
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure, stability and photocatalytic H2 production by Cr-, Mn-, Fe-, Co-, and Ni-substituted decaniobate clusters†
Jung-Ho
Son
*a,
Jiarui
Wang
 a and
William H.
Casey
*ab
a and
William H.
Casey
*ab
aDepartment of Chemistry, University of California, Davis, One Shields Ave., Davis, CA 95616, USA. E-mail: junghoson@gmail.com; whcasey@ucdavis.edu; Fax: +1 530 752 8995; Tel: +1 530 752 3211
bDepartment of Geology, University of California, Davis, One Shields Ave., Davis, CA 95616, USA
First published on 6th August 2014
Abstract
Here we report synthesis and characterization of early transition-metal(TM)-substituted decaniobates as a continuation of our previous report of tetramethylammonium (TMA) salt of FeNb9 and NiNb9: TMA6[H2CrIIINb9O28]·14H2O (1, CrNb9), TMA8[MnIIINb9O28]·29H2O (2, MnNb9) and TMA7[H2CoIINb9O28]·25H2O (3, CoNb9). Among the TM-substituted decaniobates, CoNb9 or NiNb9 exhibit a higher photocatalytic H2 evolution activity in methanol–water mixtures than others.
Early transition-metal (TM) substituted Keggin-type polyoxotungstates have been studied for decades because of their rich electrochemical, optical, magnetic and catalytic properties.1 In group 5 polyoxometalate chemistry, decametalate ions with D2v symmetry, such as decavanadate and decaniobate (Nb10) ions, are well known,2 but TM-substituted decametalates are rare, although TiIV-, FeIII-, NiII-substituted decaniobates (henceforth denoted: FeNb9 and NiNb9, respectively) and Pt-substituted decavanadate have been synthesized.3 Herein we describe the synthesis of the Cr-, Mn-, and Co-substituted decaniobates, and examine the trend in the structural, magnetic, optical, and photocatalytic H2-evolution properties of the TM-substituted decaniobates exhibit different stabilities and ease of synthesis that seem to be relatable to their structures. Moreover, the clusters show photocatalytic H2-evolution, with Ni- and Co-substituted decaniobate ions being more active than other substituted decaniobates, although the molecules partly dissociate during irradiation into the corresponding MOx and niobate. The results can aid the understanding of the factors governing the photocatalytic H2-evolution properties of TM-doped metal oxides, including titanates,4 other niobates,5 and related polyoxoniobate systems.6
Isolation of tetramethylammonium (TMA) salts of Cr-, Mn- and Co-substituted decaniobates in this paper, TMA6[H2CrIIINb9O28]·14H2O (1, CrNb9), TMA8[MnIIINb9O28]·29H2O (2, MnNb9) and TMA7[H2CoIINb9O28]·25H2O (3, CoNb9) was more challenging than our previously work on the Fe- and Ni-substituted decaniobates.3d We noticed that in the chromium-substitution reaction, CrNb9 coexisted with previously reported [Cr2(OH)4Nb10O30]8− (Cr2Nb10) in most of the syntheses.7 These structurally distinct clusters were separable by taking advantage of their slightly different solubility. Firstly, TMA salt of Cr2Nb10 was extracted with ethanol, and remaining TMA salt of CrNb9–Nb10 mixture was extracted with ethanol–methanol to yield an extract of 1. Crystallization of 2 and 3 were challenging because of the slow decomposition of MNb9 (M = Mn or Co) to Nb10 in the viscous liquid product. The color of the oily product changed from purple to brown (MnNb9) and pink to blue (CoNb9) during the crystallization attempt, concomitant with Nb10 crystal formation. This observation suggests decomposition of MNb9 cluster and oxidation of the corresponding released transition metal oxide (M = Mn or Co) by O2 in air. However, we were able to isolate decent amount of MnNb9 and CoNb9 crystals (28 and 45% yields, respectively) by cooling the concentrated ethanolic solution after extraction. Decomposition of Mn- and Co-substituted decaniobate structures during storage was avoided by prompt filtration by washing with ethanol, followed by drying and storage in vacuo. On the other hand, Cr-, Fe- or Ni-substituted decaniobate ions did not decompose noticeably either during the long crystallization step in a viscous liquid product or upon storage in air.
Electrospray-ionization mass spectrometry (ESI-MS) was used to determine the identities of substituted decaniobates [Fig. 2]. The purified samples were dissolved in water for ESI-MS analyses. ESI-MS of 1–3 shows peaks in the lower m/z region compared to Nb10 due to the substitution of one NbV site with an early TM of lower atomic mass than niobium. ESI-MS also indicates a single-site substitution, as was confirmed by X-ray crystallography (vide infra). We find no evidence of multiple site substitution, in spite of exploration of other reagent stoichiometries and/or different reaction temperatures.
Structures of the substituted decaniobate clusters with Cr, Mn and Co substituents were determined by X-ray single crystallography. The results show that the substitution occurred exclusively at the central site of the decaniobate moiety, similar to other substituted decaniobate structures (M = Ti, Fe, Ni) [Fig. 1].3 Bond-valence sum (BVS) calculations of metal centres suggest the oxidation state of the metals as CrIII (2.86 and 2.90), MnIII (3.07) and CoII (1.92) [Table S1†]. We note that some MnIV-included heteropolyniobate clusters have been reported previously.8 Numbers of TMA countercations found in the crystal structures of 1–3 are 6, 8 and 7, respectively, and these numbers agree well with the elemental analysis results. Thus the formulae of the clusters in 1–3 can be expressed as [H2CrIIINb9O28]6−, [MnIIINb9O28]8− and [H2CoIINb9O28]7−, respectively. In the CrNb9 structure, two protons are found on the two μ2-O atoms between Cr and Nb. Protons were not found in the electron-density map of the CoNb9 structure, but BVS calculation of the two μ2-O bound to Co (0.983 and 1.013) suggests that two μ2-O between Co and Nb are protonated, as in the CrNb9 molecule. In MnNb9 structure, BVS values of all Mn-bound oxygen atoms are higher than 1.5, supporting a conclusion that MnNb9 is not protonated. While CrIII and CoII retained their oxidation state from the source reagent, MnII from the reagent was oxidized to MnIII in the cluster, which might have happened in the hydrothermal synthesis condition.
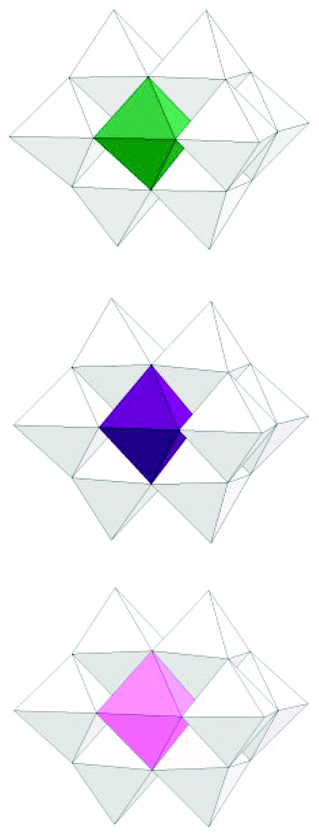 | ||
| Fig. 1 Polyhedral model of MNb9 clusters (M = CrIII, MnIII and CoII, from top to bottom) in 1–3 (white: Nb, green: Cr, purple: Mn, pink: Co). | ||
 | ||
| Fig. 2 ESI-MS of compounds 1–3 dissolved in water (from top to bottom). | ||
Enough of these MNb9 structures are now available to compare the M–O bond lengths [Fig. 3]. We find that the M–μ6-O and M–μ2-O lengths increase from Cr to Co then decrease slightly for the Ni-substituted molecule. This trend is similar to the Shannon's ionic radii of the TM ion series.9 We speculate that a discrepancy in this trend for MnNb9 is due to the disordered central site with half occupancy of Nb in the structure of MnNb9. We point out that two axial trans M–μ3-O bonds are asymmetric in Cr, Fe and Ni derivatives of the MNb9, while those in Mn- and Co-substituted MNb9 are more symmetric [Fig. 3]. Larger differences in the axial trans M–μ3-O bond lengths are observed as the group number of the substituted metal increases. (The red stretched circles are shown the same size to better indicate how asymmetry increases as one moves to the right in Fig. 3.) We note that clusters with large asymmetry in the axial M–μ3-O bonds have greater stability than clusters with symmetric M–μ3-O bonds lengths: MnNb9 and CoNb9 slowly decomposed to Nb10 in the viscous crude product, as described above. The correlation is interesting but inconclusive and but immediately suggests a useful computational study.
