
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and structural investigation of R2Si (R = Me, Ph) bridged di-N-heterocyclic carbenes†
Rajendra S.
Ghadwal
 *,
Sven O.
Reichmann
,
Elena
Carl
and
Regine
Herbst-Irmer
*,
Sven O.
Reichmann
,
Elena
Carl
and
Regine
Herbst-Irmer
Institut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstrasse 4, 37077 Göttingen, Germany. E-mail: rghadwal@uni-goettingen.de
First published on 24th July 2014
Abstract
Functionalization of the C4 carbon of an imidazol-derived N-heterocyclic carbene (NHC) may allow fine-tuning of the electronic and steric properties of the C2 carbene center. A facile route to silyl-functionalized di-N-heterocyclic carbenes (Di-NHCs) is described. Treatment of the polymeric lithiated NHC, {Li(IPrH)}n (1) (Li(IPrH) = {(N-2,6-iPr2C6H3)2CHCLi}C:) with a dichlorosilane affords monomeric silyl-functionalized Di-NHCs, R2Si(IPrH)2 (R = Ph, 2; Me, 3). Interestingly, silyl-functionalized mono-NHC, Ph2(Cl)Si(IPrH) (4) with a pendant chloro-substituent can also be exclusively isolated maintaining the reactants 1 and Ph2SiCl2 ratio. NHCs 2 and 4 readily form copper complexes, Ph2Si{(IPrH)CuCl}2 (5) and Ph2(Cl)Si{(IPrH)CuCl} (6), on reaction with CuCl. Straightforward conversion of an NHC to a Di-NHC (2 or 3) via C4 functionalization is reported for the first time. Molecular structures of 2, 4, 5 and 6 have been established by single crystal X-ray diffraction studies.
Introduction
N-heterocyclic carbenes (NHCs), which were originally considered as simple phosphine mimics, have established exceptional applications beyond ligands in organometallic and transition metal catalysis.1–5 The utility of NHCs has expanded from the stabilization of elusive compounds with a low-valent main group element3 or complexes with a metal in the high-oxidation state2 to the pharmaceuticals4 and materials science.5 The success of NHCs as ligands in catalysis is largely attributed to their strong σ-donor properties and relatively high covalent contribution to the metal–C(carbene) bond.6 A very small modification in the structure may have a significant impact on the properties of an NHC and hence derived metal complexes as well as related species.7 A unique structural feature allows fine-tuning of NHCs in more ways, therefore a variety of NHCs with variable structural and electronic properties has been prepared.8 Interesting poly-NHCs featuring two or more carbene functionalities have been reported; however, a majority of them is linked via the nitrogen atom of the imidazol ring.9–14 In addition to their applications as ligands in catalysis,9 such poly-NHCs have shown their potential as building-blocks to design a new class of structurally dynamic materials.14Among available methods of fine-tuning, functionalization of NHCs impacts the properties as well as allows access to a new class of NHCs.15–22 The prospect of direct access to functionalized NHCs from an NHC, with p-block moieties in particular, is enormous as it enables the synthesis of new ligands. Functionalization of an imidazol-2-ylidene with a phosphaalkene was first observed by Gates et al. to yield a 4-phosphino-substituted NHC (Scheme 1, I).16 Bertrand et al. developed a method for the preparation of 4-substituted imidazol-2-ylidenes (Scheme 1, II) by deprotonation of a C2-substituted imidazolium salt.17 Formation of II presumably occurs via 1,3 rearrangement of an imidazol-4-ylidene (mesoionic carbene) intermediate. Nevertheless, stable metal-free imidazol-4-ylidenes have also been reported by the same group.23 Functionalization of an NHC has also been achieved with a silylene (IPr)SiCl2, when treated with adamantyl azide (Scheme 1, III).18 Robinson et al. revealed a facile route to a lithiated NHC, Li(IPrH) (1) (Li(IPrH) = {(N-2,6-iPr2C6H3)2CHCLi}C:),19 which has facilitated the access to new molecular scaffolds with interesting properties and applications.20
 | ||
| Scheme 1 C4 functionalized mono-NHCs. | ||
Treatment of an NHC-precursor, 1-methyl-3-tert-butyl-imidazol-2-thione (Scheme 2, IV), with nBuLi in the presence of a phosphine affords phosphino-bridged di-thiones (V).22 Interestingly, in contrast to the established protocol to NHCs,24 reaction of (V) with potassium metal does not yield the expected phosphino-bridged Di-NHC (VI) but an imidazolium phosphanide zwitterion (VII).22 Silyl-functionalized mono-NHCs (Scheme 1, II and III) can be prepared starting from an NHC (Scheme 1);17–19 however, to the best of our knowledge no Di-NHC has been synthesized so far. Therefore, we became interested in developing a protocol for synthesis of Di-NHCs via direct C4-functionalization of an NHC.
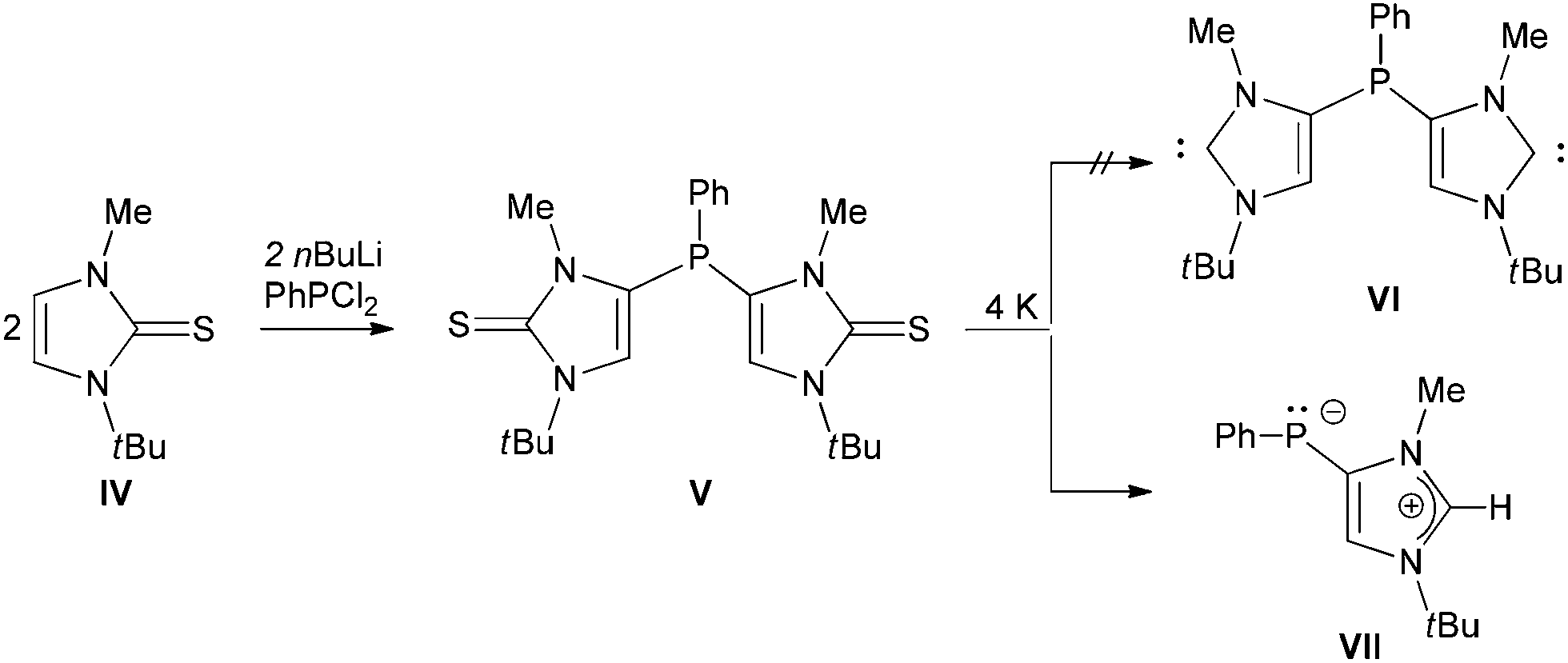 | ||
| Scheme 2 Reaction of di-thione (V) with potassium metal. | ||
Herein, we report a straightforward route to C4-bridged Di-NHCs, R2Si(IPrH)2 (R = Ph, 2; Me, 3) and a copper complex Ph2Si{(IPrH)CuCl}2 (5) (Schemes 3 and 4). A Similar protocol also allows access to silyl-functionalized mono-NHC, Ph2(Cl)Si(IPrH) (4) with a pendant chloro-substituent.
