
Ultrafast electronic and vibrational relaxations in mixed-ligand dithione–dithiolato Ni, Pd, and Pt complexes†
Abstract
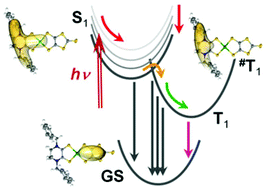
Ultrafast excited-state dynamics of planar Pt, Pd, and Ni dithione–dithiolato complexes were investigated by transient absorption spectroscopy on the femtosecond–picosecond timescale. All studied complexes show a common photobehaviour, although individual kinetics parameters and quantum yields vary with the metal, the dithione ligand and, namely the solvent (DMF, MeCN). Laser pulse irradiation at 800 nm populates the lowest singlet excited state of a dithiolato → dithione charge transfer character, 1LL′CT. The optically excited state undergoes a solvation-driven sub-picosecond electronic relaxation that enhances the dithione/dithiolato charge separation. The 1LL′CT state decays with a 1.9–4.5 ps lifetime by two simultaneous pathways: intersystem crossing (ISC) to the lowest triplet state 3LL′CT and non-radiative decay to the ground state. ISC occurs on a ∼6 ps timescale, virtually independent of the metal, whereas the rate of the non-radiative decay to the ground state decreases on going from Ni (2 ps) to Pd (3 ps) and Pt (∼10 ps). 3LL′CT is initially formed as a vibrationally excited state. Its equilibration (cooling) takes place on a picosecond timescale and is accompanied by a competitive decay to the ground state. Equilibrated 3LL′CT is populated with a quantum yield of less than 50%, depending on the metal: Pt > Pd > Ni. 3LL′CT is long-lived for Pt and Pd (≫500 ps) and short-lived for Ni (∼15 ps). Some of the investigated complexes also exhibit spectral changes due to vibrational cooling of the singlet (2–3 ps, depending on the solvent). Rotational diffusion occurs with lifetimes in the 120–200 ps range. Changing the dithione (Bz2pipdt/iPr2pipdt) as well as dithiolate/diselenolate (dmit/dsit) ligands has only small effects on the photobehavior. It is proposed that the investigated dithione–dithiolato complexes could act as photooxidants (*Eo ≈ +1.2 V vs. NHE) utilizing their lowest excited singlet (1LL′CT), provided that the excited-state electron transfer is ultrafast, competitive with the picosecond decay. On the other hand, the efficiency of any triplet-based processes would be severely limited by the low quantum yield of the triplet population.
- This article is part of the themed collection: Spectroscopy of Inorganic Excited States
 Please wait while we load your content...
Please wait while we load your content...