 | ||
| Fig. 3 The central M–O bond lengths in decaniobate and TM-substituted decaniobates. X-axis shows corresponding central atoms and their electron configurations. | ||
An ESI-MS titration of 2 mM solutions of each cluster was performed to compare the stabilities of the substituted decaniobates as a function of pH [Fig. S1–S3†]. The varying intensity of the strongest peak of each ESI-MS data (445–450 m/z) was plotted to evaluate the stability of each substituted decaniobate clusters according to pH [Fig. 4]. While 1–3 show similarly decreasing peak abundance above pH 11 in the base titration, which suggests decomposition, a different trend is evident in the acid titration. CoNb9 forms a precipitate immediately upon adding a small amount of acid, as we have found previously in the titration of FeNb9 and NiNb9.3d However, MnNb9 and CrNb9 did not readily precipitate by adding acid; titration of MnNb9 and CrNb9 with acid exhibited some buffering and significant precipitation only occurred below pH 5.3 and pH 4.7, respectively. Although the stabilities are broadly similar across the series [Fig. 4], the stability window in acidic region is CrNb9 > MnNb9 > Fe ≈ Co ≈ NiNb9.
 | ||
| Fig. 4 Normalized peak intensity (strongest peak) in ESI-MS of 1–3 as a function of pH, based on Fig. S1–S3.† | ||
A purified sample of 1 has a lighter green color relative to the dark turquoise (bluish green) of [Cr2Nb10O34]8−, both in solution and solid. Crystals of 2 are deep purple and those of 3 are violet. The UV-Vis spectra of 1–3 during titration with TMAOH solution are shown in Fig. S4–S6.† The solution of 1 shows absorption at 450 and 650 nm from 4A2g(F)→4T1g(F) and 4A2g(F)→4T2g(F) transitions, respectively [Fig. S4†].10 Different electronic transitions from light absorption are responsible for the slightly different colors of CrNb9 and Cr2Nb10, as CrNb9 is absorbing at 650 nm, while Cr2Nb10 shows absorption at 600 nm.7 During the titration of 1 with base, the two absorption maxima at 450 and 650 nm start to shift to 470 and 670 nm above pH 9 and a new absorption at 320 nm becomes evident. Titration coupled to ESI-MS indicated that significant decomposition only occurred above pH ∼ 11 [Fig. 4 and Fig. S1†]. We thus suggest that the change of spectral profile of CrNb9 above pH 9 is more likely due to deprotonation than decomposition, although this conclusion is speculative. A solution of 2 exhibits a broad absorption at 550 nm, which can be assigned to 5Eg→5T2g transition of MnIII [Fig. S5†].10 The natural pH attained by a 2 mM solution of 2 is relatively high (∼10), compared to 1 (pH 6.7) and 3 (pH 8.6), indicating a higher proton affinity. During the base titration of 2, an isosbestic point was observed around pH 11. A solution of 3 shows absorption at 500 and 545 nm (4T1g(F)→4T1g(P) transition), which is a similar feature in the [Co(H2O)6]2+ ion [Fig. S6†].10 The spectra of CoNb9 did not change significantly until pH ∼ 12, which is similar to the behavior found in UV-Vis titrations of FeNb9 and NiNb9.3d
The magnetic measurements satisfy the Curie law, as can be seen from the almost linear 1/χmvs. temperature plot of each compound [Fig. S7†]. Thus the compounds are paramagnetic, as is expected given that the clusters each contain a single isolated TM in otherwise diamagnetic niobate framework. The Curie constants derived by curve fitting χmvs. temperature plot are presented in Table S2.