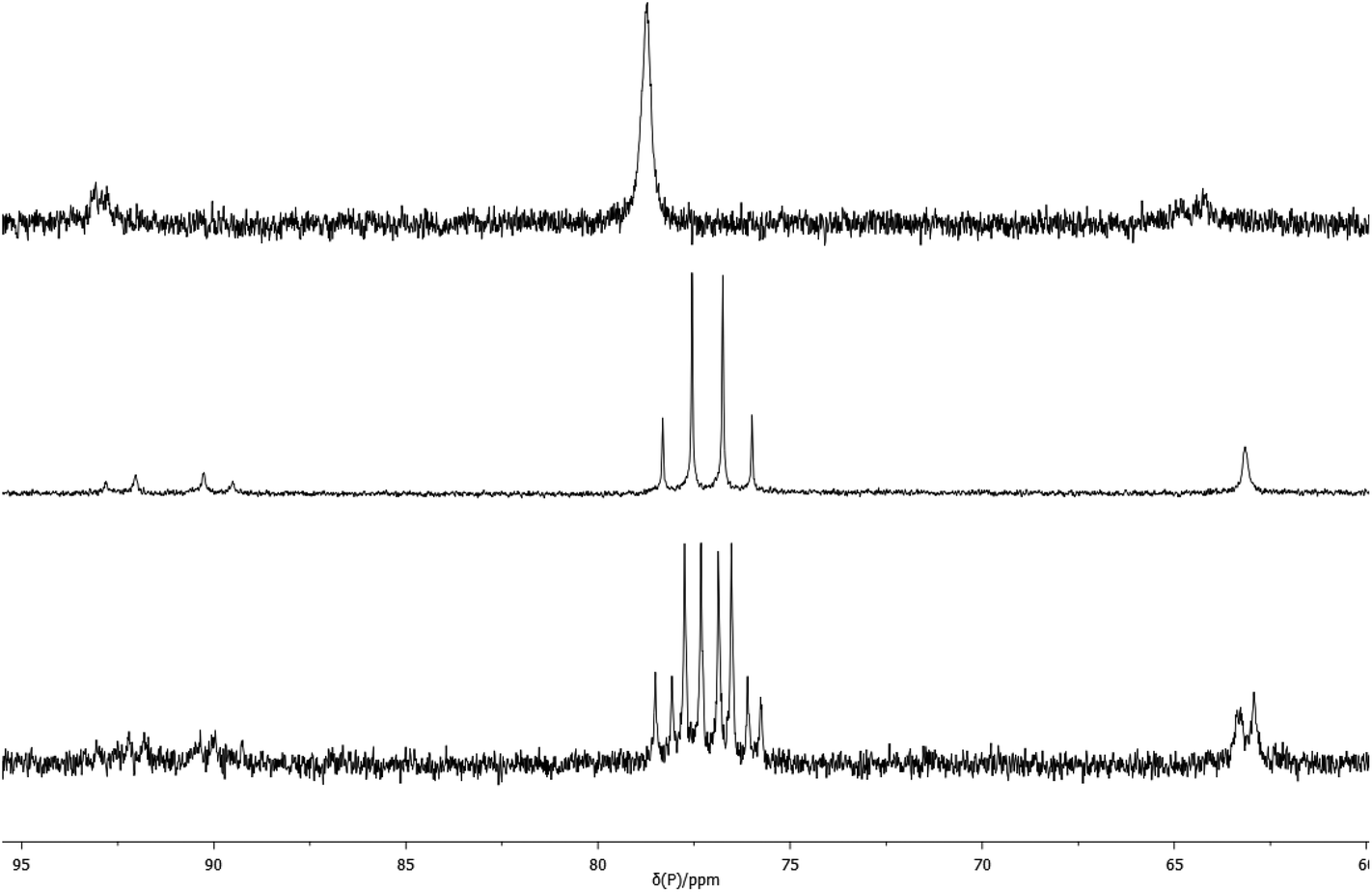 | ||
| Fig. 1 Variable temperature 31P NMR spectra: top, 2b, room temperature; middle, 2b, −90 °C; bottom, 2b*, −90 °C. | ||
The labelled compounds [Pt(L)(13CO)] (2a*, L = La; 2b*, L = Lb) were prepared in order to confirm their structures and to probe further the apparent fluxionality of 2b. Treatment of 2a and 2b with 13CO gave the labelled complexes 2a* and 2b*. The 31P NMR spectrum of 2a* at ambient temperature is a doublet with 2J(PC) = 45 Hz and its 13C NMR spectrum shows a triplet at 229 ppm with 1J(PtC) = 2096 Hz and the same 2J(PC) of 45 Hz; these spectra support the monocarbonyl structure assigned to 2a. The 31P NMR spectrum of 2b* at −90 °C showed an ABX pattern with 195Pt satellites (see Fig. 1) and its 13C NMR spectrum showed a doublet of doublets at 235 ppm with 2J(PC) = 52 and 41 Hz and 1J(PtC) = 2136 Hz; again, these spectra are consistent with the monocarbonyl structure assigned to 2b. The ambient temperature 31P and 13C NMR spectra of 2b* resembled the spectra for the unlabelled 2b, being broad and unresolved. Above room temperature, the 31P NMR spectrum of 2b* broadens progressively until at +100 °C, w1/2 ∼ 400 Hz. In the 13C NMR spectra of 2b* measured between +40 and +100 °C, no CO signal was observed at all presumably because of its broadness. When the high temperature NMR samples were cooled to ambient temperature, the 31P and 13C NMR spectra reassumed their original forms.
It is not obvious what the source of the line-broadening is in the 31P and 13C NMR spectra of 2b at ambient temperature and above. If the conformation of the 6-membered chelate in 2b is B/B′-like (see Chart 2), then 2b would exist as a pair of enantiomers (with identical 31P shifts) and therefore interchange between such conformers would not contribute to the line broadening. If intermolecular CO exchange were taking place, P–C coupling would be lost in the labelled 2b* which apparently it is, but this observation is inconclusive because the large observed NMR line-width would swamp the 2J(PC) of 52 and 41 Hz (measured at −90 °C). Nevertheless, one explanation for the NMR behaviour is that at elevated temperatures, CO exchange is taking place perhaps via an analogue of [Pt2(CO)(μ-CO)(Lg)2] (see Chart 1 for structure of Lg) reported by Mezailles et al.14 which is in equilibrium with 2b.
The dicarbonyl complexes, [PtL(CO)2] (3a, L = La; 3b, L = Lb) were generated when solutions of 2a and 2b were pressurised with 2 atm of CO. The solid state IR spectrum of 3a showed ν(CO) bands at higher frequencies (1971 and 1931 cm−1) than the 1907 cm−1 for 2a, consistent with the two CO ligands in 3a sharing the electron density from the platinum(0). The 31P NMR spectrum of 3a at room temperature showed no discernable resonances but at −90 °C, a singlet at 35.3 ppm with 1J(PPt) of 3107 Hz was observed. At +40 °C, the only 31P NMR signal observed was for 2a, showing that CO dissociation from 3a occurs readily and this explains the great broadness of the signal at ambient temperatures. The labelled complex [Pt(La)(13CO)2] (3a*) was prepared from 3a and 13CO and its 13C NMR spectrum at −90 °C had multiplets at 187.1 and 186.6 ppm with 1J(CPt) of 1832 and 1900 Hz respectively which were assigned to inequivalent Pt–CO ligands. The 2J(CP) coupling was approximately 4 Hz, leading to multiplets in the 31P and 13C NMR spectra. The inequivalence of the CO ligands detected in the low temperature 13C NMR spectrum of 3a* is consistent with an A′-type conformation of the 7-membered chelate (see Chart 2) giving rise to CO ligands being syn and anti to the phenylene of the chelate.
The solid state IR spectrum of 3b showed ν(CO) bands at higher frequencies (1961 and 1915 cm−1) than the 1898 cm−1 in 2b. The 31P NMR spectrum of 3b at ambient temperatures showed 2 sets of broad singlets at 44.4 and 42.7 ppm with 1J(PPt) = 3106 and 3818 Hz respectively. The 13C NMR spectrum of the labelled complex [Pt(Lb)(13CO)2] (3b*) had two signals at 187.1 and 186.0 ppm with 1J(CPt) of 1930 and 1850 Hz respectively; the C–P coupling was not resolved in the 31P or 13C NMR spectra of 3b*. According to solid-state IR spectroscopy (see Fig. 2), when red solid monocarbonyl 2b was subjected to a CO atmosphere, it was converted to yellow 3b and this was reversed upon application of a vacuum to powdered 3b.
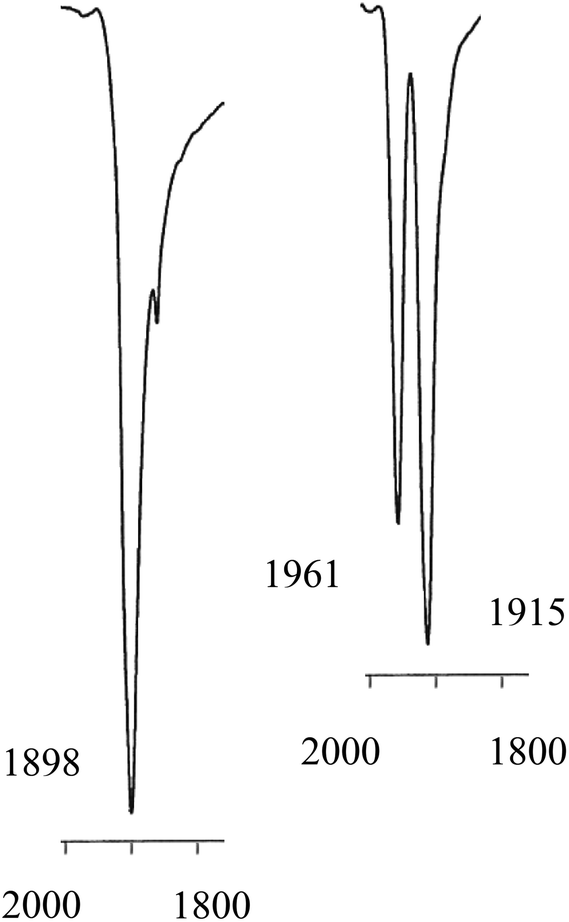 | ||
| Fig. 2 Solid state IR spectra in the ν(CO) region for monocarbonyl 2b (left) and dicarbonyl 3b (right). | ||
Dissociation of CO from the dicarbonyls 3a/3b to give 2a/2b occurred slowly when their solutions were stirred under a N2 atmosphere and rapidly when solutions were put under vacuum, presumably due to the dissolved CO being removed under the reduced pressure. The uptake of CO by 2a/2b (eqn (3)) is notable because of its rarity13 and reversibility.14 There are several examples of 18-electron complexes of the type [Pt(CO)2(PR3)2]13,15 and [Pt(CO)2(diphos)]14,16 including the complex where diphos = Lh17 (see Chart 1) which has νCO values (1912, 1960 cm−1) closely similar to those for 3b (see eqn (3)); the bulk and bite angles of Lh and Lb should be similar. However there is only one previously reported example of a 16-electron [Pt(CO)(diphos)] complex, 2f, which Roddick et al.18 reported undergoes a similar CO interchange between 2f and 3f (eqn (3)). The explanation that was given for the stability of 2f relative to 3f was that the high π-acceptor capacity of the fluorophos ligand Lf efficiently delocalises the electron-density on the Pt(0); this is supported by the high νCO values for 2f and 3f (see eqn (3)). A similar argument is not tenable for 2a/2b since La/Lb are strong σ-donors, as reflected in the low νCO values for their carbonyl complexes (see eqn (3)). The stability of the coordinatively unsaturated 2a/2b is therefore associated with the large bulk of La/Lb. This steric argument can be extended to explain why CO dissociation from 3a appears to be more facile than from 3b, since La is more sterically demanding than Lb.11 The complementary explanations for the equilibria shown in eqn (3) for La,bvs.Lf exemplify how ligands with very different stereoelectronic properties can produce complexes with similar properties.
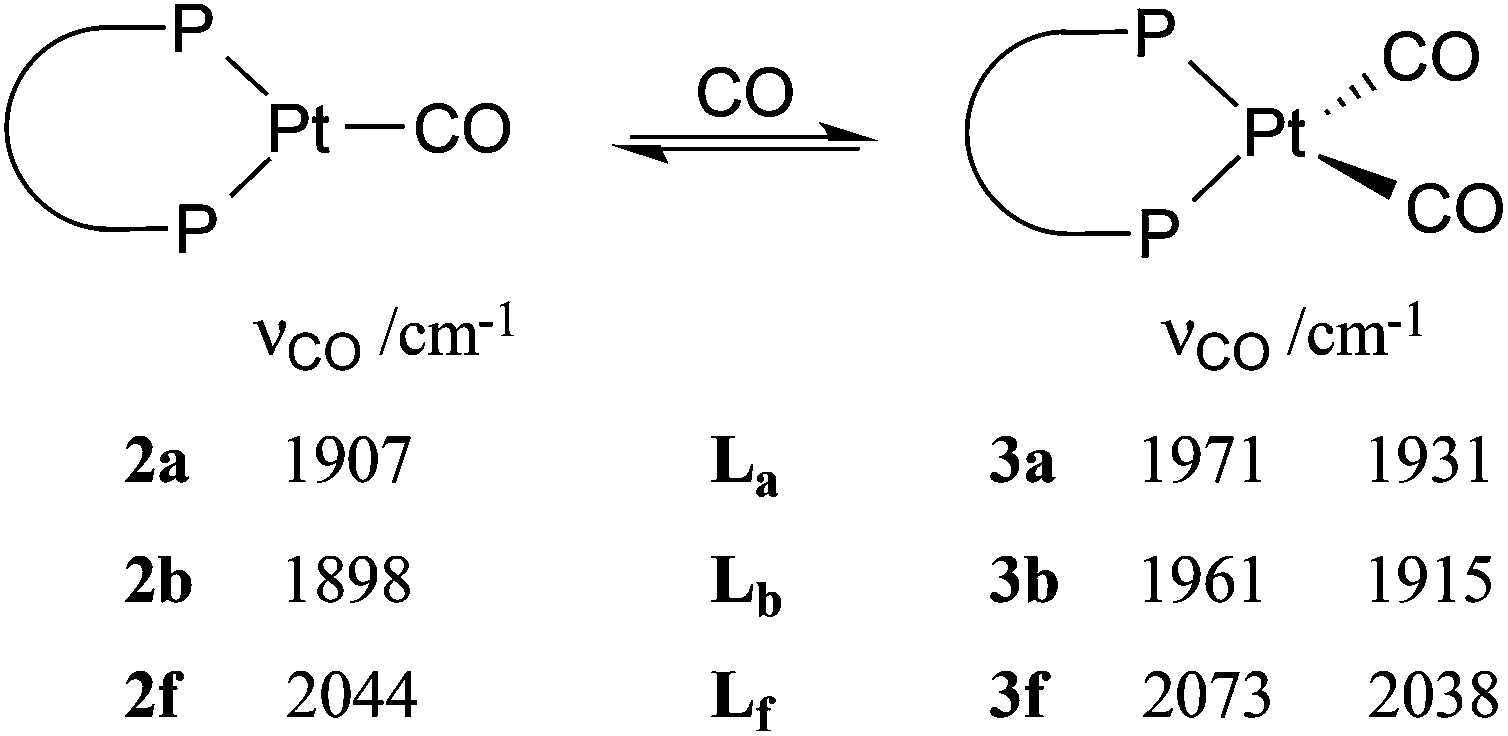 | (3) |
When a toluene solution of 2a was stirred under an atmosphere of H2 for 5 days, the cis-[PtH2(La)] was formed in approximately 50% NMR yield (along with other uncharacterised products), as shown by the close matching of the NMR data (1H: δ −3.8 ppm, JPtH = 1004 Hz; 31P: δ 45.7 ppm, JPtP = 2109 Hz) with the literature values for this compound.1
The CO ligands in 2a were displaced by ethene to give 4a by repeated vacuum/ethene-addition cycles (see Experimental) and this transformation was readily reversed by the application of a CO atmosphere to 4a (Scheme 2). Complex 4a was characterised by matching of the NMR data with those previously reported for this complex.19 Under similar conditions to those used to generate 4a, 2b reacted with ethene but the product, according to the 31P NMR spectrum at −90 °C, was a mixture containing 2b (50%), 3b (30%) and a third species (20%) whose 31P NMR parameters (60.9 with 1J(PPt) = 3288 Hz, 2J(PP) = 45 Hz; 57.9 1J(PPt) = 3288 Hz) led us to tentatively assign this minor product to 4b, although we have not isolated it.
In the carbonylation catalytic cycle shown in Scheme 1, neutral species are not involved in the core cycle because the reaction is carried out in an acidic medium. For this reason, we investigated the protonation of the monocarbonyl complex 2a. Treatment of the monocarbonyl complex 2a with 1 equivalent of [(Et2O)2H][B(C6F5)4]20 in chlorobenzene gave a product assigned structure 5a (Scheme 2) on the basis of its 1H NMR spectrum which showed a signal at −4.7 ppm with J(PtH) = 736 Hz characteristic of a hydride. A band at 2092 cm−1 in its IR spectrum is typical of a cationic Pt(II)–CO.21 The NMR spectroscopic data for 5a match well those reported by Iggo et al.7 for the triflate salt of 5a which they characterized in solution only.
Crystals of 5a suitable for X-ray crystallography grew from its CH2Cl2 solution, crystallising in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two [Pt(La)(H)(CO)][B(C6F5)4] moieties in the asymmetric unit, i.e. Z′ = 2. Both of these moieties have essentially the same geometrical conformation and selected bond lengths and angles are listed below Fig. 3. The location of the hydride was not determined directly from the data, but its position was inferred from the metal geometry and located accordingly. The P1–Pt1–P2 bite angle is 104.00(12)° and the P3–Pt2–P4 bite angle is 104.74(12)° and all four of the half cone angles are ∼117°.
with two [Pt(La)(H)(CO)][B(C6F5)4] moieties in the asymmetric unit, i.e. Z′ = 2. Both of these moieties have essentially the same geometrical conformation and selected bond lengths and angles are listed below Fig. 3. The location of the hydride was not determined directly from the data, but its position was inferred from the metal geometry and located accordingly. The P1–Pt1–P2 bite angle is 104.00(12)° and the P3–Pt2–P4 bite angle is 104.74(12)° and all four of the half cone angles are ∼117°.
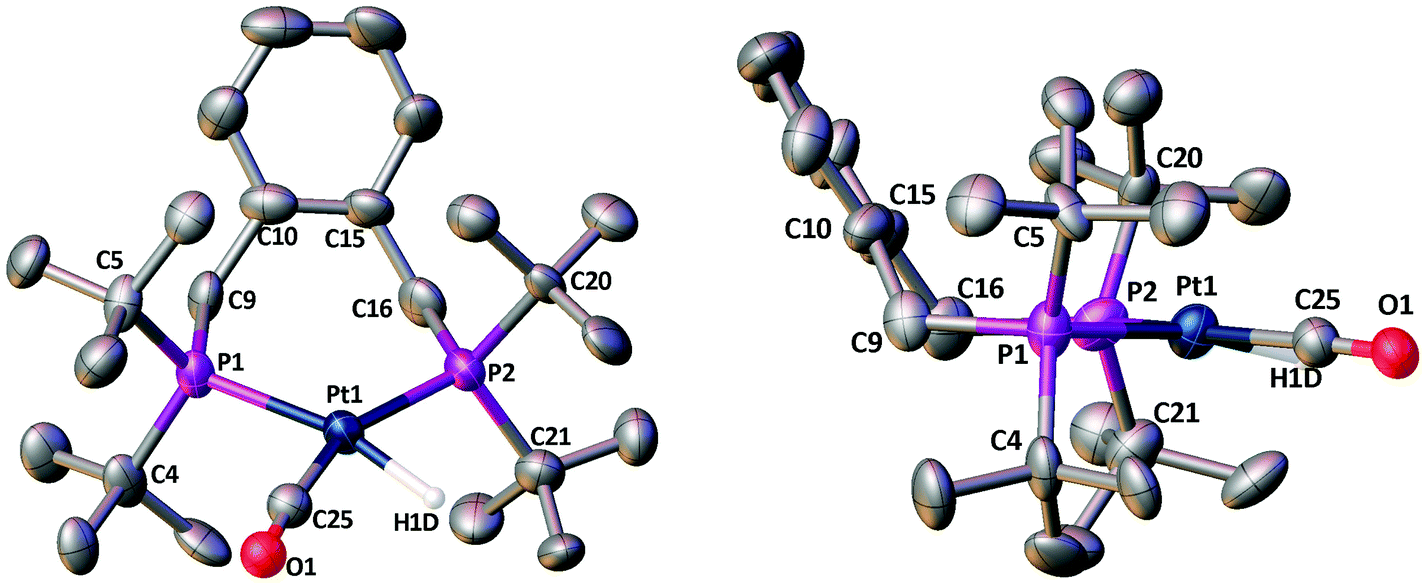 | ||
| Fig. 3 Crystal structure of [Pt(La)(H)(CO)][B(C6F5)4] (5a). The view on the right shows the chelate ring conformation. Only one of the two Pt-containing moieties in the asymmetric unit is shown and only one position of the disordered CO/H combination. For clarity all of the hydrogen atoms (apart from the hydride H1D) are omitted along with the [B(C6F5)4] counterions. Selected bond lengths (Å) and angles (°): Pt1−P1 2.366(3); Pt1−P2 2.328(3); Pt1−C25 1.900(14); Pt1−H1D 1.660(3); P1−C4 1.886(13); P1−C5 1.869(12); P1−C9 1.818(11); P2−C16 1.835(13); P2−C20 1.888(12); P2−C21 1.886(14); P1−Pt1−P2 104.00(12); C25−Pt1−P1 98.4(5); C25−Pt1−P2 157.5(5). | ||
The chelate conformations for xylenediyl diphos chelates can be described in terms of the two M–P–C–C(Ph) torsion angles. There are currently 12 Pt and 26 Pd xylenediyl diphos chelates in the Cambridge Crystallographic Database (CSD)22, 35 of which have structures where the two M–P–C–C(Ph) torsion angles have similar absolute values and the ring conformation is half-chair-like with an essentially coplanar MP2C2 component as depicted as A in Chart 2. In the remaining 3 structures (HUXCIN,23 HUXCEJ23 and IHETOF24) the chelate rings adopt a twist-boat-like conformation, with two different Pt–P–C–C(Ph) torsion angles of approximately ±50° and ±14°. The conformation of the Pt–La ring in 5a (see Fig. 3) matches the majority of those in the CSD, with Pt–P–C–C(Ph) torsion angles for Pt1 of −54.5(12)° and 65.0(11)° and for Pt2 of 57.9(11)° and −57.7(11)°.
Protonation of 2b gave a mixture of geometric isomers of hydridocarbonyl complexes where the position of the H ligand relative to the P-aryl is cis (5b) or trans (6b). Evidence for the two isomers in solution comes from the 1H NMR spectrum which showed 2 hydride signals at −2.3 and −4.3 ppm in a 1:4 ratio and the 31P NMR spectrum showed two sets of signals; the NMR data for the two species are similar and therefore it was not possible to assign the major isomer (see Experimental for the data). The solid state IR spectrum of the 5b–6b mixture shows 2 ν(CO) bands at 2102 and 2094 cm−1.
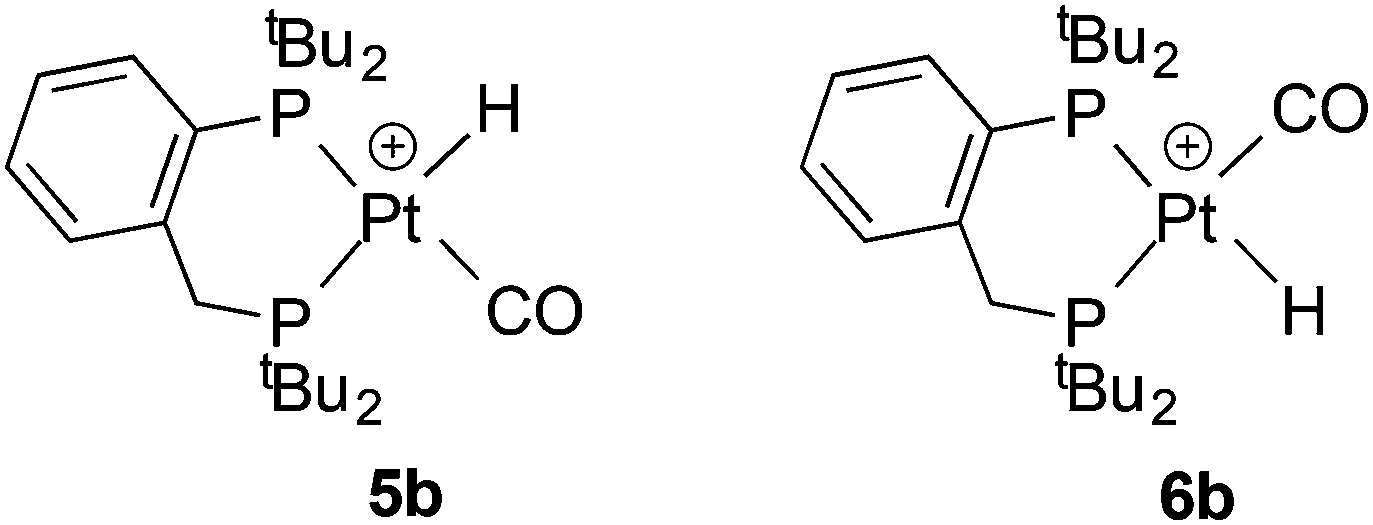
Conclusion
We have shown here that the mono- and dicarbonylplatinum(0) complexes 2a/2b and 3a/3b are readily formed and interconvert. The stability of the coordinatively unsaturated [Pt(CO)(diphos)] species 2a/2b relative to 3a/3b is associated with the steric congestion provided by the bulky diphos ligands La and Lb. The only related [Pt(CO)n(diphos)] species reported (2f/3f) feature the electron-poor fluorinated diphos ligand Lf (which has modest steric bulk)25 where the explanation given for the stability of the monocarbonyl is electronic – the π-acceptor properties of the diphos ligand destabilises the corresponding [Pt(CO)2(diphos)]. This is a textbook example of ligands with very different stereoelectronic properties producing similar outcomes in terms of the properties of their complexes.The hydromethoxycarbonylation of ethene is efficiently catalysed by Pd-complexes of La and Lb and Iggo et al.7 have shown that the analogous organoplatinum chemistry is relevant in the study of the mechanism of the catalysis. Ethene reacts with the coordinatively unsaturated 2a to give 4a presumably via the 18-electron tetrahedral intermediate X (Scheme 3). The protonation of the 2a/2b to give the cationic hydridocomplexes 5a/5b (and the geometric isomer 6b) is pertinent because the catalysis is carried out at low pH. It is possible that complexes of the type 2–5 described above are present in non-productive equilibria during the Pt-catalysed carbonylation shown in Scheme 1; their place is shown in Scheme 3.
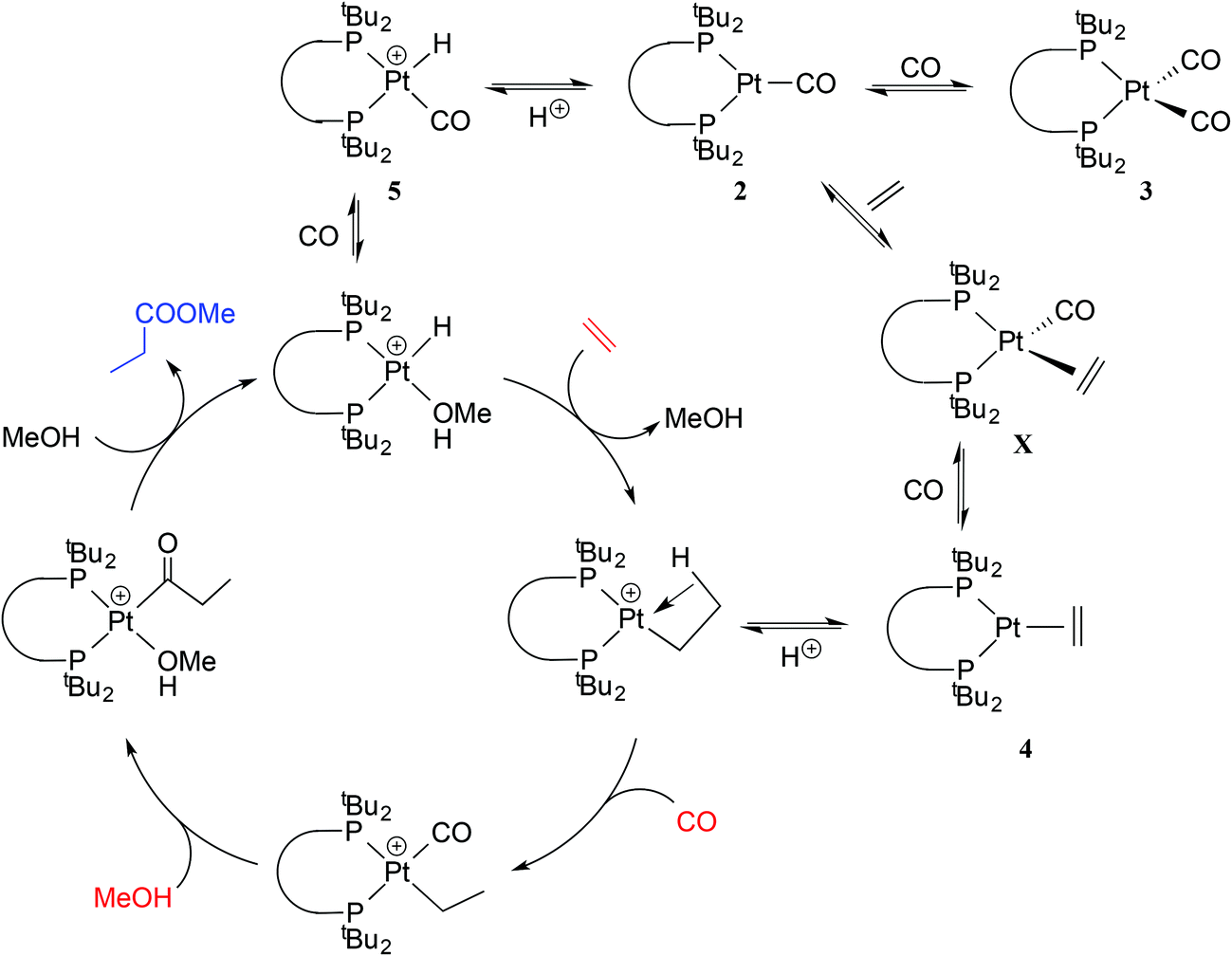 | ||
| Scheme 3 Potential role for complexes 2–5 in the Pt-catalysed methoxycarbonylation mechanism. | ||
Experimental
Unless otherwise stated, all reactions were carried out using standard Schlenk line and dry box techniques. Dry N2-saturated solvents were collected from a Grubbs system in flame and vacuum-dried glassware. Deuterated solvents were dried and distilled from CaH2. Pentane was dried over 4 Å molecular sieves and N2-saturated by repeated freeze, vacuum and thawing cycles. The complex [Pt(η2-nbe)3]26 was prepared by literature methods. Ligand La was obtained from Lucite International. Ligand Lb was prepared as previously described.11 CO, C2H4 and H2, were used as obtained from BOC and 13CO was used as obtained from Aldrich. NMR spectra were recorded on a Jeol ECP (Eclipse) 300 or a Varian VNMR S500 spectrometer. Chemical shifts are referenced relative to high frequency of Si(CH3)4 (1H or 13C), 85% H3PO4 (31P), and CF3Cl (19F). Infrared spectra were obtained using a Perkin Elmer 1600 series FTIR. Mass spectrometry was carried out by the Mass Spectrometry Service at the University of Bristol. Elemental analyses were carried out by the Microanalytical Laboratory at the University of Bristol.Preparation of [Pt(La)(η2-nbe)] (1a)
This was made according to the method of Spencer et al. and characterised by comparison of the NMR spectra with the reported data.2 The spectra at low temperature have not been previously reported. 31P{H} NMR (202 MHz, C6D5CH3, −60 °C): δ 49.2 (s, 1J(PtP) = 3251 Hz), δ 47.3 (s, 1J(PtP) = 3308 Hz).Preparation of [Pt(Lb)(η2-nbe)] (1b)
A solution of Lb (0.089 g, 0.26 mmol) in toluene (5 mL) was added in one portion to a solution of [Pt(η2-nbe)3] (0.125 g, 0.262 mmol) in toluene (5 mL) at −78 °C and the resulting mixture was stirred for 2 h, allowed to warm to room temperature and then stirred for a further 16 h. The volatiles were then removed under reduced pressure to yield an off-white solid that was dissolved in pentane (1 mL) and the product crystallized at −78 °C. The supernatant was removed by cannula and the white solid dried under vacuum (0.149 g, 0.222 mmol, 86%). Elemental analysis (calcd for C30H52P2Pt): C: 54.17 (53.80) H: 7.83 (7.39). ESI accurate mass spectrum: (calcd for C30CH52P2Pt 669.3190) Mr – 669.3184. 31P{H} NMR (202 MHz, C6D6): δ 63.9 (d, 1J(PPt) = 3157 Hz, 2J(PP) = 45 Hz), 57.6 (d, 1J(PPt) = 3192 Hz, 2J(PP) = 45 Hz). 1H NMR (500 MHz, CD2Cl2): δ 7.1–6.9 (m, 4H), 3.3 (m, 2H, PCH2), 3.1 (m, 2H, CCH), 2.5–2.2 (m, 2H, PtCH), 2.0 (m, 2H, CCHH), 1.7 (m, 2H, CCHH), 1.4 (d, 3J(PH) = 13 Hz, 9H, CCH3), 1.3 (d, 3J(PH) = 13 Hz, 9H, CCH3), 1.2 (d, 3J(PH) = 12 Hz, 9H, CCH3), 1.1 (d, 3J(PH) = 12 Hz, 9H, CCH3), 0.8 (m, 1H, CHH bridge), 0.7 (m, 1H, CHH bridge).Preparation of [Pt(La)(CO)] (2a)
A solution of La (0.158 g, 0.415 mmol) in toluene (5 mL) was added in one portion to a solution of [Pt(η2-nbe)3] (0.198 g, 0.415 mmol) in toluene (5 mL) at −78 °C, the resulting mixture was stirred for 2 h, allowed to warm to room temperature and then stirred overnight. CO was then bubbled through the solution for 30 min to give an orange solution which was filtered and then the volatiles removed under reduced pressure. The residue was redissolved in toluene (5 mL) and CO was bubbled through solution again. The vacuum/CO cycle was repeated twice more to ensure that all norbornene had been displaced. The solvent was then removed under reduced pressure and the solid extracted with pentane (3 × 5 mL). The volume of the solution was reduced to 3 mL and the product crystallised at −78 °C. The supernatant was removed by cannula and the orange solid dried under vacuum (0.146 g, 0.238 mmol, 58%). Elemental analysis (calcd for C25H44OP2Pt, 0.5C5H12): C: 50.46 (50.53) H: 7.41 (7.71). ESI accurate mass spectrum: (calcd for C25H44OP2Pt 617.2513) Mr – 617.617.2532. IR: νCO 1907 cm−1. 31P{H} NMR (162 MHz, C6D6): δ 70.2 (s, 1J(PPt) = 3647 Hz). 1H NMR (500 MHz, C6D6): δ. 7.1 (m, 2H,), 6.9 (m, 2H), 3.5 (br, 4H, CH2), 1.2 (m, 36H. CH3). 13C{H} NMR (101 MHz, C6D6): δ 137.9 (s), 133.7 (s), 125.6 (s), 36.8 (br, CH2), 32.3 (m, CCH3,), 29.9 (m, CCH3).Preparation of [Pt(La)(13CO)] (2a*)
A solution of 2a (9.4 mg, 0.015 mmol) in d6-benzene (1.2 mL) was placed in a valved NMR tube. The solution was frozen with liquid N2 and then the NMR tube was evacuated and sealed. The solution was then thawed and the NMR tube backfilled with 13CO (2 bar) and the resulting mixture allowed to react for 30 min. The vacuum/CO cycle was repeated twice more to ensure that most of the 12CO had been displaced. The solution was then saturated with N2 and the product was identified in solution by NMR only. The 1H NMR spectrum of 2a* was essentially the same as for 2a. 31P{H} NMR (121 MHz, C6D6): δ 70.2 (d, 1J(PPt) = 3647 Hz, 2J(PC) = 45 Hz). 13C{H} NMR (101 MHz, C6D6): δ 228.9 (t, 2J(CP) = 45 Hz, CO), 1J(CPt) = 2100 Hz).Preparation of [Pt(Lb)(CO)] (2b)
CO was bubbled through solution a solution of 1b (0.135 g, 0.202 mmol) in toluene (10 mL) for 30 min to give a yellow solution which was filtered and then the volatiles removed under reduced pressure. The residue was redissolved in toluene (5 mL) and CO was bubbled through the solution again. The vacuum/CO cycle was repeated twice more to ensure that all of the norbornene had been displaced. The solvent was removed under reduced pressure to give a yellow solid which turned red under prolonged exposure to vacuum. The solid was extracted with pentane (3 mL) and the product crystallized at −78 °C. The supernatant was removed via cannula and the red solid dried under vacuum (0.108 g, 0.179 mmol, 88%). Elemental analysis (calcd for C24H42OP2Pt, 0.5C5H12): C: 50.19 (49.76) H: 7.75 (7.56). ESI accurate mass spectrum: (calcd for C24H43OP2Pt 604.2434) Mr +1–604.2431. IR: νCO 1898 cm−1. 31P{H} NMR (202 MHz, C6D6): δ 78.5 (br, 1J(PPt) = 3450 Hz). δ 77.9 (d, 1J(PPt) = 3556 Hz, 2J(PP) = 93 Hz), 76.4 (d, 1J(PPt) = 3250 Hz, 2J(PP) = 93 Hz). 31P{H} NMR (121 MHz, C6D5CD3, −90 °C): δ 77.9 (dd, 1J(PPt) = 3556 Hz, 2J(PP) = 93 Hz), 76.4 (dd, 1J(PPt) = 3250 Hz, 2J(PP) = 93 Hz). 1H NMR (400 MHz, C6D6): δ 7.8 (m, 1H), 7.1–6.9 (m, 3H), 3.2 (m, 2H, CH2), 1.3–1.1 (m, 36H, CCH3). 13C{H} NMR (101 MHz, C6D6): δ 134.6 (m), 129.0 (m), 128.9 (s), 128.8 (s), 125.6 (s), 125.0 (m), 35.7 (br, CH2), 30.9 (br, CCH3), 29.3 (br, CCH3).Preparation of [Pt(Lb)(13CO)] (2b*)
A solution of 2b (11.2 mg, 0.0185 mmol) in benzene (1.2 mL) was placed in a valved NMR tube. The solution was frozen with liquid N2 and then the NMR tube was evacuated and sealed. The solution was then thawed and the NMR tube backfilled with 13CO (2 bar) and the resulting mixture allowed to react for 30 min. The vacuum/CO cycle was repeated twice more to ensure that all the 12CO had been displaced. The volatiles were removed under reduced pressure to yield a yellow solid that turned red under prolonged exposure to vacuum. The product was identified in solution by NMR only. The 1H NMR spectrum of 2b* was essentially the same as for 2b. 31P{H} NMR (121 MHz, C6D5CD3, −90 °C): δ 77.9 (dd, 1J(PPt) = 3556 Hz, 2J(PP) = 93 Hz, 2J(PC) = 52 Hz), 76.4 (dd, 1J(PPt) = 3250 Hz, 2J(PP) = 93 Hz, 2J(PC) = 41 Hz). 13C{H} NMR (75 MHz, C6D5CD3, −90 °C): δ 234.6 (dd, 1J(CPt) = 2189 Hz 2J(PC) = 41 Hz, 2J(PC) = 52 Hz, CO).Preparation of [Pt(La)(CO)2] (3a)
A solution of 2a (10.2 mg, 0.0165 mmol) in d8-toluene (1 mL) was placed in a valved NMR tube. The solution was frozen with liquid N2 and then the NMR tube was evacuated and sealed. The solution was then thawed and the NMR tube backfilled with CO (2 bar) and the resulting mixture allowed to react for 30 min. The product was not isolated because it reverted to 2a upon removal of solvent and was identified in solution by NMR. 31P{H} NMR (121 MHz, C6D5CD3, −90 °C): δ 35.3 (s, 1J(PPt) = 3107 Hz). 1H NMR (300 MHz, C6D5CD3): δ 7.1 (m, 2H), 6.9 (m, 2H), 3.3 (br, 4H, CH2), 1.2 (m, 36H, CCH3). 13C{H} NMR (100 MHz, C6D5CD3): δ 142.9 (s), 138.7 (s), 130.5 (s), 41.8 (br, CH2), 37.3 (br, CCH3), 34.8 (m, CCH3). νCO 1971, 1931 cm−1.Preparation of [Pt(La)(13CO)2] (3a*)
A solution of 2a (10.0 mg, 0.0162 mmol) in d8-toluene (1.2 mL) was placed in a valved NMR tube. The solution was frozen with liquid N2 and then the NMR tube was evacuated and sealed. The solution was then thawed and the NMR tube backfilled with 13CO (2 bar) and the resulting mixture allowed to react for 30 min. The vacuum/CO cycle was repeated twice more to ensure that most of the 12CO had been displaced. The solution was then saturated with N2 to remove the excess of 13CO. The product was not isolated because it reverted to 2a* upon removal of solvent and was identified in solution by NMR. The 1H NMR spectrum of 3a* was essentially the same as for 3a. 31P{H} NMR (121 MHz, C6D5CD3, −90 °C): δ 35.3 (m, 1J(PPt) = 3107 Hz). 13C{H} NMR (75 MHz, C6D5CD3, −90 °C): δ 187.1 (m, 1J(CPt) = 1832 Hz, CO) 186.6 (m, 1J(CPt) = 1900 Hz, CO).Preparation of [Pt(Lb)(CO)2] (3b)
A solution of 2b (8.7 mg, 0.014 mmol) in d8-toluene (1 mL) was placed in a valved NMR tube. The solution was frozen with liquid N2 and then the NMR tube was evacuated and sealed. The solution was then thawed and the NMR tube backfilled with CO (2 bar) and the resulting mixture allowed to react for 30 min. The product was not isolated because it reverted to 2b upon removal of solvent and was identified in solution by NMR. 31P{H} NMR (202 MHz, C6D6): δ 44.4 (d, 1J(PPt) = 3106 Hz, 2J(PP) = 20 Hz), 42.7 (d, 1J(PPt) = 3818 Hz, 2J(PP) = 20 Hz). 1H NMR (300 MHz, C6D5CD3): δ 7.8 (m, 1H), 7.0 (m, 2H), 6.9 (m, 1H), 3.2 (m, 2H, CH2), 1.3 (d, 3J(PH) = 13 Hz, 18H, CCH3), 1.1 (d, 3J(PH) = 13 Hz, 18H, CCH3). 13C{H} NMR (126 MHz, C6D6): δ 143.8 (d, J(CP) = 15 Hz), 134.5 (dd, J(CP) = 8 Hz, J(CP) = 5 Hz), 134.4 (d, J(CP) = 2 Hz), 129.0 (s), 125.4 (s), 124.3 (s), 36.9 (m, CCH3), 34.8 (m, CCH3), 32.4 (m, CCH3), 30.1 (br, CH2), 29.8 (m, CCH3), 28.8 (d, CCH3). νCO 1961, 1915 cm−1.Preparation of [Pt(Lb)(13CO)2] (3b*)
A solution of 2b (9.9 mg, 0.0164 mmol) in d8-toluene (1.2 mL) was placed in a valved NMR tube. The solution was frozen with liquid N2 and then the NMR tube was evacuated and sealed. The solution was then thawed and the NMR tube backfilled with 13CO (2 bar) and then the resulting mixture allowed to react for 30 min. The vacuum/CO cycle was repeated twice more to ensure that all 12CO had been displaced. The product was not isolated because it reverted to 2b* upon removal of solvent and was identified in solution by NMR. The 1H NMR spectrum of 3b* was essentially the same as for 3b. 31P{H} NMR (121 MHz, C6D5CD3, −90 °C): δ 43.0 (m, 1J(PPt) = 3046 Hz), 40.3 (m 1J(PPt) = 2788 Hz). 13C{H} NMR (75 MHz, C6D5CD3, −90 °C): δ.187.1 (m, 1J(CPt) = 1903 Hz, CO), 186.0 (m, 1J(CPt) = 1850 Hz, CO).Preparation of [Pt(La)(C2H4)] (4a)
A solution of 2a (12.2 mg, 0.0194 mmol) in d6-benzene (1.2 mL) was placed in a valved NMR tube. The solution was frozen with liquid N2 and then the NMR tube was evacuated and sealed. The solution was then thawed and the NMR tube backfilled with C2H4 (2 bar) and the resulting mixture allowed to react for 30 min. The vacuum/C2H4 cycle was repeated twice more to ensure that most of the CO had been displaced. The product was identified by comparison of its NMR spectra with the literature data for this previously reported complex.19 ESI mass spectrum: (calcd for C26H48P2Pt 617.3) Mr – 617.3. 31P{H} NMR (121 MHz, C6D6): δ 48.9 (1J(PPt) = 3548 Hz). 1H NMR (300 MHz, C6D6): δ 7.1–6.9 (m, 4H), 3.4 (br, 4H, PCH2), 2.2 (br, 2J(PtH) = 54 Hz, 4H, PtCH2), 1.3 (d, 3J(PH) = 13 Hz, 36H, CH3).Reaction of [Pt(Lb)(CO)] with ethene
A solution of 2b (10.6 mg, 0.0175 mmol) in d8-toluene (1.2 mL) was placed in a valved NMR tube. The solution was frozen with liquid N2 and then the NMR tube was evacuated and sealed. The solution was then thawed and the NMR tube backfilled with C2H4 (2 bar) and the resulting mixture allowed to react for 30 min. The solution was frozen with liquid N2 and then the NMR tube was evacuated and sealed. The solution was then thawed and the NMR tube backfilled with C2H4 (2 bar) and then the resulting mixture allowed to react for 30 min. The vacuum/C2H4 cycle was repeated twice. The product was a mixture of 2b, 3b and a species tentatively assigned to 4b on the basis of its 31P NMR parameters. 31P{H} NMR (121 MHz, C6D5CD3): 60.9 (d, 1J(PPt) = 3288 Hz, 2J(PP) 45 Hz), 57.9 (d, 1J(PPt) = 3288 Hz, 2J(PP) = 45 Hz).Preparation of [Pt(La)(H)(CO)][B(C6F5)4] (5a)
A solution of [(Et2O)2H][B(C6F5)4] (13.8 mg, 0.0166 mmol) in chlorobenzene (0.5 mL) was added in one portion to a solution of 2a (10.5 mg, 0.0169 mmol) in chlorobenzene (0.5 mL) and the resulting mixture was left for 16 h. The solution was layered with pentane (3 mL) resulting in the slow formation of colourless crystals suitable for X-ray crystallography (14.8 mg, 0.012 mmol, 71%). ESI accurate mass spectrum: (calcd for C25H44OP2Pt 618.6655) Mr – 618.2589. IR: νCO 2092 cm−1. Satisfactory C, H elemental analyses for 5a were not obtained even when a sample from the crystals used for the X-ray crystallography were submitted. 31P{H} NMR (162 MHz, CD2Cl2): δ 43.4 (d, 1J(PPt) = 2994 Hz, 2J(PP) = 19 Hz), 34.2 (d, 1J(PPt) = 2018 Hz, 2J(PP) = 19 Hz). 1H NMR (400 MHz, CD2Cl2): δ 7.4 (m, 2H), 7.3 (m, 2H), 3.9 (br, 4H, CH2), 1.4 (m, 36H, CCH3), −4.7 (dd, 2J(PH) = 145 Hz, 2J(PH) = 15 Hz, 1J(PtH) = 736 Hz, 1H, PtH). 11B{H} NMR (128 MHz, CD2Cl2): δ −17.7 (s). 19F NMR (376 MHz, CD2Cl2): δ −133.0 (m, 8F, o-C6F5), −162.3 (t, 4F, p-C6F5), 166.2 (m, 8F, m-C6F5).Preparation of [Pt(Lb)(CO)(H)][B(C6F5)4] (5b/6b)
A solution of [(Et2O)2H][B(C6F5)4] (12.5 mg, 0.0151 mmol) in PhCl (0.5 mL) was added in one portion to a solution of 2b (9.2 mg, 0.015 mmol) in PhCl (0.5 mL) and the resulting mixture was left for 16 h. The solution was layered with pentane (3 mL) to give colourless crystals (17.8 mg, 0.014 mmol, 93%). The product was a mixture of 2 geometric isomers. 31P{H} NMR (202 MHz, CD2Cl2): δ 54.9 (d, 1J(PPt) = 2948 Hz, 2J(PP) = 22 Hz), 53.5 (d, 1J(PPt) = 2978 Hz, 2J(PP) = 22 Hz) 49.8 (d, 1J(PPt) = 1879 Hz, 2J(PP) = 22 Hz), 49.7 (d, 1J(PPt) = 1918 Hz, 2J(PP) = 22 Hz). 1H NMR (500 MHz, CD2Cl2): δ 8.1 (m, 1H), 7.6 (m, 2H), 7.5 (m, 1H), 3.6 (m, 2H, CH2), overlapping tBu peaks: 1.5 (d, 3J(PH) = 15 Hz, CH3) 1.5 (d, 3J(PH) = 16 Hz, CH3) (total integration 18H), overlapping tBu peaks: 1.3 (d, 3J(PH) = 15 Hz, CH3) 1.3 (d, 3J(PH) = 16 Hz, CH3) (total integration 18H), −2.3 (dd, 1J(PtH) = 777 Hz, 2J(PH) = 147 Hz, 2J(PH) = 11 Hz, 0.2H, PtH), −4.3 (dd, 1J(PtH) = 810 Hz, 2J(PH) = 148 Hz, 2J(PH) = 10 Hz, 0.8H, PtH. 11B{H} NMR (128 MHz, CD2Cl2): δ −16.69 (s). 19F NMR (376 MHz, CD2Cl2): δ −133.16 (m, 8F, o-C6F5), −163.63 (t, 4F, p-C6F5), 167.51 (m, 8F, m-C6F5). ESI accurate mass spectrum: (calcd for C24CH43OP2Pt 604.2431) Mr – 604.2429. Elemental analysis (calcd for C48H43BF20OP2Pt, PhCl): C: 46.65 (46.45) H: 3.10 (3.47). νCO 2102, 2094 cm−1.Crystal structure determination
A single crystal of 5a was mounted on a glass fibre and X-ray diffraction data were collected at 100 K on a Bruker APEX II CCD diffractometer using graphite monochromatised Mo-Kα radiation (λ = 0.71073 Å). Absorption corrections were based on equivalent reflections using SADABS.27 The structure was solved by direct methods in SHELXS and refined by full matrix least squares on F2 in SHELXL.28 All of the non-hydrogen atoms were refined anisotropically and all of the hydrogen atoms were located geometrically and refined using a riding model with the exception of H1D, H1E, H2D, H2E. The structure showed a small amount of disorder in the positions of the CO group and hydride attached to the Pt metal centres. The occupancy of each CO group was determined by refining them against a free variable with the sum of the occupancies for the two CO sites attached to each Pt set to equal one, prior to fixing the occupancy at the refined values. The hydrogen atoms (H1D, H1E, H2D, H2E) were located on the basis of chemical knowledge and their occupancy fixed to that of the related CO occupancy. Restraints were applied to maintain chemically sensible geometries (DFIX, SADI, DANG) for the disordered sections, while the CO thermal parameters were restrained to similar values using SIMU and the H Uiso values was fixed at 1.5 × Ueq(Pt). Crystal structure and refinement data are given in Table 1.| Empirical formula | C49H45BF20OP2Pt |
| Formula weight | 1297.69 |
| Temperature/K | 100(2) |
| Crystal system | Triclinic |
| Space group |
P |
| a/Å | 14.3193(7) |
| b/Å | 15.3989(8) |
| c/Å | 23.2615(11) |
| α/° | 93.138(4) |
| β/° | 106.589(4) |
| γ/° | 90.129(4) |
| Volume/Å3 | 4907.5(4) |
| Z | 2 |
| ρ calc/g cm−3 | 1.756 |
| μ/mm−1 | 3.039 |
| F(000) | 2560.0 |
| Crystal size/mm3 | 0.10 × 0.15 × 0.17 |
| Radiation | MoKα (λ = 0.71073) |
| 2θ range for data collection/° | 3.01 to 51.36 |
| Index ranges | −17 ≤ h ≤ 17, −18 ≤ k ≤ 18, −28 ≤ l ≤ 28 |
| R int | 0.1485 |
| Reflections collected | 53729 |
| Independent reflections | 17979 |
| Data/restraints/parameters | 17979/76/1406 |
| Goodness-of-fit on F2 | 0.981 |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.0789, wR2 = 0.1490 |
| Final R indexes [all data] | R 1 = 0.1799, wR2 = 0.1877 |
| Largest diff. peak/hole/e Å−3 | 1.62/−1.61 |
References
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Chem. Commun., 1976, 365 RSC.
- N. Carr, B. J. Dunne, A. G. Orpen and J. L. Spencer, J. Chem. Soc., Chem. Commun., 1988, 926 RSC.
- L. Mole, J. L. Spencer, N. Carr and A. G. Orpen, Organometallics, 1991, 10, 49 CrossRef CAS.
- (a) R. F. M. J. Parton and M. C. C. Janssen, WO2013030344 (A1), 2013; (b) R. P. Tooze, G. R. Eastham, W. Keith and X. L. Wang, WO9619434 (A1), 1996; (c) J. M. Pearson and R. A. Hadden, WO9841495 (A1), 1998; (d) J. G. De Vries, N. Sereinig, E. W. M. Van de Vondervoort and M. C. C. Janssen, WO2012131027 (A1), 2012; (e) G. R. Eastham, R. P. Tooze, X. L. Wang and K. Whiston, US6335471 (B1), 1996; (f) W. Clegg, M. R. J. Elsegood, G. R. Eastham, R. P. Tooze, X. Lan Wang and K. Whiston, Chem. Commun., 1999, 1877 RSC.
- (a) R. I. Pugh, E. Drent and P. G. Pringle, Chem. Commun., 2001, 1476 RSC; (b) S. Doherty, E. G. Robins, J. G. Knight, C. R. Newman, B. Rhodes, P. A. Champkin and W. Clegg, J. Organomet. Chem., 2001, 640, 182 CrossRef CAS; (c) J. G. Knight, S. Doherty, A. Harriman, E. G. Robins, M. Betham, G. R. Eastham, R. P. Tooze, M. R. J. Elsegood, P. Champkin and W. Clegg, Organometallics, 2000, 19, 4957 CrossRef CAS; (d) O. V. Gusev, A. M. Kalsin, M. G. Peterleitner, P. V. Petrovskii, K. A. Lyssenko, N. G. Akhmedov, C. Bianchini, A. Meli and W. Oberhauser, Organometallics, 2002, 21, 3637 CrossRef CAS; (e) C. Bianchini, A. Meli, W. Oberhauser, M. A. Zuideveld, Z. Freixa, P. C. J. Kamer, A. L. Spek, O. V. Gusev and A. M. Kal'sin, Organometallics, 2003, 22, 2409 CrossRef CAS; (f) O. V. Gusev, A. M. Kalsin, P. V. Petrovskii, K. A. Lyssenko, Y. F. Oprunenko, C. Bianchini, A. Meli and W. Oberhauser, Organometallics, 2003, 22, 913 CrossRef CAS; (g) N. R. Vautravers and D. J. Cole-Hamilton, Dalton Trans., 2009, 2130 RSC; (h) E. Drent, J. A. M. Van Broekhoven and M. J. Doyle, J. Organomet. Chem., 1991, 417, 235 CrossRef CAS; (i) S. J. Dossett, A. Gillon, A. G. Orpen, J. S. Fleming, P. G. Pringle, D. F. Wass and M. D. Jones, Chem. Commun., 2001, 699 RSC; (j) C. Bianchini, H. M. Lee, A. Meli, S. Moneti, F. Vizza, M. Fontani and P. Zanello, Macromolecules, 1999, 32, 4183 CrossRef CAS; (k) C. Bianchini, H. Man Lee, P. Barbaro, A. Meli, S. Moneti and F. Vizza, New J. Chem., 1999, 23, 929 RSC; (l) E. Lindner, M. Schmid, J. Wald, J. A. Queisser, M. Geprägs, P. Wegner and C. Nachtigal, J. Organomet. Chem., 2000, 602, 173 CrossRef CAS; (m) P. W. N. M. van Leeuwen, M. A. Zuideveld, B. H. G. Swennenhuis, Z. Freixa, P. C. J. Kamer, K. Goubitz, J. Fraanje, M. Lutz and A. L. Spek, J. Am. Chem. Soc., 2003, 125, 5523 CrossRef CAS PubMed.
- (a) G. R. Eastham, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, Chem. Commun., 2000, 609 RSC; (b) W. Clegg, G. R. Eastham, M. R. J. Elsegood, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, Organometallics, 2002, 21, 1832 CrossRef CAS.
- J. Wolowska, G. R. Eastham, B. T. Heaton, J. A. Iggo, C. Jacob and R. Whyman, Chem. Commun., 2002, 2784 RSC.
- T. Fanjul, G. Eastham, N. Fey, A. Hamilton, A. G. Orpen, P. G. Pringle and M. Waugh, Organometallics, 2010, 29, 2292 CrossRef CAS.
- T. Fanjul, G. Eastham, M. F. Haddow, A. Hamilton, P. G. Pringle, A. G. Orpen, T. P. W. Turner and M. Waugh, Catal. Sci. Technol., 2012, 2, 937 CAS.
- V. de la Fuente, M. Waugh, G. R. Eastham, J. A. Iggo, S. Castillón and C. Claver, Chem. – Eur. J., 2010, 16, 6919 CrossRef CAS PubMed.
- T. Fanjul, G. Eastham, J. Floure, S. J. K. Forrest, M. F. Haddow, A. Hamilton, P. G. Pringle, A. G. Orpen and M. Waugh, Dalton Trans., 2013, 42, 100 RSC.
- P. G. Edwards, J. C. Knight and P. D. Newman, Dalton Trans., 2010, 39, 3851 RSC.
- S. Bertsch, H. Braunschweig, M. Forster, K. Gruss and K. Radacki, Inorg. Chem., 2011, 50, 1816 CrossRef CAS PubMed and references therein.
- E. Nicolas, G. Nocton and N. Mezailles, Eur. J. Inorg. Chem., 2013, 4000 CrossRef CAS.
- (a) P. Chini and G. Longoni, J. Chem. Soc. A, 1970, 1542 RSC; (b) G. K. Anderson, H. C. Clark and J. A. Davies, Organometallics, 1982, 1, 550 CrossRef CAS; (c) V. V. Grushin, I. S. Akbem and M. E. Vol'pin, J. Organomet. Chem., 1989, 371, 403 CrossRef CAS; (d) P. Giannocaro, A. Sacco and G. Vasapollo, Inorg. Chim. Acta, 1979, 31, L455 CrossRef; (e) R. S. Paonessa and W. C. Trogler, J. Am. Chem. Soc., 1982, 104, 1138 CrossRef CAS.
- G. K. Anderson, G. J. Lumetta and J. W. Siria, J. Organomet. Chem., 1992, 434, 253 CrossRef CAS.
- T. Yoshida, T. Yamagata, T. H. Tulip, J. A. Ibers and S. Otsuka, J. Am. Chem. Soc., 1978, 100, 2063 CrossRef CAS.
- B. L. Bennett and D. M. Roddick, Inorg. Chem., 1996, 35, 4703 CrossRef CAS.
- N. Carr, L. Mole, A. G. Orpen and J. L. Spencer, J. Chem. Soc., Dalton Trans., 1992, 2653 RSC.
- P. Jutzi, C. Muller, A. Stammler and H.-G. Stammler, Organometallics, 2000, 19, 1442 CrossRef CAS.
- D. Vuzman, E. Poverenov, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon and D. Milstein, Dalton Trans., 2007, 5692 RSC.
- F. H. Allen, Acta Cryst., 2002, B58, 380 CrossRef CAS PubMed.
- P. G. Edwards, J. C. Knight and P. D. Newman, Dalton Trans., 2010, 39, 3851 RSC.
- T. Weisheit, H. Petzold, H. Gorls, G. Mloston and W. Weigand, Eur. J. Inorg. Chem., 2009, 3545 CrossRef CAS.
- M. F. Ernst and D. M. Roddick, Organometallics, 1990, 9, 1586 CrossRef CAS.
- S. Ogoshi, M. Morita, K. Inoue and H. Kurosawa, J. Organomet. Chem., 2004, 689, 662 CrossRef CAS PubMed.
- Bruker, SADAB-2012/1, Absorption Correction, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † CCDC 1015609. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02303j |
| This journal is © The Royal Society of Chemistry 2014 |