
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLoop shaped dicarboxylate-bridged dimolybdenum(II) bisphosphine compounds – a rational synthesis†
Dominik
Höhne
,
Eberhardt
Herdtweck
,
Alexander
Pöthig
and
Fritz E.
Kühn
*
Chair of Inorganic Chemistry/Molecular Catalysis, Catalysis Research Center, Technische Universität München, Ernst-Otto-Fischer-Straße 1, D-85747 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de; Tel: +49 89 289 13096
First published on 19th August 2014
Abstract
The reaction of the tetrametallic molecular loop [(CH3CN)6Mo2(OOC-C4H6-COO)]2[BF4]4 (1) (1 equiv.) with 2 equiv. of bis(diphenylphosphino)amine (dppa), 1,2-bis(diphenylphosphino)ethane (dppe) and bis(diphenylphosphino)methane (dppm) in propionitrile leads to the formation of the still tetrametallic complexes [(CH3CH2CN)4(X)Mo2(OOC-C4H6-COO)]2[BF4]4 (X = dppa (2), dppe (3), dppm (4)), also displaying a loop structure. All three complexes are characterized by NMR spectroscopy (1H, 11B, 13C{1H}, 19F, 31P{1H}), IR spectroscopy, elemental analysis, TG-MS measurements and UV-vis spectroscopy, compounds 2 and 3 additionally by X-ray single crystal diffraction.
Introduction
A wide variety of multiply bonded homo- and heteronuclear complexes of the general type M2X4(LL)2 (M = Mo, W; LL = bridging bisphosphine ligand) is described in the literature.1–9 In the case of dimolybdenum derivatives, the well-known compound [Mo2(O2CCH3)4]10,11 often serves as a source of the Mo24+ moiety. The number of dinuclear metal complexes with a multiple metal–metal bond and bisphosphine substituents was additionally increased by introducing monocarboxylic functionalities as structural motifs, leading, amongst others, to compounds of general formula [M2(O2CR)2X2(LL)2],7,9,12,13 [M2(O2CR)2(LL)2][BF4]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14–17 or related axially substituted complexes.18,19
14–17 or related axially substituted complexes.18,19
Another possible method for synthesizing new Mo24+ containing systems is the reaction with dicarboxylates, which leads to numerous structural motifs, depending on the structure of the utilized linker. Previous experiments have shown that [Mo2(NCCH3)10][BF4]420 is an excellent precursor for the synthesis of larger coordination compounds, such as molecular dimers, loops, triangles and squares.21,22 In contrast to structurally similar compounds synthesized by Cotton et al.,23–28 who used partially protected starting materials in order to ensure controlled reaction conditions and obtain defined structures, the isolated products based on the precursor [Mo2(NCCH3)10][BF4]4 possess remaining weakly bound nitrile ligands. This might allow further substitutions.
In this work, novel bisphosphine substituted molecular loops are presented, which were synthesized from [(CH3CN)6Mo2(OOC-C4H6-COO)]2[BF4]4 (1)22 and bis(diphenylphosphino)amine or 1,2-bis(diphenylphosphino)ethane and bis(diphenylphosphino)methane, respectively. Both structural motifs are combined, namely dicarboxylate bridging of Mo24+ units and bisphosphine substitution.
Results and discussion
Syntheses
Compounds 2–4 were synthesized by treatment of 1 equiv. of precursor 1 with 2 equiv. of dppa, dppe or dppm, respectively, in propionitrile (see Scheme 1). The progress of all reactions was traced by an immediate color change of the reaction solution from pink to dark red when the respective bisphosphine ligand was added. The reaction of 1 with dppa afforded the pink complex 2 in a yield of 64%, whereas the pink compound 3 was isolated in a combined yield of 55%. Based on the molar ratio of the starting material 1, a yield of 58% was obtained for the pink product 4. The oxygen and moisture stability of complexes 2–4, which is shown by an unvarying pink color when exposed to air overnight, is worth mentioning. Decomposition of the complexes is indicated by a gradual color change to purple-brownish within approximately 2 days. | ||
| Scheme 1 Syntheses of compounds 2–4. | ||
Spectroscopic examinations
A sufficient solubility in CD3CN (∼27 g L−1 for 2, ∼29 g L−1 for 3, ∼9 g L−1 for 4) and the diamagnetic nature of the products 2–4 allow the characterization by NMR spectroscopy. The 1H NMR of 2 in CD3CN shows eight signal groups (Fig. S1†). The phenyl protons of the dppa ligands of 2 cause a broad multiplet in the range 7.71–7.52 ppm. A similar multiplet appears in the 1H NMR spectrum of free dppa in the range 7.40–7.31 ppm. A triplet representing the amine protons of the dppa substituents can be found at 6.86 ppm for 2 (triplet at 4.31 ppm for free dppa). The two signals of the cyclobutane rings of 1 with a ratio of 8:4 at 3.35 and 2.37 ppm22 are split into four multiplets due to the bisphosphine substitution, which prevents, in contrast to complex 1, the cyclobutane rings from changing its conformation (Fig. S2†). The four multiplets with a ratio of 4:4:2:2 belong to the same spin system as proven by COSY measurements (Fig. 1 and S3†). They can be found at 3.30, 2.84, 1.90 and 1.28 ppm. The latter two signals partially overlap with the signal of CD3CN at 1.94 ppm and the triplet of free propionitrile at 1.20 ppm, respectively, complicating the integration of the signals. The second signal of free propionitrile, which is released due to an exchange with CD3CN, arises at 2.36 ppm.