 | ||
| Scheme 3 Reaction synthesis of NHDCs 2 and 3 and mono-NHC 4. | ||
 | ||
| Scheme 4 Synthesis of NHC copper complexes (5) and (6). | ||
Results and discussion
Di-NHCs 2 and 3 have been prepared (Scheme 3) in almost quantitative yield on treatment of {Li(IPrH)}n (1) with an appropriate dihalosilane. Silyl-functionalized mono-NHC, Ph2(Cl)Si(IPr) (4) is also accessible on treatment of 1 with one equiv. of Ph2SiCl2 (Scheme 3) in 82% yield. Compounds 2–4 are colorless crystalline solids, which are soluble in common organic solvents (benzene, toluene, and THF). They are stable under an inert atmosphere of nitrogen or argon gas. The EI-mass spectrum of 2 exhibits the molecular ion peak at 956.6 amu. A comparison of the NMR spectral data of 2–4 with those of the unfunctionalized NHC, IPr (IPr = {(N-2,6-iPr2C6H3)CH}2C:) exhibits significant differences. For instance, imidazol back-bone protons (NCH) of 2 appear as a singlet at δ 7.65 ppm (in C6D6), which are shifted by δ 1.03 ppm down field when compared with IPr (δ 6.62 ppm in C6D6).25 This significant chemical shift may be attributed due to the presence of electron withdrawing phenyl groups on the silicon atom. As expected, NCH protons of 3 exhibit a singlet at δ 7.16 ppm (in THF-d8), which are shifted slightly up field than those of IPr (δ 7.18 ppm, in THF-d8). The 1H NMR spectrum of 2 shows a remarkably high field signal for two isopropyl groups (δ 0.65 ppm, 12H, HCMe2), which may be inferred as the shielding contributions from the π-current density of adjacent phenyl substituents of the SiPh2 moiety. This is consistent with the solid state structure of 2 and thus the structure is maintained in the solution state. This feature is not available in 3, therefore HCMe2 protons appear in the expected region. The 13C NMR spectrum of 2 and 3 each exhibits resonances consistent with their 1H NMR spectral data. Each of the NHCs 2 and 3 shows a signal at δ 225.19 and 225.12 ppm for the carbene carbon atom. Each of the compounds 2 (δ −31.17) and 3 (δ −22.14 ppm) exhibits a signal in the 29Si{1H} NMR spectrum, which is consistent with those observed for four-coordinate organosilicon compounds.17–19,26 The 1H and 13C NMR signals of 4 are fully in agreement with its solid state structure. Compound 4 exhibits a signal at δ −9.19 ppm in the 29Si{1H} NMR spectrum.26aThe molecular structure of 2 was established by a single crystal X-ray diffraction study, which clearly reveals the formation of a Di-NHC (Fig. 1). Compound 2 crystallizes in the monoclinic space group P21/c. The silicon atom in 2 is four-fold coordinated and bridges two flanking NHC moieties. Each of the imidazol-rings features a delocalized π-electron system with short single and long double bonds. The N1–C2–N3 bond angle of 101.65(13) further supports the carbene character of the C2 carbon.8 The Si1–C4 (imidazol) and Si1–C59 (Ph) (av. 1.87 Å) bond lengths are equal to the sum of the covalent radii of Si and C.27
 | ||
| Fig. 1 Molecular structure of Di-NHC 2. Isopropyl groups and hydrogen atoms have been omitted for clarity; anisotropic displacement parameters are depicted at the 50% probability level. Selected bond lengths [Å] and angles [°]: N1–C2 1.367(2), N1–C5 1.385(2), N3–C2 1.3669(19), C4–C5 1.358(2), Si1–C4 1.8791(16), Si1–C59 1.8760(17); N1–C2–N3 101.65(13), N3–C4–C5 103.50(13), C4–Si1–C59 107.08(7), C4–Si1–C9 104.24(7). | ||
Compound 4 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The molecular structure of 4 shows functionalization of the C4 carbon atom with Si(Cl)Ph2 (Fig. 2). The Si1–Cl1 bond length of 2.0701(8) Å is consistent with those measured for the compounds with four coordinate silicon atoms.26a,27 The four-fold coordinated silicon atom adopts a distorted tetrahedral geometry.
. The molecular structure of 4 shows functionalization of the C4 carbon atom with Si(Cl)Ph2 (Fig. 2). The Si1–Cl1 bond length of 2.0701(8) Å is consistent with those measured for the compounds with four coordinate silicon atoms.26a,27 The four-fold coordinated silicon atom adopts a distorted tetrahedral geometry.
 | ||
| Fig. 2 Molecular structure of mono-NHC 4. Hydrogen atoms have been omitted for clarity; anisotropic displacement parameters are depicted at the 50% probability level. Selected bond lengths [Å] and angles [°]: N1–C2 1.3732(19), N1–C5 1.3799(19), N3–C2 1.3678(19), C4–C5 1.355(2), Si1–Cl1 2.0701(8), Si1–C4 1.8575(15), Si1–C36 1.8562(16); N1–C2–N3 100.79(12), N3–C4–C5 103.82(12), C4–Si1–C36 117.75(7), C4–Si1–Cl1 101.93(6). | ||
NHCs 2 and 4 each reacts with CuCl and affords complexes Ph2Si{(IPrH)CuCl}2 (5) and Ph2(Cl)Si{(IPrH)CuCl} (6), respectively (Scheme 4). Complexes 5 and 6 are colorless crystalline solids, soluble in THF and CH2Cl2, and stable under an inert gas atmosphere. Formation of 5 and 6 is supported by the appearance of the corresponding molecular ion peak in the EI-mass spectrum. The 1H NMR spectrum of each of compounds 5 and 6 shows signals for NHC moieties. Appearance of a new signal in the 13C NMR spectrum of 5 at δ 186.3 ppm is assigned for the carbene carbon atom (C–Cu). Complexes 5 and 6 show a 29Si{1H} NMR resonance at δ −30.10 and −10.95 ppm.17–19,26
Complex 5 crystallizes in the orthorhombic space group Pna21.25 The molecular structure of 5 reveals that each of the carbene centers coordinates to a CuCl moiety (Fig. 3). The Cu–C(carbene) bond length of 1.886 Å (av.) and slightly distorted linear geometry with C–Cu–Cl angles of 174.14° (av.) are consistent with those of reported NHC-copper complexes.17b,28 The Cu–Cl bond length of 2.11 Å (av.) may be assigned for the terminal CuCl.17b,28 The four-fold coordinated silicon atom adopts distorted tetrahedral geometry with a significant decrease in the C4–Si1–C9 bond angle of 98.63(14). The molecular structure of 6 is shown in Fig. 4. Complex 6 crystallizes in the monoclinic space group P21/c. The copper atom in 6 features a distorted linear geometry with C2–Cu1–Cl1 bond angle of 174.02(5)°. As expected for both complexes 5 and 6, the N–C–N bond angle increases by 2.9° (av.) on coordination of the carbene carbon to CuCl.
 | ||
| Fig. 3 Molecular structure of dicopper complex 5. Isopropyl groups and hydrogen atoms have been omitted for clarity; anisotropic displacement parameters are depicted at the 50% probability level. Selected bond lengths [Å] and angles [°]: Cu1–C2 1.898(5), Cu1–Cl1 2.1116(15), Cu2–C7 1.875(5), Cu2–Cl2 2.1152(14), N1–C2 1.362(6), N1–C5 1.367(7), N3–C2 1.350(6), C4–C5 1.350(7), Si1–C4 1.888(5), Si1–C59 1.858(5); C2–Cu1–Cl1 174.84(15), N1–C2–N3 104.5(4), N3–C4–C5 104.6(4), C4–Si1–C59 108.9(2), C4–Si1–C9 98.63(14). | ||
 | ||
| Fig. 4 Molecular structure of complex 6. Hydrogen atoms have been omitted for clarity; anisotropic displacement parameters are depicted at the 50% probability level. Selected bond lengths [Å] and angles [°]: Cu1–C2 1.8779(16), Cu1–Cl2 2.1076(5), N1–C2 1.3610(19), N1–C5 1.376(2), N3–C2 1.357(2), C4–C5 1.360(2), Si1–Cl1 2.0527(6), Si1–C4 1.8615(16), Si1–C36 1.8623(17); C2–Cu1–Cl2 174.02(5), N1–C2–N3 103.73(12), N3–C4–C5 104.31(13), C4–Si1–C36 106.12(7), C4–Si1–Cl1 110.09(5). | ||
Experimental
General
All synthesis and manipulations were carried out under an inert atmosphere of dry argon gas using Schlenk line techniques or Glove-box. C6D6 and THF-d8 were dried over K-benzophenone ketyl and distilled under dry argon atmosphere prior to use. CD2Cl2 was dried over CaH2 and distilled under dry argon prior to use. All other solvents were dried and purified by a MBRAUN solvent purification system (MB SPS 800). 1H, 13C, and 29Si NMR spectra were recorded using a Bruker Avance III 300 spectrometer. EI-mass spectra were recorded with MAT 95 (70 eV). Elemental analyses were performed at the Institute for Inorganic Chemistry, Universität Göttingen. Ph2SiCl2 and Me2SiCl2 were distilled prior to use. Anhydrous CuCl (Aldrich) was used as received. Compound {Li(IPrH)}n (1) was prepared according to the reported method.19a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture and stored at 4 °C for two days to yield colorless crystals of Ph2Si(IPrH)2 (2). Yield: 6.48 g, 84%. Anal calcd for C66H80N4Si (957): C 82.79, H 8.42, N 5.85; found C 82.72, H 8.44, N 5.66. 1H NMR (300 MHz, C6D6, 25 °C): δ 0.65 (d, 12H, J = 6.82 Hz, HCMe2); 1.19 (d, 12H, J = 6.97 Hz, HCMe2); 1.22, 125 (dd, 24H, J = 6.74 Hz, 6.79 Hz, HCMe2); 2.64 (sept, 4H, J = 6.79 Hz, HCMe2); 3.14 (sept, 4H, J = 6.90 Hz, HCMe2); 6.76 (d, 4H, J = 7.76 Hz, m-C6H3); 6.91 (d, J = 7.45 Hz, m-C6H3); 7.00 (m, 6H, o-, p-C6H5); 7.10–7.19 (m, 4H, m-C6H5); 7.29 (t, 4H, p-C6H3); 7.65 (s, 2H, NCH) ppm. 13C{1H} NMR (75 MHz, C6D6, 25 °C): δ 21.02, 24.10, 24.23, 26.18 (HCMe2); 28.62, 29.01 (HCMe2); 122.84, 123.70, 130.10, 134.54, 136.54, 138.19, 139.12 (CHCH, C6H3, C6H5), 146.02 (ipso-C6H3), 225.19 (NCN) ppm. 29Si{1H} NMR (59 MHz, C6D6, 25 °C): δ −31.17 ppm. EI-MS: m/z (%): 956.6 (85) [M+], 387.4 (100) [IPrH+].
:1) mixture and stored at 4 °C for two days to yield colorless crystals of Ph2Si(IPrH)2 (2). Yield: 6.48 g, 84%. Anal calcd for C66H80N4Si (957): C 82.79, H 8.42, N 5.85; found C 82.72, H 8.44, N 5.66. 1H NMR (300 MHz, C6D6, 25 °C): δ 0.65 (d, 12H, J = 6.82 Hz, HCMe2); 1.19 (d, 12H, J = 6.97 Hz, HCMe2); 1.22, 125 (dd, 24H, J = 6.74 Hz, 6.79 Hz, HCMe2); 2.64 (sept, 4H, J = 6.79 Hz, HCMe2); 3.14 (sept, 4H, J = 6.90 Hz, HCMe2); 6.76 (d, 4H, J = 7.76 Hz, m-C6H3); 6.91 (d, J = 7.45 Hz, m-C6H3); 7.00 (m, 6H, o-, p-C6H5); 7.10–7.19 (m, 4H, m-C6H5); 7.29 (t, 4H, p-C6H3); 7.65 (s, 2H, NCH) ppm. 13C{1H} NMR (75 MHz, C6D6, 25 °C): δ 21.02, 24.10, 24.23, 26.18 (HCMe2); 28.62, 29.01 (HCMe2); 122.84, 123.70, 130.10, 134.54, 136.54, 138.19, 139.12 (CHCH, C6H3, C6H5), 146.02 (ipso-C6H3), 225.19 (NCN) ppm. 29Si{1H} NMR (59 MHz, C6D6, 25 °C): δ −31.17 ppm. EI-MS: m/z (%): 956.6 (85) [M+], 387.4 (100) [IPrH+].
X-ray crystallography
Suitable single crystals were selected from the mother liquor in the Schlenk flask and covered with perfluorinated polyether oil on a microscope slide, which was cooled with a nitrogen gas flow using the X-Temp2 device.29 The diffraction data of compound 2 were collected at 100 K on a Bruker D8 three circle diffractometer equipped with a SMART APEX II CCD detector and a microfocus source30 with INCOATEC Quazar mirror optics (λ = 0.71073 Å). The diffraction data for compounds 4, 5, and 6 were collected at 100 K on a Bruker TXS-Mo rotating anode with mirror optics and a Smart Apex II Ultra detector. The data were integrated with SAINT31 and a multi-scan absorption correction with SADABS32 was applied.The structures were solved by direct methods (SHELXS-97)33 and refined against all data by full-matrix least-squares methods on F2 (SHELXL2013)34,35 within the SHELXLE GUI.36 The hydrogen atoms were refined isotropically on calculated positions using a riding model with their Uiso values constrained to 1.5Ueq of their pivot atoms for terminal sp3 carbon atoms and 1.2 times for all other carbon atoms. All non-hydrogen-atoms were refined with anisotropic displacement parameters. Disordered moieties were refined using distance restraints (SAME) and anisotropic displacement parameter restraints (SIMU and RIGU).35
837 reflections measured, 12433 independent (Rint = 0.0485), R1 = 0.0449 [I > 2σ(I)], wR2 = 0.1170 (all data), residual density peaks: 0.343 to −0.259 e Å−3.
303 reflections measured, 9200 independent (Rint = 0.0418), R1 = 0.0410 [I > 2σ(I)], wR2 = 0.1034 (all data), residual density peaks: 0.354 to −0.346 e Å−3.
192 reflections measured, 14390 independent (Rint = 0.0340), R1 = 0.0352 [I > 2σ(I)], wR2 = 0.0968 (all data), residual density peaks: 0.824 to −0.561 e Å−3.
304 reflections measured, 8799 independent (Rint = 0.0519), R1 = 0.0288 [I > 2σ(I)], wR2 = 0.0740 (all data), residual density peaks: 0.367 to −0.272 e Å−3.
Conclusions
In conclusion, we have reported a convenient method which allows direct functionalization of an NHC to afford Di-NHCs 2 and 3 in high yield. Mono-NHC 4 with a pendant chloro-substituent is also accessible using a similar protocol. Compound 4 offers new promises to prepare novel hybrid ligands by further functionalization. Di-NHCs (2 and 3), mono-NHC (4), and copper complexes (5 and 6) are fully characterized. We believe that this method can be applied to other new functionalized Di-NHCs with different bridging moieties, such as VI. Investigation on potential application of Di-NHCs as ligands in homogeneous catalysis and as organocatalysts is currently under way to examine the scope of new Di-NHC scaffolds.Acknowledgements
The authors are thankful to Prof. Dr Dietmar Stalke for his generous support and encouragement.Notes and references
- (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed; (b) W. A. Herrmann, Angew. Chem., 2002, 114, 1342–1363 ( Angew. Chem., Int. Ed. , 2002 , 41 , 1290–1309 ) CrossRef; (c) E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., 2007, 119, 2824–2870 ( Angew. Chem., Int. Ed. , 2007 , 46 , 2768–2813 ) CrossRef PubMed; (d) F. E. Hahn and M. C. Jahnke, Angew. Chem., 2008, 120, 3166–3216 ( Angew. Chem., Int. Ed. , 2008 , 47 , 3122–3172 ) CrossRef PubMed; (e) S. Díez–González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (f) R. Wolf and W. Uhl, Angew. Chem., 2009, 121, 6905–6907 ( Angew. Chem., Int. Ed. , 2009 , 48 , 6774–6776 ) CrossRef PubMed.
- (a) N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (b) N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, Berlin, 2007 Search PubMed; (c) N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, Heidelberg, 2011 Search PubMed.
- (a) R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444–456 CrossRef CAS PubMed; (b) R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., 2009, 121, 5793–5796 ( Angew. Chem., Int. Ed. , 2009 , 48 , 5683–5686 ) CrossRef PubMed; (c) A. C. Filippou, O. Chernov and G. Schnakenburg, Angew. Chem., 2009, 121, 5797–5800 ( Angew. Chem., Int. Ed. , 2009 , 48 , 5687–5690 ) CrossRef PubMed; (d) H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420–1422 CrossRef CAS PubMed; (e) A. Higelin, S. Keller, C. Göhringer, C. Jones and I. Krossing, Angew. Chem., 2013, 125, 5041–5044 ( Angew. Chem., Int. Ed. , 2013 , 52 , 4941–4944 ) CrossRef PubMed; (f) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. von R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 CrossRef CAS PubMed; (g) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. von R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069–1071 CrossRef CAS PubMed.
- (a) A. Gautier and F. Cisnetti, Metallomics, 2012, 4, 23–32 RSC; (b) W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC; (c) L. Oehninger, R. R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269–3284 RSC.
- L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC.
- (a) T. Dröge and F. Glorius, Angew. Chem., 2010, 122, 7094–7107 ( Angew. Chem., Int. Ed. , 2010 , 49 , 6940–6952 ) CrossRef PubMed; (b) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- (a) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC; (b) M. Albrecht, Chem. Commun., 2008, 3601–3610 RSC.
- (a) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed; (b) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., 2010, 122, 8992–9032 ( Angew. Chem., Int. Ed. , 2010 , 49 , 8810–8849 ) CrossRef PubMed; (c) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS PubMed.
- (a) A. B. Powell, C. W. Bielawski and A. H. Cowley, J. Am. Chem. Soc., 2010, 132, 10184–10194 CrossRef CAS PubMed; (b) J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723–1732 RSC.
- R. Maity, A. Rit, C. Schulte to Brinke, J. Kösters and F. E. Hahn, Organometallics, 2013, 32, 6174–6177 CrossRef CAS.
- M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS PubMed.
- D. M. Khramov, A. J. Boydston and C. W. Bielawski, Angew. Chem., 2006, 118, 6332–6335 ( Angew. Chem., Int. Ed. , 2006 , 45 , 6186–6189 ) CrossRef PubMed.
- M. Nussbaum, O. Schuster and M. Albrecht, Chem. – Eur. J., 2013, 19, 17517–17527 CrossRef CAS PubMed.
- B. M. Neilsona, A. G. Tennysona and C. W. Bielawski, J. Phys. Org. Chem., 2012, 25, 531–543 CrossRef PubMed.
- O. Kühl, Functionalised N-Heterocyclic Carbene Complexes, John Wiley & Sons Ltd, 2010 Search PubMed.
- J. Bates, P. Kennepohl and D. P. Gates, Angew. Chem., 2009, 121, 10028–10031 ( Angew. Chem., Int. Ed. , 2009 , 48 , 9844–9847 ) CrossRef PubMed.
- (a) D. Mendoza-Espinosa, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 7264–7265 CrossRef CAS PubMed; (b) D. Mendoza-Espinosa, B. Donnadieu and G. Bertrand, Chem. – Asian J., 2011, 6, 1099–1103 CrossRef CAS PubMed.
- R. S. Ghadwal, H. W. Roesky, M. Granitzka and D. Stalke, J. Am. Chem. Soc., 2010, 132, 10018–10020 CrossRef CAS PubMed.
- (a) Y. Wang, Y. Xie, M. Y. Abraham, P. Wei, H. F. Schaefer III, P. von R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2010, 132, 14370–14372 CrossRef CAS PubMed; (b) Y. Wang, M. Y. Abraham, R. J. Gilliard Jr., P. Wei, J. C. Smith and G. H. Robinson, Organometallics, 2012, 31, 791–793 CrossRef CAS.
- (a) S. Kronig, E. Theuergarten, C. G. Daniliuc, P. G. Jones and M. Tamm, Angew. Chem., 2012, 124, 3294–3298 ( Angew. Chem., Int. Ed. , 2012 , 51 , 3240–3244 ) CrossRef PubMed; (b) Y. Wang, Y. Xie, M. Y. Abraham, R. J. Gilliard Jr., P. Wei, C. F. Campana, H. F. Schaefer III, P. von R. Schleyer and G. H. Robinson, Angew. Chem., 2012, 124, 10320–10323 ( Angew. Chem., Int. Ed. , 2012 , 51 , 10173–10176 ) CrossRef PubMed.
- For other functionalized NHCs and derivatives: (a) P. K. Majhi, S. Sauerbrey, G. Schnakenburg, A. J. Arduengo III and R. Streubel, Inorg. Chem., 2012, 51, 10408–10416 CrossRef CAS PubMed; (b) J. Ruiz and A. F. Mesa, Chem. – Eur. J., 2012, 18, 4485–4488 CrossRef CAS PubMed; (c) A. Solovyev, E. Lacôte and D. P. Curran, Org. Lett., 2011, 13, 6042–6045 CrossRef CAS PubMed.
- P. K. Majhi, G. Schnakenburg, Z. Kelemen, L. Nyulaszi, D. P. Gates and R. Streubel, Angew. Chem., 2013, 125, 10264–10267 ( Angew. Chem., Int. Ed. , 2013 , 52 , 10080–10083 ) CrossRef PubMed.
- E. Aldeco-Perez, A. J. Rosenthal, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Science, 2009, 326, 556–559 CrossRef CAS PubMed.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561–562 CrossRef CAS PubMed.
- See ESI† for details.
- (a) R. S. Ghadwal, S. O. Reichmann, F. Engelhardt, D. M. Andrada and G. Frenking, Chem. Commun., 2013, 49, 9440–9442 RSC; (b) R. S. Ghadwal, R. Azhakar, K. Pröpper, B. Dittrich and M. John, Chem. Commun., 2013, 49, 5987–5989 RSC.
- (a) S. K. Liew, S. M. I. Al-Rafia, J. T. Goettel, P. A. Lummis, S. M. McDonald, L. J. Miedema, M. J. Ferguson, R. McDonald and E. Rivard, Inorg. Chem., 2012, 51, 5471–5480 CrossRef CAS PubMed; (b) F. Popp, J. B. Natscher, J. O. Daiss, C. Burschka and R. Tacke, Organometallics, 2007, 26, 6014–6028 CrossRef CAS; (c) A. C. T. Kuate, S. K. Mohapatra, C. G. Daniliuc, P. G. Jones and M. Tamm, J. Organomet. Chem., 2012, 696, 4281–4292 CrossRef PubMed.
- (a) H. Kaur, F. K. Zinn, E. D. Stevens and S. P. Nolan, Organometallics, 2004, 23, 1157–1160 CrossRef CAS; (b) A. Szadkowska, A. Makal, K. Wozniak, R. Kadyrov and K. Grela, Organometallics, 2009, 28, 2693–2700 CrossRef CAS; (c) J. Chun, I. G. Jung, H. J. Kim, M. Park, M. S. Lah and S. U. Son, Inorg. Chem., 2009, 48, 6353–6355 CrossRef CAS PubMed; (d) S. Díez-González, E. C. Escudero-Adán, J. Benet-Bucholz, E. D. Stevens, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 7595–7606 RSC.
- (a) D. Stalke, Chem. Soc. Rev., 1998, 27, 171–178 RSC; (b) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef CAS.
- SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2000.
- G. M. Sheldrick, SADABS, Universität Göttingen, Germany, 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. Müller, R. Herbst–Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, in Crystal Structure Refinement–A Crystallographer's Guide to SHELXL, IUCr Texts on Crystallography, ed. P. Müller, Oxford University Press, Oxford, UK, vol. 8, 2006 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of 2–6 and X-ray crystallographic information on 2, 4, 5 and 6 are provided. CCDC 982101 (2), 982103 (4), 982104 (5) and 982102 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt01681e |
| This journal is © The Royal Society of Chemistry 2014 |