†Fig. 5 shows the effective magnetic moments (μeff) of the series as a function of temperature. The μeff values of each compound at their maxima are close to typical experimental μeff values for compounds with single corresponding TM ion in high-spin configuration (i.e. 3.8, 4.9, 5.9, 4.8, 3.2 for CrIII, MnIII, FeIII, CoII, NiII, respectively),11 confirming the single-site substitution, the assigned oxidation states and high-spin states of the heterometals. However, the μeff values of all compounds slightly decrease with increasing temperature, which might be due to the spin disorder at higher temperatures. We note that FeNb9 and CrNb9 have maximum μeff around 7 K. On the other hand, NiNb9, MnNb9 and CoNb9 show maxima at 20 K, 35 K and 120 K, respectively. The sharp decrease of μeff of NiII and MnIII compounds in the low-temperature region is known to be due to zero-field splitting.12 The gradual decrease of μeff for the CoII-substituted decaniobate at lower temperatures has been observed for other CoII compounds, and is generally attributed to spin–orbit coupling.12
 | ||
| Fig. 5 μ eff vs. temperature for TMA salts of TM-substituted decaniobates. | ||
TM-doped polyoxometalate clusters have recently been shown to possess electrocatalytic properties for water oxidation.13 To test the ability of the niobate clusters to act as photocatalysts for H2 evolution, irradiation tests were conducted. For the experiment, 50 mg of each compound was dissolved in mixed solution of methanol and water (50 mL, 20% v/v, methanol as sacrificial oxidant). Visible-light irradiation by using UV filter (cut-off wavelength <400 nm) on the sample solutions showed no appreciable H2 evolution, indicating that the electron–hole pairs created by excitation of the TMs by visible light are not accessible for redox reactions on the cluster surface. However, irradiation with the full spectrum of the Xe lamp produced significant amounts of H2. NiNb9 and CoNb9 showed ∼4 times higher H2 evolution than Nb10 [Fig. S8†]. FeNb9, MnNb9 and CrNb9 showed similar or lower activity than Nb10. ESI-MS of the solutions after irradiation indicated that a significant amount of each cluster had decomposed to hexaniobate and Nb10 during irradiation. To explain the nature of active photocatalyst, we carried out H2-evolution experiments using higher cluster concentrations.
When the H2-evolution experiment was performed with four-times higher cluster concentration (i.e. with 0.2 g of sample in 50 mL MeOH–water, 20% v/v; 1.6 to 2.0 mM), similar trends were found, with NiNb9 (217 μmol g−1 h−1) and CoNb9 (214 μmol g−1 h−1) showing higher H2-evolution activity than the other clusters, which was generally similar or lower than Nb10 (59 μmol g−1 h−1) [Fig. 6]. The non-linearly increasing H2-evolution rate of both NiNb9 and CoNb9 suggests formation of photocatalytically active forms from consumption or dissociation of original cluster. The color of the solutions changed after irradiation [Fig. S9†] and the solution exhibited scattering of laser light by colloids. Overall absorbance in UV-Vis spectra of the solutions increased after irradiation, which is also consistent with the formation of metal–oxide colloids upon irradiation [Fig. S10†]. ESI-MS indicated that a large portion of the MNb9 clusters in solutions decomposed after irradiation to hexaniobate and Nb10, but some MNb9 still remained [Fig. S11†]. Similarly, we observed photodecomposition of the Te-substituted Lindqvist-type niobate clusters into hexaniobate and metallic tellurium nanowires, which showed high H2-evolution activity.14 High H2-evolution activity from NiNb9 is not surprising, since Ni-doped K4Nb6O17 showed much higher H2-evolution activity compared to other early TM- (from Cr to Cu) doped K4Nb6O17.5 The high H2-evolution activity of Ni-loaded K4Nb6O17 was attributed to segregated NiO nanoparticles on K4Nb6O17 sheets.15 Thus the high activity of NiNb9 could similarly be attributed to formation of Ni0 or NiOx particles and their interaction with niobates. Niobate will generate electron–hole pairs upon UV light irradiation16 and Ni0/NiOx particle will reduce protons, producing H2. We note that whether Ni0, or NiOx, or both, are the active cocatalyst is controversial; we cannot contribute to this discussion here.17