 | ||
| Fig. 1 Excerpt from the COSY spectrum of 2 in CD3CN. | ||
The presence of BF4 anions in 2 can be deduced from the singlet at −1.10 ppm in the 11B NMR and two singlets with a ratio of 1:4, which can be explained by the natural abundance of the boron isotopes (10B/11B = 19.9%/80.1%), at −151.42 and −151.47 ppm in the 19F NMR of 2. The 13C{1H} NMR spectrum of 2 shows a peak at 188.1 ppm, which can be assigned to the carboxylic C atoms. The aromatic C atoms of the phenyl rings of 2 are represented by multiple peaks in the range 134.0–130.1 ppm. Due to the coupling with phosphorus, an overlap of signals and different chemical environments of the phenyl rings, the signals cannot be correlated to the corresponding aromatic C atoms of 2. Three signals at 122.4, 11.3 and 10.8 ppm prove the presence of free propionitrile. Additional three peaks can be observed at 60.5, 32.8 and 17.0 ppm and are caused by the different C atoms of the cyclobutane rings of 2. The explicit correlation of proton and carbon signals of 2 is done with HSQC measurements (Fig. S4†). The coordinating nature of the dppa ligands of 2 can be visualized with the help of 31P{1H} NMR measurements and the comparison with the respective signal of free dppa. One singlet at 82.23 ppm occurs in the corresponding spectrum for 2 underlining that all P atoms are chemically equivalent (singlet at 43.20 ppm for free dppa).
A broad multiplet in the range 7.59–7.41 ppm occurs in the 1H NMR spectrum of 3 in CD3CN (Fig. S5†). It can be assigned to the aromatic protons of the dppe ligands. The different proton species of the dppe ligands and the cyclobutane rings in 3 (Fig. S6†) cause signals, which partially overlap. With the help of COSY measurements (Fig. S7 and S8†), two corresponding spin systems can be identified. The two different species of ethylene protons of the dppe bridge in 3 are responsible for two multiplets with a ratio of 4:4 at 3.53 and 3.07 ppm. These two species are generated due to the bisphosphine substitution of 1. The 1H NMR spectrum of free dppe shows a sharp signal at 7.32 ppm representing the phenyl protons and one signal at 2.06 ppm caused by the equivalent ethylene protons of dppe (ratio 40:8). The other spin system with four multiplets at 3.42, 3.07, 2.24 and 1.98 ppm having a ratio of 4:4:2:2 can be attributed to the four different types of cyclobutane protons originating again from the fixed conformation of the cyclobutane rings due to the bulkiness of the phenyl rings of the coordinating dppe ligands. Two additional signals in the 1H NMR of 3 at 2.36 and 1.20 ppm (ratio 2:3) represent free propionitrile. One singlet at −1.10 ppm in the 11B NMR and two singlets at −151.41 and −151.46 ppm in the 19F NMR prove the existence of BF4 anions in 3. The signals of the 13C{1H} NMR spectrum of 3 can be assigned with the help of HSQC measurements (Fig. S9†) and are comparable to those of complex 2. One additional signal at 22.5 ppm appears and originates from the equivalent ethylene carbon atoms of the dppe ligands. The signal at 20.34 ppm in the 31P{1H} NMR spectrum of 3 in CD3CN proves the coordinating nature of dppe (one signal at −13.82 ppm for free dppe).
The 1H NMR spectrum of 4 in CD3CN (Fig. S10†) exhibits similar signals and shifts for the aromatic protons of the bisphosphine ligands dppm and for the cyclobutane rings (Fig. S11†) as the spectra of complexes 2 and 3. The couplings of the different proton species of 4 are again proven by COSY measurements (Fig. S12 and S13†). The fact that the signal of the two methylene protons in free dppm (2.92 ppm in CD3CN) is split into two multiplets with half the intensity at 5.53 and 5.20 ppm in 4 has to be highlighted and can be explained by different chemical environments of the methylene protons generated due to the coordination of dppm to the Mo2 centers of 1. The quartet at 2.36 and the triplet at 1.20 ppm (ratio 2:3) represent free propionitrile. The latter compound is released when an excess of CD3CN is added to 4. The presence of BF4 anions is again proven by a singlet at −1.03 ppm in the 11B NMR and two singlets with a ratio of 1:4 at −151.16 and −151.21 ppm in the 19F NMR of 4. The 13C{1H} NMR spectrum of 4 shows comparable signals and shifts to those of compounds 2 and 3. The assignment of the respective signals is again supported by HSQC measurements (Fig. S14†). This is particularly necessary for the identification of the methylene carbon signal of the dppm substituents at 37.9 ppm. The latter peak is very weak due to the coupling with phosphorus. The coordinating nature of the dppm ligands of 4 can be visualized by 31P{1H} NMR spectroscopy in CD3CN. The singlet at −23.14 ppm in free dppm is low field shifted to 23.85 ppm in 4.
Table 1 emphasizes, for all compounds 2–4, the general trend to low field shifted peaks in the 1H and 31P{1H} NMR spectra when compared to the signals for the free bisphosphine ligands.
| Compound | 1H (δ [ppm]) | 31P (δ [ppm]) | |
|---|---|---|---|
| Cyclobutane ring (Int.) | Bisphosphine ligand (Int.) | ||
| 1 | 3.35, 2.37 (8:4)22 |
— | — |
| Free linker in 1 | 2.48, 1.94 (8:4)22 |
— | — |
| 2 | 3.30, 2.84, 1.90, 1.28 (4:4:2:2) |
7.71–7.52, 6.86 (40:2) |
82.23 |
| Free dppa | — | 7.40–7.31, 4.31 (40:2) |
43.20 |
| 3 | 3.42, 3.07, 2.24, 1.98 (4:4:2:2) |
7.59–7.41, 3.53, 3.07 (40:4:4) |
20.34 |
| Free dppe | — | 7.32, 2.06 (40:8) |
−13.82 |
| 4 | 3.58, 3.39, 2.21, 1.86 (4:4:2:2) |
7.65–7.32, 5.53, 5.20 (40:2:2) |
23.85 |
| Free dppm | — | 7.49–7.32, 2.92 (40:4) |
−23.14 |
Additional characterization of complexes 1–4 was done by IR spectroscopy. Typical shifts as well as the corresponding vibrations of compounds 1–4 and the free bisphosphine ligands are summarized in Table 2, respectively. The IR spectrum of complex 2 shows a ν(NH) band at 3224 cm−1 with low intensity. An almost identical signal is found in the IR spectrum of the free ligand dppa at 3225 cm−1 proving that the NH bond strength of dppa is not influenced by the state of coordination. Similar peaks with comparable shifts for compound 2 and free dppa are also found for ν(CHarom.) and ν(CCarom.) signals. The same applies to complexes 3 and 4 and the corresponding free bisphosphine ligands, illustrating again that the bonding conditions of the bisphosphines are almost identical, regardless of whether they coordinate or not. The comparison of the ν(COOasym.) and ν(COOsym.) bands of the starting material [(CH3CN)6Mo2(OOC-C4H6-COO)]2[BF4]4 (1) and complexes 2–4, which can be found in the range of 1520–1513 and 1382–1380 cm−1, respectively, reveals the structural similarity of all compounds. Bands with a comparable shift to these vibrations and the ν(BF) vibration of compounds 1–4 can be found in the literature for the dicarboxylate-bridged Mo2 complexes [(CH3CN)4Mo2(OOC-Fc-COO)]4[BF4]8, [(CH3CH2CN)4Mo2(OOC-C6F4-COO)]4[BF4]8 and [(CH3CN)6Mo2(m-bdc-F)]3[BF4]6.21 The presence of nitrile ligands in compounds 1–4 is further supported by ν(CN) bands at around 2280 cm−1. The acetonitrile containing educt 1 shows an additional signal at 2318 cm−1 with less intensity.
| Comp. | ν (NH) | ν (CHarom.) | ν (CH) | ν (CN) | ν (COO) | ν (CCarom.) | ν (BF) |
|---|---|---|---|---|---|---|---|
| 1 | — | — | 2942 | 2318, 2288 | 1520, 1382 | — | 1023 |
| 2 | 3224 | 3057 | 2950 | 2281 | 1513, 1380 | 1436 | 1057 |
| Free dppa | 3225 | 3050 | — | — | — | 1431 | — |
| 3 | — | 3055 | 2968, 2949 | 2278 | 1520, 1381 | 1434 | 1050 |
| Free dppe | — | 3067 | 2927 | — | — | 1432 | — |
| 4 | — | 3058 | 2959, 2905 | 2275 | 1519, 1381 | 1434 | 1057 |
| Free dppm | — | 3052 | 2897, 2859 | — | — | 1430 | — |
TG-MS measurements were carried out for compounds 2–4. The decomposition of 2 starts with the loss of propionitrile ligands in the temperature range 100–300 °C. Further decomposition of 2 leads to stepwise release of CO2, which is generated during the decomposition process of the dicarboxylate linker. The MS curve of CO2+ reveals peak maxima at 178, 280, 355 and 652 °C. A fragmentation of the bisphosphine ligands dppa can be observed in the temperature range 300–600 °C. It can be assigned to dissociated phenyl rings. The residual mass of ca. 26% can be predominantly explained by elemental molybdenum (Fig. S15†). The removal of the propionitrile ligands of 3 takes place in the temperature range 175–250 °C (Fig. S16†). Apparently, the propionitrile ligands in 3 are stronger bound than those in 2, since a higher temperature is needed to remove them. The decomposition of the dicarboxylate linker of 3 occurs stepwise, as is the case for 2. The MS curve of CO2+ displays three maxima at 220, 407 and 611 °C. The comparison with 2, where the decomposition of the dicarboxylate linker starts at lower temperatures, shows again that 3 is more temperature-stable. Phenyl rings, which are an indication of the decomposition of the bisphosphine ligands dppe, can be detected in the temperature range 300–600 °C. This observation is in agreement with the results obtained for compound 2. Complex 4 loses its propionitrile ligands between 100 and 300 °C (Fig. 2). Hence, the temperature necessary to start the decomposition is the same as that for complex 2. The dicarboxylate linker in 4 again decomposes in steps. This is proven by four maxima in the corresponding MS curve at 152, 247, 369 and 605 °C. The temperature range where phenyl groups, generated during the decomposition of the dppm ligands, can be detected is 300–600 °C. This range is the same for all three complexes 2–4. The starting material 1, however, loses the bulk of its nitrile ligands gradually between 75 and 500 °C. Accordingly, the temperature range in this case is significantly larger than that for complexes 2–4. The acetonitrile ligands in 1 are apparently more weakly bound than the propionitrile ligands in 2–4, since less temperature is needed to remove them. For complex 1 four peak maxima in the MS curve of CO2+ can be found at 119, 233, 398 and 565 °C. The onset of detection of CO2+ is at about 100 °C. It is therefore very similar to the decomposition onsets observed for compounds 2 and 4 (∼100 °C and ∼125 °C, respectively), but considerably lower compared to complex 3 (∼175 °C).
 | ||
| Fig. 2 TGA (blue) and MS curves (green for propionitrile, red for CO2 and black for phenyl group) of compound 4. | ||
The UV-vis spectra of the pink compounds 2–4 measured in CH3CN feature absorption maxima at 534, 531 and 538 nm, respectively. This is in agreement with the pink educt 1 having an absorption maximum at 533 nm.22 Hence, the reaction of 1 with the bisphosphines dppa, dppe and dppm causes a color change of the initial reaction solution from pink to dark red. However, the products 2–4 obtained by drying of their light red crystals or precipitates under vacuum are pink, indicating that the dark color is a consequence of the formation of byproducts, which is also reflected in the product yields.
Cyclic voltammetry measurements were additionally carried out for compounds 2–4. A reversible oxidation/reduction process can be excluded for all three complexes (see ESI for measurement parameters and Fig. S17–S22† for cyclic voltammograms of 2–4).
Crystallographic examinations
Crystals suitable for single-crystal X-ray diffraction could be obtained for complexes 2 and 3 by layering a saturated propionitrile solution with n-pentane. Several attempts to grow crystals of compound 4 failed. Changing the solvent from propionitrile to acetonitrile did not lead to crystals of 4 either. The crystallographic data of compounds 2 and 3 are summarized in Table 3.| 2 | 3 | |
|---|---|---|
| a R 1 = ∑(||Fo| − |Fc||)/∑|Fo|; wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2; GOF = {∑[w(Fo2 − Fc2)2]/(n–p)}1/2. | ||
| Formula | C84H94B4F16Mo4N10O8P4 | C94H110B4F16Mo4N10O8P4 |
| Formula weight | 2226.57 | 2362.81 |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21/c (no. 14) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2) (no. 2) |
| a (Å) | 18.183(2) | 12.4049(5) |
| b (Å) | 15.535(2) | 18.0187(7) |
| c (Å) | 18.873(2) | 24.6268(10) |
| α (°) | 90 | 74.092(2) |
| β (°) | 116.427(3) | 88.172(2) |
| γ (°) | 90 | 81.785(2) |
| V (Å3) | 4774.0(10) | 5239.3(4) |
| Z | 2 | 2 |
| T (K) | 123 | 123 |
| d calcd (g cm−3) | 1.549 | 1.498 |
| μ (mm−1) | 0.669 | 0.614 |
| No. of independent reflections | 8821 | 19171 |
| No. of observed reflections (I > 2σ(I)) | 8337 | 15070 |
| No. of data/restraints/parameters | 8821/0/590 | 19171/189/1374 |
| R 1/wR2 (I > 2σ(I))a | 0.0361/0.1008 | 0.0533/0.1247 |
| R 1/wR2 (all data)a | 0.0389/0.1036 | 0.0717/0.1408 |
| GOF (on F2)a | 1.034 | 1.066 |
Fig. 3 shows the molecular structure of 2. The two Mo24+ units of 2 are orientated parallel to each other and the Mo centers exhibit an octahedral coordination sphere, as was found earlier for 1 (weakly bound axial nitrile ligands are neglected in Fig. 3).22 Compared to the starting material 1 with a Mo–Mo bond length of 2.1529(2) Å,22 the metal–metal distance in 2 (2.1477(6) Å) is slightly shorter. This can be explained by the electron donating nature of the bisphosphine ligands dppa in 2. The literature known complex [Mo2(O2CCH3)2(dppa)2(CH3CN)2][BF4]2 exhibits two dppa ligands coordinating to one Mo24+ center and displays consequently a short Mo–Mo bond length (2.133(1) Å).15 The Mo–O bond length in 2 is about 2.10 Å (2.086(2)–2.112(2) Å). This is slightly larger than that in the starting material [(CH3CN)6Mo2(OOC-C4H6-COO)]2[BF4]4 (1) (2.079(1)–2.093(1) Å)22 and similar to that of [Mo2(O2CCH3)2(dppa)2(CH3CN)2][BF4]2 (2.082(5)–2.105(5) Å).15 The Mo–Neq bond length in compound 1 (2.150(2)–2.159(2) Å)22 is comparable to that in the propionitrile substituted complex 2 (2.143(3)–2.146(3) Å), indicating similar coordination properties of both types of nitrile ligands. The Mo–P bond length of ∼2.55 Å (2.5530(9)–2.553(1) Å) in 2 is insignificantly smaller than that in the dppa substituted compound [Mo2(O2CCH3)2(dppa)2(CH3CN)2][BF4]2 (2.573(2)–2.580(2) Å).15 The O2–Mo1–O4, O1–Mo2–O3, N1–Mo1–P1 and N2–Mo2–P2 angles in 2 are 87.0(1), 87.1(1), 84.9(1) and 88.34(9)°, respectively. Thus, the assumption of an octahedral coordination sphere of the Mo centers is justified.
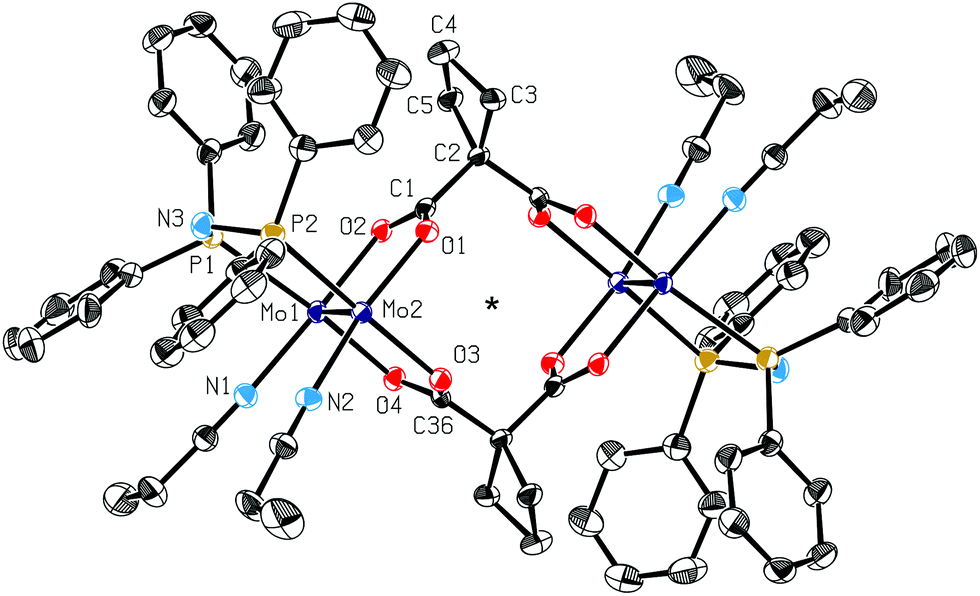 | ||
| Fig. 3 ORTEP style plot of the cationic part of compound 2 in the solid state. Thermal ellipsoids are drawn at a 50% probability level. The centrosymmetric molecule of compound (2) is generated by a crystallographic inversion centre (symmetry code _a: −x, −y, −z), indicated by a star. H atoms are omitted for clarity. Selected bond distances (Å), angles (°): Mo1–Mo2 2.1477(6), Mo1–O2 2.086(2), Mo1–O4 2.111(2), Mo2–O1 2.088(2), Mo2–O3 2.112(2), Mo1–N1 2.143(3), Mo2–N2 2.146(3), Mo1–P1 2.553(1), Mo2–P2 2.5530(9), C1–C2 1.520(4), C2–C36_a 1.518(4); O2–Mo1–O4 87.0(1), O1–Mo2–O3 87.1(1), N1–Mo1–P1 84.9(1), N2–Mo2–P2 88.34(9), P1–N3–P2 117.4(2), C1–C2–C36_a 105.2(3). | ||
The molecular structure of 3 is shown in Fig. 4. Besides the parallel alignment of the Mo24+ units and the octahedral coordination sphere of all Mo centers, the Mo–Mo bond length in 3 (2.1453(5)–2.1470(5) Å) is almost the same as that in compound 2. However, it is significantly larger than that observed for the literature known complex [Mo2(O2CCH3)2(dppe)2][BF4]2 (2.0928(7) Å),14 which possesses similar structural motifs, such as carboxylate and dppe substitution of a Mo24+ unit and BF4 counterions. The same applies to the Mo–O bond length (2.082(3)–2.124(3) Å) and the Mo–Neq bond length (2.135(4)–2.146(4) Å) for compound 3. Compared to complex 2, the Mo–P bond length in 3 is larger (2.560(1)–2.604(1) Å), which implies that dppe is a weaker electron donor than dppa. Typical angles are 86.2(1), 86.4(1), 87.4(1) and 86.0(1)° for O1–Mo1–O3, O2–Mo2–O4, N1–Mo1–P1 and N2–Mo2–P2, respectively. These values are in good agreement with an octahedral coordination sphere of the Mo centers.
 | ||
| Fig. 4 ORTEP style plot of the cationic part of compound 3 in the solid state. Thermal ellipsoids are drawn at a 30% probability level. H atoms are omitted for clarity. Selected bond distances (Å), angles (°): Mo1–Mo2 2.1453(5), Mo1–O1 2.082(3), Mo1–O3 2.122(3), Mo2–O2 2.092(3), Mo2–O4 2.113(3), Mo1–N1 2.135(4), Mo2–N2 2.138(4), Mo1–P1 2.560(1), Mo2–P2 2.604(1), C1–C2 1.527(7), C2–C6 1.517(6), Mo3–Mo4 2.1470(5), Mo3–O6 2.124(3), Mo3–O8 2.085(4), Mo4–O5 2.122(3), Mo4–O7 2.095(3), Mo3–N4 2.146(4), Mo4–N3 2.141(4), Mo3–P4 2.564(1), Mo4–P3 2.600(1), C8–C12 1.520(8), C7–C8 1.514(7); O1–Mo1–O3 86.2(1), O2–Mo2–O4 86.4(1), N1–Mo1–P1 87.4(1), N2–Mo2–P2 86.0(1), C1–C2–C6 105.0(4), O6–Mo3–O8 86.5(1), O5–Mo4–O7 86.2(1), N4–Mo3–P4 86.8(1), N3–Mo4–P3 88.3(1), C7–C8–C12 106.1(4). | ||
Conclusion
The reaction of [(CH3CN)6Mo2(OOC-C4H6-COO)]2[BF4]4 (1) with two equivalents of different bisphosphines results in the isolation of three new molecular loop structures 2–4, displaying one bisphosphine ligand per dimolybdenum moiety. The loop structure is not destroyed, as the bridging carboxylates are not forced to adopt trans position as is often found in the case of quadruply bonded dimolybdenum species.12,13,15,29,30 The synthesis of compounds 2–4 demonstrates that the acetonitrile ligands of 1 are still reactive and can be utilized for further substitution reactions. Furthermore, the reaction leads to a well defined 1:1 coordination of one bisphosphine and one dimolybdenum unit. The structures of the products therefore show that it might very well be possible to build up larger structural motifs by combining molecules 2–4, e.g. via di-bisphosphines or via other linkers replacing the still remaining equatorial nitrile ligands. The application of compounds 2–4 as potential building units for defined metal–organic frameworks appears therefore possible. Whether this also applies to other previously published structures, such as molecular triangles and squares, is currently under investigation.
Experimental section
General information
All reactions and sample preparations were carried out using standard Schlenk techniques and argon as an inert gas. The solvents were dried according to conventional procedures and stored over molecular sieves (3 Å, 4 Å). The ligands bis(diphenylphosphino)amine (98%), 1,2-bis(diphenylphosphino)ethane (97%) and bis(diphenylphosphino)methane (99%) were purchased from ABCR and used as received. The precursor [(CH3CN)6Mo2(OOC-C4H6-COO)]2[BF4]4 (1) was synthesized according to literature procedures.22 NMR measurements were recorded on an AVANCE DPX 400 or AVANCE DRX 400 spectrometer manufactured by Bruker. The chemical shifts were given in ppm and referenced to the solvent CD3CN as an internal standard for 1H and 13C{1H} measurements (1H: 1.94 ppm, 13C: 118.26 and 1.32 ppm). IR spectra were obtained using a Varian FTIR-670 spectrometer. The shifts of the signals were given in cm−1. UV-vis spectra were recorded on a Shimadzu UV-160 spectrophotometer using a quartz cell with a path length of 1 cm and acetonitrile as a solvent. The microanalytical laboratory of TU München was responsible for elemental analyses. Thermogravimetric analyses coupled with mass spectrometry for fragment detection (TGA-MS) were carried out using a Netzsch-STA 409PC/PG machine. Around 2–5 mg of the samples were heated from 30 to 1000 °C with a heating rate of 10 °C min−1.Synthesis of [(CH3CH2CN)4(dppa)Mo2(OOC-C4H6-COO)]2[BF4]4 (2)
A propionitrile solution (5 mL) of 2 equiv. of bis(diphenylphosphino)amine (30.7 mg, 0.0796 mmol) is added to a propionitrile solution (8 mL) of 1 equiv. of 1 (60.0 mg, 0.0398 mmol) within 10 minutes, resulting in a color change from pink to red. After stirring for 20 h at room temperature, the reaction solution is layered with 7 mL of n-pentane leading to red crystals and a pink precipitate within 2 days. The supernatant is filtered off and the residue is dried under vacuum, leading to 56.8 mg of 2 (0.0255 mmol, 64% yield) as a pink solid. 1H NMR (400 MHz, CD3CN): δ = 7.71–7.52 (m, aromat. H, 40H), 6.86 (t, NH, 2H), 3.30 (m, Ha(/b), 4H), 2.84 (m, Hb(/a), 4H), 2.36 (q, CH2, 12H), 1.90 (m, Hc(/d), ca. 2H), 1.28 (m, Hd(/c), ca. 2H), 1.20 (t, CH3, ca. 18 H). 11B NMR (128 MHz, CD3CN): δ = −1.10 (s, BF4). 13C{1H} NMR (101 MHz, CD3CN): δ = 188.1 (COO), 134.0–130.1 (Caromat.), 122.4 (CN), 60.5 (Cquat.Cyclobut.), 32.8 (C2Cyclobut.), 17.0 (C3Cyclobut.), 11.3 (CH2), 10.8 (CH3). 19F NMR (377 MHz, CD3CN): δ = −151.42 (s, BF4), −151.47 (s, BF4). 31P{1H} NMR (162 MHz, CD3CN): δ = 82.23 (s, dppa). UV-vis (CH3CN): λmax (nm) 534. Anal. Calcd for C84H94B4F16Mo4N10O8P4 (= 2): C, 45.31; H, 4.26; N, 6.29. Found: C, 44.89; H, 4.27; N, 6.15. Selected IR (cm−1): 3224 (w), 3057 (w), 2950 (w), 2281 (w), 1513 (m), 1436 (m), 1380 (m), 1057 (s).Synthesis of [(CH3CH2CN)4(dppe)Mo2(OOC-C4H6-COO)]2[BF4]4 (3)
A propionitrile solution (5 mL) of 2 equiv. of 1,2-bis(diphenylphosphino)ethane (31.7 mg, 0.0796 mmol) is added to a propionitrile solution (8 mL) of 1 equiv. of 1 (60.0 mg, 0.0398 mmol) within 10 minutes, whereupon the color of the reaction solution immediately changes from pink to dark red. After stirring for 20 h at room temperature, the reaction solution is layered with 7 mL of n-pentane leading to red crystals within one week. The solution is filtered off, concentrated and layered again with 4 mL of n-pentane, which affords 20.0 mg of compound 3 as a pink powder. Drying of the crystals under vacuum results in the isolation of 28.9 mg of 3 in the form of a pink powder (0.0217 mmol, 55% combined yield). 1H NMR (400 MHz, CD3CN): δ = 7.59–7.41 (m, aromat. H, 40H), 3.53 (m, He(/f), 4H), 3.42 (m, Ha(/b), 4H), 3.07 (m, Hf(/e), Hb(/a), 8H (each with 4H)), 2.36 (q, CH2, 8H), 2.24 (m, Hc(/d), 2H), 1.98 (m, Hd(/c), 2H), 1.20 (t, CH3, 12H). 11B NMR (128 MHz, CD3CN): δ = −1.10 (s, BF4). 13C{1H} NMR (101 MHz, CD3CN): δ = 188.0 (COO), 134.4–130.0 (Caromat.), 122.4 (CN), 60.3 (Cquat.Cyclobut.), 32.9 (C2Cyclobut.), 22.5 (CH2dppe), 17.4 (C3Cyclobut.), 11.3 (CH2), 10.8 (CH3). 19F NMR (377 MHz, CD3CN): δ = −151.41 (s, BF4), −151.46 (s, BF4). 31P{1H} NMR (162 MHz, CD3CN): δ = 20.34 (s, dppe). UV-vis (CH3CN): λmax (nm) 531. Anal. Calcd for C79H85B4F16Mo4N5O8P4 = [(CH3CH2CN)2.5(dppe)Mo2(OOC-C4H6-COO)]2[BF4]4 (= 3 − 3EtCN): C, 45.45; H, 4.10; N, 3.35. Found: C, 45.29; H, 4.10; N, 3.23. Selected IR (cm−1): 3055 (w), 2968 (w), 2949 (w), 2278 (w), 1520 (m), 1434 (m), 1381 (m), 1050 (s).Synthesis of [(CH3CH2CN)4(dppm)Mo2(OOC-C4H6-COO)]2[BF4]4 (4)
A propionitrile solution (5 mL) of 2 equiv. of bis(diphenylphosphino)methane (30.6 mg, 0.0796 mmol) is added to a propionitrile solution (8 mL) of 1 equiv. of 1 (60.0 mg, 0.0398 mmol) within 10 minutes, leading to a color change from pink to dark red. After stirring for 20 h at room temperature, a little amount of a pink solid precipitates. Addition of 7 mL of n-pentane to the reaction solution causes a further precipitation of a pink solid, which is isolated by filtration and dried under vacuum, leading to 51.4 mg of 4 (0.0231 mmol, 58% yield) as a pink powder. 1H NMR (400 MHz, CD3CN): δ = 7.65–7.32 (m, aromat. H, 40H), 5.53 (m, He(/f), 2H), 5.20 (m, Hf(/e), 2H), 3.58 (m, Ha(/b), 4H), 3.39 (m, Hb(/a), 4H), 2.36 (q, CH2, 8H), 2.21 (m, Hc(/d), 2H), 1.86 (m, Hd(/c), 2H), 1.20 (t, CH3, 12H). 11B NMR (128 MHz, CD3CN): δ = −1.03 (s, BF4). 13C{1H} NMR (101 MHz, CD3CN): δ = 188.1 (COO), 134.8–130.0 (Caromat.), 122.4 (CN), 60.8 (Cquat.Cyclobut.), 37.9 (CH2dppm), 33.2 (C2Cyclobut.), 17.5 (C3Cyclobut.), 11.3 (CH2), 10.8 (CH3). 19F NMR (377 MHz, CD3CN): δ = −151.16 (s, BF4), −151.21 (s, BF4). 31P{1H} NMR (162 MHz, CD3CN): δ = 23.85 (s, dppm). UV-vis (CH3CN): λmax (nm) 538. Anal. Calcd for C80H86B4F16Mo4N6O8P4 = [(CH3CH2CN)3(dppm)Mo2(OOC-C4H6-COO)]2[BF4]4 (= 4 − 2EtCN): C, 45.44; H, 4.10; N, 3.97. Found: C, 45.14; H, 4.05; N, 3.85. Selected IR (cm−1): 3058 (w), 2959 (w), 2905 (w), 2275 (w), 1519 (m), 1434 (m), 1381 (m), 1057 (s).Single-crystal structure determinations
Data were collected on an X-ray single crystal diffractometer equipped with a CCD detector (Bruker APEX II, κ-CCD), a rotating anode (Bruker AXS, FR591) with MoKα radiation (λ = 0.71073 Å), and a graphite monochromator using the SMART software package.31 The measurements were performed on single crystals coated with perfluorinated ether. The crystals were fixed on the top of a cactus prickle (Opuntia ficus-indica) and transferred to the diffractometer. The crystals were frozen under a stream of cold nitrogen. A matrix scan was used to determine the initial lattice parameters. Reflections were merged and corrected for Lorentz and polarization effects, scan speed, and background using SAINT.32 Absorption corrections, including odd and even ordered spherical harmonics, were performed using SADABS.32 Space group assignments were based upon systematic absences, E statistics, and successful refinement of the structures. Structures were solved by direct methods with the aid of successive difference Fourier maps, and were refined against all data using WinGX33 based on SIR-9234 or SIR-9735 in conjunction with SHELXL-97.36 Unless otherwise noted, methyl hydrogen atoms were refined as part of rigid rotating groups, with a C–H distance of 0.98 Å and Uiso(H) = 1.5Ueq(C). Other H atoms were placed in calculated positions and refined using a riding model, with aromatic C–H distances of 0.95 Å and with methylene C–H distances of 0.99 Å, and Uiso(H) = 1.2Ueq(C). For compound 2, the N–H distance was fixed to 0.88 Å and Uiso(H) = 1.2Ueq(N). Unless mentioned otherwise, non-hydrogen atoms were refined with anisotropic displacement parameters. Full-matrix least-squares refinements were carried out by minimizing ∑w(Fo2 − Fc2)2 with the SHELXL-9736 weighting scheme. Neutral atom scattering factors for all atoms and anomalous dispersion corrections for the non-hydrogen atoms were taken from the International Tables for Crystallography.37 Images of the crystal structures were generated using PLATON.38 For detailed information see ESI.† CCDC 989256 (2) and CCDC 989257 (3) contain the supplementary crystallographic data for these compounds.Acknowledgements
D. H. thanks the TUM graduate school for financial support, M. Anthofer for helpful discussions concerning the interpretation of NMR data and F. Kirchberger for valuable contributions regarding the experimental work.References
- E. H. Abbott, K. S. Bose, F. A. Cotton, W. T. Hall and J. C. Sekutowski, Inorg. Chem., 1978, 17, 3240–3245 CrossRef CAS.
- F. L. Campbell, F. A. Cotton and G. L. Powell, Inorg. Chem., 1984, 23, 4222–4226 CrossRef CAS.
- F. A. Cotton, K. R. Dunbar, B. Hong, C. A. James, J. H. Matonic and J. L. C. Thomas, Inorg. Chem., 1993, 32, 5183–5187 CrossRef CAS.
- F. A. Cotton, K. R. Dunbar and R. Poli, Inorg. Chem., 1986, 25, 3700–3703 CrossRef CAS.
- F. A. Cotton, L. R. Falvello, W. S. Harwood, G. L. Powell and R. A. Walton, Inorg. Chem., 1986, 25, 3949–3953 CrossRef CAS.
- F. A. Cotton and C. A. James, Inorg. Chem., 1992, 31, 5298–5307 CrossRef CAS.
- J. L. Eglin, L. T. Smith, R. J. Staples, E. J. Valente and J. D. Zubkowski, J. Organomet. Chem., 2000, 596, 136–143 CrossRef CAS.
- H.-F. Lang, P. E. Fanwick and R. A. Walton, Inorg. Chim. Acta, 2001, 322, 17–22 CrossRef CAS.
- Y.-Y. Wu, J.-D. Chen, L.-S. Liou and J.-C. Wang, Inorg. Chim. Acta, 1997, 258, 193–199 CrossRef CAS.
- A. B. Brignole and F. A. Cotton, Inorg. Synth., 1972, 13, 87–89 CrossRef PubMed.
- D. Lawton and R. Mason, J. Am. Chem. Soc., 1965, 87, 921–922 CrossRef CAS.
- F. A. Cotton, F. E. Kühn and A. Yokochi, Inorg. Chim. Acta, 1996, 252, 251–256 CrossRef CAS.
- D. I. Arnold, F. A. Cotton and F. E. Kühn, Inorg. Chem., 1996, 35, 4733–4737 CrossRef CAS.
- F. A. Cotton, J. L. Eglin and K. J. Wiesinger, Inorg. Chim. Acta, 1992, 195, 11–23 CrossRef CAS.
- F. A. Cotton and F. E. Kühn, Inorg. Chim. Acta, 1996, 252, 257–264 CrossRef CAS.
- L. J. Farrugia, A. McVitie and R. D. Peacock, Inorg. Chem., 1988, 27, 1257–1260 CrossRef CAS.
- T. Tanase, T. Igoshi and Y. Yamamoto, Inorg. Chim. Acta, 1997, 256, 61–67 CrossRef CAS.
- W.-M. Xue, F. E. Kühn, G. Zhang, E. Herdtweck and G. Raudaschl-Sieber, J. Chem. Soc., Dalton Trans., 1999, 4103–4110 RSC.
- W.-M. Xue, F. E. Kühn, G. Zhang and E. Herdtweck, J. Organomet. Chem., 2000, 596, 177–182 CrossRef CAS.
- F. A. Cotton and K. J. Wiesinger, Inorg. Chem., 1991, 30, 871–873 CrossRef CAS.
- X.-M. Cai, D. Höhne, M. Köberl, M. Cokoja, A. Pöthig, E. Herdtweck, S. Haslinger, W. A. Herrmann and F. E. Kühn, Organometallics, 2013, 32, 6004–6011 CrossRef CAS.
- M. Köberl, M. Cokoja, B. Bechlars, E. Herdtweck and F. E. Kühn, Dalton Trans., 2011, 40, 11490–11496 RSC.
- F. A. Cotton, J. P. Donahue, C. Lin and C. A. Murillo, Inorg. Chem., 2001, 40, 1234–1244 CrossRef CAS PubMed.
- F. A. Cotton, C. Lin and C. A. Murillo, Inorg. