
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphine complexes of aluminium(III) halides – preparation and structural and spectroscopic systematics†
Jennifer
Burt
,
William
Levason
*,
Mark E.
Light
and
Gillian
Reid
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: wxl@soton.ac.uk
First published on 8th August 2014
Abstract
Six-coordinate pseudo-octahedral complexes trans-[AlX2(L–L)2][AlX4] (X = Cl, Br or I; L–L = o-C6H4(PMe2)2, Me2P(CH2)2PMe2) are produced from reaction of AlX3 with the diphosphine in CH2Cl2 (X = Cl) or toluene (X = Br or I) solution. Four-coordinate dimers [Cl3Al(μ-L′–L′)AlCl3] (L′–L′ = Me2P(CH2)2PMe2, Cy2P(CH2)2PCy2), and the tetrahedral cation [AlCl2{o-C6H4(PPh2)2}][AlCl4] were also obtained. Both four- and five-coordinate complexes [AlX3(PMe3)] and [AlX3(PMe3)2] could be isolated with PMe3 depending upon the ratio of reagents used. These extremely moisture sensitive complexes have been characterised by microanalysis, IR and multinuclear NMR (1H, 31P{1H} and 27Al) spectroscopy. X-ray crystal structures are reported for [AlCl2{o-C6H4(PMe2)2}2][AlCl4], [AlCl2{Me2P(CH2)2PMe2}2][AlCl4], [Cl3Al{μ-Me2P(CH2)2PMe2}AlCl3], [Cl3Al{μ-Cy2P(CH2)2PCy2}AlCl3], [AlCl3(PMe3)], [AlCl3(PMe3)2], and for the six-coordinate cation complex [AlCl2{o-C6H4(PPh2)2}2][AlCl4], although a bulk sample of the last could not be isolated. Tertiary arsines (AsPh3 or AsEt3) form only 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes even with excess arsine present. The unstable [AlCl2{o-C6H4(AsMe2)2}][AlCl4] is also described, and shown to decompose rapidly in CH2Cl2 solution to form the diquaternised diarsine cation [o-C6H4(AsMe2)2(CH2)][AlCl4]2, which was fully characterised. Comparisons are drawn with the corresponding gallium(III) systems (Cheng et al., Inorg. Chem., 2007, 46, 7215–7223) and with AlX3 complexes of Group 16 ligands (George et al., Dalton Trans., 2014, 43, 3637–3648), and it is concluded that the differences between the Al and Ga systems reflect the higher Lewis acidity of aluminium(III) towards soft donor ligands.
:1 complexes even with excess arsine present. The unstable [AlCl2{o-C6H4(AsMe2)2}][AlCl4] is also described, and shown to decompose rapidly in CH2Cl2 solution to form the diquaternised diarsine cation [o-C6H4(AsMe2)2(CH2)][AlCl4]2, which was fully characterised. Comparisons are drawn with the corresponding gallium(III) systems (Cheng et al., Inorg. Chem., 2007, 46, 7215–7223) and with AlX3 complexes of Group 16 ligands (George et al., Dalton Trans., 2014, 43, 3637–3648), and it is concluded that the differences between the Al and Ga systems reflect the higher Lewis acidity of aluminium(III) towards soft donor ligands.
Introduction
Aluminium chloride is an archetypal Lewis acid, widely used in industry and in the laboratory as a catalyst for condensation, polymerisation and isomerisation reactions. It is perhaps best known as a Friedel–Crafts catalyst for alkylations or acylations where the strong affinity for chloride generates incipient carbocations in combination with [AlCl4]− anions.1,2 Typically, AlBr3 and AlI3 are weaker Lewis acids, but chemically similar. Solid AlCl3 contains six-coordinate aluminium, but in the melt or when dissolved in inert solvents, dimeric tetrahedral molecules [Cl2Al(μ-Cl)2AlCl2] are present. Solid AlBr3 is also dimeric, [Br2Al(μ-Br)2AlBr2], whereas AlI3 is a chain polymer [{I2Al(μ-I)}n].3 The extensive coordination chemistry of these three halides, mostly with hard N- or O-donor ligands, continues to attract much effort.3 In contrast, AlF3 is an inert polymer containing six-coordinate aluminium; few complexes are known.4 Complexes of aluminium halides with neutral soft-donor ligands have received much less attention and current knowledge is not systematic.3 We recently reported5 complexes with thio-, seleno- and telluro-ether ligands, including examples of four-coordinate [X3Al(ER2)] (X = Cl, Br or I; E = S, Se or Te; R = alkyl) and [(AlCl3)2{o-C6H4(CH2SEt)2}], as well as six-coordinate [AlX2{MeE(CH2)2EMe}2][AlX4] (E = S or Se). The isolation of the latter was unexpected since gallium(III) halides gave only four-coordinate complexes with the bidentate chalcogenoethers.6 Both Al and Ga can achieve six-coordination with thia- and selena-macrocycles.6,7 There are a substantial number of phosphine and arsine complexes of trialkylaluminiums, and even some examples with stibine and bismuthine ligands; all contain four-coordinate aluminium with a single Group 15 donor per Al centre, even when polydentate ligands are used.8 In contrast, complexes with AlX3 are few, and almost all are of the type [X3Al(E′R3)] (E′R3 = PPh3,9 AsPh3,10 AsEt3,10 AsMe3,10 P(mesityl)311a or P(SiMe3)311b) containing tetrahedral aluminium centres.8 A rare example of a higher coordination number (five) is found in the trigonal bipyramidal [AlI3(PEt3)2].12 Surprisingly, no examples with bidentate phosphine or arsine ligands have been prepared. In a previous study with gallium(III) halides,13 four-coordination was dominant, either as neutral dimers [X3Ga{μ-Et2P(CH2)2PEt2}GaX3] or [X3Ga{μ-Ph2As(CH2)2AsPh2}GaX3], or tetrahedral cations [GaX2(L–L)]+ (L–L = o-C6H4(PPh2)2 or o-C6H4(AsMe2)2). Octahedral cations, [GaX2{o-C6H4(PMe2)2}2]+, are only known with o-C6H4(PMe2)2, which is a very strong σ-donor ligand, has small steric demands and is pre-organised for chelation.13 The larger In(III) centre forms both four-coordinate [InX2(L–L)]+ and six-coordinate [InX2(L–L)2]+ cations, as well as neutral six-coordinate dimers, [In2X6(L–L)2], and very rare six-coordinate iodoanions [InI4(L–L)]− (L–L = o-C6H4(PMe2)2, o-C6H4(AsMe2)2).8,14
There has also been much recent interest in exploring the relative Lewis acidity among the Group 13 halides. DFT calculations have predicted that Lewis acidity generally falls AlCl3 > AlBr3 ≫ AlI3 and AlCl3 > AlBr3 > GaCl3 > GaBr3 (gas phase) (ref. 8,9,15 and references therein) and this is supported by experimental data, although in some cases intermolecular interactions or solvate molecules can mask these trends in the solid state.9 Here we report systematic studies of AlX3 (X = Cl, Br or I) complexes with some diphosphine and diarsine ligands and detailed comparisons with the gallium(III) analogues.
Experimental section
All preparations were carried out under rigorously anhydrous conditions via a dry dinitrogen atmosphere and standard Schlenk and glove-box techniques. Anhydrous grade aluminium trihalides were obtained commercially (Aldrich) and used as received. The ligands were obtained commercially (Strem or Aldrich), apart from o-C6H4(PMe2)2 and o-C6H4(AsMe2)2 which were made by the literature methods.16 Solvents were dried by distillation from CaH2 (CH2Cl2, MeCN) or sodium benzophenone ketyl (hexane, toluene). IR spectra were recorded as Nujol mulls between CsI plates using a Perkin Elmer Spectrum 100 spectrometer over the range 4000–200 cm−1. 1H and 31P{1H} NMR spectra were recorded using a Bruker AV300 or DPX400 spectrometer and referenced to the residual solvent resonance and external 85% H3PO4 respectively. 27Al NMR spectra were recorded with a Bruker DPX400 spectrometer and referenced to external [Al(H2O)6]3+. Microanalytical measurements were performed by Medac Ltd or London Metropolitan University.trans-[AlCl2{o-C6H4(PMe2)2}2][AlCl4]
To a suspension of AlCl3 (0.067 g, 0.51 mmol) in CH2Cl2 (5 mL) was added o-C6H4(PMe2)2 (0.100 g, 0.51 mmol) in CH2Cl2 (5 mL) to form a colourless solution, which was allowed to stir for 1.5 h. The solvent volume was reduced to about 3 mL in vacuo. The resulting white precipitate was isolated by filtration and dried in vacuo to give a white powder. Yield: 0.071 g (43%). Small colourless blocks suitable for single crystal X-ray diffraction study were grown from a CH2Cl2 solution containing AlCl3 and o-C6H4(PMe2)2 in a 2:1 mol ratio kept at −18 °C. Anal. Calc. for C20H32Al2Cl6P4: C, 36.2; H, 4.9. Found: C, 36.0; H, 4.8%. 1H NMR (CD2Cl2, 295 K): δ = 1.74 (s, [24H], CH3), 7.67–7.77 (m, [8H], C6H4). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −42.8 (sextet, 1JPAl = 155 Hz); (253 K): −42.6 (sextet). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 103.3 (s, AlCl4–), 0.7 (quintet, 1JAlP = 155 Hz); (253 K): 103.4 (s), 0.4 (quintet). IR (Nujol): ν = 488 (vs, AlCl4–), 410 (s, AlCl) cm−1.
