
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal complexes containing the S(NtBu)42− tetraimidosulfate dianion†‡
Julia
Matussek
,
Regine
Herbst-Irmer
,
Ina
Objartel
and
Dietmar
Stalke
*
Institut für Anorganische Chemie der Georg-August-Universität Göttingen, Tammannstr. 4, 37077 Göttingen, Germany. E-mail: dstalke@chemie.uni-goettingen.de; Fax: +49 551 39 33373; Tel: +49 551 39 33000
First published on 9th September 2014
Abstract
Three novel metal complexes [(acac)2Cu2(NtBu)4S] (3), [Li(thf)4]2[I4Cd2(NtBu)4S] (4) and [(thf)2Li{(SiMe3)2N}Zn(NtBu)4S] (5) are prepared from the intended transmetalation of the dilithium complex of N,N′,N′′,N′′′-tetrakis(tert-butyl)tetraimidosulfate [(thf)4Li2(NtBu)4S] (1). The two lithium cations are replaced by either the cationic (acac)Cu(II) moiety, the neutral I2Cd(II) residue or only a single lithium cation is substituted by the cationic (Me3Si)2NZn(II) fragment. The complexes show two main results: first the S(NtBu)42− tetrahedron can serve as a ligand to transition metals from the soft Cu(II) to the harder Zn(II) at opposite sides and second the S–N bond distances vary only marginally in response to the various metals and the four distances constantly sum up to 6.38(2) Å. Hence the electropositive sulfur atom responds by internal shift to the metal-polarized negative charge at the outside of the S(NR)42− tetrahedron.
Isovalent electronic replacement of the oxygen atoms in the classic SOnm− molecules and ions by NR imido groups yields the polyimido sulfur species S(NR)nm− (n = 2, 3, 4 and m = 0, 2).1–8 By introducing organic substituents to the chelating nitrogen atoms, the polyanion becomes more lipophilic, thus, the resulting complexes are frequently soluble in non-polar hydrocarbons and stay in the molecular regime rather than aggregate like their S–O counterparts.6 Due to the large variety of coordination modes as found for sulfate anions, these polyimido compounds hold interesting electronic and stereochemical properties.9–14 In 1997 we first synthesized the starting material to the current paper, dilithium-N,N′,N′′,N′′′-tetrakis(tert-butyl)tetraimido sulfate (1) (Scheme 1) in a dual addition reaction of first lithium amide to S(NtBu)2 to give the product S(NtBu)3 upon oxidation with bromine.15,16 Subsequently another equivalent of lithium tert-butylamide is added to the sulfurtriimide to give 1 (Scheme 1). Once the S(NR)42− scaffold was synthesized in the following year the barium complex [(thf)4Ba2{N(SiMe3)2}2{(NtBu)4S}] (2) could be obtained (Scheme 2) by first protonating 1 with tert-butylammonium chloride to give (tBuNH)2S(NtBu)2 and subsequent metallation with [Ba{N(SiMe3)2}2].17
