
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron transfer dynamics and excited state branching in a charge-transfer platinum(II) donor–bridge-acceptor assembly†
Paul A.
Scattergood‡
a,
Milan
Delor‡
a,
Igor V.
Sazanovich
ab,
Oleg V.
Bouganov
c,
Sergei A.
Tikhomirov
c,
Alexander S.
Stasheuski
c,
Anthony W.
Parker
b,
Gregory M.
Greetham
b,
Michael
Towrie
b,
E. Stephen
Davies
d,
Anthony J. H. M.
Meijer
 *a and
Julia A.
Weinstein
*a
*a and
Julia A.
Weinstein
*a
aDepartment of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. E-mail: Julia.Weinstein@sheffield.ac.uk; A.Meijer@sheffield.ac.uk
bCentral Laser Facility, Research Complex at Harwell, Rutherford Appleton Laboratory, Research Complex at Harwell, Harwell Science and Innovation Campus, Chilton, Oxfordshire OX11 0QX, UK
cB.I. Stepanov Institute of Physics, 68 Nezavisimosti Ave., Minsk, 220072, Belarus
dSchool of Chemistry, The University of Nottingham, Nottingham NG7 2RD, UK
First published on 31st October 2014
Abstract
A linear asymmetric Pt(II) trans-acetylide donor–bridge-acceptor triad designed for efficient charge separation, NAP![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Pt(PBu3)2Ph–CH2–PTZ (1), containing strong electron acceptor and donor groups, 4-ethynyl-N-octyl-1,8-naphthalimide (NAP) and phenothiazine (PTZ) respectively, has been synthesised and its photoinduced charge transfer processes characterised in detail. Excitation with 400 nm, ∼50 fs laser pulse initially populates a charge transfer manifold stemming from electron transfer from the Pt-acetylide centre to the NAP acceptor and triggers a cascade of charge and energy transfer events. A combination of ultrafast time-resolved infrared (TRIR) and transient absorption (TA) spectroscopies, supported by UV-Vis/IR spectroelectrochemistry, emission spectroscopy and DFT calculations reveals a self-consistent photophysical picture of the excited state evolution from femto- to milliseconds. The characteristic features of the NAP-anion and PTZ-cation are clearly observed in both the TRIR and TA spectra, confirming the occurrence of electron transfer and allowing the rate constants of individual ET-steps to be obtained. Intriguingly, 1 has three separate ultrafast electron transfer pathways from a non-thermalised charge transfer manifold directly observed by TRIR on timescales ranging from 0.2 to 14 ps: charge recombination to form either the intraligand triplet 3NAP with 57% yield, or the ground state, and forward electron transfer to form the full charge-separated state 3CSS (3[PTZ+–NAP−]) with 10% yield as determined by target analysis. The 3CSS decays by charge-recombination to the ground state with ∼1 ns lifetime. The lowest excited state is 3NAP, which possesses a long lifetime of 190 μs and efficiently sensitises singlet oxygen. Overall, molecular donor–bridge-acceptor triad 1 demonstrates excited state branching over 3 different pathways, including formation of a long-distant (18 Å) full charge-separated excited state from a directly observed vibrationally hot precursor state.
Pt(PBu3)2Ph–CH2–PTZ (1), containing strong electron acceptor and donor groups, 4-ethynyl-N-octyl-1,8-naphthalimide (NAP) and phenothiazine (PTZ) respectively, has been synthesised and its photoinduced charge transfer processes characterised in detail. Excitation with 400 nm, ∼50 fs laser pulse initially populates a charge transfer manifold stemming from electron transfer from the Pt-acetylide centre to the NAP acceptor and triggers a cascade of charge and energy transfer events. A combination of ultrafast time-resolved infrared (TRIR) and transient absorption (TA) spectroscopies, supported by UV-Vis/IR spectroelectrochemistry, emission spectroscopy and DFT calculations reveals a self-consistent photophysical picture of the excited state evolution from femto- to milliseconds. The characteristic features of the NAP-anion and PTZ-cation are clearly observed in both the TRIR and TA spectra, confirming the occurrence of electron transfer and allowing the rate constants of individual ET-steps to be obtained. Intriguingly, 1 has three separate ultrafast electron transfer pathways from a non-thermalised charge transfer manifold directly observed by TRIR on timescales ranging from 0.2 to 14 ps: charge recombination to form either the intraligand triplet 3NAP with 57% yield, or the ground state, and forward electron transfer to form the full charge-separated state 3CSS (3[PTZ+–NAP−]) with 10% yield as determined by target analysis. The 3CSS decays by charge-recombination to the ground state with ∼1 ns lifetime. The lowest excited state is 3NAP, which possesses a long lifetime of 190 μs and efficiently sensitises singlet oxygen. Overall, molecular donor–bridge-acceptor triad 1 demonstrates excited state branching over 3 different pathways, including formation of a long-distant (18 Å) full charge-separated excited state from a directly observed vibrationally hot precursor state.
Introduction
Photoinduced electron transfer, an elementary process, underpins a variety of applications, especially in light-harvesting, photocatalysis, and optoelectronics. The process of electron transfer can be extremely fast, occurring on sub-picosecond timescales and as such has been widely studied by ultrafast spectroscopic methods to follow excited state dynamics in real time.1 The mechanism of intramolecular photoinduced electron transfer usually involves the formation of a charge-separated excited state (CSS).2,3 A CSS is formed upon a shift of electron density from an electron donor D to an electron acceptor A, often via a connecting “bridging” unit which can play a major role in mediating charge transfer. Extensive assemblies designed on this principle, which contain multiple donor and acceptor units of gradually increased D/A strength, act as model systems for investigating charge transfer events, similar to those observed in photosynthesis. Transition metal donor–bridge-acceptor (DBA) molecular triads and their multi-donor/acceptor relatives have proven to be an effective design motif in which a photoinduced CSS can be attained.2,4–10There is currently much interest in understanding the behaviour and energy relaxation processes that occur immediately after light absorption. Electron transfer from a non-thermalised manifold, which competes with vibrational relaxation in the excited state, is one such process.11 Time-resolved broadband infrared spectroscopy (TRIR), a method where an excited state is optically prepared by a UV/Vis pulse and probed by a broadband IR pulse, is particularly suited to investigate such dynamics. Recent developments in ultrafast TRIR permit broad spectral coverage and simultaneous observation of the dynamics of several infrared reporting groups, expanding significantly the range of excited state processes which can be followed directly.12–15 This paper demonstrates how selective strong IR chromophores within Pt(II) based molecular assemblies permit characterisation of ultrafast electron transfer, and direct observation of an effect of excited state branching from vibrationally hot electronic states.
The use of Pt(II) containing bridging units is particularly attractive for ultrafast ET investigations owing to the square planar coordination environment around the metal centre which provides synthetic versatility for the specific attachment of ligands, and enables control over the directionality of electron transfer.14–16 The utilisation of donor acetylide ligands in particular facilitates the formation of more stable, longer-lived charge-separated states due to strong-field acetylides raising the energy of a deactivating dd-state, and led to a large family of cis bis-acetylide Pt(II) complexes employing redox-active diimine acceptor ligands.16–21
Here we investigate light-induced processes in a linear, asymmetric trans acetylide donor–acceptor Pt(II) triad 1, Fig. 1.22 Our design employs a neutral co-ligand phosphine,23–32 which lacks low lying π* orbitals hence does not engage in the charge-transfer processes between the trans configured donor and acceptor ligands, ensuring that the light-harvesting system has a pre-conditioned charge transfer directionality. A number of Pt(II) bis-phosphine complexes with a trans arrangement of the two identical acetylides9,33–44 has been reported, largely driven by their applications in optoelectronics and optical power limiting. Examples of asymmetric [donor-CC–Pt–CC-acceptor] motifs include complexes incorporating a triarylamine electron donor and an oxadiazole based electron acceptor,23 ethynyl-naphthalimide electron acceptor and phenyl-acetylene co-ligand,31 a (diphenylamino)-2,7-fluorenylene (DPAF) electron donor and a naphthalene diimide (NDI) electron acceptor,45 and some more elaborate assemblies.34,46,47
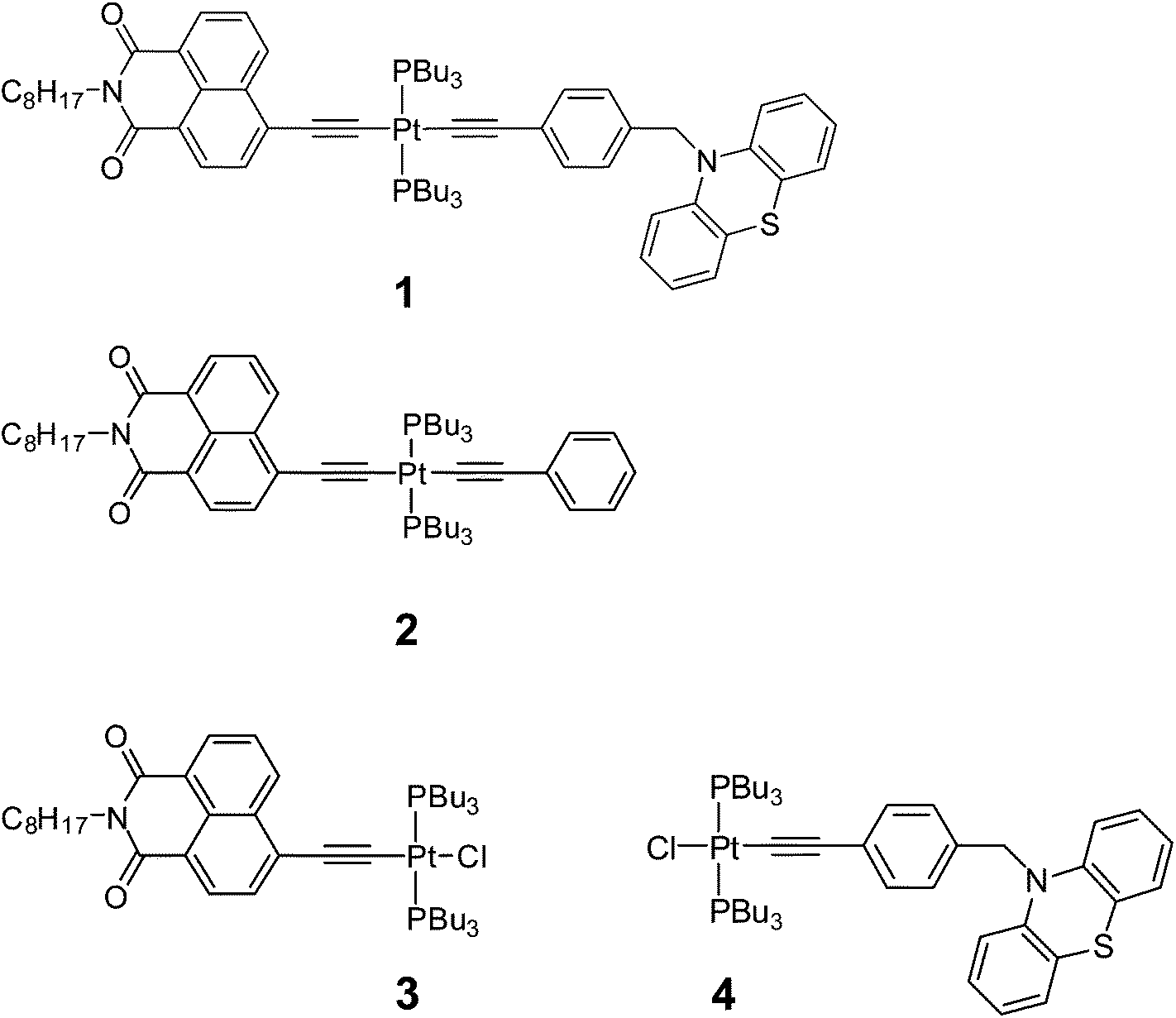 | ||
| Fig. 1 The Pt(II) acetylide complexes investigated in this study: the donor–bridge-acceptor triad NAPPt(PBu3)2Ph–CH2–PTZ (‘NAP–Pt–PTZ’, 1), and its analogs and precursors NAPPt(PBu3)2Ph (‘NAP–Pt–Ph’, 2), NAPPt(PBu3)2–Cl (‘NAP–Pt–Cl’, 3), and Cl–Pt(PBu3)2Ph–CH2–PTZ (‘Cl–Pt–PTZ’, 4). | ||
The triad 1 combines a strong electron acceptor 4-ethynyl-N-octyl-1,8-naphthalimide (NAP) and an electron donor phenothiazine (PTZ). Aromatic acid imide acceptor groups have been used in diverse charge-transfer assemblies21,48–50 owing to their low reduction potentials, and the ability to form stable radical-anions which possess distinct and intense absorptions in both UV-Vis and IR spectral ranges.48,51,52 Likewise, the donor PTZ is readily oxidised, forming a stable radical cation which has a distinct absorption in the visible range.53–56 To enable a step-wise charge-transfer process, the donor moiety in 1 is connected to the Pt-acetylide bridge through a saturated linker. The elucidation of light-induced processes in 1 was assisted by a detailed study of the “building blocks” 2–4, which contain only the acceptor fragment (3), the acceptor and the bis-acetylide Ph–CC–Pt–CC-bridge (2), and only the donor fragment (4), Fig. 1.