 | ||
| Fig. 6 H2-evolution upon Xe-lamp irradiation of 0.2 g of TM-substituted decaniobate TMA salts in 50 mL MeOH–H2O solution (20% v/v). | ||
Upon irradiation, CoNb9 solutions exhibited H2-evolution curves that were similar to NiNb9 solution [Fig. 6]. ESI-MS spectrum after irradiation indicated that a solution of CoNb9 is still dominated by the CoNb9 ion, but the UV-Vis spectra had changed, indicating some decomposition [Fig. S10 and S11†]. The appreciable activity of CoNb9 in photocatalytic H2-evolution is interesting because Co-doped K4Nb6O17 showed much lower H2-evolution activity compared to Ni-doped K4Nb6O17.5 Hill et al. remarked that the distinction between homogeneous and heterogeneous catalysis is elusive for their Co-doped polyoxotungstate catalytic systems, and that is certainly also true here for the substituted niobates.18
The H2-evolution activity of the cluster solution depends on pH. A large amount of light grey precipitate formed after irradiating the NiNb9 solution when the pH was lowered before irradiation, and this solution showed enhanced H2-evolution activity upon irradiation, with a distinct nonlinear curve (986 μmol g−1 h−1) [Fig. S12†]. No clusters remained in the solution after irradiation, as indicated by ESI-MS. The pH after irradiation was 5.8, much lower than natural pH of a solution formed from freshly dissolved solid, and is instead consistent with extensive hydrolysis reactions upon irradiation, leading to proton release and precipitation. Transmission-electron microscopy (TEM) images of the precipitate showed agglomerated nanoparticles (<10 nm), and the composition is about Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Nb = 1:8.7, as determined by energy-dispersive X-ray spectroscopy (EDS) [Fig. S13†], which is close to the cluster composition. This result suggests that the nanoparticles are composed of NiOx and NbOx, but phase distinction was not possible due to resolution limit. Powder X-ray diffraction of this precipitate indicated no crystallinity. One hypothesis is that, by forcing precipitation at low pH, the NiNb9 system exhibited a higher H2-evolution because the colloids were catalytic. Interestingly, CoNb9 exhibited an opposite trend [Fig. S12†]. The H2-evolution activity of CoNb9 was nearly lost after precipitate formed by lowering the pH, which suggests that H2 evolution in CoNb9 solution may be from cluster ions and not the colloids.
:Nb = 1:8.7, as determined by energy-dispersive X-ray spectroscopy (EDS) [Fig. S13†], which is close to the cluster composition. This result suggests that the nanoparticles are composed of NiOx and NbOx, but phase distinction was not possible due to resolution limit. Powder X-ray diffraction of this precipitate indicated no crystallinity. One hypothesis is that, by forcing precipitation at low pH, the NiNb9 system exhibited a higher H2-evolution because the colloids were catalytic. Interestingly, CoNb9 exhibited an opposite trend [Fig. S12†]. The H2-evolution activity of CoNb9 was nearly lost after precipitate formed by lowering the pH, which suggests that H2 evolution in CoNb9 solution may be from cluster ions and not the colloids.
Conclusions
Early TM-substituted (from group 6 to 10) decaniobate ions have differences in their solid-state structure that can be related to their stability and properties. The range of widely varying stabilities of the clusters is a key challenge in the synthesis and purification of this series of polyoxoniobates. We suggest that the higher H2-evolution activity from the Ni- and Co-substituted decaniobate ions arises via separate heterogeneous (Ni) and homogeneous (Co) routes, but in any case is only evident during UV irradiation. Their increased activity is attributed to cocatalysis from the photodecomposition products, most likely as Ni0 and Ni oxide, and Co hydroxide, with amorphous Nb2O5 or with the niobate cluster in solution. CoNb9 is interesting because a relatively larger amount of the cluster ions survive irradiation.Experimental details
Synthesis of 1 (CCDC 990475)
Hydrous niobium oxide (5 g, 80% w/w) was mixed with 0.89 g of CrCl3·6H2O in a 23 mL capacity PTFE-lined autoclave and 5.5 g of TMAOH·6H2O was added. The mixture was reacted at 110 °C for 4 days. Reaction mixture solution was washed with isopropanol in a plastic centrifuge tube (50 mL) several times until the sticky product remained. The product was extracted with ethanol until extract was nearly colorless. Ethanol extract was discarded and remaining green precipitate was extracted with methanol–ethanol (ca. 1:1) solution. Crystalline product was obtained after evaporation. Yield = 1.9 g (28%). Elemental analysis Found: C 14.34, H 5.19, N 4.06, Cr 2.34, Nb 38.40. Calcd for C24H102N6CrNb9O42: C 14.15, H 5.05, N 4.13, Cr 2.56, Nb 41.09.
Synthesis of 2 (CCDC 990476)
Hydrous niobium oxide (5 g, 80% w/w) was mixed with 0.66 g of MnCl2·4H2O in a 23 mL capacity PTFE-lined autoclave and 5.5 g of TMAOH·6H2O was added. The mixture was reacted at 110 °C for 4 days. Reaction mixture solution was washed with isopropanol in a plastic centrifuge tube (50 mL) several times until the sticky product remained. The product was extracted with ethanol (ca. 200 mL). The ethanolic solution was concentrated to less than 50 mL by using rotary evaporator and kept in a freezer. Dark purple rod-like crystals formed. The product crystals were quickly filtered on a frit and washed with minimum amount of ethanol, and dried in vacuo. Yield = 2.3 g (28%). Elemental analysis Found: C 15.39, H 5.99, N 4.41, Mn 2.19, Nb 34.10. Calcd for C32H154N8MnNb9O57: C 15.65, H 6.32, N 4.56, Mn 2.24, Nb 34.07.Synthesis of 3 (CCDC 990477)
Hydrous niobium oxide (5 g, 80% w/w) was mixed with 0.8 g of CoCl2·6H2O in a 23 mL capacity PTFE-lined autoclave and 5.5 g of TMAOH·6H2O was added. The mixture was reacted at 110 °C for 4 days. Reaction mixture solution was washed with isopropanol in a plastic centrifuge tube (50 mL) several times until the sticky product remained. The product was extracted with ethanol (ca. 200 mL). The ethanolic solution was concentrated to less than 50 mL by using rotary evaporator and kept in a freezer. Pale violet needle-like crystals formed were quickly filtered on a frit and washed with minimum amount of ethanol and dried in vacuo. Yield = 3.5 g (45%). Elemental analysis Found: C 14.53, H 6.01, N 4.44, Co 2.50, Nb 34.30. Calcd for C28H136N7CoNb9O53: C 14.52, H 5.92, N 4.24, Co 2.55, Nb 36.14.Crystal data
(1) CCDC 990475. C24H93N6Cr1.03Nb8.98O42.44, M = 2032.17, monoclinic, a = 16.6016(8), b = 17.2436(8), c = 24.0263(11) Å, β = 106.121(1)°, U = 6607.6(5) Å3, T = 93 K, space group P21/n (no. 14), Z = 4, 66308 reflections measured, 13488 unique (Rint = 0.0218) which were used in all calculations. The final wR(F2) was 0.0656 (all data). (2) CCDC 990476. C33.60H60N8Mn1.04Nb8.96O56.90, M = 2376.48, monoclinic, a = 22.846(6), b = 13.767(4), c = 17.970(5) Å, β = 129.353(4)°, U = 4370(2) Å3, T = 93 K, space group C2/m (no. 12), Z = 2, 23936 reflections measured, 5199 unique (Rint = 0.0138) which were used in all calculations. The final wR(F2) was 0.1285 (all data). (3) CCDC 990477. C28H72N7CoNb9O53, M = 2250.05, monoclinic, a = 25.543(2), b = 13.8124(12), c = 23.383(2) Å, β = 104.026(1)°, U = 8003.9(12) Å3, T = 88 K, space group P21/c (no. 14), Z = 4, 126087 reflections measured, 24416 unique (Rint = 0.0315) which were used in all calculations. The final wR(F2) was 0.1700 (all data).
Acknowledgements
This work was supported by an NSF CCI grant through the Center for Sustainable Materials Chemistry, number CHE-1102637. We thank Prof. Frank E. Osterloh for useful discussions. We thank Aimee Brian and Peter Klavins for magnetic property measurements, and Prof. Kirill Kovnir for discussion about magnetism data.Notes and references
- (a) L. C. W. Baker, V. S. Baker, K. Eriks, M. T. Pope, M. Shibata, O. W. Rollins, J. H. Fang and L. L. Koh, J. Am. Chem. Soc., 1966, 88, 2329 CrossRef CAS; (b) L. C. W. Baker and J. S. Figgis, J. Am. Chem. Soc., 1970, 92, 3794–3797 CrossRef CAS; (c) T. J. R. Weakley, J. Chem. Soc., Dalton Trans., 1973, 341–346 RSC; (d) C. L. Hill and R. B. Brown Jr., J. Am. Chem. Soc., 1986, 108, 536–538 CrossRef CAS PubMed; (e) M. Faraj and C. L. Hill, J. Chem. Soc., Chem. Commun., 1987, 1487–1489 RSC; (f) J. Hu and R. C. Burns, J. Mol. Catal. A: Chem., 2002, 184, 451–464 CrossRef CAS; (g) J.-H. Choi, J. K. Kim, D. R. Park, T. H. Kang, J. H. Song and I. K. Song, J. Mol. Catal. A: Chem., 2013, 371, 111–117 CrossRef CAS PubMed.
- M. T. Pope, Heteropoly and Isopolyoxometalates, Springer Verlag, Berlin, 1983 Search PubMed.
- (a) M. Nyman, L. J. Criscenti, F. Bonhomme, M. A. Rodriguez and R. T. Cygan, J. Solid State Chem., 2003, 176, 111–119 CrossRef CAS; (b) U. Lee, H.-C. Joo, K.-M. Park, S. S. Mal, U. Kortz, B. Keita and L. Nadjo, Angew. Chem., Int. Ed., 2008, 47, 793–796 CrossRef CAS PubMed; (c) C. A. Ohlin, E. M. Villa, J. C. Fettinger and W. H. Casey, Dalton Trans., 2009, 2677–2678 RSC; (d) J.-H. Son, C. A. Ohlin and W. H. Casey, Dalton Trans., 2013, 42, 7529–7533 RSC.
- (a) A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS; (b) D. W. Hwang, H. G. Kim, J. S. Jang, S. W. Bae, S. M. Ji and J. S. Lee, Catal. Today, 2004, 93, 845–850 CrossRef PubMed; (c) R. Niishiro, H. Kato and A. Kudo, Phys. Chem. Chem. Phys., 2005, 7, 2241–2245 RSC; (d) P. D. Tran, L. Xi, S. K. Batabyal, L. H. Wong, J. Barber and J. S. C. Loo, Phys. Chem. Chem. Phys., 2012, 14, 11596–11599 RSC; (e) H. Yu, S. Ouyang, S. Yan, Z. Li, T. Yua and Z. Zou, J. Mater. Chem., 2011, 21, 11347–11351 RSC; (f) R. Dholam, N. Patel, M. Adami and A. Miotello, Int. J. Hydrogen Energy, 2009, 34(13), 5337–5346 CrossRef CAS PubMed.
- (a) K. Domen, A. Kudo, A. Shinozaki, A. Tanaka, K. Maruya and T. Onishi, J. Chem. Soc., Chem. Commun., 1986, 356–357 RSC; (b) A. Kudo, A. Tanaka, K. Domen, K. Maruya, K. Aika and T. Onishi, J. Catal., 1988, 111, 67–76 CrossRef CAS; (c) Y. Miseki and A. Kudo, ChemSusChem, 2011, 4, 245–251 CAS.
- (a) Z. Zhang, Q. Lin, D. Kurunthu, T. Wu, F. Zuo, S.-T. Zheng, C. J. Bardeen, X. Bu and P. Feng, J. Am. Chem. Soc., 2011, 133, 6934–6937 CrossRef CAS PubMed; (b) P. Huang, C. Qin, Z.-M. Su, Y. Xing, X.-L. Wang, K.-Z. Shao, Y.-Q. Lan and E.-B. Wang, J. Am. Chem. Soc., 2012, 134, 14004–14010 CrossRef CAS PubMed; (c) Z.-L. Wang, H.-Q. Tan, W.-L. Chen, Y.-G. Li and E.-B. Wang, Dalton Trans., 2012, 41, 9882–9884 RSC.
- J.-H. Son, C. A. Ohlin and W. H. Casey, Dalton Trans., 2012, 41, 12674–12677 RSC.
- (a) B. W. Dale and M. T. Pope, Chem. Commun., 1967, 792 RSC; (b) B. W. Dale, J. M. Buckley and M. T. Pope, J. Chem. Soc. A, 1969, 301–304 RSC; (c) C. M. Flynn Jr. and G. D. Stucky, Inorg. Chem., 1969, 8, 332–334 CrossRef; (d) C. M. Flynn Jr. and G. D. Stucky, Inorg. Chem., 1969, 8, 335–344 CrossRef; (e) J.-H. Son and W. H. Casey, Dalton Trans., 2013, 42, 13339–13342 RSC.
- (a) R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 925–945 CrossRef CAS; (b) R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley-Interscience, New York, 6th edn, 1999 Search PubMed.
- J. M. D. Coey, Magnetism and Magnetic Materials, Cambridge University Press, 2010 Search PubMed.
- H. Liu, C. J. Gómez-García, J. Peng, J. Sha, Y. Li and Y. Yan, Dalton Trans., 2008, 6211–6218 RSC.
- Q. S. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- J.-H. Son, J. Wang, F. E. Osterloh, P. Yu and W. H. Casey, Chem. Commun., 2014, 50, 836–838 RSC.
- A. Kudo, K. Sayama, A. Tanaka, K. Asakura, K. Domen, K. Maruya and T. Onishi, J. Catal., 1989, 120, 337–352 CrossRef CAS.
- (a) A. Furube, T. Shiozawa, A. Ishikawa, A. Wada, K. Domen and C. Hirose, J. Phys. Chem. B, 2002, 106, 3065–3072 CrossRef CAS; (b) A. G. S. Prado, L. B. Bolzon, C. P. Pedroso, A. O. Moura and L. L. Costa, Appl. Catal., B, 2008, 82, 219–224 CrossRef CAS PubMed.
- T. K. Townsend, N. D. Browning and F. E. Osterloh, Energy Environ. Sci., 2012, 5, 9543–9550 CAS.
- J. W. Vickers, H. Lv, J. M. Sumliner, G. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc., 2013, 135, 14110–14118 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-MS and UV-Vis titration data, magnetism data, detailed H2 evolution data with change of solution speciation by ESI-MS, UV-Vis, and TEM/EDS data of the colloids after irradiation. CCDC 990475, 990476 and 990477. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02020k |
| This journal is © The Royal Society of Chemistry 2014 |