 | ||
| Scheme 1 Preparation of dilithium-N,N′,N′′,N′′′-tetrakis(tert-butyl)tetraimidosulfate (1).15,16 | ||
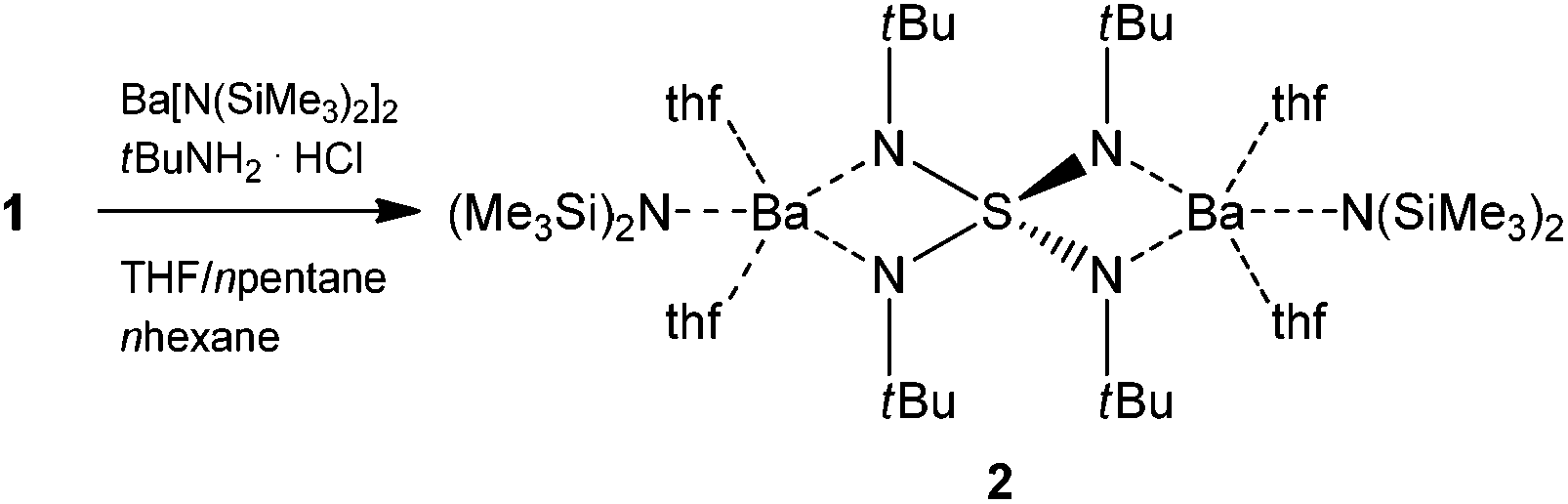 | ||
| Scheme 2 Preparation of [(thf)4Ba2{N(SiMe3)2}2{(NtBu)4S}] (2).17 | ||
Thus, these previous results indicate that S(NR)42− can be coordinated by main group metals such as lithium and barium, but the coordination to transition metals remained unknown. Hence we now embarked to synthesize d-block metal complexes, selecting late transition metals first as they resemble alkaline and alkaline earth metal properties best. By the preparation and isolation of [(acac)2Cu2(NtBu)4S] (3), [Li(thf)4]2[I4Cd2(NtBu)4S] (4) and [(thf)2Li(N(SiMe3)2)Zn(NtBu)4S] (5) presented herein we show for the first time that the lithium cations in 1 can be replaced by the transition metals copper(II), zinc(II) and cadmium(II). Apart from their similarity to s-block metals these metals were picked because of their divalent character, availability and potential application in catalysis. They will considerably widen the scope of the established sulfur imido chemistry.8,18
Results and discussion
Synthetic and structural studies
The copper complex [(acac)2Cu2(NtBu)4S] (3) can be isolated from the reaction of copper acetylacetonate with [(thf)4Li2(NtBu)4S] (1) according to Scheme 3. Two equivalents of lithium acetylacetonate precipitate and are removed by filtration. After one week in THF at −24 °C colorless blocks, suitable for X-ray structure determination, were obtained at a yield of 59%. 3 crystallizes in the monoclinic space group P2/n with half of the molecule and one THF molecule in the asymmetric unit. | ||
| Scheme 3 Synthesis of [(acac)2Cu2(NtBu)4S] (3). | ||
Each copper(II) atom is fourfold coordinated by the two oxygen atoms of the planar chelating acetylacetonate anion and by two nitrogen atoms of two opposite sides of the S(NtBu)42− tetrahedron. The fourfold coordination at the Cu(II) atom can be described as a nearly square planar environment (O1–Cu1–N1: 169.79°, O2–Cu1–N2: 169.33°). This differs considerably from the nearly tetrahedral N2O2-coordination of the lithiated starting material 1 (Fig. 1 and 2).
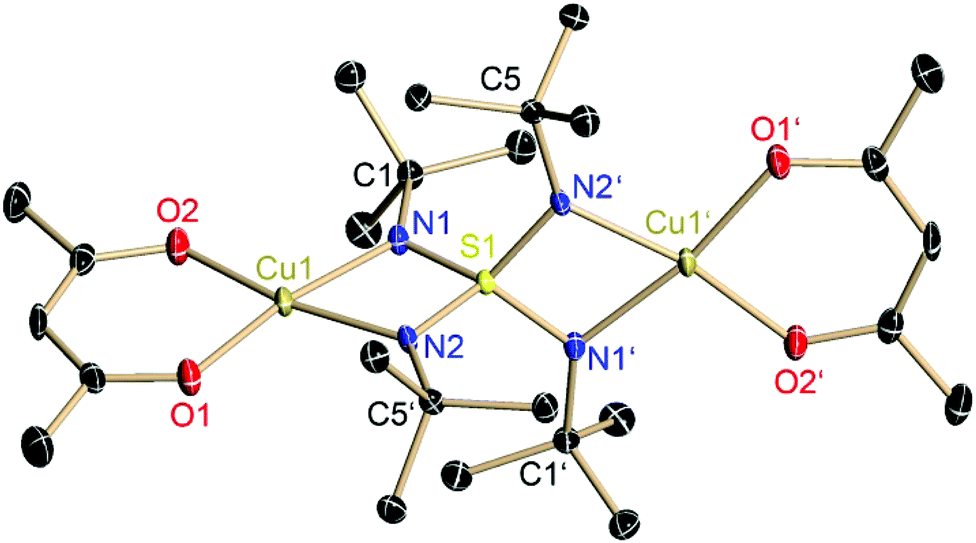 | ||
| Fig. 1 Crystal structure of [(acac)2Cu2(NtBu)4S] (3). Hydrogen atoms are omitted for clarity. Anisotropic displacement parameters are depicted at the 50% probability level. Selected bond lengths [Å] and angles [°]: S1–N1 1.5867(14), S1–N2 1.547(14), N1–Cu1 1.9598(15), N2–Cu1 1.9555(15), N1–S1–N2 93.47(7), N1–Cu1–N2 72.29(6). | ||
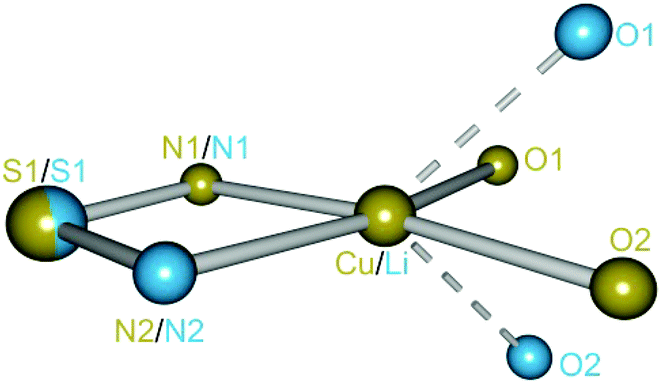 | ||
| Fig. 2 Superposition plot of 1 (Li, light blue) and 3 (Cu, brass). The atoms S1, N1 and N2 are projected onto each other with a deviation of 0.0161 Å. | ||
The two crystallographically independent S–N bond lengths in 3 (1.59 Å) do not differ significantly from the S–N bond lengths in 1 (1.60 Å) and are half-way between the values normally quoted for a typical S–N single bond (1.69 Å)19 and a S–N double bond (1.52 Å).19 However, for none of the investigated S–N bonds in methyl(diimido)sulfinic acid H(NtBu)2SMe (1.68 and 1.58 Å), methylene-bis(triimido)sulfonic acid H2C{S(NtBu)2(NHtBu)}2 (1.52 to 1.65 Å), sulfurdiimide S(NtBu)2 (1.54 and 1.53 Å), and sulfurtriimide S(NtBu)3 (1.51 Å), a classical double bond formulation could be supported from charge density investigations.20 This was further substantiated by the NBO/NRT approach. Valence expansion to more than eight electrons at the sulfur atom can definitely be excluded to explain the bonding.17,21 The same was shown recently for the sulfate anion, SO42−, as well by charge density based both on experimental and theoretical methods.22
Due to the similar bond lengths, equal distribution of the two negative charges over the four nitrogen atoms of the S(NR)42−-ligand is assumed. Furthermore, the N⋯M coordination (3/1: 1.96 Å) and the angles N⋯M⋯N (3: 72.29°, 1: 73.95°) and N–S–N (3: 93.47°, 1: 94.60°) are comparable. This can be explained by the similar cationic radius of Cu(II) and Li(I) (Cu2+: 0.71 Å, Li+: 0.73 Å).23 In published complexes with coordinated metal acetylacetonate at the nitrogen atom the N⋯M distances are 2.17 Å on average but the Cu(acac)+ cation in a fourfold coordination sphere attains N⋯M distances of 1.96 Å24 which is in excellent agreement with this result.
In the reaction of [(thf)4Li2(NtBu)4S] (1) with cadmium iodide surprisingly [Li(thf)4]2[I4Cd2(NtBu)4S] (4) is obtained according to Scheme 4. The anticipated transmetalation and salt elimination of LiI, which should be the driving force for the reaction, did not occur. Instead, the solvent separated ion pair [Li(thf)4]2[I4Cd2(NtBu)4S] (4) is found, where two equivalents of cadmium(II) iodide are coordinated by two opposite sides of the S(NtBu)42− tetrahedron, resulting in the [I4Cd2(NtBu)4S]2− dianion. Two tetrahedrally solvent coordinated [Li(thf)4]2 cations provide electro neutrality. This phenomenon might be explained by considering the various lattice energies of the involved metal halide salts. The lattice energy of CdI2 with 2455 kJ mol−1 (ref. 25) is overwhelmingly larger than the lattice energy of lithium iodide (2 × 746 kJ mol−1 (ref. 25)). Thus, considering the energetic balance, elimination of LiI is unfavorable and a higher solvation enthalpy for the lithium ions with THF is accepted.
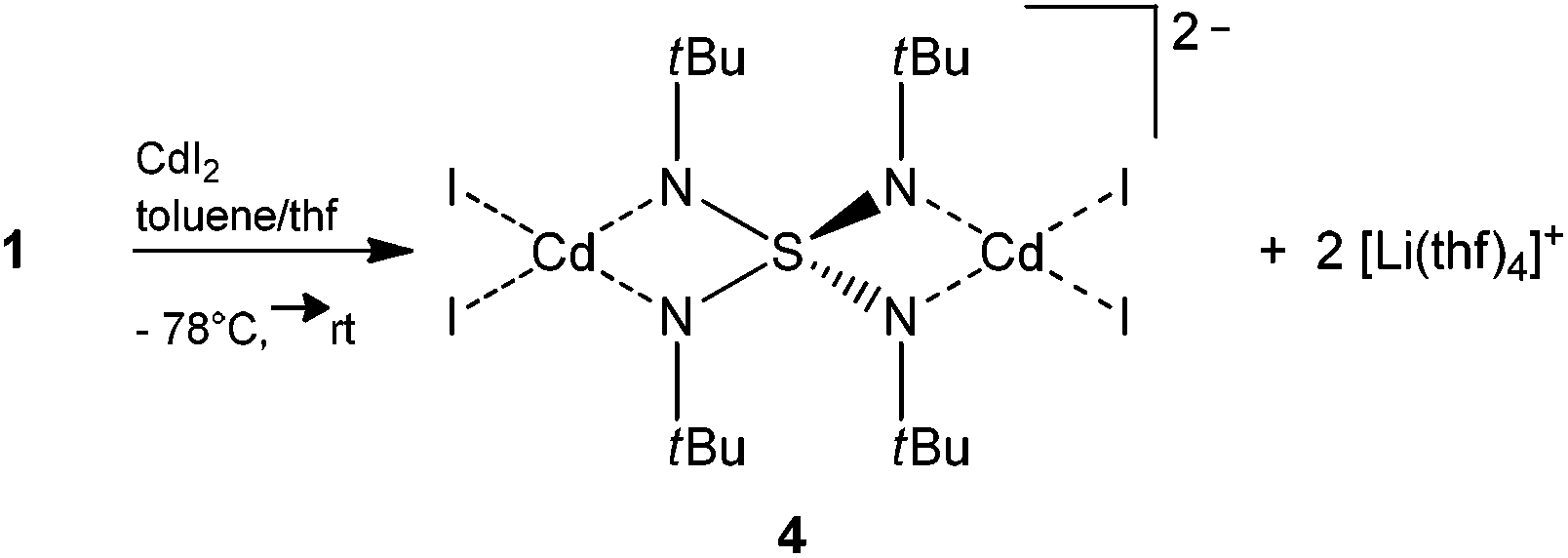 | ||
| Scheme 4 Synthesis of [Li(thf)4]2[I4Cd2(NtBu)4S] (4). | ||
4 crystallizes from THF–toluene within 4 weeks at −24 °C as colorless blocks in the monoclinic space group P21/c in a yield of 22%. The asymmetric unit contains one dianion, two cations and one THF molecule. The two cadmium atoms are tetrahedrally coordinated by two iodine atoms and two nitrogen atoms of the ligand. The central sulfur atom of the ligand possesses a distorted tetrahedral environment (95.3°–116.9°).
It is interesting to note that all three S–N bonds in all known metal complexes of the S-alkyltriimidosulfonates [RS(NR)3]− (M = Li, Ba, Al, Zn) and in the triimidosulfonic acid MeS(NtBu)2NHtBu constantly sum up to 4.70(2) Å. The SN3 unit responds flexibly to different electronic requirements induced by either different metal cations or conjugated S-substituents in terms of the sulfur atom being shifted relative to an otherwise fixed N3 environment. This seems to be valid for the S(VI)–N bonds as well and experimentally emphasizes the predominantly ionic S–N bonding rather than valence expansion and d-orbital participation in bonding.26 The four crystallographically independent S–N bond lengths average to 1.59 Å in 4 (Fig. 3).
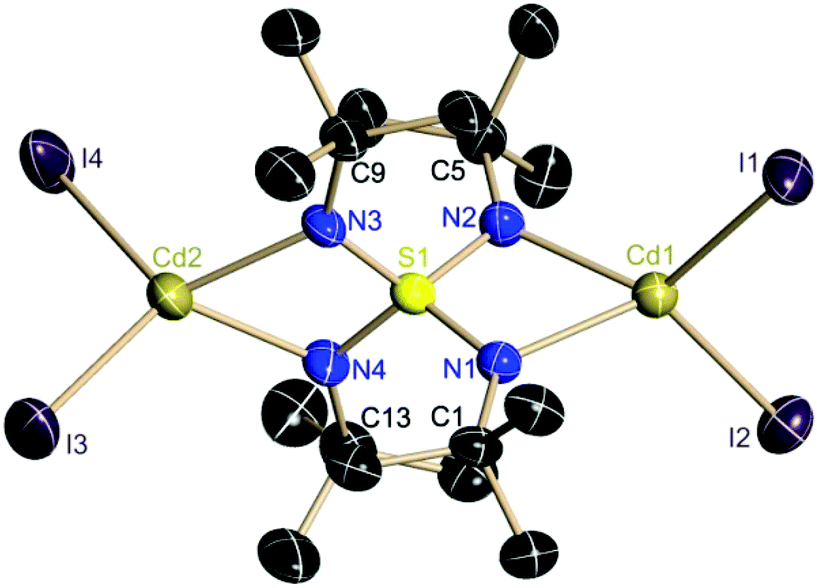 | ||
| Fig. 3 Crystal structure of the dianion in the solvent separated ion pair [Li(thf)4]2[I4Cd2(NtBu)4S][thf] (4). Hydrogen atoms are omitted for clarity. Anisotropic displacement parameters are depicted at the 50% probability level. Selected bond lengths [Å] and angles [°]: S1–N1 1.585(5), S1–N2 1.595(5), S1–N3 1.592(5), S1–N4 1.589(5), N1–Cd1 2.227(5), N2–Cd1 2.215(5), N3–Cd2 2.227(5), N4–Cd2 2.217(5), Cd1–I1 2.7416(7), Cd1–I2 2.7489(7), Cd2–I3 2.7531(9), Cd2–I4 2.7372(8), N1–S1–N2 95.8(3), N3–S1–N4 95.2(3), N1–Cd1–N2 64.16(18), N3–Cd2–N4 63.81(18), I1–Cd1–I2 108.40(2), I3–Cd2–I4 109.34(3). | ||
While [(thf)4Ba2{N(SiMe3)2}2{(NtBu)4S}] (2) is the product of a transmetalation using barium-hexamethylsilylamide from [(thf)4Li2(NtBu)4S] (1) via a hydrogenation first (Scheme 2)17 we now report a transmetalation without the previous generation of a protonated species. Firstly, lithium hexamethylsilylamide and zinc chloride were reacted under elimination of lithium chloride to give the assumed intermediate Zn(Cl)N(SiMe3). Subsequently, this intermediate gives the first isolated heterobimetallic compound [(thf)2Li{(SiMe3)2N}Zn(NtBu)4S] (5) after addition of 1 (Scheme 5).
 | ||
| Scheme 5 Synthesis of [(thf)2Li{(SiMe3)2N}Zn(NtBu)4S] (5). | ||
5 crystallizes from THF–toluene within four days at −24 °C, to give colorless blocks in a 34% yield, which were suitable for X-ray structure analysis. The compound crystallizes in the monoclinic space group C2/c with half a molecule per asymmetric unit. At one site of the S(NtBu)42− dianion a lithium ion remains coordinated like in the starting material and at the other site the zinc atom is complexed in a trigonal planar fashion by the two chelating nitrogen atoms of the ligand and on additional N(SiMe3)2 amide group. In 5 the S1–N1 bond (1.5661(14) Å) is shorter than the S–N bond (1.59 Å) of the starting material because of the electron withdrawing effect of ZnN(SiMe3)2+. While the harder Zn2+ cation claims more negative charge from the two imide groups than the softer lithium cation the zinc-coordinated imide groups remain less attractive to the positively polarized sulfur atom (S1–N2 1.6312(14) Å), which compensates for its part at the lithium coordinated imide groups. The N1′–S1–N1 angle (96.80(11)) is wider than the N2′–S1–N2 angle (91.38(10)) presumably due to the higher steric demand of the (thf)2Li moiety compared to the N(SiMe3)2 anion. The Li–N distance of 1.988(3) Å is typical for Li–N bonds.27 Published distances between a lithium ion which is coordinated by two THF molecules and two nitrogen atoms, are on average 2.066 Å. The Zn–N(amide) distance is 1.880(2) Å which is only marginally shorter than the mean average of Zn–amide bonds in the CCDC (Fig. 4).28
 | ||
| Fig. 4 Crystal structure of [(thf)2Li{(SiMe3)2N}Zn(NtBu)4S] (5). Hydrogen atoms are omitted for clarity. Anisotropic displacement parameters are depicted at the 50% probability level. Selected bond lengths [Å] and angles [°]: Li1–N1 1.988(3), N1–S1 1.5661(14), N2–S1 1.6312(14), N2–Zn1 1.9580(14), N3–Zn1 1.880(2), N2–S1–N2′ 91.38(10), N1–S1–N1′ 96.80(11), N1–Li1–N1′ 72.18(15), S1–N1–Li1 95.51(10), S1–N2–Zn1 97.71(7), N2–Zn1–N3 143.41(4), N2–Zn1–N2′ 73.19(8). | ||
Structural comparison
From the comparison of the three compounds it is interesting to note that in 2 the Cd–N bonds are longer than the M–N bonds in the other complexes, even longer than anticipated by the higher radius. They are further widened due to the lower electrostatic interactions to the neutral CdI2 moieties compared to the otherwise cationic parts. Nevertheless, the complex to be formed indicates a certain binding affinity of the S(NR)42− ligand even to neutral residues.As in the metal S-alkyltriimidosulfonates RS(NR)3− also in the metal tetraimidosulfates S(NR)42− the sum of the S–N bond distances seems to be almost invariant to the metal coordination (4.70(2) in the first and 6.38(2) Å in the latter). In the rigid framework of the four electron-rich imido nitrogen atoms the electropositive sulfur atom is pulled towards the nitrogen atoms coordinated by the Li+ because they remain more attractive as the Li+ loses the competition for imide nitrogen density against the Zn2+ and there remains more density at the LiN2 site of the SN4 tetrahedron. Hence the sulfur atom inside the N4-cage responds to the metal-polarized negative charge at the outside of the S(NR)42− dianion. This again emphasises predominantly ionic S–N bonding, reminiscent to S–O bonding in sulfate.22,29
Experimental section
General procedure
All experiments were performed either in an inert gas atmosphere of purified dry argon with standard Schlenk techniques30,31 or in an argon glove box. The glassware was dried at 130 °C, assembled hot and cooled under reduced pressure. All solvents were dried over appropriate alkali metals, distilled and degassed prior to use. All NMR spectra were either recorded on a Bruker Avance DPX 300 MHz or Bruker Avance DRX 500 MHz spectrometer using TMS (1H, 13C and 29Si) and LiCl (7Li) as external reference and the protons of the deuterated solvents as internal standard. The spectra were measured at room temperature if not stated otherwise. Elemental analyses (C, H, N and S) were carried out at the Mikroanalytisches Labor, Institut für Anorganische Chemie, Universität Göttingen.Single-crystal structural analysis
Single crystals were selected from a Schlenk flask under argon atmosphere and covered with perfluorated polyether oil on a microscope slide, which was cooled with a nitrogen gas flow supplied by the X-TEMP2 device.32 An appropriate crystal was selected using a polarizing microscope, fixed on the tip of a MiTeGen© MicroMount, transferred to a goniometer head, and shock cooled by the crystal cooling device. The data for 3, 4, and 5 were collected from these shock-cooled crystals at 100(2) K. The data for 3 and 4 were collected on an Incoatec Mo microfocus source33 equipped with Helios mirror optics and an APEX II detector at a D8 goniometer. The data for 5 were measured on a Bruker TXS Mo rotating anode with Helios mirror optics and an APEX II detector at a D8 goniometer. Important data are summarized in Table 1. Both diffractometers used Mo Kα radiation, λ = 0.71073 Å. The data for all structures were integrated with SAINT,34 and an empirical absorption correction (SADABS)35 was applied. The structures were solved by direct methods (SHELXS-97)36 and refined by full-matrix least-squares methods against F2 (SHELXL-97)19,37 within the SHELXLE GUI.38 All non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were refined isotropically on calculated positions using a riding model with their Uiso values constrained to equal 1.5 times the Ueq of their pivot atoms for terminal sp3 carbon atoms and 1.2 times for all other carbon atoms. Disordered moieties were refined using bond lengths and angles restraints and anisotropic displacement parameter restraints. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre. The CCDC numbers, crystal data and experimental details for the X-ray measurements are listed in Table 1.| Parameters | 3 | 4 | 5 |
|---|---|---|---|
| CCDC no. | 1011527 | 1011528 | 1011529 |
| Empirical formula | C34H66Cu2N4O6S | C52H108Cd2I4Li2N4O9S | C30H70LiN5O2SSi2Zn |
| Formula weight | 786.04 | 1711.76 | 693.46 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P2/n | P21/c | C2/c |
| a/Å | 12.546(2) | 15.428(2) | 15.507(3) |
| b/Å | 9.281(2) | 22.442(3) | 15.390(3) |
| c/Å | 16.950(3) | 20.869(2) | 18.081(3) |
| β/° | 93.85(2) | 103.56(2) | 113.090(10) |
| V/Å3, Z | 1969.2(6), 2 | 7024.2(16), 4 | 3969.4(13), 4 |
| Density (calcd) | 1.326 g cm−3 | 1.619 g cm−3 | 1.160 g cm−3 |
| Absorption coefficient | 1.178 mm−1 | 2.440 mm−1 | 0.762 mm−1 |
| F (000) | 840 | 3400 | 1512 |
| Crystal size/mm | 0.08 × 0.08 × 0.01 | 0.10 × 0.10 × 0.05 | 0.05 × 0.05 × 0.02 |
| θ range for data collection | 1.958 to 26.371° | 1.353 to 25.521° | 1.947 to 27.222° |
| Limiting indices | −15 ≤ h ≤ 15; −11 ≤ k ≤ 11; −21 ≤ l ≤ 21 | −18 ≤ h ≤ 18; −27 ≤ k ≤ 27; −25 ≤ l ≤ 25 | −19 ≤ h ≤ 19; −19 ≤ k ≤ 19; −23 ≤ l ≤ 23 |
| Reflections collected | 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 841 841 |
284967 |
41968 |
| Independent reflections | 4036 (Rint = 0.0350) | 12944 (Rint = 0.1384) |
4420 (Rint = 0.0822) |
| Completeness to θ | 100% (θ = 25.242°) | 100.0% (θ = 25.242°) | 100% (θ = 25.242°) |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 4036/558/311 | 12944/2738/883 |
4420/0/201 |
| Goodness-of-fit on F2 | 1.045 | 1.062 | 1.053 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0270, wR2 = 0.0641 | R 1 = 0.0510, wR2 = 0.1090 | R 1 = 0.0308, wR2 = 0.0742 |
| R indices (all data) | R 1 = 0.0367, wR2 = 0.0673 | R 1 = 0.0724, wR2 = 0.1226 | R 1 = 0.0438, wR2 = 0.0797 |
| Largest diff. peak and hole/e Å−3 | 0.416 and −0.259 | 1.886 and −1.933 | 0.384 and −0.353 |
Conclusion
The three metal complexes [(acac)2Cu2(NtBu)4S] (3), [Li(thf)4]2[I4Cd2(NtBu)4S] (4) and [(thf)2Li{(SiMe3)2N}Zn(NtBu)4S] (5) show that transition metal complexes containing the tetraimidosulfate dianion are feasible. With the right metal moiety at opposite sides of the tetrahedron they are stable and not subject to ligand scrambling. 4 can be envisaged as an intermediate on the metathesis reaction or a co-complex between CdI2 and the lithium precursor.39 Like in the intriguing structure of [(thf)2Li{(NtBu)3SMe}·ZnMe2]26 the S–N bonds vary considerably in the heterobimetallic complex [(thf)2Li{(SiMe3)2N}Zn(NtBu)4S] (5). The electropositive sulfur atom inside the imido nitrogen tetrahedron responds to the polarization induced by the coordinated metals. The more the N atoms lose density to the most electropositive metal at the outside the less they are attractive to the sulfur and the longer the S–N bonds get. The electropositive sulfur in the inside mirrors the electron density distribution on the outside.Acknowledgements
The support of the Danish National Research Foundation (DNRF93) funded Center for Materials Crystallography (CMC) for partial funding is acknowledged. JM thanks Sebastian Bachmann for help with the NMR spectroscopy.Notes and references
- H. W. Roesky, Angew. Chem., Int. Ed. Engl., 1979, 18, 91–97 CrossRef.
- T. Chivers, Chem. Rev., 1985, 85, 341–356 CrossRef CAS.
- R. Mews, P. G. Watson and E. Lork, Coord. Chem. Rev., 1997, 158, 233–273 CrossRef CAS.
- R. Fleischer and D. Stalke, Coord. Chem. Rev., 1998, 176, 431–450 CrossRef CAS.
- D. Stalke, Proc. Indian Acad. Sci., 2000, 112, 155–170 CrossRef CAS.
- J. K. Brask and T. Chivers, Angew. Chem., Int. Ed., 2001, 40, 3960–3976 CrossRef CAS.
- F. T. Edelmann, S. Blaurock, V. Lorenz and T. Chivers, Z. Anorg. Allg. Chem., 2008, 634, 413–415 CrossRef.
- D. Stalke, Chem. Commun., 2012, 48, 9559–9573 RSC.
- M. M. Meinholz, E. Carl, E. Kriemen and D. Stalke, Chem. Commun., 2011, 47, 10948–10950 RSC.
- M. M. Meinholz, S. K. Pandey, S. M. Deuerlein and D. Stalke, Dalton Trans., 2011, 40, 1662–1671 RSC.
- M. M. Meinholz and D. Stalke, Eur. J. Inorg. Chem., 2011, 2011, 4578–4584 CrossRef CAS.
- M. M. Meinholz and D. Stalke, Z. Anorg. Allg. Chem., 2011, 637, 2233–2238 CrossRef CAS.
- M. M. Meinholz, E. Carl, E. Kriemen and D. Stalke, Chem. Commun., 2011, 47, 10948–10950 RSC.
- M. M. Meinholz and D. Stalke, Z. Naturforsch., B: Chem. Sci., 2011, 66, 981 CrossRef CAS PubMed.
- R. Fleischer, S. Freitag and D. Stalke, J. Chem. Soc., Dalton Trans., 1998, 193–198 RSC.
- R. Fleischer, A. Rothenberger and D. Stalke, Angew. Chem., Int. Ed. Engl., 1997, 36, 1105–1107 CrossRef CAS.
- R. Fleischer, B. Walfort, A. Gbureck, P. Scholz, W. Kiefer and D. Stalke, Chem. – Eur. J., 1998, 4, 2266–2274 CrossRef CAS.
- T. Chivers and J. Konu, Comments Inorg. Chem., 2009, 30, 131 CrossRef CAS.
- P. Müller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, in Crystal Structure Refinement – A Crystallographer's Guide to SHELXL, ed. P. Müller, Oxford University Press, Oxford, England, 2006 Search PubMed.
- D. Leusser, J. Henn, N. Kocher, B. Engels and D. Stalke, J. Am. Chem. Soc., 2004, 126, 1781–1793 CrossRef CAS PubMed.
- J. Henn, D. Ilge, D. Leusser, D. Stalke and B. Engels, J. Phys. Chem. A, 2004, 108, 9442–9452 CrossRef CAS.
- M. S. Schmøkel, S. Cenedese, J. Overgaard, M. R. V. Jørgensen, Y.-S. Chen, C. Gatti, D. Stalke and B. B. Iversen, Inorg. Chem., 2012, 51, 8607–8616 CrossRef PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–768 CrossRef.
- K. Heinze and A. Reinhart, Inorg. Chem., 2006, 45, 2695–2703 CrossRef CAS PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, Taylor & Francis, Boca Raton, 2011–2012, vol. 92 Search PubMed.
- B. Walfort, A. P. Leedham, C. R. Russell and D. Stalke, Inorg. Chem., 2001, 40, 5668–5674 CrossRef CAS PubMed.
- (a) D. R. Armstrong, D. Barr, W. Clegg, R. E. Mulvey, D. Reed, R. Snaith and K. Wade, J. Chem. Soc., Chem. Commun., 1986, 869 RSC; reviews: (b) K. Gregory, P. v. R. Schleyer and R. Snaith, Adv. Inorg. Chem., 1991, 37, 47 CrossRef CAS; (c) R. E. Mulvey, Chem. Soc. Rev., 1991, 20, 167 RSC; (d) R. E. Mulvey, Chem. Soc. Rev., 1998, 27, 339 RSC.
- Cambridge Structural Database, v5.35 (November 2013), Cambridge Crystallographic Data Centre, Cambridge, 2013 Search PubMed.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- (a) W. Schlenk and A. Thal, Ber. Dtsch. Chem. Ges., 1913, 46, 2840–2854 CrossRef; (b) W. Schlenk, J. Appenrodt, A. Michael and A. Thal, Ber. Dtsch. Chem. Ges., 1914, 47, 473–490 CrossRef CAS.
- http://www.stalke.chemie.uni-goettingen.de/virtuelles_labor/de.html .
- (a) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef; (b) T. Kottke, R. J. Lagow and D. Stalke, J. Appl. Crystallogr., 1996, 29, 465–468 CrossRef CAS; (c) D. Stalke, Chem. Soc. Rev., 1998, 27, 171–178 RSC.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef CAS.
- SAINT v7.68A in Bruker APEX v2011.9, Bruker AXS Inst. Inc., Madison, USA, 2008 Search PubMed.
- G. M. Sheldrick, SADABS 2008/2, Universität Göttingen, Germany, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- (a) E. Hevia, J. Z. Chua, P. Garzía-Álvarez, A. R. Kennedy and M. D. McCall, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5294–5299 CrossRef CAS PubMed; (b) D. R. Armstrong, W. Clegg, P. Garzía-Álvarez, A. R. Kennedy, M. D. McCall, L. Russo and E. Hevia, Chem. – Eur. J., 2011, 17, 8333–8341 CrossRef CAS PubMed.
Footnotes |
| † In memory of Professor Ken Wade. |
| ‡ Electronic supplementary information (ESI) available: Three X-ray files in CIF format. CCDC 1011527–1011529. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt01995d |
| This journal is © The Royal Society of Chemistry 2014 |