The presence of strong IR reporters – the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the imide electron acceptor as well as ν(CC) positioned on both D and A molecular fragments – enable the use of ultrafast TRIR for monitoring excited state dynamics and intermediate states. The combination of vibrational spectroscopy with time-resolved electronic transient absorption spectroscopy (TA) along with UV/Vis/IR (spectro)electrochemical and computational methods resolves the rich photophysics of the charge-transfer assemblies, involving multiple excited states evolving across the femto-to-millisecond time range.
O) of the imide electron acceptor as well as ν(CC) positioned on both D and A molecular fragments – enable the use of ultrafast TRIR for monitoring excited state dynamics and intermediate states. The combination of vibrational spectroscopy with time-resolved electronic transient absorption spectroscopy (TA) along with UV/Vis/IR (spectro)electrochemical and computational methods resolves the rich photophysics of the charge-transfer assemblies, involving multiple excited states evolving across the femto-to-millisecond time range.
Results and discussion
Synthesis
1 and 2 were prepared through a stepwise approach via the “acceptor-bridge” dyad 3. In turn, 3 and the “donor-bridge” dyad 4 were prepared from cis-Pt(PBu3)2Cl2. All four complexes contain a common trans-Pt(II) bis phosphine core. The Pt-containing starting material cis-Pt(PBu3)2Cl2 was prepared via the reaction of PtCl2 with a slightly sub-stoichiometric quantity of PnBu3 at r.t. to yield the four-coordinate complex in moderate yields (60–70%) with absolute cis stereochemistry. This route allows one to perform the reaction on a smaller scale in comparison to the procedure employing K2PtCl4 where the use of small quantities of reagents was reported to decrease the product yield.57It should be noted that the commonly used copper-assisted Hagihara type coupling results in predominantly bis substitution at the Pt centre to give symmetrical trans acetylide products in a statistical mixture. Therefore, amine mediated high temperature reaction conditions were employed instead to produce asymmetric mono-substituted Pt(II) acetylides24,31,58 which then can be functionalised with another acetylide ligand. Thus, [trans-acetylide-Pt–Cl] dyads 3 and 4 were formed in a reaction of cis-Pt(PBu3)2Cl2 with 4-ethynyl-N-octyl-1,8-naphthalimide59 and N-(4-ethynylbenzyl)-phenothiazine,60 respectively, in boiling iPr2NH in the absence of CuI.
1 and 2 were formed directly from 3 utilising the CuI assisted cross coupling procedure61,62 to coordinate either phenylacetylene or an ethynyl moiety containing N-(4-ethynylbenzyl)-phenothiazine. Both syntheses proceeded with moderate to high yields, affording bright yellow coloured products which were purified via column chromatography and satisfactorily characterised through NMR and mass spectrometry. 31P NMR yields JPt–P values in the region of 2300 Hz that are indicative of the trans geometry of the phosphine groups at the metal centre. Conversely, significantly larger coupling (JPt–P ≈ 3500 Hz) is noted for cis phosphine configurations such as that present in the Pt(PBu3)2Cl2 precursor material.63 We highlight that despite starting from a cis phosphine precursor complex, 1–3 were found to be formed with trans stereochemistry. No cis isomers were isolated nor has any isomerisation been observed either in solution or after exposure to visible light.
Electrochemistry
In order to determine the nature and the energy of the frontier orbitals, cyclic voltammetry studies were performed for solutions of 1–4 in CH2Cl2 across the potential window from −2.2 to +1.3 V (Fig. S1a,†Table 1).| Complex | Reduction [V] | Oxidation [V] |
|---|---|---|
| a Irreversible oxidation process, the anodic peak potential is quoted. b Process found to be reversible when isolated across the potential range −0.5 to + 0.7 V. c In CH2Cl2 containing 0.4 M [NBu4][BF4]; anodic/cathodic peak separation for the Fc+/Fc couple used as the internal standard was 0.08 V. | ||
| 1 | −1.83 (0.14) | +0.34 (0.09)b, +0.94a, +1.13a |
| 2 | −1.83 (0.13) | +0.87a |
| 3 | −1.83 (0.13) | +1.14a |
| 4 | — | +0.35 (0.08), +1.07a |
Complexes 1–3 show one electrochemically quasi-reversible process at −1.83 V assigned to reduction of the naphthalimide group,53 an assignment supported by the results of DFT calculations (vide infra). The potential of this reduction is unaffected by the group positioned mutually trans to the NAP functionality indicating a lack of electronic communication through the metal-acetylide centre in the electronic ground state. Compound 4 displays no reduction processes within the potential range studied.
Complexes 1 and 4 each display an electrochemically reversible process at ca. +0.35 V assigned as an oxidation based on the phenothiazine (PTZ) functionality.54,55,60,64,65 This value is consistent with a potential of +0.37 V found for the PTZ containing ethynyl ligand (‘PTZ-H’) and the iodide terminated synthetic precursor (‘PTZ-I’) (see ESI†).
A single, irreversible oxidation observed for 2 and 3 at +0.87 and +1.14 V, respectively, is assigned in both complexes to oxidation of a central Pt-acetylide unit, an electrochemical feature which has been readily observed in a number of previously reported Pt(II) acetylide complexes.19,54,60,66–68 The PTZ-containing compounds 1 and 4 also have additional oxidation processes in the range from +0.94 V to +1.13 V (Table 1), which could not be assigned with confidence due to their electrochemically irreversible nature. From the CV data, the HOMO and LUMO for 1 are based on the PTZ and NAP groups, respectively, allowing an estimate of 2.17 V for the energy of the lowest charge-separated excited state [PTZ+–NAP−].
UV/Vis absorption spectroscopy
The ground-state absorption spectra of complexes 1–4 in CH2Cl2 (Fig. 2) in the region from 240 to 350 nm are dominated by high energy intraligand π–π* transitions. Comparison of the spectra of the individual molecular units shows that the transition centered at 320 nm is present in the spectra of 1, 2 and 4 but not of 3 and therefore we assign this spectral component to phenyl and PTZ π–π* transitions, also observed in the starting PTZ-H ligand. The lowest energy transition in 3, observed at 424 nm, is assigned to a metal-to-ligand charge transfer (MLCT) transition from the Pt/acetylide center to the NAP moiety.31,49 The relative broadness of this band suggests that there are several charge-transfer transitions contributing to the absorption envelope. In 1 and 2, this band is broader and slightly red-shifted, to 430 nm. The absorption spectra for 1 and 2 in the visible region are virtually identical, suggesting that the lowest excited state corresponds to a mixed-metal-ligand-to-ligand charge transfer (ML/L′CT) manifold which involves the phenylacetylide fragment and the Pt center, but does not involve the PTZ fragment in 1. These band assignments are confirmed by DFT calculations (vide infra), which show that in 1 and 2, there are at least three transitions within the ∼430 nm absorption envelope, all predominantly due to electron density shift from the central bridge CC–Pt–CC–Ph to the NAP moiety. The extinction coefficients at the maximum of the charge transfer bands are the same for 1–3 within instrumental error, (42 ± 1) × 103 dm3 mol−1 cm−1.
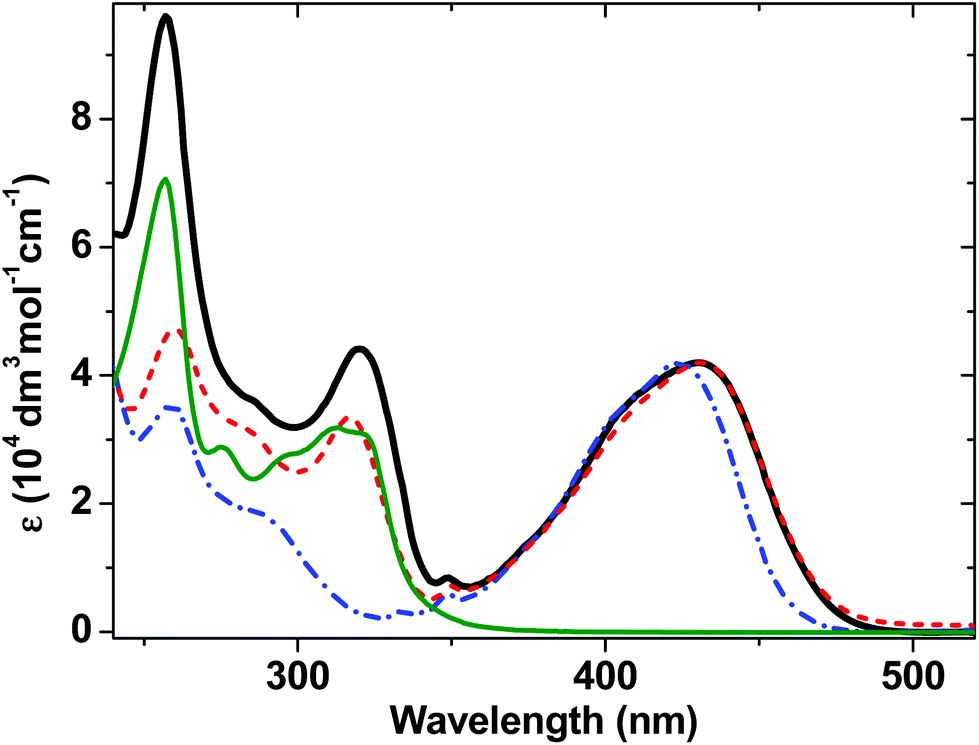 | ||
| Fig. 2 UV/Vis absorption spectra for 1 (solid black line), 2 (red dashed line) and 3 (blue dot-dashed line), and 4 (thin dark green line) in CH2Cl2 at r.t. | ||
As the absorption spectrum of the Cl–Pt–PTZ model complex 4 does not have any bands at wavelengths longer than 350 nm, it follows that electronic excitation of 1 with >370 nm will exclusively populate a bridge-to-NAP charge transfer excited state manifold without initially affecting the PTZ part of the complex. Since this paper focuses on investigating the light-induced properties of DBA system 1 using wavelengths >400 nm, 4 which does not absorb at these wavelengths was not investigated further.
Emission spectroscopy (Table 2)
Complexes 1–3 display dual emission. A high energy structureless emission band is centered at 518 nm for 1, 522 nm for 2 and 498 nm for 3. A lower energy emission band has a clearly defined vibronic structure, with a first component at ∼640 nm in CH2Cl2 at r.t. (Fig. 3). These dual emission observations are consistent with those reported for the very close analogues of 2 and 3,31 and other trans-Pt(bis)acetylides.41,69–71 The structured spectral profile of the low-energy emission band closely corresponds to the phosphorescence from the intraligand triplet localized on the NAP moiety (3NAP).31,49,72 This low-energy emission process is efficiently quenched by oxygen and is absent in aerated solution (Fig. S2†), which, along with its lack of solvatochromism, 190 microsecond lifetime, and the data from flash photolysis (Fig. S3†) and TRIR (see below), confirms its assignment to a 3NAP state. The high-energy emission band maximum shifts from 518 to 481 nm (1) and from 498 to 461 nm (3) when the solvent is changed from CH2Cl2 to the less polar toluene (Fig. S2†), suggesting that this emission emanates from a charge-transfer state.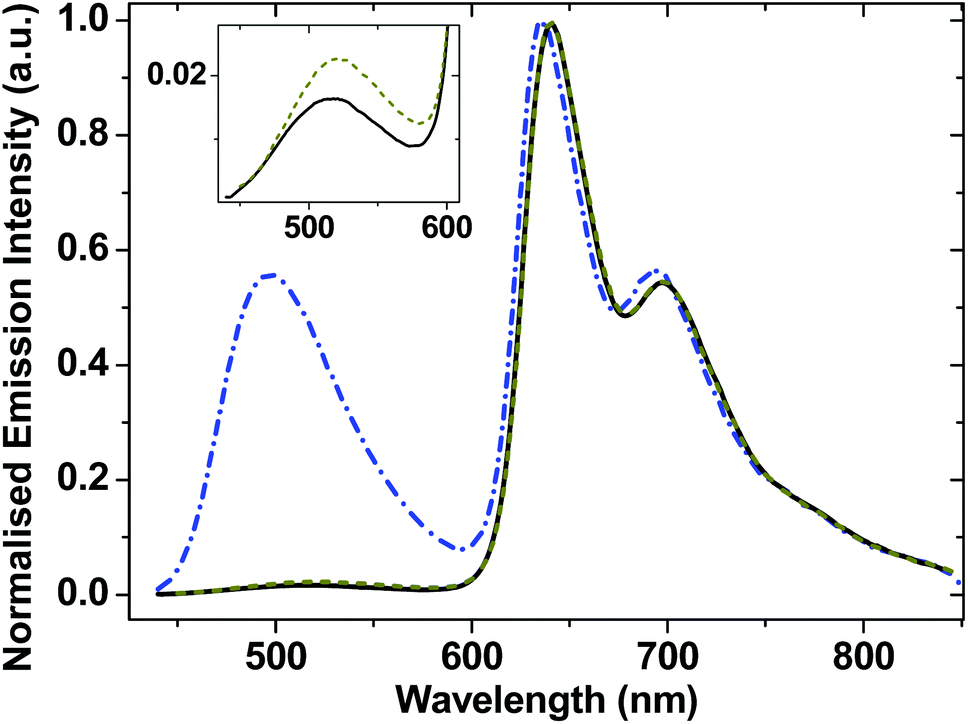 | ||
| Fig. 3 Normalised emission spectra for degassed solutions of 1 (solid black line), 2 (dark yellow dashed line), and 3 (blue dot-dashed line) in CH2Cl2 and at r.t. The inset shows an expansion of the high-energy profiles for complexes 1 and 2. | ||
| Complex | λ abs [nm] | λ em [nm] | τ [ps] | τ [μs] |
ϕ
em![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (%) c (%) |
1O2d (%) |
|---|---|---|---|---|---|---|
| a Values averaged from the results of ultrafast transient absorption and TRIR. b From emission lifetime measurements under 424 nm excitation for 3 and 430 nm excitation for 1 and 2. c Relative to a solution of [Ru(bpy)3]Cl2 in H2O, ϕem = 0.028.73 d Singlet oxygen quantum yields measured against reference compound perinaphthenone in CH2Cl2. e Vibronic progression indicated in brackets. f From ref. 31. g Same value as in ref. 31. | ||||||
| 1 | 430 | 518 | 14 ± 1 | 0.1 | ||
| 640 (696, 775)e | 190 ± 10 | 5.5 | 57 ± 7 | |||
| 2 | 430 | 522 | 20 ± 1 | 0.2 | ||
| 640 (696, 775)e | 190 ± 10 | 7.4 | 82f | |||
| 3 | 424 | 498 | 209 ± 9 | 2.6 | ||
| 635 (693, 775)e | 190 ± 10 | 4.9 | 90 ± 10g | |||
Emission lifetimes of the high energy emission band (estimated using the Edinburgh Instrument mini-τ, instrument response function ≈80 ps) is ≈200 ± 50 ps for 3, and instrument-limited for 1 and 2. The short lifetime of the first emissive state is attributed to an ultrafast conversion into low-lying states which will be described in later sections. The lifetime of the emissive state responsible for the low-energy emission band is 190 μs (Fig. S3†).
Compounds 1–3 efficiently sensitise singlet oxygen under visible light, confirming the triplet nature of the lowest excited sate as 3NAP. Singlet oxygen quantum yields (90% for 3, 57% for 1, Table 2) provide lower limit for the yield of the 3NAP product state.
Fourier transform IR (FTIR) and IR spectroelectrochemistry
Ground state FTIR spectra of 1, 2 and 3 in CH2Cl2 in the spectral range from 1475 to 2200 cm−1 (Fig. 4) are dominated by NAP ring breathing modes ν(NAP) at 1583 cm−1 and asymmetric/symmetric combinations of the NAP-localised carbonyl vibrations ν(CO) at 1652/1691 cm−1 in the fingerprint region. The presence of two acetylide moieties in 1 gives rise to asymmetric/symmetric combinations of the bridge-localised acetylide vibrations ν(CC) at 2087 (ε ∼ 1800 dm3 mol−1 cm−1) and 2109 cm−1 in the high frequency region, whilst 3 only has a single acetylide stretch mode at 2100 cm−1. All assignments are supported by DFT calculations.
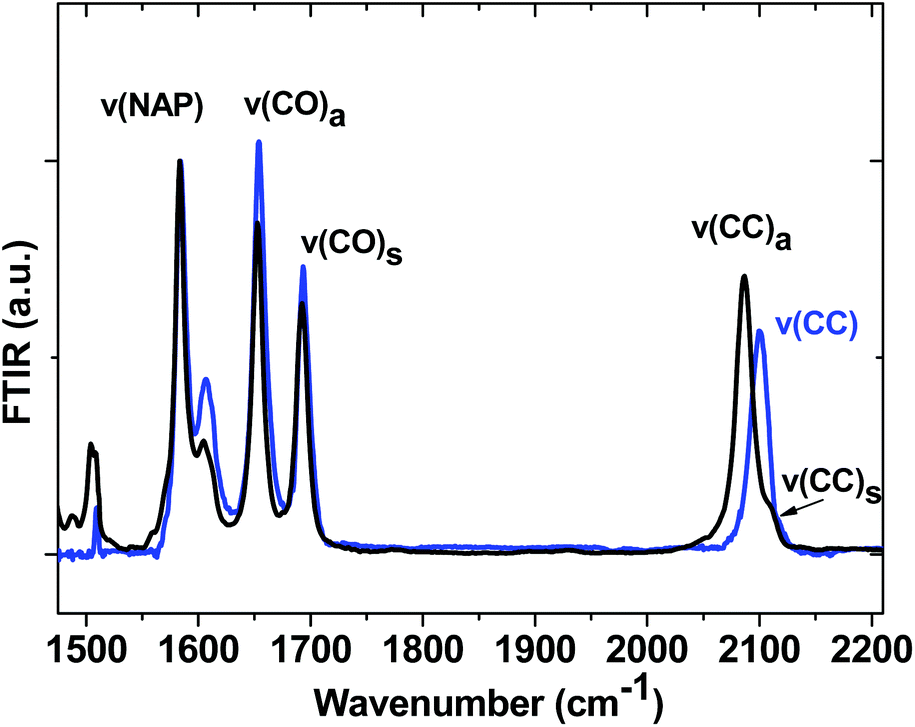 | ||
| Fig. 4 Normalised Fourier-Transform IR (FTIR) spectra for 1 (black) and 3 (blue) in CH2Cl2 in the 1475 cm−1 to 2200 cm−1 region. The spectrum of 2 follows that of 1 very closely and is omitted for clarity. | ||
The spectrum of 3−˙ produced by one-electron reduction of 3 in an IR OTTLE cell is given in Fig. S4.† The main IR bands at 1510 cm−1, 1555 cm−1 and 1607 cm−1 are assigned to the NAP-anion due to the NAP-localised LUMO.
The ultrafast excited state dynamics of 1, 2 and 3 were investigated by a combination of transient infrared and electronic transient absorption spectroscopies using a 50 fs, 400 nm excitation pulse. Excitation with 400 nm light is expected to populate a singlet excited state manifold of several charge-transfer states, which in 1 and 2 will be of [Ph–CC–Pt–CC]-to-NAP charge transfer nature (see DFT section). Both TRIR and TA experimental data were analysed using global fit analysis in order to obtain component spectra and related lifetimes for the transient states. The vibrational spectra of the several excited states involved were assigned with the aid of DFT calculations and IR spectroelectrochemistry.
Time-resolved infrared spectroscopy
Fig. 5 shows TRIR spectra for complexes 3, 1 and 2 (top, middle and bottom, respectively) in CH2Cl2 at r.t. at representative delay times after the laser excitation, along with kinetic traces (Fig. 5, bottom) at selected frequencies.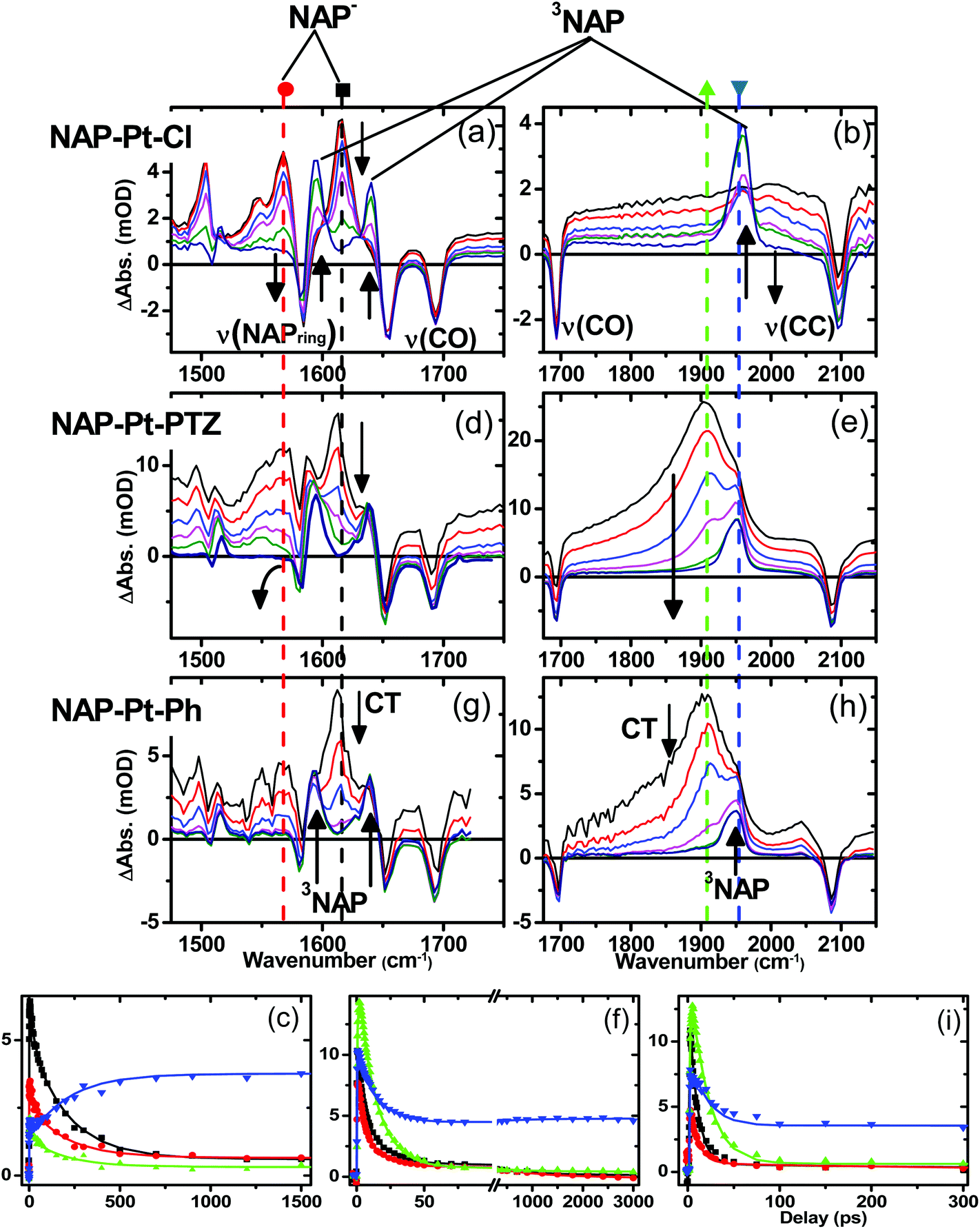 | ||
| Fig. 5 Time-resolved IR (TRIR) spectra following 400 nm excitation for 3 (NAP–Pt–Cl, a–c), 1 (NAP–Pt–PTZ, d–f) and 2 (NAP–Pt–Ph, g–i) in CH2Cl2 and at r.t. The TRIR spectra are shown at representative delay times: 3, 10, 32, 100, 300 and 1200 ps for 3; 3, 5, 10, 20, 100, and 3000 ps for 1, and 5, 10, 20, 50, 100 and 200 ps for 2. The black and red arrows on (d) and (e) represent the major spectral evolution present on all three complexes. The kinetic traces (c, f and i for 3, 1 and 2, respectively) are shown for single pixel data at 1613 cm−1 (black squares), 1560 cm−1 (red circles), 1900 cm−1 (green triangles) and 1950 cm−1 (blue inverted triangles). The solid lines on the kinetic data were obtained by global fit analysis with instrument response deconvolution. See text for details of the fitting parameters. | ||
The well-defined IR transient signals at 1500 cm−1, 1560 cm−1 and 1613 cm−1 grow in within ≈500 fs and decay with a lifetime of 212 ± 7 ps. These band positions correspond to ν(NAP) and ν(CO) modes in the reduced form of the NAP–CC– fragment (see IR spectrum of the electrochemically generated 3−˙, Fig. S4†).48 In the high frequency region (Fig. 5b) a small signal with a maximum at 2000 cm−1 (superimposed upon the broad transient electronic absorption band mentioned above), also decays with a lifetime of 212 ps. This band is attributed to the ν(CC) mode in the Pt-to-NAP charge transfer state, shifted to lower energies in comparison to its ground state position due to increased electron density on the antibonding orbital.