trans-[AlBr2{o-C6H4(PMe2)2}2][AlBr4]
AlBr3 (0.135 g, 0.51 mmol) was dissolved in toluene (5 mL) to form a yellow solution. To this was added o-C6H4(PMe2)2 (0.100 g, 0.51 mmol) in toluene (5 mL) which immediately led to the solution turning colourless, with evidence of a large amount of white precipitate. The reaction was stirred for 2 h, then the white powder was isolated by filtration and dried in vacuo. Yield: 0.213 g (91%). Anal. Calc. for C20H32Al2Br6P4: C, 25.8; H, 3.5. Found: C, 26.0; H, 3.3%. 1H NMR (CD2Cl2, 295 K): δ = 1.80 (s, [24H], CH3), 7.71–7.78 (m, [8H], C6H4). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −42.6 (br s); (223 K): −42.6 (br s). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 80.5 (s, AlBr4–), −12.0 (br s); (253 K): 80.5 (s), −11.4 (br s). IR (Nujol): ν = 398 (vs, AlBr4–), 326 (m, AlBr) cm−1.trans-[AlI2{o-C6H4(PMe2)2}2][AlI4]
This was made similarly from AlI3 (0.166 g, 0.41 mmol) and o-C6H4(PMe2)2 (0.080 g, 0.41 mmol) in toluene (10 mL). Yield: 0.231 g (94%). Anal. Calc. for C20H32Al2I6P4: C, 19.8; H, 2.7. Found: C, 20.0; H, 2.8%. 1H NMR (CD2Cl2, 295 K): δ = 1.90 (s, [24H], CH3), 7.79–7.89 (m, [8H], C6H4). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −42.3 (br s); (223 K): −42.2 (br s). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = −26.5 (s, AlI4–); (223 K): −23.9 (s). IR (Nujol): ν = 334 (vs, AlI4–) 281 (m, AlI) cm−1.trans-[AlCl2{Me2P(CH2)2Me2}2][AlCl4]
To a suspension of AlCl3 (0.090 g, 0.67 mmol) in CH2Cl2 (5 mL) was added Me2P(CH2)2PMe2 (0.100 g, 0.67 mmol) in CH2Cl2 (5 mL). The resulting solution was stirred for 2 h, then the solvent volume was reduced to about 2 mL in vacuo and layered with hexane (2.5 mL), whereupon small colourless blocks suitable for single crystal X-ray diffraction study grew. The crystalline material was then isolated by filtration and dried in vacuo to give a white solid. Yield: 0.145 g (77%). Anal. Calc. for C12H32Al2Cl6P4: C, 25.4; H, 5.7. Found: C, 25.2; H, 5.9%. 1H NMR (CD2Cl2, 295 K): δ = 1.44 (br s, [24H], CH3), 2.00 (br s, [8H], CH2). 31P{1H} NMR (CD2Cl2, 295 K): δ = −40.6 (sextet, 1JPAl = 164 Hz). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 103.3 (s, AlCl4–), 1.2 (quintet, 1JAlP = 164 Hz). IR (Nujol): ν = 478 (vs, br, AlCl4–), 377 (s, AlCl) cm−1.trans-[AlBr2{Me2P(CH2)2Me2}2][AlBr4]
AlBr3 (0.133 g, 0.50 mmol) was dissolved in toluene (5 mL) to form a yellow solution. To this was added Me2P(CH2)2PMe2 (0.075 g, 0.50 mmol) in toluene (5 mL) which caused the immediate precipitation of a white solid. The reaction was stirred for 1 h, then the white powder was isolated by filtration and dried in vacuo. Yield: 0.176 g (85%). Anal. Calc. for C12H32Al2Br6P4: C, 17.3; H, 3.9. Found: C, 17.2; H, 3.9%. 1H NMR (CD2Cl2, 295 K): δ = 1.52 (br s, [24H], CH3), 2.04 (br s, [8H], CH2). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −38.0 (br s). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 80.4 (s, AlBr4–), −12.3 (br s). IR (Nujol): ν = 394 (vs, AlBr4–), 326 (m, AlBr) cm−1.trans-[AlI2{Me2P(CH2)2Me2}2][AlI4]
AlI3 (0.204 g, 0.50 mmol) was dissolved in toluene (5 mL) to form a yellow solution. To this was added Me2P(CH2)2PMe2 (0.078 g, 0.51 mmol) in toluene (5 mL) which caused the immediate precipitation of a white solid. The reaction was stirred for 2 h, then the white powder was isolated by filtration and dried in vacuo. Yield: 0.270 g (97%). Anal. Calc. for C12H32Al2I6P4: C, 12.9; H, 2.9. Found: C, 13.0; H, 3.0%. 1H NMR (CD2Cl2, 295 K): δ = 1.63 (br s, [24H], CH3), 2.09 (br s, [8H], CH2). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −39.1 (br s); (203 K): −37.7 (br s). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = −27.1 (s, AlI4–), −33.9 (br s); (203 K): −26.1 (s). IR (Nujol): ν = 337 (vs, AlI4–), 285 (s, AlI) cm−1.[(AlCl3)2{μ-Me2P(CH2)2PMe2}]
To a suspension of AlCl3 (0.178 g, 1.33 mmol) in CH2Cl2 (5 mL) was added Me2P(CH2)2PMe2 (0.099 g, 0.66 mmol) in CH2Cl2 (5 mL). The resulting solution was stirred for 1 h, then the solvent volume was reduced to about 4 mL in vacuo whereupon a white solid precipitated out. The solid was isolated by filtration and dried in vacuo. Yield: 0.055 g. The solid is a mixture of trans-[AlCl2{Me2P(CH2)2Me2}2][AlCl4] and [(AlCl3)2{μ-Me2P(CH2)2PMe2}]. Small colourless blocks suitable for single crystal X-ray diffraction study were grown from a CH2Cl2 solution containing AlCl3 and Me2P(CH2)2PMe2 in a 1.5:1 ratio kept at −18 °C. Spectroscopic data for this complex obtained from the mixture, the resonances of the trans-[AlCl2{Me2P(CH2)2Me2}2][AlCl4] present were identical to those listed above: 1H NMR (CD2Cl2, 295 K): δ = 1.90 (m, [12H], CH3), 2.17 (d, [4H], CH2). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −31.6 (br s). 27Al NMR (CH2Cl2–CD2Cl2, 298 K): δ = 109.3 (br s); (253 K): 108.9 (s).
[AlCl2{o-C6H4(PPh2)2}][AlCl4]
To a solution of o-C6H4(PPh2)2 (0.075 g, 0.17 mmol) in CH2Cl2 (10 mL) was added AlCl3 (0.045 g, 0.34 mmol). The resulting solution was stirred for 1.5 h, then the solvent volume was reduced to about 3 mL in vacuo and hexane (3 mL) was added. The resulting white precipitate was isolated by filtration and dried in vacuo. Yield = 0.035 g (29%). Anal. Calc. for C30H24Al2Cl6P2: C, 50.5; H, 3.4. Found: C, 50.5; H, 3.7%. 1H NMR (CD2Cl2, 295 K): δ = 7.22–7.53 (aromatics). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −8.3 (v br); (180 K): −12.4 (v br). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 103.3 (s, AlCl4–). IR (Nujol): ν = 483 (br, s, AlCl4–) cm−1. Some small crystals of [AlCl2{o-C6H4(PPh2)2}2][AlCl4] were obtained from a similar preparation by careful hexane layering.[(AlCl3)2{μ-Cy2P(CH2)2PCy2}]
To a suspension of AlCl3 (0.063 g, 0.47 mmol) in CH2Cl2 (5 mL) was added a solution of Cy2P(CH2)2PCy2 (0.101 g, 0.24 mmol) in CH2Cl2 (5 mL). The resulting solution was stirred for 1 h, then the solvent volume was reduced to about 3 mL in vacuo. Small colourless blocks suitable for single crystal X-ray diffraction study grew upon cooling the solution to −18 °C. Anal. Calc. for C26H48Al2Cl6P2: C, 45.3; H, 7.0. Found: C, 45.1; H, 7.0%. 1H NMR (CD2Cl2, 295 K): δ = 1.32–1.89 (m [44H]), 2.53 (m, [4H], 2JPH = 13 Hz). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −0.5 (s); (213 K): −1.4 (br). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 110.6 (s); (213 K): 111.3 (s). IR (Nujol): ν = 478 (vs, br), 375 (w, br, AlCl) cm−1.[AlCl3(PMe3)]
To a suspension of AlCl3 (0.177 g, 1.33 mmol) in CH2Cl2 (5 mL) was added PMe3 (0.100 g, 1.31 mmol) in CH2Cl2 (5 mL). The resulting colourless solution was stored at −18 °C and over a few days large, colourless, temperature-sensitive crystals formed. These were separated and dried. The solvent was then removed in vacuo and the resulting white solid was washed with hexane (5 mL), and dried in vacuo. Yield: 0.174 g (63%). Anal. Calc. for C3H9AlCl3P: C, 17.2; H, 4.3. Found: C, 17.2; H, 4.4%. 1H NMR (CD2Cl2, 295 K): δ = 1.50 (d, 2JHP = 10 Hz, CH3). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −42.6 (br s); (248 K): −42.3 (sextet); (223 K): −42.3 (sextet, 1JPAl = 275 Hz). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 108.8 (s); (223 K): 111.4 (d, 1JAlP = 275 Hz). IR (Nujol): ν = 482 (vs, br, AlCl) cm−1.trans-[AlCl3(PMe3)2]
To a suspension of AlCl3 (0.133 g, 1.00 mmol) in CH2Cl2 (5 mL) was added PMe3 (0.154 g, 2.02 mmol) in CH2Cl2 (5 mL). The resulting solution was stirred for 1.5 h, then the solvent was removed in vacuo and the resulting white solid was washed with hexane (6 mL). The hexane was decanted off and the white powder was dried in vacuo. Yield: 0.191 g (67%). Small colourless crystals suitable for single crystal X-ray diffraction study were grown from a concentrated CH2Cl2 reaction solution kept at −18 °C. 1H NMR (CD2Cl2, 295 K): δ = 1.28 (d, 2JHP = 8.0 Hz, CH3). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −37.9 (s); (253 K): −38.6 (br s); (183 K): −39.7 (broad, ill-defined coupling). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 66.0 (s); (253 K): 61.3 (s). IR (Nujol): ν = 488 (vs, br, AlCl) cm−1.[AlBr3(PMe3)]
To a yellow solution of AlBr3 (0.175 g, 0.66 mmol) in toluene (3 mL) was added PMe3 (0.050 g, 0.66 mmol) in toluene (3 mL). The resulting colourless solution was stirred for 1.5 h, then the solvent was removed in vacuo to yield a white powder, which was dried in vacuo. Yield: 0.186 g (83%). Anal. Calc. for C3H9AlBr3P: C, 10.5; H, 2.6. Found: C, 10.6; H, 2.6%. 1H NMR (CD2Cl2, 295 K): δ = 1.39 (d, 2JHP = 9.6 Hz, CH3). 31P{1H} NMR (toluene-d8 toluene, 295 K): δ = −40.9 (sextet, 1JAlP = 242 Hz); (CH2Cl2–CD2Cl2, 193 K): δ = −37.1 (sextet, 1JPAl = 259 Hz). 27Al NMR (toluene-d8 toluene, 295 K): δ = 101.5 (d, 1JAlP = 242 Hz); (CH2Cl2–CD2Cl2, 193 K): δ = 102.7 (d, 1JAlP = 258 Hz). IR (Nujol): ν = 414 (m), 393 (s, AlBr) cm−1.[AlI3(PMe3)]
This was made similarly from AlI3 (0.282 g, 0.69 mmol) and PMe3 (0.053 g, 0.69 mmol) in toluene (8 mL). Yield: 0.264 g (78%). Anal. Calc. for C3H9AlI3P: C, 7.4; H, 1.9. Found: C, 7.5; H, 2.0%. 1H NMR (CD2Cl2, 295 K): δ = 1.45 (d, 2JHP = 10.0 Hz, CH3). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −42.8 (br s); (193 K): −40.8 (sextet, 1JPAl = 205 Hz). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 50.2 (br s); (193 K): 52.6 (d, 1JAlP = 209 Hz). IR (Nujol): ν = 367 (s), 340 (s, AlI) cm−1.[AlBr3(PMe3)2]
To a yellow solution of AlBr3 (0.173 g, 0.65 mmol) in toluene (2 mL) was added PMe3 (0.100 g, 1.31 mmol) in toluene (3 mL) which immediately led to the solution turning colourless, with formation of a large amount of white precipitate. The reaction was stirred for 45 min, then the solvent volume was reduced to about 1 mL in vacuo. A white powder was isolated by filtration and dried in vacuo for 15 min. Yield: 0.182 g (66%). Anal. Calc. for C6H18AlBr3P2: C, 17.2; H, 4.3. Found: C, 17.1; H, 4.4%. 1H NMR (CD2Cl2, 295 K): δ = 1.25 (d, 2JHP = 7.0 Hz, CH3). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −37.4 (br s); (223 K): −32.6 (br s); (193 K): −30.3 (s). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 44.5 (br s); (193 K): no resonance. IR (Nujol): ν = 373 (s, AlBr) cm−1.[AlI3(PMe3)2]
This was made similarly from AlI3 (0.135 g, 0.33 mmol) and PMe3 (0.050 g, 0.66 mmol) in toluene (10 mL). Yield: 0.090 g (49%). Anal. Calc. for C6H18AlI3P2: C, 12.9; H, 3.2. Found: C, 12.8; H, 3.3%. 1H NMR (CD2Cl2, 295 K): δ = 1.33 (d, 2JHP = 9.2 Hz, CH3). 31P{1H} NMR (CH2Cl2–CD2Cl2, 295 K): δ = −31.6 (br s); (223 K): −28.1 (br s); (193 K): −27.2 (s). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 4.4 (br s); (193 K): no resonance. IR (Nujol): ν = 324 (s, AlI) cm−1.The two [AlI3(AsR3)] (R = Et or Ph)10 were made by reaction of a 1:1 molar ratio of AlI3 and AsR3 in toluene. [AlI3(AsPh3)]: 27Al NMR (toluene–toluene-d8, 295 K): δ = 33.1 (br s); (203 K): no resonance; [AlI3(AsEt3)]: 27Al NMR (toluene–toluene-d8, 295 K): δ = 31.9 (s); (243 K): 32.4 (s); (203 K): 31.4 (br s).
[AlCl2{o-C6H4(AsMe2)2}][AlCl4]
To a suspension of AlCl3 (0.093 g, 0.70 mmol) in toluene (5 mL) was added o-C6H4(AsMe2)2 (0.101 g, 0.35 mmol) in toluene (5 mL), resulting in a colourless solution with a white solid suspended in it. After the reaction was stirred for 1.5 h the solid was isolated by filtration, washed with hexane and dried in vacuo to give a sticky white solid. Anal. Calc. for C10H16Al2As2Cl6: C, 21.7; H, 2.9. Found: C, 21.6; H, 3.1%. 1H NMR (CD2Cl2, 295 K): δ = 1.76 (s, [12H], CH3), 7.62–7.70 (m, [4H], C6H4). 27Al NMR (CH2Cl2–CD2Cl2, 295 K): δ = 103.2 (s, AlCl4–); (203 K): 103.2 (s). IR (Nujol): ν = 490 (s, br, AlCl) cm−1.[o-C6H4(AsMe2)2(CH2)][AlCl4]2
To a suspension of AlCl3 (0.094 g, 0.70 mmol) in CH2Cl2 (5 mL) was added o-C6H4(AsMe2)2 (0.100 g, 0.35 mmol) in CH2Cl2 (5 mL). Initially a colourless solution formed, with a white solid precipitating out after 15 min stirring. The reaction was stirred for 2.5 h and then allowed to sit overnight, whereupon small colourless crystals suitable for single crystal X-ray diffraction study grew. Over the period of a week the white precipitate slowly redissolved and more colourless crystals grew, which were isolated and dried in vacuo to give a white powder. Yield: 0.052 g (23%). Anal. Calc. for C11H18Al2As2Cl8: C, 20.7; H, 2.8. Found: C, 20.6; H, 2.7%. ESI+ (CH3CN): m/z 191.2 [M + CH3CN]2+. 1H NMR (CD3CN, 295 K): δ = 2.43 (s, [12H], CH3), 3.43 (s, [2H], CH2), 8.02–8.15 (m, [4H], C6H4). 27Al NMR (CH3CN–CD3CN, 295 K): δ = 103.5 (s, AlCl4–). IR (Nujol): ν = 478 (vs, br, AlCl4–) cm−1.X-Ray experimental
Details of the crystallographic data collection and refinement parameters are given in Table 1. Crystals suitable for single crystal X-ray analysis were obtained as described above. Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (70 μm focus) with the crystal held at 100 K (N2 cryostream). Structure solution and refinements were performed with either SHELX(S/L)97 or SHELX(S/L)201317 and were straightforward, except where detailed below. H atoms bonded to C were placed in calculated positions using the default C–H distance and refined using a riding model. Distance restraints were used in [AlCl3(PMe3)] to stop hydrogen atoms disordering across the mirror plane. [o-C6H4(AsMe2)2(CH2)] [AlCl4]2 was treated as a racemic twin with the batch scale of 0.241.| Compound | [AlCl3(PMe3)] | [AlCl3(PMe3)2] | [AlCl2{o-C6H4(PMe2)2}2][AlCl4] | [AlCl2{o-C6H4(PPh2)2}2][AlCl4] |
|---|---|---|---|---|
| Formula | C3H9AlCl3P | C6H18AlCl3P2 | C20H32Al2Cl6P4 | C60H48Al2Cl6P4 |
| M | 209.40 | 285.47 | 663.00 | 1159.52 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Triclinic |
| Space group | P21/m (no. 11) | Pnma (no. 62) | C2/c (no. 15) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2) (no. 2) |
| a/Å | 6.414(3) | 9.970(3) | 13.278(4) | 10.024(3) |
| b/Å | 10.292(4) | 10.599(4) | 14.545(4) | 11.752(4) |
| c/Å | 7.649(4) | 13.562(4) | 16.554(5) | 24.438(8) |
| α/° | 90 | 90 | 90 | 81.926(14) |
| β/° | 113.698(12) | 90 | 104.126(6) | 85.52(2) |
| γ/° | 90 | 90 | 90 | 82.25(2) |
| U/Å3 | 462.4(4) | 1433.1(8) | 3100.2(16) | 2818.9(16) |
| Z | 2 | 4 | 4 | 2 |
| μ(Mo-Kα/mm−1 | 1.174 | 0.883 | 0.828 | 0.489 |
| F(000) | 212 | 592 | 1360 | 1192 |
| Total no. reflns | 4220 | 14433 |
14366 |
24430 |
| Unique reflns | 1113 | 1486 | 3544 | 12194 |
| R int | 0.056 | 0.140 | 0.032 | 0.076 |
| No. of params, restraints | 44, 9 | 66, 0 | 151, 0 | 652, 0 |
| R 1, wR2 [I > 2σ(I)]b | 0.052, 0.124 | 0.065, 0.087 | 0.030, 0.065 | 0.079, 0159 |
| R 1, wR2 (all data) | 0.055, 0.125 | 0.088, 0.091 | 0.035, 0.067 | 0.135, 0.193 |
| Compound | [(AlCl3)2{μ-Me2P(CH2)2PMe2}] | [AlCl2{Me2P(CH2)2PMe2}2][AlCl4] | [(AlCl3)2{μ-Cy2P(CH2)2PCy2}] | [o-C6H4(AsMe2)2(CH2)][AlCl4]2 |
|---|---|---|---|---|
| a Common items: T = 100 K; wavelength (Mo-Kα) = 0.71073 Å; θ(max) = 27.5°. b R 1 = ∑||Fσ| − |Fc||/∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2/∑wFo2]1/2. | ||||
| Formula | C6H16Al2Cl6P2 | C12H32Al2Cl6P4 | C26H48Al2Cl6P2 | C11H18Al2As2Cl8 |
| M | 416.82 | 566.92 | 689.24 | 637.65 |
| Crystal system | Monoclinic | Orthorhombic | Triclinic | Orthorhombic |
| Space group | P21/c (no. 14) | Pbca (no. 61) |
P (no. 2) |
Pna21 (no. 33) |
| a/Å | 10.738(6) | 19.631(5) | 8.251(2) | 14.7537(10) |
| b/Å | 7.948(4) | 16.157(4) | 9.830(2) | 14.9361(10) |
| c/Å | 11.772(6) | 34.831(8) | 11.921(3) | 10.9633(8) |
| α/° | 90 | 90 | 113.905(8) | 90 |
| β/° | 113.938(9) | 90 | 91.329(6) | 90 |
| γ/° | 90 | 90 | 103.390(7) | 90 |
| U/Å3 | 918.2(8) | 11048(5) |
852.4(4) | 2415.9(3) |
| Z | 2 | 16 | 1 | 4 |
| μ(Mo-Kα/mm−1 | 1.182 | 0.916 | 0.666 | 3.719 |
| F(000) | 420 | 4672 | 362 | 1248 |
| Total no. reflns | 10169 |
92232 |
8428 | 19941 |
| Unique reflns | 2093 | 10858 |
3884 | 4813 |
| R int | 0.062 | 0.1169 | 0.114 | 0.049 |
| No. of params, restraints | 73, 0 | 449, 0 | 163, 0 | 213, 1 |
| R 1, wR2 [I > 2σ(I)]b | 0.051, 0.076 | 0.0503, 0.0985 | 0.0859, 0.1751 | 0.0409, 0.0954 |
| R 1, wR2 (all data) | 0.065, 0.081 | 0.0871, 0.1148 | 0.1743, 0.2276 | 0.0499, 0.1002 |
Results and discussion
All syntheses, manipulations and spectroscopic measurements on the aluminium pnictogen complexes require rigorous exclusion of moisture, which otherwise causes displacement of the phosphorus donor from the aluminium by water, and generation of phosphonium salts, [R2P(H) ∩ PR2][AlX4] or [R2P(H) ∩ PR2(H)][AlX4]2. The latter are readily identified by 1H, 31P{1H} and 31P NMR spectroscopy and during the course of this work X-ray structures were obtained on several examples (see ESI†). The diarsine is very easily displaced from the aluminium by moisture, but protonates less readily. We also found that while CH2Cl2 can be used to prepare the complexes with AlCl3, attempts to use CH2Cl2 for the synthesis of AlBr3 or AlI3 complexes result in fast Cl/X exchange and incorporation of substantial amounts of chloride. This arises from the ability of the aluminium halides to promote reaction with CH2Cl2, as seen in other systems.5,18 In these cases toluene was used for the syntheses. Once isolated, the pure complexes with AlBr3 and AlI3 react only slowly with CH2Cl2, and it remains the NMR solvent of choice since it is very weakly coordinating and freezes at 176 K, with data collected immediately from freshly prepared solutions.Diphosphine complexes
The reaction of o-C6H4(PMe2)2 with AlCl3 in CH2Cl2, irrespective of the mol ratio of reactants used (1:1 or 1:2), formed white crystals with a 1:1 o-C6H4(PMe2)2–AlCl3 stoichiometry (see Scheme 1). These were shown by the X-ray structure determination to be trans-[AlCl2{o-C6H4(PMe2)2}2][AlCl4], containing a pseudo-octahedral cation (Fig. 1) and the familiar tetrahedral anion. Even with a four-fold excess of AlCl3, the 31P{1H} and 27Al NMR spectra showed no evidence for the presence of a pseudo-tetrahedral cation, [AlCl2{o-C6H4(PMe2)2}]+. The crystals are isomorphous with trans-[GaCl2{o-C6H4(PMe2)2}2][GaCl4],13 and the cell dimensions, d(M–P) (M = Al or Ga), <Cl–M–P and <P–M–P are very similar, while d(M–Cl) is ∼0.05 Å longer for M = Ga. We return to these comparisons later. The corresponding reactions with AlBr3 and AlI3 also formed only [AlX2{o-C6H4(PMe2)2}2][AlX4]. They precipitate as fine powders from toluene and slow reaction with CH2Cl2 (see above) precludes using this solvent for crystallization for the bromo and iodo complexes, although it was used satisfactorily in the less reactive gallium systems.13 The complexes are also decomposed by MeCN with partial displacement of the phosphine. The spectroscopy of the three complexes show that all contain six-coordinate trans isomers; the first examples of six-coordination in aluminium phosphine complexes.8
 | ||
| Scheme 1 The types of Al(III) phosphine complexes formed. | ||
 | ||
| Fig. 1 The structure of the centrosymmetric cation trans-[AlCl2{o-C6H4(PMe2)2}2]+ showing the atom numbering scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Symmetry operation: a = −x, −y, 1 − z. Selected bond lengths (Å) and angles (°): Al1–Cl1 = 2.2692(7), Al1–P1 = 2.4745(8), Al1–P2 = 2.4832(7), Cl1–Al1–P1 = 87.703(14), Cl1–Al1–P2 = 87.46(3), P1–Al1–P2 = 81.415(13). | ||
The far-IR spectra contain strong peaks due to the t2 mode of [AlX4]− at 488 (X = Cl), 398 (X = Br) or 334 (X = I) cm−1 (ref. 19) and weaker features at lower energy assigned to the a2u modes in the cations (with local D4h symmetry) at 410 (X = Cl), 326 (X = Br) or 281 (X = I) cm−1.
Their 1H NMR spectra in CD2Cl2 at 295 K each show single broad δ(Me) resonances without resolved 2JPH couplings, confirming that only the trans forms are present. Similarly, the 31P{1H} NMR spectra of all three complexes show resonances with modest high frequency coordination shifts which differ very little with X (Table 2). For X = Cl, the 31P{1H} resonance shows a well-defined six line pattern due to coupling to the quadrupolar 27Al nucleus (27Al, I = 5/2, 100%), but for X = Br the coupling is not clearly resolved, and for X = I only a broad singlet (w1/2 ∼ 500 Hz) is present. The 27Al NMR spectrum of trans-[AlCl2{o-C6H4(PMe2)2}2][AlCl4] shows a very sharp singlet at δ = 103.4, due to [AlCl4]− (ref. 20) and a binomial quintet at δ = +0.7 from coupling to four equivalent P atoms in the cation (Fig. 2).
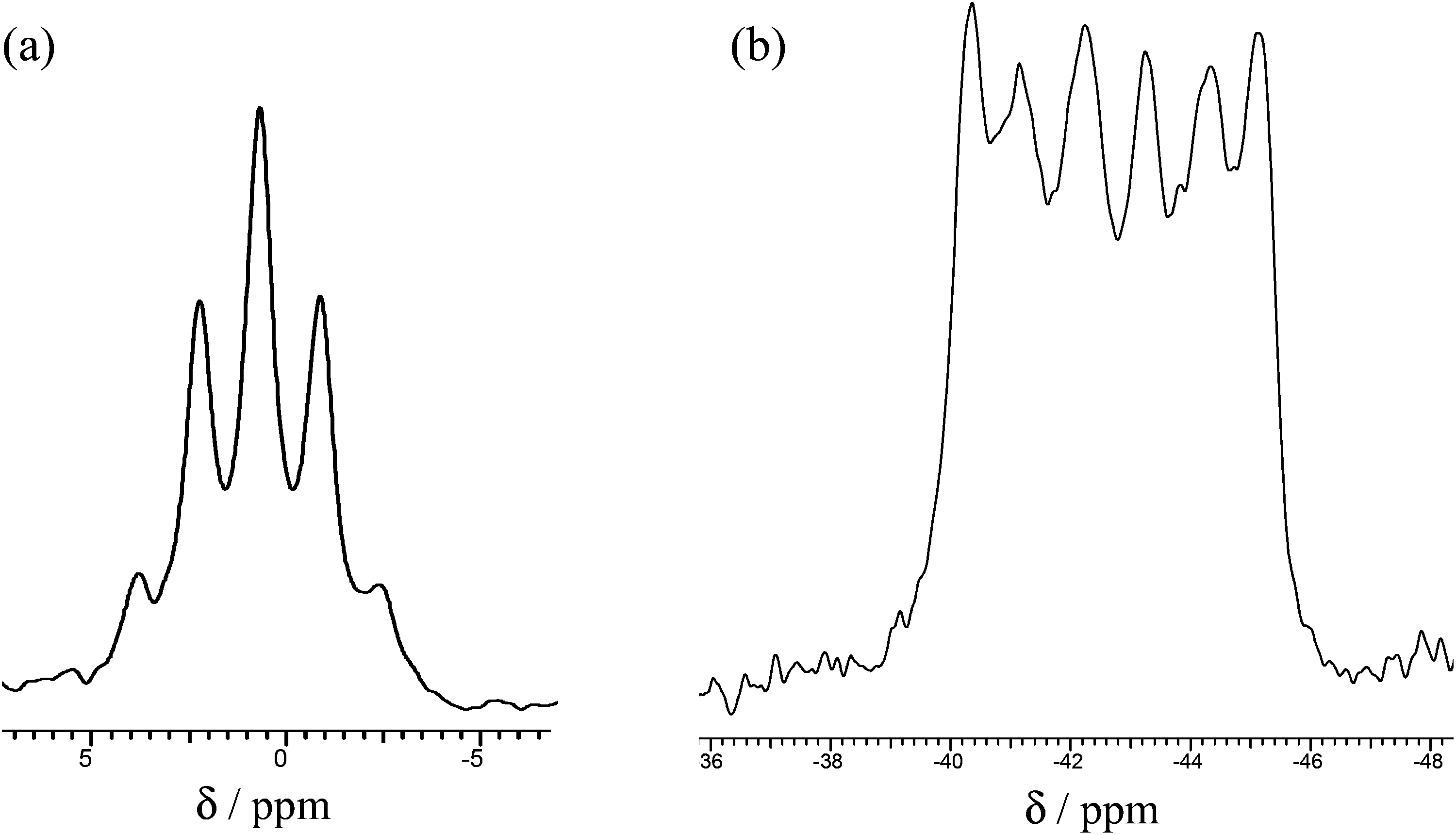 | ||
| Fig. 2 (a) 27Al NMR of [AlCl2{o-C6H4(PMe2)2}2]+ and (b) 31P{1H} NMR of [AlCl2{o-C6H4(PMe2)2}2]+ in CH2Cl2 at 295 K. | ||
| Complex | δ(31P{1H}) 295 Ka (1JAl–P/Hz) | Δ = δ(31Pcomplex − δ(31Pligand)a (295 K) | δ (27Al)a,b (295 K) |
|---|---|---|---|
| a CH2Cl2–CD2Cl2 except c., s = singlet, br s = broad singlet, v br = very broad. b 27Al resonances of [AlX4]− anions not listed; δ([AlCl4]−) = 103, δ([AlBr4]−) = 80.5, δ([AlI4]−) = −26.5. c Toluene/d8 toluene. | |||
| [AlCl2{o-C6H4(PMe2)2}2][AlCl4] | −42.8 (sextet, 155) | +12.2 | 0.7 (quintet) |
| [AlBr2{o-C6H4(PMe2)2}2][AlBr4] | −42.6 (s) | +12.4 | −12.0 (s) |
| [AlI2{o-C6H4(PMe2)2}2][AlI4] | −42.3(s) | +12.7 | – |
| [AlCl2{Me2P(CH2)2Me2}2] [AlCl4] | −40.6 (sextet, 164) | +6.4 | 1.2 (quintet) |
| [AlBr2{Me2P(CH2)2Me2}2] [AlBr4] | −38.0 (s) | +9 | −12.3 (s) |
| [AlI2{Me2P(CH2)2Me2}2] [AlI4] | −39.1 (br s) | +8 | −33.9 (br s) |
| [(AlCl3)2{μ-Me2P(CH2)2PMe2}] | −31.6 (br s). | +15.4 | 109.3 (s) |
| [AlCl2{o-C6H4(PPh2)2}][AlCl4] | −8.3 (v br) | ∼+5 | — |
| [(AlCl3)2{μ-Cy2P(CH2)2PCy2}] | −0.5 (br, s) | −2.7 | 110.6 (s) |
| [AlCl3(PMe3)] | −42.6 (br s) | +19.4 | 108.8 (s) |
| [AlCl3(PMe3)2] | −37.9 (s) | +24 | 66.0 (s) |
| [AlBr3(PMe3)] | −40.9 (sextet, 240)c | +21 | 101.5 (d)c |
| [AlBr3(PMe3)2] | −37.4 (s) | +24.5 | 44.5 (s) |
| [AlI3(PMe3)] | −42.8 (br s) | +19 | 50.2 (s) |
| [AlI3(PMe3)2] | −31.6 (br s); | +30.5 | 4.4 (s) |
| [AlI3(AsEt3)] | — | — | 31.9 (s)c |
| [AlI3(AsPh3)] | — | — | 33.1 (s)c |
The NMR spectra are essentially invariant over the temperature range 295–233 K, in contrast to those of the corresponding thioether or selenoether complexes, which show fast dissociative ligand exchange is present at ambient temperatures.5trans-[AlBr2{o-C6H4(PMe2)2}2][AlBr4] similarly shows 27Al resonances at δ = 80.5 (s) assigned to [AlBr4]− and a broad singlet at δ −12.0 (s) from the cation, whereas for trans-[AlI2{o-C6H4(PMe2)2}2][AlI4] only the 27Al resonance of the anion (δ = −26.5 (s)) is observed. The loss of coupling and then the loss of the cation resonance along the series X = Cl → Br → I can be ascribed to increasing quadrupolar relaxation rates as the electric field gradients around the Al nucleus increase with the changing donor sets.
The reaction of AlCl3 with the sterically bulkier o-C6H4(PPh2)2 in either toluene or CH2Cl2 using a variety of conditions and molar ratios, invariably produced white powders with an AlCl3–o-C6H4(PPh2)2 stoichiometry of 2:1. These showed a strong band in the IR spectrum at 483 cm−1 indicative of [AlCl4]−, leading to the proposed composition, [AlCl2{o-C6H4(PPh2)2}][AlCl4], containing a pseudo-tetrahedral cation. The 27Al NMR spectrum shows only the sharp resonance for the anion over the range 295 to 180 K, whilst the 31P{1H} NMR spectrum (CH2Cl2 solution) at ambient temperatures is a very broad feature at δ ∼ −8. On cooling the solution, the latter drifts to low frequency, reaching δ ∼ −12 at 180 K, but remaining broad. Addition of o-C6H4(PPh2)2 to the solution shows a second resonance at δ = −13.3 (295 K) which is the value for the free diphosphine, showing intermolecular exchange is slow on the NMR timescale at room temperature. Gallium(III) halides form only the [GaX2{o-C6H4(PPh2)2}][GaX4] complexes with this ligand, irrespective of the reaction conditions or ratio of reagents used.13,21 The reason for the broad 31P{1H} NMR resonance in the aluminium complex is not entirely clear; the X-ray crystal structures of [SnCl2{o-C6H4(PPh2)2}]22 and [GeCl2{o-C6H4(PPh2)2}]23 show the diphosphine coordinated in a very asymmetric manner, probably best described as κ1, and these complexes also exhibit broad 31P{1H} NMR resonances at all temperatures 295–190 K, which were attributed to intramolecular exchange between the ‘free’ and coordinated phosphorus donor groups in the diphosphine. The 31P{1H}NMR spectrum of the aluminium complex also rules out other than occasional traces of the phosphonium species [o-C6H4(PPh2)(PPh2H)]+ or [o-C6H4(PPh2H)2]2+.24 The preparation of [AlCl2{o-C6H4(PPh2)2}][AlCl4] produced a small number of colourless crystals, which were found to be trans-[AlCl2{o-C6H4(PPh2)2}2][AlCl4], containing a pseudo-octahedral cation, although the bulk product was reproducibly [AlCl2{o-C6H4(PPh2)2}][AlCl4], based upon microanalysis and spectroscopic data. We assume that the trans-[AlCl2{o-C6H4(PPh2)2}2][AlCl4] crystallised preferentially from a solution in which it was a minor constituent. The structure of the cation is shown in Fig. 3. The d(Al–P) are ∼0.1 Å longer than those in trans-[AlCl2{o-C6H4(PMe2)2}2]+, reflecting the relatively weaker donor power of the diphenylphosphino groups, and possible steric hindrance. All attempts to isolate the six-coordinate complex as a bulk product have failed, but its existence was very surprising given the presence of two very bulky ligands chelating to the small aluminium centre, and the absence of similar gallium complexes.
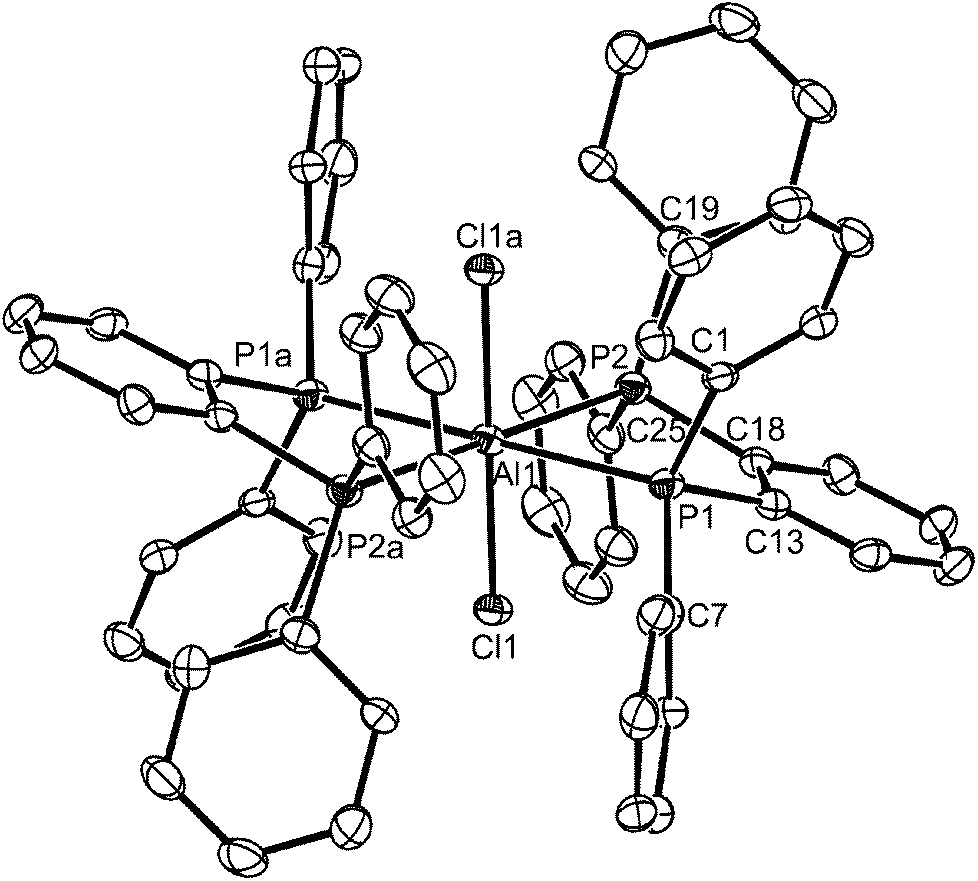 | ||
| Fig. 3 The structure of the centrosymmetric Al1 centred cation in trans-[AlCl2{o-C6H4(PPh2)2}2][AlCl4] showing the atom labelling scheme. The C6 rings are numbered cyclically with only the ipso-carbon atoms labelled and H atoms are excluded for clarity. Ellipsoids are drawn at the 50% probability level. The other cation is similar. Symmetry operation: a = 2 − x, 1 − y, 1 − z. Selected bond lengths (Å) and angles (°): Al1–Cl1 = 2.2222(13), Al1–P1 = 2.5574(15), Al1–P2 = 2.5843(15), P1–Al1–P2 = 78.69(4), Cl1–Al1–P1 = 87.80(5), Cl1a–Al1–P1 = 92.20(5), Cl1–Al1–P2 = 87.13(5), Cl1a–Al1–P2 = 92.87(5). | ||
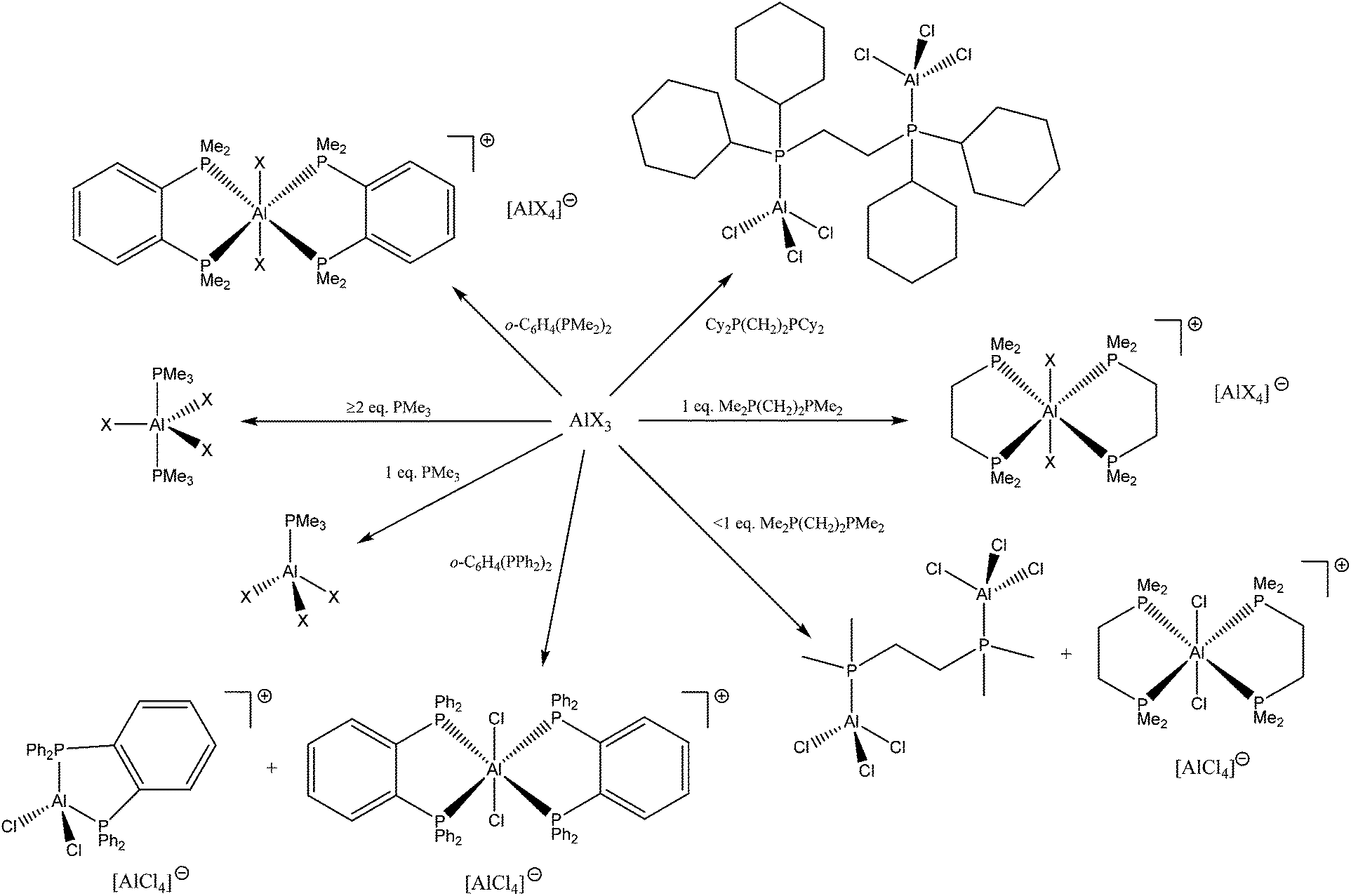
The reaction of AlX3 (X = Cl, Br or I) with Me2P(CH2)2PMe2 in a 1:1 mol ratio produces trans-[AlX2{Me2P(CH2)2PMe2}2][AlX4], which contrasts with the corresponding GaX3 complexes of diphosphinoethanes which produce only the dimers [X3Ga{μ-R2P(CH2)2PR2}GaX3].13 The presence of six-coordinate cations and [AlX4]− was confirmed by the X-ray structure of trans-[AlCl2{Me2P(CH2)2PMe2}2][AlCl4] (Fig. 4).
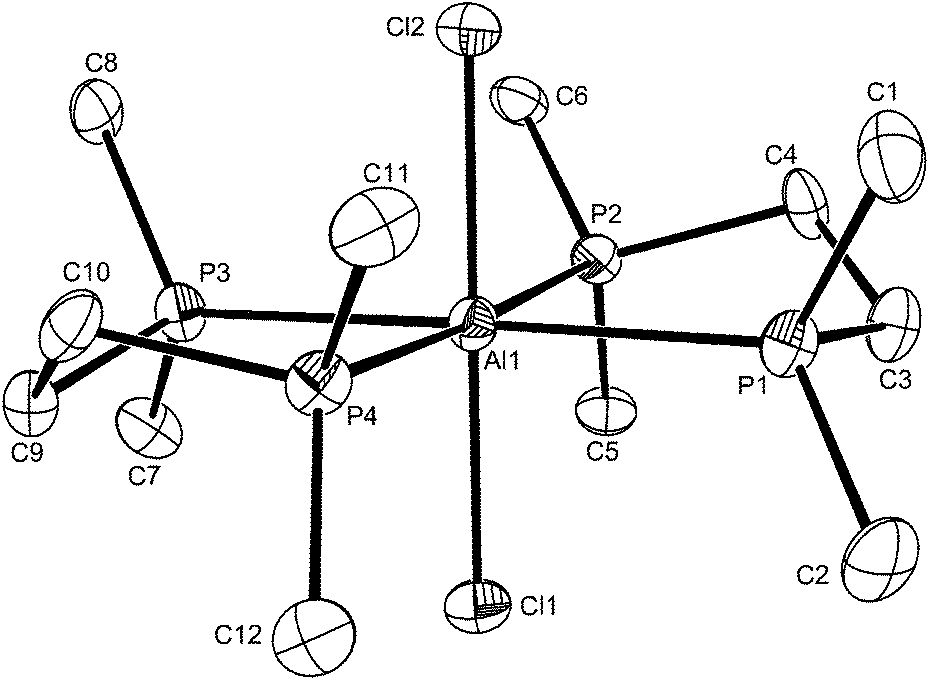 | ||
| Fig. 4 The structure of the cation in trans-[AlCl2{Me2P(CH2)2PMe2}2][AlCl4] showing the atom labelling scheme and with H atoms excluded for clarity. Ellipsoids are drawn at the 50% probability level. The other cation is similar. Selected bond lengths (Å) and angles (°): Cl1–Al1 = 2.2911(16), Cl2–Al1 = 2.2959(16), P1–Al1 = 2.4929(16), P2–Al1 = 2.4773(16), P3–Al1 = 2.4863(16), P4–Al1 = 2.4899(16), P3–Al1–P4 = 83.56(5), P2–Al1–P1 = 83.51(5), Cl1–Al1–Cl2 = 179.83(8), Cl1–Al1–P2 = 91.96(6), Cl2–Al1–P2 = 87.88(5), Cl1–Al1–P1 = 89.50(5), Cl2–Al1–P1 = 90.44(5), Cl1–Al1–P3 = 89.03(5), Cl1–Al1–P4 = 91.35(5), Cl2–Al1–P4 = 88.81(5), Cl2–Al1–P3 = 91.02(5). | ||
The multinuclear NMR data obtained for the three trans-[AlX2{Me2P(CH2)2PMe2}2][AlX4] (Table 2 and Experimental section) show these to be the only forms present in CH2Cl2 solution, with the chemical shifts and coupling patterns similar to those discussed above for the complexes of o-C6H4(PMe2)2. Notably, for trans-[AlI2{Me2P(CH2)2PMe2}2][AlI4] the 27Al NMR resonance of the cation, δ = −33.9, was observed as a broad singlet, illustrating how the presence or absence of a resonance due to the quadrupolar nucleus varies with very small changes in the coordination environment, which clearly affect the electric field gradient and hence the relaxation rate. The solution NMR data showed no evidence for cis-isomers, whereas [AlX2{MeS(CH2)2SMe}2]+ show mixtures of cis and trans forms in solution at low temperatures, both of which were crystallographically characterised in trans-[AlCl2{MeS(CH2)2SMe}2][AlCl4] and cis-[AlI2{MeS(CH2)2SMe}2][AlI4].5
Reaction of Me2P(CH2)2PMe2 with AlCl3 in a 1:≤2 mol ratio in CH2Cl2 or toluene resulted in a mixture containing varying amounts of trans-[AlCl2{Me2P(CH2)2PMe2}2][AlCl4] and a second complex identified by a combination of 1H, 31P{1H} and 27Al NMR spectroscopy as [Cl3Al{μ-Me2P(CH2)2PMe2}AlCl3]. Despite many attempts, we have been unable to isolate a pure bulk sample of the diphosphine-bridged complex, and similar studies with AlBr3 or AlI3 showed that only the [AlX2{Me2P(CH2)2PMe2}2][AlX4] type formed, even with excess AlX3, although the solids containing excess aluminium halide were extremely moisture sensitive, and prone to forming [Me2P(CH2)2PMe2H][AlX4]. A few colourless crystals formed from one reaction mixture containing AlCl3–Me2P(CH2)2PMe2 in a 1.5:1 mol ratio were found to be of the bridged complex, thus confirming its identity (Fig. 5).
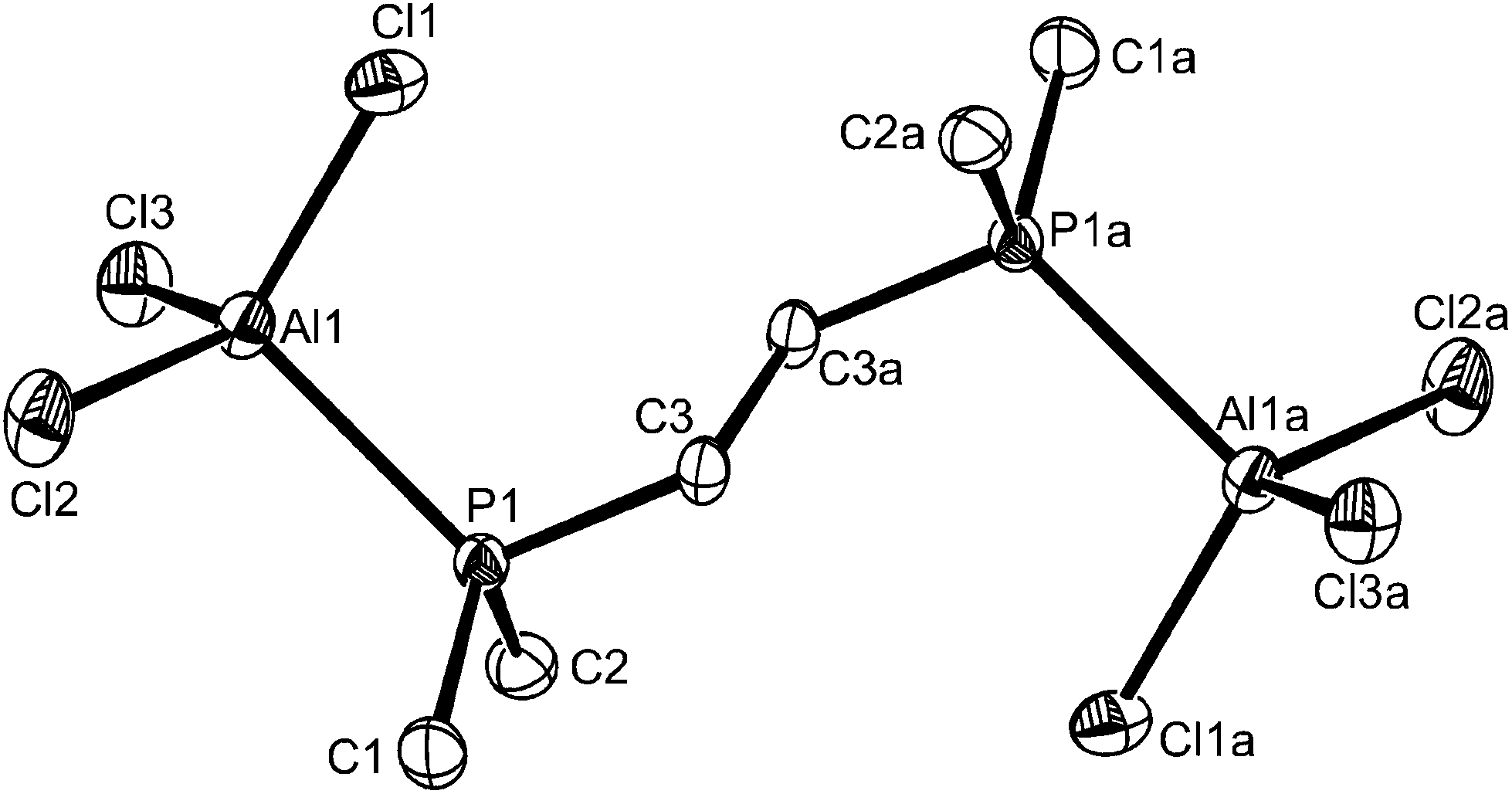 | ||
| Fig. 5 The structure of centrosymmetric [(AlCl3)2{μ-Me2P(CH2)2PMe2}] showing the atom labelling scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Symmetry operation: a = −x, 1 − y, −z. Selected bond lengths (Å) and angles (°): Al1–Cl2 = 2.1166(14), Al1–Cl3 = 2.1203(15), Al1–Cl1 = 2.1284(15), Al1–P1 = 2.4059(14), Cl2–Al1–Cl3 112.15(6), Cl2–Al1–Cl1 = 112.54(6), Cl3–Al1–Cl1 = 110.78(5), Cl2–Al1–P1 = 106.34(5), Cl3–Al1–P1 = 109.97(6), Cl1–Al1–P1 = 104.68(5). | ||
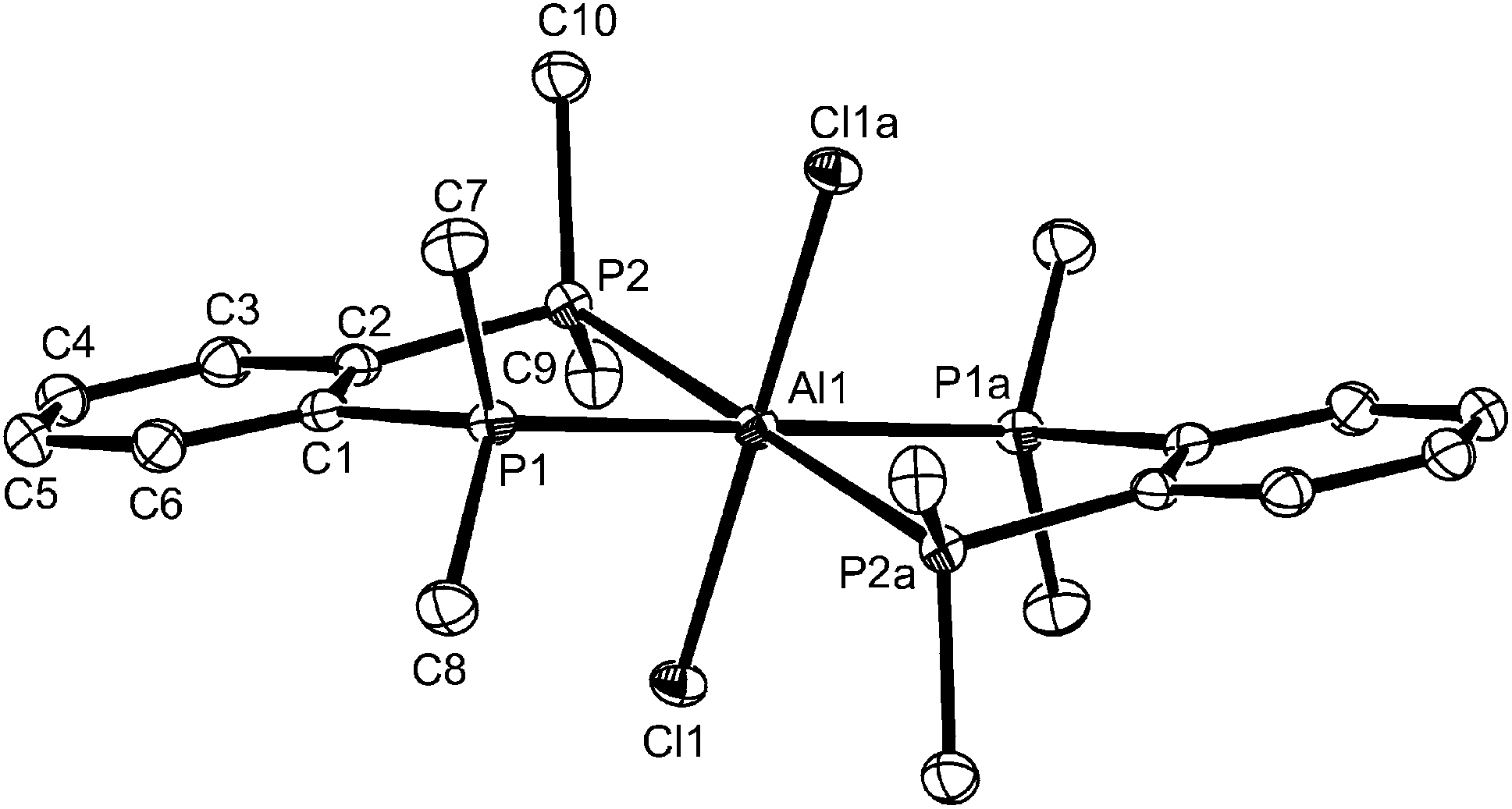
The molecule is centrosymmetric with rather shorter d(Al–P) and d(Al–Cl) than in the corresponding six-coordinate complexes described above, due to the decreased coordination number. Although not isomorphous, it is also generally similar to the structures reported13 for [(GaX3)2{Et2P(CH2)2PEt2}] (X = Br or I). The formation of the ligand-bridged dimer in the chloride system alone shows that steric factors from the increased radius of the halide co-ligands are not the driving force, but increasing the cone angle at phosphorus would be expected to favour a lower coordination number at Al. To explore this we examined the AlCl3 complexes of Et2P(CH2)2PEt2 and Cy2P(CH2)2PCy2. The former gave a similar mixture of six- and four-coordinate species to that described and these complexes were not studied further, but the more bulky cyclohexyl-substituted ligand gave only the dimer, [(AlCl3)2{μ-Cy2P(CH2)2PCy2}].
The [(AlCl3)2{μ-Cy2P(CH2)2PCy2}] is extremely sensitive and very readily decomposed by trace moisture, and quite quickly by standing in CH2Cl2 solution at room temperature, generating [AlCl4]− and phosphonium cations. The X-ray structure (Fig. 6) was obtained from a very small crystal, and confirms the centrosymmetric dimer. The d(Al–P), d(Al–Cl), <Cl–Al–Cl and <Cl–Al–P are similar to those in the dimer shown in Fig. 5, showing the bulky cyclohexyl groups do not exert any great steric effects at the four-coordinate aluminium centre.
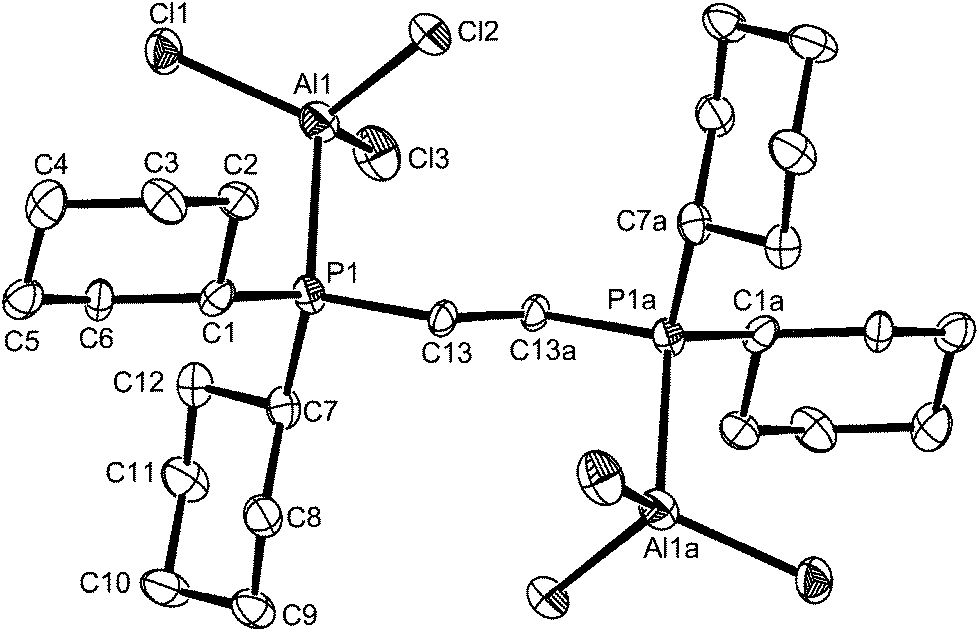 | ||
| Fig. 6 The structure of centrosymmetric [(AlCl3)2{μ-Cy2P(CH2)2PCy2}] showing the atom labelling scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Symmetry operation: a = 2 − x, 1 − y, 1 − z. Selected bond lengths (Å) and angles (°): Al1–Cl3 = 2.114(3), Al1–Cl1 = 2.123(3), Al1–Cl2 = 2.129(3), Al1–P1 = 2.407(3), Cl3–Al1–Cl1 = 112.98(12), Cl3–Al1–Cl2 = 113.23(12), Cl1–Al1–Cl2 112.00(12), Cl3–Al1–P1 = 106.20(11), Cl–Al1–P1 = 108.76(11), Cl2–Al1–P1 = 102.90(10). | ||
The 27Al NMR (CH2Cl2–CD2Cl2, 295 K) of this complex shows a singlet at δ = 110.6, consistent with a tetrahedral aluminium centre, whilst the 31P{1H} NMR is a singlet at δ = −0.5. The coordination shift (Δ) for this complex is very small and negative (−0.3) which contrasts with the small positive values of Δ seen for the other diphosphine complexes. This observation parallels those observed in trimethylaluminium–phosphine complexes, where small phosphines exhibit small positive coordination shifts, but as the steric bulk of the phosphine increases the Δ values become negative, attributed to steric effects.25
Monophosphine complexes
The reaction of AlCl3 with PMe3 in a 1:1 molar ratio in anhydrous CH2Cl2 forms four-coordinate [AlCl3(PMe3)], whilst using a 1:≥2 molar ratio produced five-coordinate trans-[AlCl3(PMe3)2]. The X-ray structures of both were determined (Fig. 7 and 8) and reveal the expected C3v and D3h geometries respectively. The d(Al–Cl) and d(Al–P) distances are ∼0.07 Å longer in the five-coordinate complex, attributed to the increased coordination number. The d(Al–P) in trans-[AlCl3(PMe3)2] is also shorter (by 0.06 Å) than that in trans-[AlI3(PEt3)2],12 consistent with the rather weaker Lewis acidity expected for aluminium iodide.
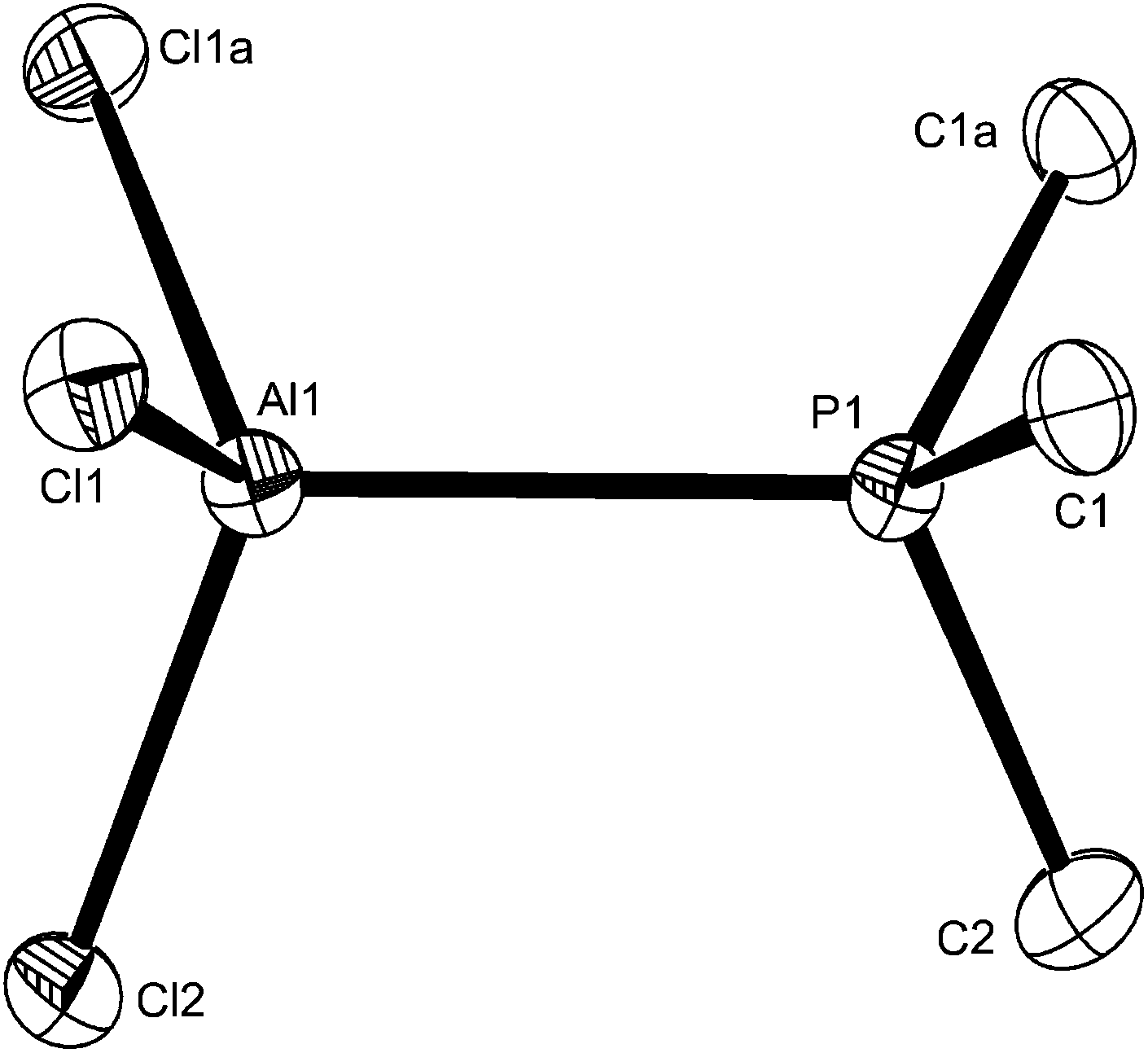 | ||
| Fig. 7 The structure of [AlCl3(PMe3)] showing the atom labelling scheme. The molecule has a mirror plane. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Symmetry operation: a = x, 1/2 − y, z. Selected bond lengths (Å) and angles (°): Al1–Cl1 = 2.1291(15), Al1–Cl2 = 2.130(2), Al1–P1 = 2.392(2), Cl1–Al1–Cl1a = 111.15(10), Cl1–Al1–Cl2 = 111.06(6), Cl1–Al1–P1 = 107.22(6), Cl2–Al1–P1 = 108.94(9). | ||
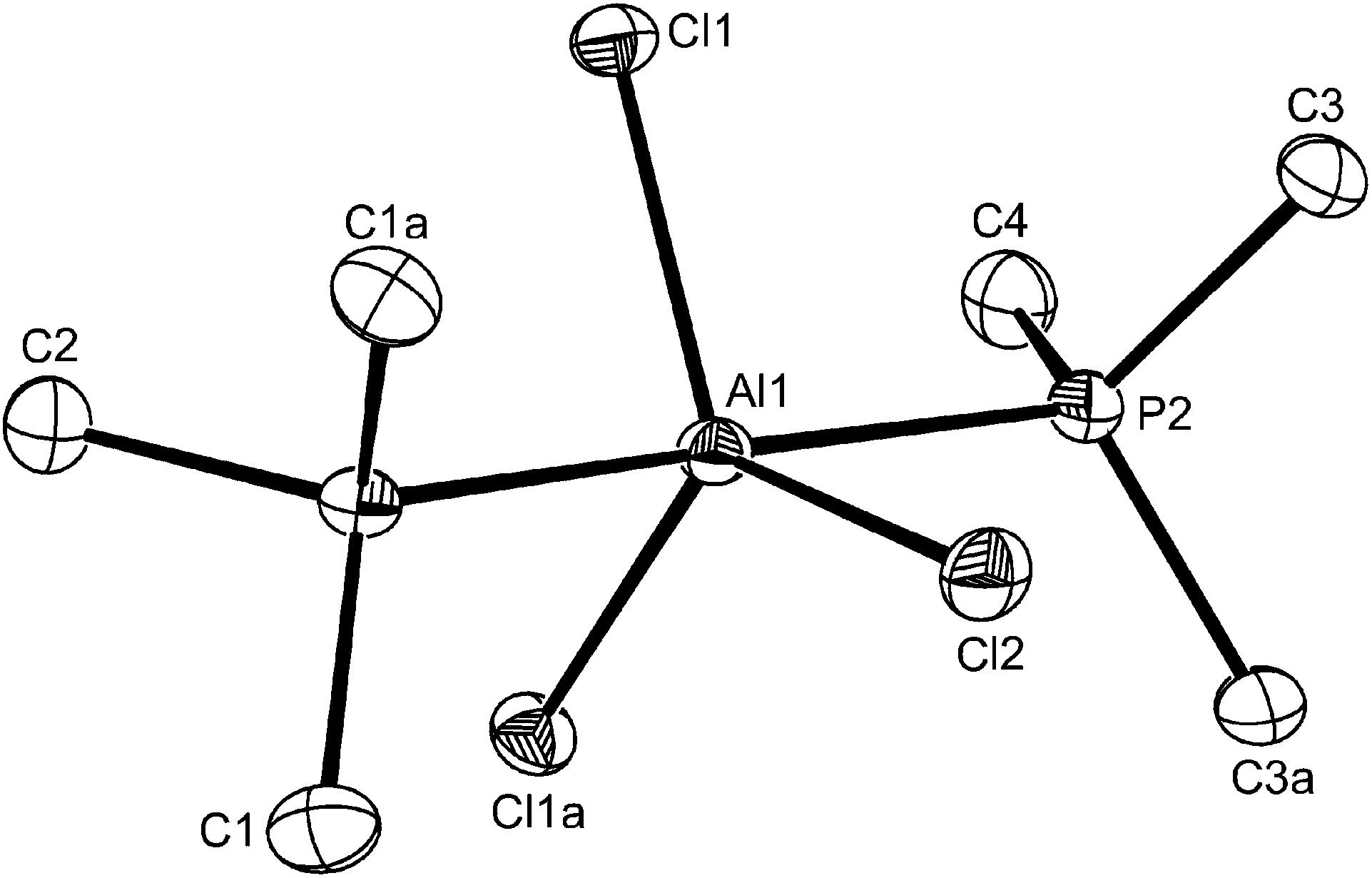 | ||
| Fig. 8 The structure of trans-[AlCl3(PMe3)2] showing the atom numbering scheme. The molecule lies on a mirror plane going through C4, P2, Al1, Cl2, P1, C1. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Symmetry operation: a = x, 1/2 − y, z. Selected bond lengths (Å) and angles (°): Al1–Cl2 = 2.203(2), Al1–Cl1 = 2.2029(14), Al1–P1 = 2.452(2), Al1–P2 = 2.459(2), Cl2–Al1–Cl1 = 121.22(5), Cl1–Al1–Cl1 = 117.56(9), P1–Al1–P2 = 177.31(9), Cl2–Al1–P1 88.79(8), Cl1–Al1–P1 = 90.44(6), Cl2–Al1–P2 88.52(8), Cl1–Al1–P2 = 90.96(6). | ||
The 31P{1H} NMR spectrum of [AlCl3(PMe3)] at 295 K in CH2Cl2–CD2Cl2 exhibits a broad resonance at δ = −42.6, which on cooling the solution, resolves into a six-line coupling pattern with 1JPAl = 275 Hz. The corresponding 27Al NMR spectrum at 295 K is a singlet at δ = 108.8, which splits into a doublet with the same 1JPAl on cooling. In contrast, the 31P{1H} NMR spectrum of [AlCl3(PMe3)2] shows ill-defined coupling even at low temperatures, whilst the 27Al NMR spectrum is a singlet at δ = 66.0 (295 K). Examination of the 31P{1H} spectra of either complex in CH2Cl2 solution containing added PMe3, shows singlets at room temperature due to fast exchange between the ‘free’ and coordinated phosphine, but on cooling the exchange slows and separate resonances for the complex and PMe3 are observed. Even with a large excess of PMe3 there was no evidence for the formation of a six-coordinate species such as [AlCl3(PMe3)3] or [AlCl2(PMe3)4]+.
The corresponding reactions of PMe3 with AlX3 (X = Br or I) were carried out in toluene, since in CH2Cl2 rapid formation of aluminium chloro-species was observed, as discussed. Depending on the PMe3:AlX3 ratio used, either [AlX3(PMe3)] or [AlX3(PMe3)2] were isolated as very moisture sensitive white powders. The pure complexes dissolve in CD2Cl2 and NMR spectra can be obtained from the freshly prepared solutions without significant Cl/X exchange. However, if some AlX3 is present (or added to the solution) rapid halide scrambling occurs. The spectroscopic properties of the bromide and iodide complexes (Experimental section) are much as expected and the NMR spectra (Table 2) show systematic chemical shift changes with the aluminium coordination number and the halogen present.
Diarsine complexes
A series of arsine complexes [AlX3(AsR3)] (X = Cl, Br or I; R = Me, Et or Ph) have been described, along with the X-ray structures of [AlX3(AsPh3)] (X = Cl or I).10 These show the presence of the expected pseudo-tetrahedral molecules. In passing we note that 27Al NMR spectra reported (obtained from in situ reaction mixtures in CH2Cl2) for X = Br or I, show resonances in the regions typical of chloro-species (δ(27Al) = 100–112), indicating Cl/X exchange with the solvent had most likely occurred. To confirm this we obtained 27Al NMR spectra for [AlI3(AsR3)] prepared and recorded in toluene solution, which showed δ = 33.1 (R = Ph) or δ = 31.9 (s) (R = Et), similar to the values reported for the phosphine complexes (above). We also found that addition of AsEt3 to a toluene solution of [AlI3(AsEt3)] failed to generate any new resonance, indicating that a five-coordinate complex did not form, which is in contrast to the phosphine systems.The reaction of AlCl3 with the diarsine, o-C6H4(AsMe2)2, in toluene solution gave a slightly sticky white solid with an AlCl3:o-C6H4(AsMe2)2 composition of 2:1, which we formulate as [AlCl2{o-C6H4(AsMe2)2}][AlCl4] based upon IR and 27Al NMR spectroscopic evidence for the presence of the anion. Storing the CH2Cl2 solution produced from reacting AlCl3 with o-C6H4(AsMe2)2 in a 2:1 molar ratio for a few days formed a modest yield (23%) of colourless crystals. These were identified as [o-C6H4(AsMe2)2(CH2)][AlCl4]2, in which the diarsine has been diquaternized by the solvent, yielding a dication with a CH2 bridge between the arsenic centres. Spectroscopic data for the cation are reported in the Experimental section and the structure is shown in Fig. 9. The dimensions are unexceptional, but the formation of this species again shows that the reactivity of the aluminium halide systems is not limited to ligand coordination. The diphosphine analogue, [o-C6H4(PMe2)2(CH2)]2+ was obtained in (failed) attempts to make phosphine complexes of SnF2, and may be synthesised from the diphosphine and CH2I2 in toluene.22 A related diarsine cation with an oxido bridge, [o-C6H4(AsMe2Cl)(μ-O)AsMe2]+ is formed by hydrolysis of [VCl4{o-C6H4(AsMe2)2}x] (x = 1 or 2) or [VOCl2{o-C6H4(AsMe2)2}] in thf solution.26
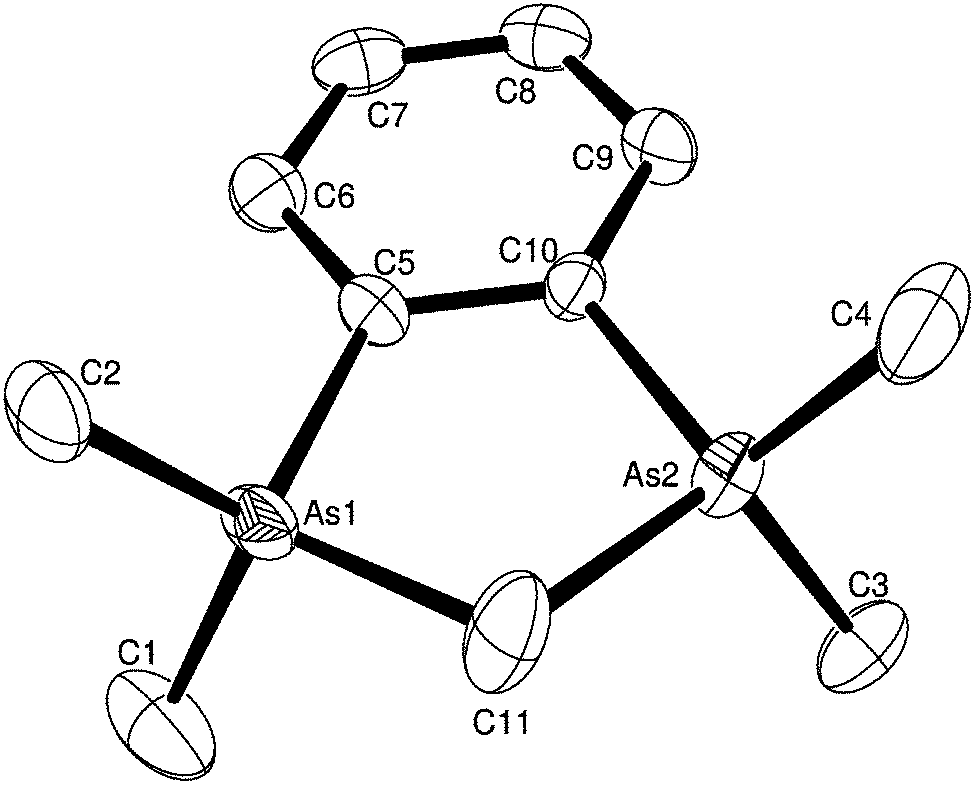 | ||
| Fig. 9 The structure of the cation in [o-C6H4(AsMe2)2(CH2)][AlCl4]2 showing the atom numbering scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): As1–C2 = 1.907(6), As1–C5 = 1.910(5), As1–C1 = 1.910(7), As1–C11 = 1.968(7), As2–C3 = 1.906(6), As2–C10 = 1.907(5), As2–C4 = 1. 910(7), As2–C11 = 1.969(7), As1–C11–As2 = 104.2(3). | ||
Comparisons with gallium systems
Gallium(III) halides form four-coordinate [GaX3(PR3)], [X3Ga(μ-L–L)GaX3] or [GaX2(L–L)]+ with most phosphines and diphosphines (L–L), with six-coordination observed only with o-C6H4(PMe2)2, in the cations [GaX2{o-C6H4(PMe2)2}2]+.8,13 In contrast, as described above, aluminium(III) halides can produce four-, five- or six-coordinate complexes depending upon the phosphine present. In p-block chemistry coordination numbers and geometries are controlled by a combination of steric and electronic effects, and the structural and composition data reported above, combined with literature data,8–13,21 should allow some elucidation of the factors involved. Considering steric effects first, the single bond covalent radii of Al(III) and Ga(III) are quoted in standard texts as very similar (∼1.25 Å), although the ionic radius of Ga3+ is ∼0.07 Å larger than that of Al3+.27 In a study of complexes with Group 16 donor ligands, we found that for comparable complexes, d(M–X) and d(M–E) (E = S, Se, Te) were very similar for M = Al or Ga and typically ∼0.2 Å longer for M = In.5 DFT calculations predict that the bond lengths involving neutral ligands for a fixed metal (Al, Ga or In) generally increase (for gas phase molecules) Cl < Br < I, reflecting decreasing Lewis acidity down the series.5,7–9 However, in some cases solid state effects (packing effects, intermolecular interactions, hydrogen bonding or interaction with solvent molecules) can mask this trend.9 In order to explore the issue of size between Al and Ga centres, we compared d(M–X) (X = Cl, Br or I) for the four-coordinate [MX4]− ions, with data taken from the Cambridge Structural Database.28 The results are shown in Table 3 and as histograms in the ESI† and show that the d(M–X) are essentially the same for gallium and aluminium. The fact that aluminium(III) has the same covalent radius to gallium(III) allows us to rule out steric effects as an explanation for the ability of aluminium to achieve a higher coordination number than gallium in many of the phosphine systems described in the present work. The similar size of Al and Ga is explained as being due to the “3d block contraction”, i.e. the increased nuclear charge resulting from insertion of the 3d transition metals, not completely offset by the screening effect of the 3d electrons.| [MX4]− | X = Cl | X = Br | X = I |
|---|---|---|---|
| M = Al | 2.126 ± 0.018 | 2.289 ± 0.015 | 2.527 ± 0.012 |
| M = Ga | 2.166 ± 0.018 | 2.321 ± 0.016 | 2.544 ± 0.018 |
| M = In | 2.337 ± 0.022 | 2.484 ± 0.018 | 2.702 ± 0.014 |
The trend to higher coordination numbers found for aluminium(III) is thus an electronic effect, resulting from higher Lewis acidity towards phosphine ligands, as predicted by the DFT studies.7,15
Conclusions
The synthesis, structures and spectroscopic characteristics of a range of four-, five, and six-coordinate aluminium complexes with phosphine donor ligands have been described. The contrasts with gallium(III) phosphine chemistry have been highlighted and shown to be due to greater Lewis acidity of the aluminium centre in these systems, consistent with computational predictions.9,15 In contrast to many d-block metals where analogous phosphine and arsine ligands produce very similar chemistries, the affinity of AlX3 for arsines is markedly less, and only four-coordinate complexes were identified. This discrimination is also seen in complexes of other light p-block elements; SiX4 (X = Cl, Br or I) form phosphine complexes but do react with AsMe3 or o-C6H4(AsMe2)2,29 whilst GeCl4 produces the structurally authenticated trans-[GeCl4(AsR3)2] (R = Me or Et), but does not form a complex with o-C6H4(AsMe2)2.30 The high reactivity of AlX3 also contributes to the synthetic challenges in this area, with their ability to activate chlorocarbon solvents toward reaction at the P- or As-centres.Acknowledgements
We thank EPSRC for support (EP/I033394/1) and Dr M. Webster for helpful discussions. The SCFED Project (http://www.scfed.net) is a multidisciplinary collaboration of British universities investigating the fundamental and applied aspects of supercritical fluids.References
- (a) S. Dagorne and S. Bellemin-Laponnaz, in The group 13 metals aluminium, gallium, indium and thallium, ed. S. Aldridge and A. J. Downs, Wiley, NY, 2011, p. 654 Search PubMed; (b) J. A. Miller, in The chemistry of aluminium, gallium, indium and thallium, ed. A. J. Downs, Blackie, London, 1993, p. 372 Search PubMed.
- (a) Comprehensive organic synthesis, ed. B. M. Tost, Pergamon, Oxford, 1991, ch. 3, vol. 2, p. 702 Search PubMed; (b) T. Ooi and K. Maruoka, in Lewis acids in organic synthesis, ed. H. Yamamoto, Wiley VCH, Weinheim, Germany, 2000, p. 191 Search PubMed.
- (a) G. H. Robinson, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, vol. 3, p. 347 Search PubMed; (b) S. Aldridge, in The group 13 metals aluminium, gallium, indium and thallium, ed. S. Aldridge and A. J. Downs, Wiley, NY, 2011, p. 75 Search PubMed.
- S. L. Benjamin, W. Levason and G. Reid, Chem. Soc. Rev., 2013, 42, 1460 RSC.
- K. George, M. Jura, W. Levason, M. E. Light and G. Reid, Dalton Trans., 2014, 43, 3637 RSC.
- C. Gurnani, M. Jura, W. Levason, R. Ratnani, G. Reid and M. Webster, Dalton Trans., 2008, 6274 RSC.
- K. George, M. Jura, W. Levason, M. E. Light, L. P. Ollivere and G. Reid, Inorg. Chem., 2012, 51, 2231 CrossRef CAS PubMed.
- J. Burt, W. Levason and G. Reid, Coord. Chem. Rev., 2014, 260, 65 CrossRef CAS PubMed.
- A. Y. Timoshkin, M. Bodensteiner, T. N. Sevastianova, A. S. Lisovenko, E. I. Davydova, M. Scheer, C. Grassi and A. V. Butlak, Inorg. Chem., 2012, 51, 11602 CrossRef CAS PubMed.
- E. Conrad, J. Pickup, N. Burford, R. McDonald and M. J. Ferguson, Can. J. Chem., 2010, 88, 797 CrossRef CAS PubMed.
- (a) G. Menard and D. W. Stephan Stephan, J. Am. Chem. Soc., 2010, 132, 1796 CrossRef CAS PubMed; (b) R. L. Wells, A. T. McPhail, J. A. Laske and P. S. White, Polyhedron, 1994, 13, 2737 CrossRef CAS.
- A. Ecker and H. Schnoeckel, Z. Anorg. Allg. Chem., 1998, 624, 813 CrossRef CAS.
- F. Cheng, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Inorg. Chem., 2007, 46, 7215 CrossRef CAS PubMed.
- F. Cheng, S. I. Friend, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Inorg. Chem., 2008, 47, 9691 CrossRef CAS PubMed.
- F. Bessac and G. Frenking, Inorg. Chem., 2006, 45, 6956 CrossRef CAS PubMed.
- (a) R. D. Feltham, A. Kasenally and R. S. Nyholm, J. Organomet. Chem., 1967, 7, 285 CrossRef CAS; (b) E. P. Kyba, S. T. Liu and R. L. Harris, Organometallics, 1983, 2, 1877 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- (a) R. T. Tjahjanto, M. F. Peintinger, T. Bredow and J. Beck, Eur. J. Inorg. Chem., 2112, 3625 Search PubMed; (b) N. Burford, B. W. Royan, R. E. vH. Spence and T. S. Cameron, J. Chem. Soc., Dalton Trans., 1990, 1521 RSC.
- K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, Wiley, NY, 4th edn, 1986 Search PubMed.
- J. Mason, Multinuclear NMR, Plenum, NY, 1987 Search PubMed.
- M. Sigl, A. Schier and H. Schmidbaur, Eur. J. Inorg. Chem., 1998, 203 CrossRef CAS.
- C. Gurnani, A. L. Hector, E. Jager, W. Levason, D. Pugh and G. Reid, Dalton Trans., 2013, 42, 8364 RSC.
- F. Cheng, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Inorg. Chem., 2010, 49, 752 CrossRef CAS PubMed.
- (a) M. Sigl, A. Schier and H. Schmidbaur, Z. Naturforsch., B: Chem. Sci., 1998, 53, 1313 CAS; (b) M. Jura, W. Levason, E. Petts, G. Reid, M. Webster and W. Zhang, Dalton Trans., 2010, 10264 RSC.
- A. R. Barron, J. Chem. Soc., Dalton Trans., 1988, 3047 RSC.
- B. M. Gray, A. L. Hector, W. Levason, G. Reid, M. Webster, W. Zhang and M. Jura, Polyhedron, 2010, 29, 1630 CrossRef CAS PubMed.
- (a) M. J. Taylor and P. J. Brothers, in The chemistry of aluminium, gallium, indium and thallium, ed. A. J. Downs, Blackie, London, 1993, p. 111 Search PubMed; (b) B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- Cambridge Structural Database (CSD version 5.34; latest update = May 2013), accessed on the 21st January 2014, http://www.ccdc.cam.ac.uk.
- W. Levason, D. Pugh and G. Reid, Inorg. Chem., 2013, 52, 5185 CrossRef CAS PubMed.
- (a) M. F. Davis, W. Levason, G. Reid, M. Webster and W. Zhang, Dalton Trans., 2008, 353 Search PubMed; (b) S. M. Godfrey, I. Mushtaq and R. Pritchard, Dalton Trans., 1999, 1319 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The X-ray structural data for [o-C6H4(PMe2)(PHMe2)]2[AlX4][X] (X = Cl or Br) and [Cy2P(H)(CH2)2PCy2(H)][AlCl4]2 and an analysis of M–X bond lengths in [MX4]− (M = Al, Ga, I) based upon data taken from the Cambridge Structural Database, with derived histograms, are also given. CCDC 1005860–1005870. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02051k |
| This journal is © The Royal Society of Chemistry 2014 |