The first excited state in 3 that can be detected in the TRIR experiments is assigned primarily to a charge-transfer state manifold involving electron transfer from the metal-acetylide center to the NAP acceptor moiety, hereon referred to as CT. A 1.3 ps component was also extracted from the global analysis and is assigned as an average value for initial excited state evolution following 400 nm excitation; however as IR frequencies are sensitive to charge distribution in the molecules, and the major spectral positions do not evolve on this timescale, it is inferred that there is little or no charge redistribution prior to the 212 ps relaxation. Concomitant with the decay of the CT state, three distinct transient bands at 1590 cm−1, 1640 cm−1 and 1950 cm−1, which are characteristic of the 3NAP state,48 grow in and persist without decay until the 3 ns limit of the setup. The 1590 cm−1 band is assigned as a combination of ν(CO)as and ν(NAP), the 1640 cm−1 band as ν(CO)s and the 1950 cm−1 band as ν(CC) in the 3NAP state. These assignments concur with the observed 3NAP nature of the lowest excited state obtained from flash photolysis, emission spectroscopy and other studies on analogous compounds.31,49
C–Pt–CC]-to-NAP charge transfer manifold which ultimately decays to 3NAP and to the ground state is observed for 1 (Fig. 5d–f) and 2 (Fig. 5g–i). The IR band assignments remain the same as for 3. In the first 20 ps the decay of the TRIR signal is complex and appears to have contributions from several charge transfer states convolved with their relaxation processes. In the low frequency region, the NAP-anion signal at 1613 cm−1 decays in a multiexponential fashion with lifetimes of up to 14 ps for 1 and up to 20 ps for 2; this decay is concomitant with the formation of 3NAP bands at 1590 and 1640 cm−1. Differently to 2 and 3, the NAP-anion decay in 1 is accompanied by the appearance of additional bands at 1605 cm−1 and 1550 cm−1, with the resulting spectrum strongly resembling that of the NAP-anion in 3−˙ and in the CT excited state of 1–3, but with a slight shift to lower frequencies. This observation indicates the formation of another excited state in 1, which also incorporates a NAP-anion but has different electron density distribution in other parts of the molecule compared to the precursor CT state. The formation of these two transient bands and associated dynamics (see Fig. 6) are suggestive of electron transfer from the PTZ donor to the oxidised Pt-acetylide centre, forming a full charge-separated state, 3[PTZ+–NAP−], hereon referred to as 3CSS. This 3CSS decays to the ground state with a lifetime of 1.0 ± 0.1 ns. No grow-in of 3NAP bands on nanosecond timescales was observed, setting the upper limit for a 3CSS decay pathway leading to formation of the lower-lying 3NAP to approximately 2%.
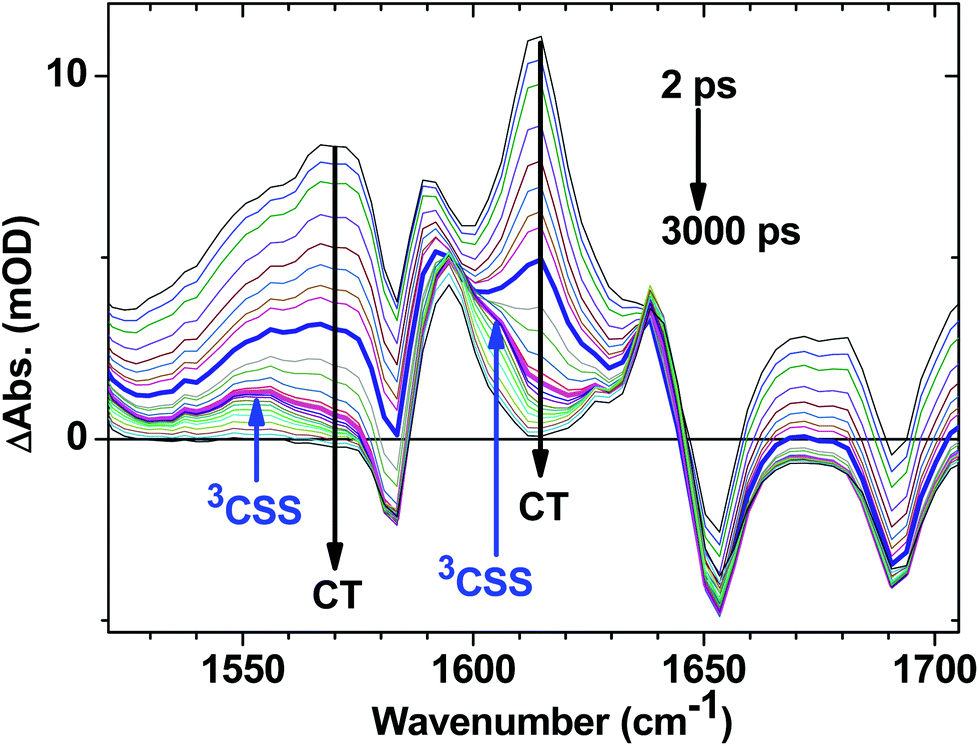 | ||
| Fig. 6 Expanded fingerprint region of the TRIR data for 1 (Fig. 5d) with delay times spanning from 2 ps to 3 ns following 400 nm, ∼50 fs excitation. Arrows indicate the bands associated with the decay of the CT state, and grow in of 3CSS. The highlighted spectra are at delay times of 10 ps (thick blue) and 45 ps (magenta); the latter contains the most pronounced features of 3CSS. | ||
In the high frequency region, the ν(CC) acetylide stretch signal is different for 1 and 2 compared to 3, due to the presence of two strongly coupled acetylide groups. A broad, intense transient signal is observed in the TRIR spectra of 1 and 2 between 1710 cm−1 and 1910 cm−1, with a maximum at 1908 cm−1, attributed to the CT manifold. The ν(CC) band for its successor state 3CSS appears as a small but clearly defined shoulder to the high frequency side of the ground state acetylide bleach (Fig. S5 and S6†), at 2105 cm−1.
Early time dynamics observed in 1 and 2 with TRIR
A satisfactory fit for the full dynamics of 1 was achieved with a four-exponential function and a constant (representing contribution of 3NAP state which has ∼190 μs lifetime and does not decay on the timescale of the ultrafast experiments), with lifetimes of τ1 = 1.1 ± 0.3 ps, τ2 = 3.1 ± 1 ps, τ3 = 14 ± 1 ps (all 3 contributing to the decay of the CT manifold and the formation of 3CSS, 3NAP, and ground state recovery) and τ4 = 1 ± 0.1 ns (decay of the 3CSS state to the ground state). For 2, a fit comprising a triple-exponential function and a constant, with τ1 = 1.0 ± 0.2 ps, τ2 = 3.2 ± 0.3 ps, and τ3 = 20 ± 1 ps (all three contributing to the decay of the CT manifold and the formation of 3NAP and ground state recovery) was sufficient.The UV pump at 400 nm (3.1 eV) contains excess energy compared to the initially populated manifold of CT states. It is therefore expected that electronic evolution within this manifold is convolved with vibrational cooling creating effectively a continuum of processes on the ultrafast time scale. Therefore, the global analysis values obtained in the 1–3 ps range are averaged representations of multiple convoluted ultrafast relaxation processes across the spectrum.
Fig. 7 shows selected kinetic traces plotted at 10 cm−1 intervals throughout the broad acetylide signal from 1710 to 1908 cm−1 for 1. The kinetic behaviour across the acetylide IR band is wavenumber-dependent, with the rise and decay times systematically increasing as the energy increases. The fastest component is instrument-limited (instrument response function ≈200 fs), whilst the slowest is 14 ps, which may be assigned to the lifetime of the relaxed, “thermalised”, lowest CT state. Structural reorganisation and the fact that the high frequency acetylide mode is anharmonically coupled to surrounding low-frequency vibrationally excited intramolecular modes contributes to the broadening of the band, and to the kinetics shown in Fig. 7.75 Furthermore, the non-exponential ground state recovery and 3NAP formation, which occur with a broad range of timescales matching the lifetime range (≈200 fs–14 ps) of the broad IR transient band, is evidence of electron transfer (ET) occurring from non-thermalised excited states.76,77 Vibrational-electronic (vibronic) interactions in these systems are therefore expected to play an important role in ET rates.1,78–80
 | ||
| Fig. 7 The initial part of normalised selected TRIR kinetic traces recorded in the range from 1710 cm−1 to 1908 cm−1 for 1 in CH2Cl2 following 400 nm, ∼50 fs excitation. | ||
The effect of excess excitation energy on excited state dynamics was further investigated using different pump wavelengths. Fig. 8 shows the TRIR signal for 2 in the acetylide region 3 ps after initial excitation at 400 nm (3.10 eV, blue line), 450 nm (2.76 eV, green line) and 470 nm (2.64 eV, red line), all within the ML/L′CT absorption manifold. Using progressively longer wavelength excitation pulse (i.e., photons of lower energy) results in the narrowing, and some blue-shift, of the dominant ν(CC) band. There is also no evidence of the near-instantaneous population of the 3NAP state when exciting with energies lower than 400 nm as the characteristic shoulder at 1950 cm−1 (Fig. 8, blue) is not observed in the experiment under 470 nm excitation (Fig. 8, red). 3NAP formation is therefore accelerated in the presence of excess energy in its precursor states. Importantly, whilst the ET dynamics are affected by the excitation wavelength, the yield of the final 3NAP state in 1–3 ultimately remains unchanged, as determined by the constant phosphorescence yield observed in steady-state emission spectroscopy under excitation ranging from 360 to 470 nm. This indicates that ET rates are uniformly accelerated across all three pathways in the presence of excess energy.
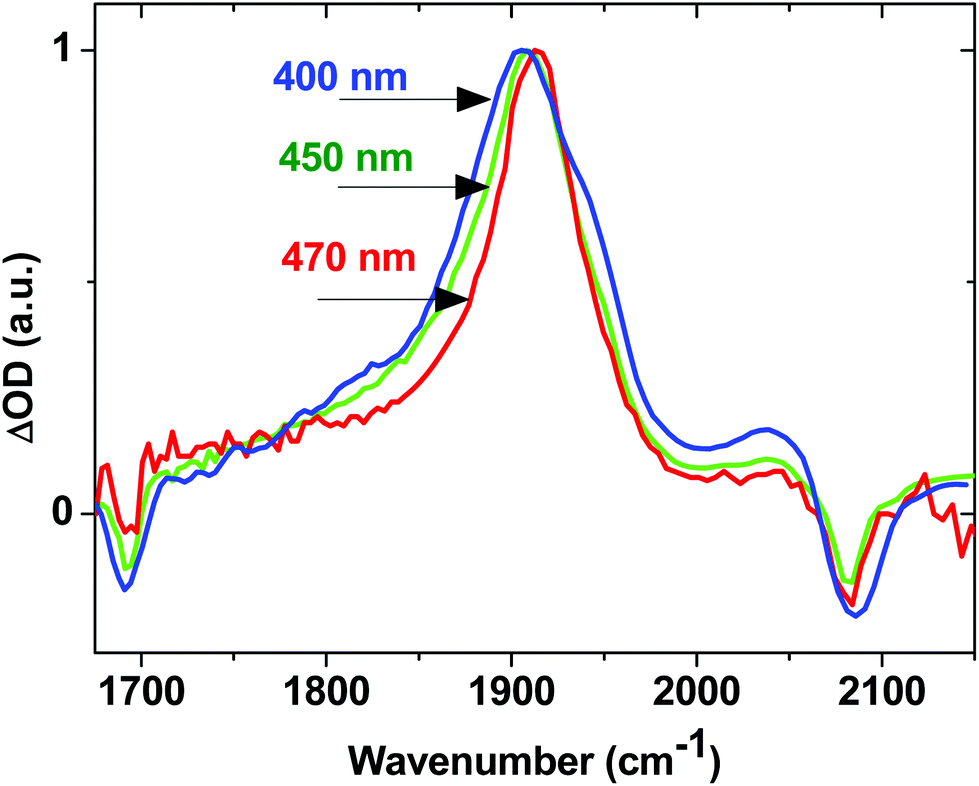 | ||
| Fig. 8 Normalised TRIR spectra for 2 in CH2Cl2, 3 ps after excitation with 400 nm (blue), 450 nm (green) and 470 nm (red) light. | ||
Estimation of the yields of product states for 1 – target analysis
The initial CT manifold in 1 (main IR bands at 1560, 1613 and 1908 cm−1) decays over three separate electron transfer pathways: (i) forward ET to form the full 3CSS, 3[PTZ+–NAP−] (1550, 1605, 2105 cm−1), and charge recombination to form either (ii) the intraligand triplet 3NAP (1590, 1640, 1950 cm−1) or (iii) the ground state. Since the lifetimes of the states involved differ by several orders of magnitude from one another, it is possible to use target analysis to determine the ideal branching model that will accurately reproduce the concentration profile of the ground state recovery. Fig. 9a shows the model assumed for 1, which takes into account electron transfer from non-thermalized excited states, including instrument-limited processes occurring in less than 200 fs, evident in the ultrafast recovery of the ground state (Fig. 9b inset) and 3NAP formation (Fig. 6) observed in the data. The 3NAP yield is fixed to that estimated by singlet oxygen yield measurements (57%), and the 3CSS and ground state yields are varied with 2% intervals between 4–16% and 39–27%, respectively.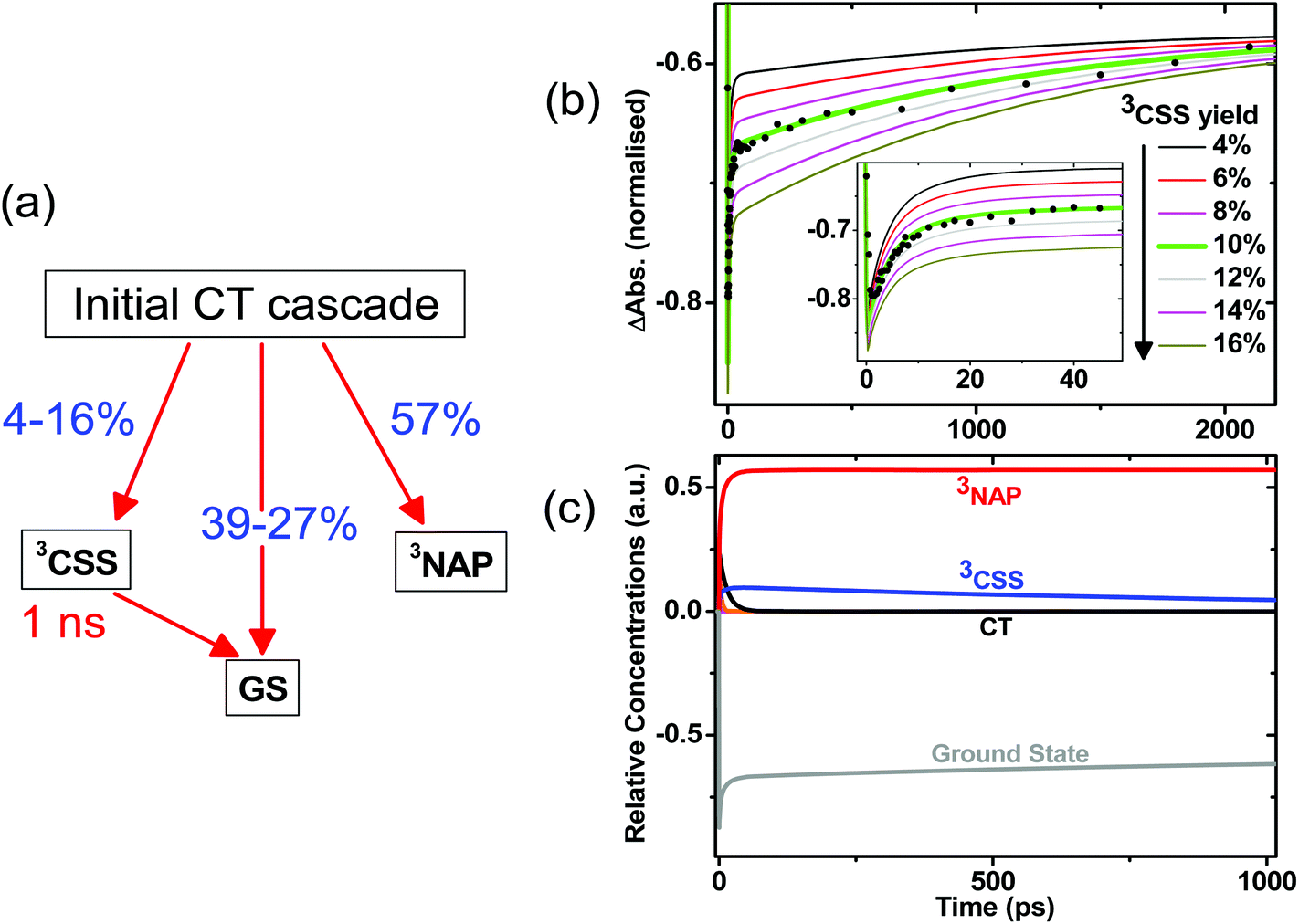 | ||
| Fig. 9 (a) Model used in target analysis of 1, where the branching ratio between the charge separated state 3CSS and GS from the initial excited states is varied. The four components used to model the ultrafast CT cascade and reproduce the dynamics satisfactorily are 0.2, 1.1, 3.2 and 14 ps. (b) Calculated concentration profiles of the ground state as a function of time for different branching parameters. The ground state ν(CO) bleach at 1690 cm−1 from the TRIR data is plotted for comparison (black circles). The best fit achieved is with 10% 3CSS and 33% GS yields, highlighted in green. Inset: early-time data. (c) Calculated concentration profiles of all modelled states up to 1 ns with a 3CSS yield of 10% and GS yield of 33%. | ||
Fig. 9b plots the calculated ground state concentration profiles for all modelled branching parameters, and compares the outputs with the ground state bleach recovery extracted from the TRIR data at 1690 cm−1, i.e. the ν(CO) band free of any overlapping transient (the broad offset is subtracted as background for this analysis). The best fit to the data is the green line, where the branching parameters are 10%, 33% and 57% to the 3CSS, ground state and 3NAP, respectively. From this analysis, the yield of the 3CSS is estimated as 10 ± 2%. The resulting concentration profile for all modelled states is shown in Fig. 9c; additional detail are given in Fig. S7.†
Ultrafast transient absorption spectroscopy
Ultrafast electronic transient absorption spectroscopy data for 1, 2 and 3 in CH2Cl2 are shown in Fig. 10. Three prominent features are common to all compounds over femto- to nanosecond timescale: the transient band around 460 nm assigned to the NAP anion in CT state(s) and partially overlapped with the ground state bleach; stimulated emission giving rise to the negative going features in the range 480–600 nm; and at late time-delays a broad feature which corresponds well to the 3NAP spectra obtained by nanosecond flash photolysis. The positions of the stimulated emission correspond to that obtained from steady-state emission spectroscopy (ca. 500 nm, Table 2) assigned to charge-transfer-to-NAP excited states. Stimulated emission obscures any absorption bands that may occur in its spectral region.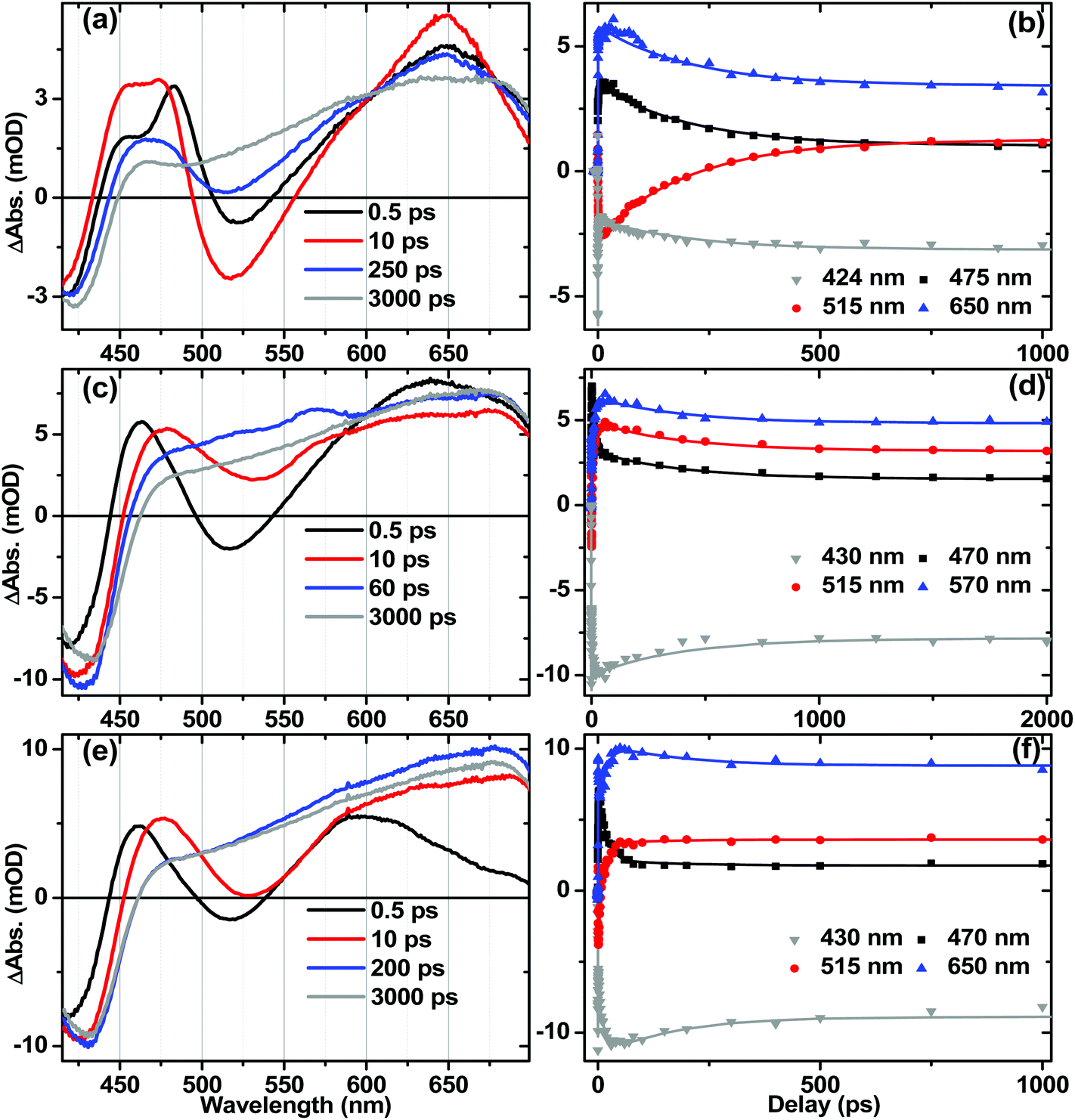 | ||
| Fig. 10 Representative UV-Visible transient absorption spectra and corresponding kinetics following 400 nm excitation for 3, NAP–Pt–Cl (a, b); 1, NAP–Pt–PTZ (c, d); and 2, NAP–Pt–Ph (e, f), in CH2Cl2 and at r.t. Solid lines on kinetic traces are the results of global analysis with instrument response deconvolution. See text for details of the fitting parameters. | ||
In 3, electronic state evolution and excess energy dissipation immediately following the 50 fs, 400 nm excitation is reflected in the initial spectral dynamics appearing as changes in the 480 nm-region and grow-in of stimulated emission at 520 nm (τ1 = 0.9 ± 0.3 ps). Such spectral evolution at early times is similar in all three compounds (see decay associated spectra in Fig. S8–S10†). Following this initial evolution, in the transient spectra of 3 the stimulated emission at 520 nm and the main transient feature centred at 460 nm decay uniformly with the formation of 3NAP with τ2 = 206 ± 5 ps, which matches well the 212 ± 7 ps lifetime obtained for this process from TRIR.
For 1 and 2, the position of the stimulated emission signal evolves with time, decreasing progressively in energy over tens of picoseconds. This is consistent with stimulated emission taking place from a continuum of vibronic states: higher-energy emission occurs at early times as higher-lying states are populated soon after the UV pump; the emission energy then decreases as electronic state evolution, IVR and solvent equilibration take place. Therefore, as in the TRIR analysis above, the lifetimes obtained from global analysis for the earliest events (<3 ps) do not necessarily represent discrete excited states, but should be considered an ensemble average of the initial concomitant charge transfer and cooling processes which lead to continuously evolving multi-exponential kinetics.
The overall dynamics of 2 (Fig. 10f) were modelled satisfactorily using τ1 = 0.6 ± 0.3 ps, τ2 = 2.2 ± 0.6 ps, τ3 = 19 ± 2 ps and a constant representing 3NAP contribution; a minor component decay of τ4 = 160 ± 30 ps was also required for the fit; this component was not reliably extracted from TRIR analysis. In 2, following initial evolution represented by τ1 and τ2, stimulated emission and the band at 470 nm decay with 19 ± 2 ps, concurring with the 20 ± 1 ps lifetime obtained for these processes from TRIR measurements.
In the DBA triad 1, the overall dynamics (Fig. 10d) can be satisfactorily modelled using τ1 = 0.5 ± 0.3 ps, τ2 = 1.8 ± 0.3 ps, τ3 = 13 ± 1 ps, τ4 = 800 ± 200 ps and a constant. Stimulated emission has fully decayed by 60 ps (with major components τ2 = 1.8 ps and τ3 = 13 ± 1 ps), by which time the presence of an additional intermediate state (τ4 = 800 ± 200 ps) overlapped with 3NAP is clearly evident by small additional transient bands at 516 nm and 565 nm, not detected in the spectra of 2 or 3. From TRIR studies we expect this state to be the full charge separated state, 3[PTZ+–NAP−]. Due to overlap of the UV-Vis absorption bands for the ground state, NAP anion, PTZ cation and 3NAP, as well as the relatively low yield of the 3CSS state, this state is not strongly pronounced in the TA data. However, the very different timescales over which these overlapping electronic states decay allow us to deconvolve the pure spectrum of the intermediate state from other electronic states: the spectrum of the intermediate state can be simply obtained by subtracting the 3 ns spectrum (3NAP spectrum) from that at 60 ps, or extracting the 800 ps lifetime spectrum from the DAS resulting from global analysis (Fig. S9†).
In order to ascertain the absorption features of NAP–CC-anion and PTZ-cation anticipated in the full charge-separated 3[PTZ+–NAP−] state of 1, UV/Vis spectroelectrochemical studies were performed on model compounds 3 and 4 (Fig. 11b). The spectrum of 3−˙ obtained upon one-electron reduction of 3 displays NAP-anion absorbances at 460, 525 and 565 nm (Fig. 11b, black spectrum). The spectrum of 4+˙ obtained as a result of one-electron oxidation of 4 displays absorption bands attributed to PTZ-cation at 516 nm (Fig. 11b, blue spectrum). Comparison of the transient absorption spectrum of the intermediate state (at 60 ps) of 1 with the spectra of the electrochemically generated radical ion species clearly show that the PTZ cation band at 516 nm and the NAP-anion band at 565 nm are observed. Thus the spectral profile of the intermediate state matches that expected for the full charge separated state 3[PTZ+–NAP−], a result that concurs with the assignment from TRIR analysis.
 | ||
| Fig. 11 (a) Transient absorption spectrum of the intermediate state, 3[PTZ+–NAP−], free from contributions of the initial CT state and the 3NAP-state (obtained by subtracting the 3NAP spectrum from the transient absorption spectrum at 60 ps). The data points around 400 nm are omitted due to strong scattering of the excitation pulse in this region. (b) UV-Vis absorption spectra of electrochemically generated radical-anion 3−˙ containing the NAP-anion (black, at 243 K), and radical-cation 4+˙ containing the PTZ-cation (blue, at 243 K), overlapped with the spectrum of the ground state of 1 (red). | ||
The photophysical pictures obtained from the TA and TRIR measurements are mutually consistent, and indicate that excitation of the DBA triad 1 with 400 nm light populates a manifold of CT states that decay over three electron transfer pathways, with the formation of a full CSS, 3NAP-state, and reformation of the ground state.
Density functional theory (DFT)
DFT calculations have been performed for 1–3, the –C8H17 group was replaced by –C2H5, and the butyl chains by methyl groups for computational efficiency. The correctness of this assumption was tested for 2 (Fig. S12†); the calculated IR spectra display almost identical positions for the major IR bands regardless of whether C8H17, CH3, C2H5, or butyl was used, showing that such replacements are not crucial to our understanding of the properties of these compounds.Fig. S13a† demonstrates a very good agreement between the calculated and the experimental FTIR spectra of 1 in CH2Cl2. Analysis of the bands confirms the assignments made above from the experimental data: the peaks at 1583 cm−1, 1653 cm−1, 1692 cm−1 are predominantly the NAP ring stretch mode and carbonyl modes (antisymmetric and symmetric), respectively; the peak at 2087 cm−1 with a shoulder at 2109 cm−1 is due to antisymmetric and symmetric combinations of the acetylide stretching vibrations. Other minor bands in the region shown are due to ring stretch and bending modes on the NAP, Phenyl or PTZ fragments. A very good agreement between the late-time experimental TRIR spectrum and the calculated TRIR spectrum (obtained by subtracting singlet ground state spectrum from the spectrum of the lowest triplet state using the scaling factors for the singlet ground state), allows us to assign the IR bands conclusively (Fig. S13b†).
Using the optimized structures for both ground singlet and triplet states, the electronic absorption spectra of both states were obtained using time-dependent DFT (TD-DFT). The resultant singlet UV-Vis spectra together with simulated TA spectra are given in Fig. S14.† The major transitions for 1 are summarized in Table 3, and the molecular orbitals involved in these transitions presented in Fig. 12. The calculations show that the absorption band centered at 430 nm in 1 comprises several transitions from the orbitals delocalized over the central bridge CC–Pt–CC–Ph (HOMO−3, HOMO−2 and HOMO−1) to the NAP-localized LUMO. TD-DFT calculations for 2 confirm that these transitions arise primarily from the same orbitals (Fig. S17, Table S5†). This result agrees well with the nearly identical experimental UV/Vis spectra (Fig. 2) for 1 and 2 in the 430 nm region.
 | ||
| Fig. 12 Orbitals involved in the major electronic transitions of 1 (see Table 3). | ||
| Transition | Wavelength (nm) | Osc. strength | Major contribs |
|---|---|---|---|
| 1 | 463.316 | 0.1204 | H−1 → LUMO (56%) HOMO → LUMO (38%) |
| 3 | 439.876 | 0.2276 | H−3 → LUMO (10%); H−2 → LUMO (78%) |
| 4 | 406.117 | 0.3257 | H−3 → LUMO (82%); H−2 → LUMO (14%) |
The nature of the multiple excited states involved can be further elucidated by electrostatic potential (ESP) maps for the singlet and triplet states of 1, Fig. 13. To simplify the discussion, only a few discreet excited states in the singlet and triplet manifolds are analysed here. This is a simplification given the strong spin–orbit coupling and the large number of excited states in both singlet and triplet manifolds that are involved in the excited state evolution; a more accurate representation requires the inclusion of spin–orbit coupling in the calculations to model the singlet/triplet mixing expected in such systems. This is however computationally prohibitively expensive for such large transition metal complexes.
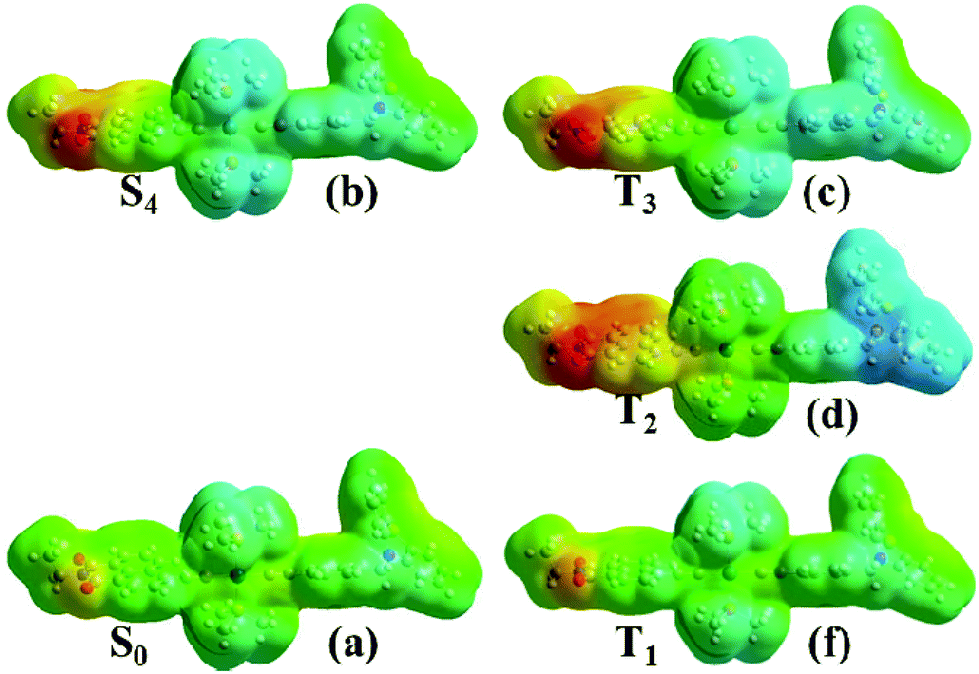 | ||
| Fig. 13 Electron density maps calculated for the initially populated singlet ground state (S0, a), singlet excited state (S4, b), second triplet excited state (T3, c), first triplet excited state (T2, d) and lowest triplet state (T1, e) for 1 in CH2Cl2. Colors correspond to charges with red being −0.1 and blue being +0.1. | ||
The singlet excited state populated with the 400 nm pulse used in the pump-probe experiments was assumed to correspond to the 4th transition from Table 3 due to its proximity to the pulse energy as well as its large oscillator strength. This state is populated instantaneously, and therefore singlet ground state geometry is preserved. It is further assumed that CT states of triplet character are populated on an ultrafast timescale and thus the initial triplet excited state T3 will still retain the geometry of the singlet ground state. T2 is a full charge separated state 3CSS with 1 ns lifetime (vide supra) and is populated over several picoseconds, and therefore its geometry is likely to be different from that of T3. The structure for T2 was thus optimized starting from the triplet ground state structure, T1. It is clear from comparing panels (a) and (b) that the initial excitation (S0→S4) induces electron transfer primarily from the central bridge, [Pt(PBu3)2Ph] to the NAP moiety. These potentials do not change significantly in the second step of the process, S4→T3 (comparing panel (b) and (c) from Fig. 13). After this the process may evolve into full charge separation when the hole transfers to the PTZ donor, as shown by the electrostatic density map of panel (d) for the first triplet excited state, T2. The lowest triplet state T1 resembles the singlet ground state in terms of electrostatic density, which concurs with its assignment as an intraligand excited state. Thus the nature of the ESPs of the excited states as obtained from our calculations agrees well with assignments inferred from spectroscopic data.
Summary of the excited state processes
The above results can be summarized as follows. In all three NAP-containing complexes 1–3, the 400 nm, ∼50 fs excitation pulse populates a manifold of charge-transfer states involving electron transfer from the Pt-acetylide centre to the NAP acceptor. The identical absorption maxima for 1 and 2, which are only slightly different compared to the Cl precursor 3, and the absence of any >350 nm absorbances in the NAP-free building block 4, confirm the lack of PTZ involvement in the initial excitation event. The early time dynamics in 1–2 immediately following excitation involve several electronic excited states, which due to the ultrafast nature of the processes involved are vibrationally hot, and therefore electronic relaxation is convolved with intramolecular vibrational redistribution, internal conversion, solvent relaxation, and intersystem crossing. Following an initial ultrafast electronic evolution on the 0.5–1 ps timescale, strong stimulated emission is observed at the same spectral position as that assigned to the emission from a [Ph–CC–Pt–CC]-to-NAP charge transfer excited state in the steady-state emission spectra. Its estimated radiative lifetime of 8–14 ns is somewhat longer than the 1–2 ns anticipated for pure spin allowed transitions, but significantly shorter than those expected for spin-forbidden transitions.81 The decay of the stimulated emission is multiexponential, and is accompanied by the formation of triplet states and recovery of the singlet ground state. Intersystem crossing in 1–3 is expected to occur on ultrafast timescales due to strong spin–orbit coupling promoted by the heavy-atom effect of the Pt centre, and indeed we observe the instantaneous rise of 3NAP signals in the transient spectra immediately following excitation. Pt(II) complexes are reported to have ISC rates ranging from 70 ± 40 fs, measured by fluorescence upconversion in a close analogue of 1 and 2, Ph–CC–Pt(PBu3)2–CC–Ph, to hundreds of picoseconds.36,43,81–85 Direct population of the triplet NAP via a direct ligand-localised excitation which would lead to slower ISC rates86 is highly unlikely as this ligand is coupled to the Pt centre through the rigid acetylide linker. It has been established that ISC rates in transition metal complexes, in addition to spin–orbit coupling, are influenced by the energy difference between the singlet and triplet states involved, by the degree of metal orbital contribution, and other factors such as transient distortion along the reaction coordinate promoting ISC.81,87–90 In 1, excitation with 400 nm light populates a manifold of S1–S4 singlet states, all of charge-transfer character and all within a 0.4 eV energy range. The energies, geometries and electron density distribution in the triplet states T3–T5 match closely to the singlet manifold, satisfying the requirements for efficient ISC. Thus, in the model assuming pure spin states, the triad 1 has a pool of nearly iso-energetic charge-transfer excited states, including at least three singlets and three triplets. Strong mixing81,91 of these states is highly likely, and may account for the simultaneous observation of an intense stimulated emission and ultrafast formation of 3NAP (the lowest triplet state which is widely separated in energy from the rest of the triplet manifold). Thus, the early time dynamics in 1 are assumed to proceed through a cascade of spin–orbit states, a behaviour similar to that reported for several other transition metal complexes.89,92,93
Using the transient absorption and infrared spectroscopic data, and extracting energy levels of the electronic states from cyclic voltammetry and emission data, one can construct an energy level diagram for 1 as presented in Fig. 14.
 | ||
| Fig. 14 Summary of electronic transitions and excited state lifetimes for 1, NAP–Pt–PTZ, following 400 nm excitation in CH2Cl2 at r.t. The energy values are derived from absorption, emission and cyclic voltammetry data. Electron transfer from the CT manifold occurs from non-thermalized states. The % relative yields of product states are indicated. | ||
Upon excitation of 1–3 with ∼50 fs, 400 nm light, a manifold of charge transfer states is populated where electron density is shifted from the central Pt-containing bridge to the NAP moiety. The initially formed CT excited state branches along three concurrent decay pathways: back electron transfer leading to ground state recovery and 3NAP-state formation (57% yield), and forward electron transfer from the PTZ-unit leading to complete charge separation with the formation of the [NAP−–PTZ+] full charge-separated state, 3CSS (10% yield, 1 ns lifetime). The yields of product states were obtained using singlet oxygen yield measurements, target analysis and ground state bleach recovery dynamics from the TRIR data.
Conclusions
This study contributes to our understanding of the rich photophysics of Pt(II) trans-acetylide donor–acceptor systems by presenting an asymmetric donor–bridge-acceptor triad containing NAP as an electron acceptor and phenothiazine as an electron donor, NAPPt(PBu3)2Ph–CH2–PTZ (1). Its photophysical properties were extensively characterized by electronic and vibrational spectroscopies on timescales ranging from ∼100 fs to milliseconds. The comparison with the photophysical behavior of the constituent “building blocks” 2–4, along with the results of UV/Vis/IR spectroelectrochemical studies and DFT calculations, assisted in assigning the nature of several excited states involved, as well as in elucidating the complex excited states dynamics and pathways in 1. The initial excitation of 1 with visible light leads to the formation of a manifold of charge-transfer states where electron density shifts from the Pt-acetylide bridge to the NAP acceptor. This charge-transfer manifold undergoes excited state branching on the ultrafast (0.2–14 ps) timescale over three pathways to form a long-lived (190 μs) acceptor-localized triplet state, to recombine back to the ground state, and to engage in forward electron transfer with the formation of the full charge-separated state. Thus the ultrafast formation of a full CSS in 1 has been established where the distance between the charges is ∼18 Å. The DBA molecular triad 1 displays intriguing dynamics where ultrafast electron transfer occurs from a manifold of non-thermalized charge transfer excited states. This direct observation may be of importance to other transition metal complexes utilised in light-harvesting and molecular electronics. Such studies are directly related to building efficient artificial systems which emulate natural photosynthesis and undergo rapid and efficient energy and electron transfer over significant distances.
Experimental section
Synthesis
Synthesis of 4-bromo-N-octyl-1,8-naphthalimide, 4-ethynyl-N-octyl-1,8-naphthalimide, N-(4-ethynylbenzyl)-phenothiazine, and cis-Pt(PnBu3)2Cl2 are given in the ESI.†:9 hexane–CH2Cl2). The product eluted as the first of two yellow/orange coloured bands, with removal of the solvent yielding the bright yellow title complex. Yield = 64 mg (0.051 mmol, 44%). 1H NMR (CDCl3): 0.84–0.92 (m, 21H), 1.20–1.47 (m, 22H), 1.58–1.67 (m, 12H), 1.67–1.76 (m, 2H), 2.05–2.20 (m, 12H), 4.15 (t, J = 7.64 Hz, 2H), 5.03 (s, 2H), 6.63 (d, J = 8.16 Hz, 2H), 6.85 (td, J = 0.88, 7.44 Hz, 2H), 6.97 (td, J = 1.48, 7.86 Hz, 2H), 7.07 (dd, J = 1.48, 7.56 Hz, 2H), 7.15 (d, J = 8.20 Hz, 2H), 7.24 (d, J = 8.20 Hz, 2H), 7.59 (d, J = 7.76 Hz, 1H), 7.66 (t, J = 7.84 Hz, 1H), 8.44 (d, J = 7.72 Hz, 1H), 8.56 (dd, J = 0.92, 7.24 Hz, 1H), 8.77 (dd, J = 0.96, 8.28 Hz, 1H). 31P NMR (CDCl3): 3.80 (JPt–P = 2332 Hz). AP-MS: 1244.5 (M+).
:3 CH2Cl2–hexane). The product was initially obtained as a clear oil, which after thorough drying in vacuo afforded a white solid. Yield = 37 mg (0.039 mmol, 36%) 1H NMR (CDCl3): 0.91 (t, J = 7.24 Hz, 18H), 1.38–1.49 (m, 12H), 1.50–1.61 (m, 12H), 1.93–2.07 (m, 12H), 5.01 (s, 2H), 6.62 (dd, J = 0.84, 8.12 Hz, 2H), 6.85 (td, J = 1.08, 7.44 Hz, 2H), 6.96 (td, J = 1.52, 7.84 Hz, 2H), 7.07 (dd, J = 1.52, 7.56 Hz, 2H), 7.13 (d, J = 8.28 Hz, 2H), 7.19 (d, J = 8.24 Hz, 2H). 31P NMR (CDCl3): 6.80 (JPt–P = 2374 Hz). MALDI-MS: m/z = 946.5 (M+), 749.4 (M+ − PTZ).
Equipment
UV/vis spectroscopy studies were carried out using a Cary-50 Bio spectrophotometer. Emission spectroscopy studies were carried out with a Fluoromax-4 spectrofluorometer. Luminescence lifetimes were measured with a Mini-τ fluorimeter (Edinburgh Instruments) containing a picosecond diode laser (410 nm, 80 ps) as an excitation source. Experimental uncertainties are estimated as 20% for emission quantum yields and 15% for the lifetimes. Solutions of the complexes were degassed in quartz cells with 3× freeze–pump–thaw cycles unless otherwise stated. The UV-Vis absorption spectra were regularly checked for stability during the experiments.Nanosecond Flash Photolysis studies were conducted on a home-built setup based on a tuneable Ti:Sapph laser, time resolution ca. 25 ns.
Picosecond TRIR studies were performed in the Ultrafast Spectroscopy Laboratory, Rutherford Appleton Laboratory, UK, ULTRA94 facility. The IR spectrometer comprised two synchronized 10 kHz, 8 W, 40 fs and 2 ps Ti:Sapph oscillator/regenerative amplifiers (Thales) which pump a range of optical parametric amplifiers (TOPAS). A portion of the 40 fs Ti:Sapph beam was used to generate tuneable mid-IR probe light with ca. 400 cm−1 bandwidth. The instrumental response function for TRIR measurements is approx. 250 fs. The probe and pump beam diameters at the sample were ca. 70 and 120 μm, resp., the pump energy at the sample was 1 to 1.5 μJ. Changes in IR absorption spectra were recorded by three HgCdTe linear-IR array detectors on a shot-by-shot basis. All experiments were carried out in Harrick cells with 2 mm thick CaF2 windows and 500 to 950 μm path length; typical optical density of 0.5 to 1 at 400 nm. All samples were mounted on a 2D-raster stage and solutions were flowed to ensure photostability.
Femtosecond TA experiments were performed at ULTRA94 facility (data shown in the text) and at the B. I. Stepanov Institute of Physics, Minsk95,96 (not shown). The data obtained on both setups are in good qualitative agreement. The details are given in the ESI.†
Analysis of ultrafast time-resolved data was performed using the open-source software Glotaran v1.3.97 A combination of Singular Value Decomposition (SVD), global and target analysis was used to extract kinetic rate constants and associated spectral contributions for each system. Full descriptions of these methods can be found elsewhere.87,97–99 More details on the results of this analysis can be found in the ESI.†
DFT
All calculations were performed using Gaussian 09, version C.01,100 compiled using Portland compiler v 8.0–2 with the Gaussian-supplied versions of ATLAS and BLAS,101 using the B3LYP functional of DFT.102 In all cases an extensive basis set was used, consisting of 6-311G**103 on all elements apart from Pt, which was described using a Stuttgart–Dresden pseudo-potential.104 Previous work shown that this approach results in a reasonably accurate description of transition-metal complexes and their properties,14,105 allowing for semi-quantitative comparison with experiments. Our calculations on 1 used 1212 basis functions for 394 electrons. The bulk solvent was described using PCM,106 with the standard parameters for CH2Cl2 as supplied by Gaussian. All integrals were done ultrafine.Acknowledgements
We thank the EPSRC, E-Futures DTC, the University of Sheffield, the STFC (access to beam time at the Lasers for Science Facility), and the Royal Society for financial support, and Dr P Portius for collaboration. All calculations were performed on the “Jupiter” cluster of the Theoretical Chemistry Group of the University of Sheffield.References
- P. F. Barbara, T. J. Meyer and M. A. Ratner, J. Phys. Chem., 1996, 100, 13148–13168 CrossRef.
- J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802–6827 CrossRef PubMed.
- R. Eisenberg, Science, 2009, 324, 44–45 CrossRef CAS PubMed.
- D. Hanss, M. E. Walther and O. S. Wenger, Coord. Chem. Rev., 2010, 254, 2584–2592 CrossRef PubMed.
- G. A. Parada, L. A. Fredin, M. Santoni, R. Lomoth, O. Johansson, P. Persson and S. Ott, Inorg. Chem., 2013, 52, 5128–5137 CrossRef CAS PubMed.
- E. C.-H. Kwok, M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Chem. – Eur. J., 2014, 20, 3142–3153 CrossRef CAS PubMed.
- B. Ventura, A. Barbieri, A. Zanelli, F. Barigelletti, J. B. Seneclauze, S. Diring and R. Ziessel, Inorg. Chem., 2009, 48, 6409–6416 CrossRef CAS PubMed.
- A. Barbieri, B. Ventura and R. Ziessel, Coord. Chem. Rev., 2012, 256, 1732–1741 CrossRef CAS PubMed.
- A. Vacher, F. Barrière, F. Camerel, J.-F. Bergamini, T. Roisnel and D. Lorcy, Dalton Trans., 2013, 42, 383–394 RSC.
- P. Jarosz, J. Thall, J. Schneider, D. Kumaresan, R. Schmehl and R. Eisenberg, Energy Environ. Sci., 2008, 1, 573–583 CAS.
- N. Damrauer, G. Cerullo, A. Yeh, T. Boussie, C. Shank and J. McCusker, Science, 1997, 275, 54–57 CrossRef CAS.
- J. M. Butler, M. W. George, J. R. Schoonover, D. M. Dattelbaum and T. J. Meyer, Coord. Chem. Rev., 2007, 251, 492–514 CrossRef CAS PubMed.
- E. A. Glik, S. Kinayyigit, K. L. Ronayne, M. Towrie, I. V. Sazanovich, J. A. Weinstein and F. N. Castellano, Inorg. Chem., 2008, 47, 6974–6983 CrossRef CAS PubMed.
- J. Best, I. V. Sazanovich, H. Adams, R. D. Bennett, E. S. Davies, A. J. H. M. Meijer, M. Towrie, S. A. Tikhomirov, O. V. Bouganov, M. D. Ward and J. A. Weinstein, Inorg. Chem., 2010, 49, 10041–10056 CrossRef CAS PubMed.
- C. J. Adams, N. Fey, Z. A. Harrison, I. V. Sazanovich, M. Towrie and J. A. Weinstein, Inorg. Chem., 2008, 47, 8242–8257 CrossRef CAS PubMed.
- S. Archer and J. A. Weinstein, Coord. Chem. Rev., 2012, 256, 2530–2561 CrossRef CAS PubMed.
- S. Chakraborty, T. J. Wadas, H. Hester, R. Schmehl, R. Eisenberg and J. Thaddeus, Inorg. Chem., 2005, 44, 6865–6878 CrossRef CAS PubMed.
- C.-W. Chan, L.-K. Cheng and C.-M. Che, Coord. Chem. Rev., 1994, 132, 87–97 CrossRef CAS.
- M. Hissler, W. B. Connick, D. K. Geiger, J. E. McGarrah, D. Lipa, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2000, 39, 447–457 CrossRef CAS.
- V. W. Yam, R. P. Tang, K. M. Wong and K.-K. Cheung, Organometallics, 2001, 20, 4476–4482 CrossRef CAS.
- M. L. Muro, A. A. Rachford, X. Wang and F. N. Castellano, Top. Organomet. Chem., 2010, 29, 159–191 CAS.
- J. A. Weinstein, M. Delor, I. V. Sazanovich, M. Towrie and P. A. Scattergood, in XXIV IUPAC symposium on photochemistry, 2012, p. 120 [PO415] Search PubMed.
- G. Zhou, W.-Y. Wong, S.-Y. Poon, C. Ye and Z. Lin, Adv. Funct. Mater., 2009, 19, 531–544 CrossRef CAS.
- P. Nguyen, G. Lesley, T. B. Marder, I. Ledoux and J. Zyss, Chem. Mater., 1997, 9, 406–408 CrossRef CAS.
- T. M. Cooper, D. M. Krein, A. R. Burke, D. G. McLean, J. E. Rogers and J. E. Slagle, J. Phys. Chem. A, 2006, 110, 13370–13378 CrossRef CAS PubMed.
- L. Kondrachova, K. E. Paris, P. C. Sanchez, A. M. Vega, R. Pyati and C. D. Rithner, J. Electroanal. Chem., 2005, 576, 287–294 CrossRef CAS PubMed.
- E. Glimsdal, M. Carlsson, T. Kindahl, M. Lindgren, C. Lopes and B. Eliasson, J. Phys. Chem. A, 2010, 114, 3431–3442 CrossRef CAS PubMed.
- T. M. Cooper, B. C. Hall, A. R. Burke, J. E. Rogers, D. G. Mclean, J. E. Slagle and P. A. Fleitz, Chem. Mater., 2004, 16, 3215–3217 CrossRef CAS.
- A. Mohammed, B. Minaev, H. Ågren, M. Lindgren and P. Norman, Chem. Phys. Lett., 2009, 481, 209–213 CrossRef CAS PubMed.
- S. C. Jones, V. Coropceanu, S. Barlow, T. Kinnibrugh, T. Timofeeva, J.-L. Brédas and S. R. Marder, J. Am. Chem. Soc., 2004, 126, 11782–11783 CrossRef CAS PubMed.
- L. Liu, D. Huang, S. M. Draper, X. Yi, W. Wu and J. Zhao, Dalton Trans., 2013, 42, 10694–10706 RSC.
- E. Glimsdal, I. Dragland, M. Carlsson, B. Eliasson, T. B. Melø and M. Lindgren, J. Phys. Chem. A, 2009, 113, 3311–3320 CrossRef CAS PubMed.
- M. S. Khan, M. R. A. Al-Mandhary, M. K. Al-Suti, A. K. Hisahm, P. R. Raithby, B. Ahrens, M. F. Mahon, L. Male, E. A. Marseglia, E. Tedesco, R. H. Friend, A. Köhler, N. Feeder and S. J. Teat, J. Chem. Soc., Dalton Trans., 2002, 1358–1368 RSC.
- C.-H. Tao, N. Zhu and V. W.-W. Yam, Chem. – Eur. J, 2005, 11, 1647–1657 CrossRef CAS PubMed.
- G. Ramakrishna, T. I. Goodson, J. E. Rogers-haley, T. M. Cooper, D. G. Mclean and A. Urbas, J. Phys. Chem. C, 2009, 113, 1060–1066 CAS.
- M.-H. Nguyen and J. H. K. Yip, Organometallics, 2012, 31, 7522–7531 CrossRef CAS.
- J. M. Keller, K. D. Glusac, E. O. Danilov, S. McIlroy, P. Sreearuothai, A. R. Cook, H. Jiang, J. R. Miller and K. S. Schanze, J. Am. Chem. Soc., 2011, 133, 11289–11298 CrossRef CAS PubMed.
- R. T. Farley, Q. Zheng, J. A. Gladysz and K. S. Schanze, Inorg. Chem., 2008, 47, 2955–2963 CrossRef CAS PubMed.
- F. Guo, K. Ogawa, Y.-G. Kim, E. O. Danilov, F. N. Castellano, J. R. Reynolds and K. S. Schanze, Phys. Chem. Chem. Phys., 2007, 9, 2724–2734 RSC.
- R. Berenguer, J. Ferna, E. Lalinde and S. Sa, Organometallics, 2013, 32, 835–845 CrossRef.
- C.-J. Lin, C.-Y. Chen, S. K. Kundu and J.-S. Yang, Inorg. Chem., 2014, 53, 737–745 CrossRef CAS PubMed.
- P. Lind, D. Boström, M. Carlsson, A. Eriksson, E. Glimsdal, M. Lindgren and B. Eliasson, J. Phys. Chem. A, 2007, 111, 1598–1609 CrossRef CAS PubMed.
- W. M. Kwok, D. L. Phillips, P. K. Yeung and V. W. Yam, J. Phys. Chem. A, 1997, 101, 9286–9295 CrossRef CAS.
- C. L. Choi, Y. F. Cheng, C. Yip, D. L. Phillips and V. W. Yam, Organometallics, 2000, 19, 3192–3196 CrossRef CAS.
- C. Liao, J. E. Yarnell, K. D. Glusac and K. S. Schanze, J. Phys. Chem. B, 2010, 114, 14763–14771 CrossRef CAS PubMed.
- R. Packheiser, P. Ecorchard, T. Ru and H. Lang, Organometallics, 2008, 27, 3534–3546 CrossRef CAS.
- W. Wu, J. Zhao, J. Sun, L. Huang and X. Yi, J. Mater. Chem. C, 2013, 1, 705–716 RSC.
- I. V. Sazanovich, M. A. H. Alamiry, A. J. H. M. Meijer, M. Towrie, E. S. Davies, R. D. Bennett and J. A. Weinstein, Pure Appl. Chem., 2013, 85, 1331–1348 CrossRef CAS.
- H. Guo, M. L. Muro-Small, S. Ji, J. Zhao and F. N. Castellano, Inorg. Chem., 2010, 49, 6802–6804 CrossRef CAS PubMed.
- F. N. Castellano, Dalton Trans., 2012, 41, 8493–8501 RSC.
- D. Gosztola, M. P. Niemczyk, W. Svec, A. S. Lukas and M. R. Wasielewski, J. Phys. Chem. A, 2000, 104, 6545–6551 CrossRef CAS.
- I. V. Sazanovich, M. A. H. Alamiry, J. Best, R. D. Bennett, O. V. Bouganov, E. S. Davies, V. P. Grivin, A. J. H. M. Meijer, V. F. Plyusnin, K. L. Ronayne, A. H. Shelton, S. A. Tikhomirov, M. Towrie and J. A. Weinstein, Inorg. Chem., 2008, 47, 10432–10445 CrossRef CAS PubMed.
- J. E. Rogers and L. A. Kelly, J. Am. Chem. Soc., 1999, 121, 3854–3861 CrossRef CAS.
- J. E. McGarrah and R. Eisenberg, Inorg. Chem., 2003, 42, 4355–4365 CrossRef CAS PubMed.
- P. Chen, T. D. Westmoreland, E. Danielson, K. S. Schanze, D. Anthon, P. E. Neveux and T. J. Meyer, Inorg. Chem., 1987, 26, 1116–1126 CrossRef CAS.
- T. Takada, K. Kawai, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2006, 128, 11012–11013 CrossRef CAS PubMed.
- G. B. Kauffman, L. A. Teter and J. E. Huheey, Inorg. Synth., 1963, 7, 245 CrossRef.
- R. D'Amato, A. Furlani, M. Colapietro, G. Portalone and M. Casalboni, J. Organomet. Chem., 2001, 627, 13–22 CrossRef.
- S. Lee, K. R. J. Thomas, S. Thayumanavan and C. J. Bardeen, J. Phys. Chem. A, 2005, 109, 9767–9774 CrossRef CAS PubMed.
- J. E. McGarrah, Y.-J. Kim, M. Hissler and R. Eisenberg, Inorg. Chem., 2001, 40, 4510–4511 CrossRef CAS.
- K. Sonogashira, T. Yatake, Y. Tohda, S. Takahashi and N. Hagihara, J. Chem. Soc., Chem. Commun., 1977, 291–292 RSC.
- K. Sonogashira, Y. Fujikura, T. Yatake, N. Toyoshima, S. Takahashi and N. Hagihara, J. Organomet. Chem., 1978, 145, 101–108 CrossRef CAS.
- G. G. Mather, A. Pidcock and G. J. N. Rapsey, J. Chem. Soc., Dalton Trans., 1973, 2095–2099 RSC.
- E. Danielson, C. M. Elliott, J. W. Merkert and T. J. Meyer, J. Am. Chem. Soc., 1987, 109, 2519–2520 CrossRef CAS.
- P. Jarosz, P. Du, J. Schneider, S.-H. Lee, D. McCamant and R. Eisenberg, Inorg. Chem., 2009, 48, 9653–9663 CrossRef CAS PubMed.
- M. Hissler, J. E. McGarrah, W. B. Connick, D. K. Geiger, S. D. Cummings and R. Eisenberg, Coord. Chem. Rev., 2000, 208, 115–137 CrossRef CAS.
- N. M. Shavaleev, H. Adams, J. Best and J. A. Weinstein, J. Organomet. Chem., 2007, 692, 921–925 CrossRef CAS PubMed.
- J. M. Keller and K. S. Schanze, Organometallics, 2009, 28, 4210–4216 CrossRef CAS.
- N. Chawdhury, A. Kohler, R. H. Friend, M. Younus, N. J. Long, P. R. Raithby and J. Lewis, Macromolecules, 1998, 31, 722–727 CrossRef CAS.
- B. El Hamaoui, F. Laquai, S. Baluschev, J. Wu and K. Müllen, Synth. Met., 2006, 156, 1182–1186 CrossRef CAS PubMed.
- K. S. Schanze, E. E. Silverman and X. Zhao, J. Phys. Chem. B, 2005, 109, 18451–18459 CrossRef CAS PubMed.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, Inorg. Chem., 2011, 50, 11446–11460 CrossRef CAS PubMed.
- K. Nakamaru, Bull. Chem. Soc. Jpn., 1982, 55, 2697–2705 CrossRef CAS.
- A. A. Rachford, S. Goeb, R. Ziessel and F. N. Castellano, Inorg. Chem., 2008, 47, 4348–4355 CrossRef CAS PubMed.
- T. Elsaesser and W. Kaiser, Annu. Rev. Phys. Chem., 1991, 42, 83–107 CrossRef CAS.
- K. G. Spears, X. Wen and R. Zhang, J. Phys. Chem., 1996, 100, 10206–10209 CrossRef CAS.
- Y. Huang, C. T. Rettner, D. J. Auerbach and A. M. Wodtke, Science, 2000, 290, 111–114 CrossRef CAS.
- M. J. Potasek and J. J. Hopfield, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 3817–3820 CrossRef CAS.
- A. Troisi, M. A. Ratner and A. Nitzan, J. Chem. Phys., 2003, 118, 6072–6082 CrossRef CAS PubMed.
- A. Nitzan, Annu. Rev. Phys. Chem., 2001, 52, 681–750 CrossRef CAS PubMed.
- M. Chergui, Dalton Trans., 2012, 41, 13022–13029 RSC.
- S. Lentijo, J. A. Miguel and P. Espinet, Inorg. Chem., 2010, 49, 9169–9177 CrossRef CAS PubMed.
- T. Cardolaccia, A. M. Funston, M. E. Kose, J. M. Keller, J. R. Miller and K. S. Schanze, J. Phys. Chem. B, 2007, 111, 10871–10880 CrossRef CAS PubMed.
- K. Kim, S. Liu, M. E. Kose and K. S. Schanze, Inorg. Chem., 2006, 45, 2509–2519 CrossRef CAS PubMed.
- E. O. Danilov, I. E. Pomestchenko, S. Kinayyigit, P. L. Gentili, M. Hissler, R. Ziessel and F. N. Castellano, J. Phys. Chem. A, 2005, 109, 2465–2471 CrossRef CAS PubMed.
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. G. Williams, Inorg. Chem., 2011, 50, 3804–3815 CrossRef CAS PubMed.
- R. M. van der Veen, A. Cannizzo, F. van Mourik, A. Vlček and M. Chergui, J. Am. Chem. Soc., 2011, 133, 305–315 CrossRef CAS PubMed.
- C. Bressler, C. Milne, V.-T. Pham, A. ElNahhas, R. M. van der Veen, W. Gawelda, S. Johnson, P. Beaud, D. Grolimund, M. Kaiser, C. N. Borca, G. Ingold, R. Abela and M. Chergui, Science, 2009, 323, 489–492 CrossRef CAS PubMed.
- A. Cannizzo, A. M. Blanco-Rodríguez, A. El Nahhas, J. Sebera, S. Zális, A. Vlcek and M. Chergui, J. Am. Chem. Soc., 2008, 130, 8967–8974 CrossRef CAS PubMed.
- J. N. Schrauben, K. L. Dillman, W. F. Beck and J. K. McCusker, Chem. Sci., 2010, 1, 405–410 RSC.
- A. C. Bhasikuttan, M. Suzuki, S. Nakashima and T. Okada, J. Am. Chem. Soc., 2002, 124, 8398–8405 CrossRef CAS PubMed.
- A. El Nahhas, A. Cannizzo, F. van Mourik, A. M. Blanco-Rodríguez, S. Zális, A. Vlcek and M. Chergui, J. Phys. Chem. A, 2010, 114, 6361–6369 CrossRef CAS PubMed.
- R. Heydová, E. Gindensperger, R. Romano, J. Sýkora, A. Vlček, S. Záliš and C. Daniel, J. Phys. Chem. A, 2012, 116, 11319–11329 CrossRef PubMed.
- G. Greetham, P. Burgos, Q. Cao, I. Clark, P. Codd, R. Farrow, M. George, M. Kogimtzis, P. Matousek, A. Parker, M. Pollard, D. Robinson, Z.-J. Xin and M. Towrie, Appl. Spectrosc., 2010, 64, 1311–1319 CrossRef CAS PubMed.
- A. Blokhin, M. Gelin, O. Buganov, V. Dubovskii, S. Tikhomirov and G. B. Tolstorozhev, J. Appl. Spectrosc., 2003, 70, 66–72 Search PubMed.
- A. J. Taylor, E. S. Davies, J. A. Weinstein, I. V. Sazanovich, O. V. Bouganov, S. A. Tikhomirov, M. Towrie, J. McMaster and C. D. Garner, Inorg. Chem., 2012, 51, 13181–13194 CrossRef CAS PubMed.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Software, 2012, 49, 1–22 Search PubMed.
- K. M. Solntsev, S. P. Laptenok and P. Naumov, J. Am. Chem. Soc., 2012, 134, 16452–16455 CrossRef CAS PubMed.
- M. Kullmann, A. Hipke, P. Nuernberger, T. Bruhn, D. C. G. Götz, M. Sekita, D. M. Guldi, G. Bringmann and T. Brixner, Phys. Chem. Chem. Phys., 2012, 14, 8038–8050 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, J. M. A. Robb, R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, A. J. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, 2009.
- R. C. Whaley and A. Petitet, Software: Pract. Exper., 2005, 35, 101–121 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS PubMed.
- X. Cao and M. Dolg, J. Chem. Phys., 2001, 115, 7348–7355 CrossRef CAS PubMed.
- S. P. Foxon, C. Green, M. G. Walker, A. Wragg, H. Adams, J. A. Weinstein, S. C. Parker, A. J. H. M. Meijer and J. A. Thomas, Inorg. Chem., 2012, 51, 463–471 CrossRef CAS PubMed.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detail on synthesis, NMR, electrochemistry, CV for all compounds. Emission data for 1 and 3 in toluene. IR spectroelectrochemistry for 3. Further ultrafast spectroscopy analysis, including decay associated spectra resulting from global analysis. Full computational details including IR spectra for the non-truncated structure of 2, all structures, coordinates, electronic transitions and molecular orbitals. Details of ultrafast TRIR and transient absorption setups. See DOI: 10.1039/c4dt01682c |
| ‡ These authors contributed equally to the work presented herein. |
| This journal is © The Royal Society of Chemistry 2014 |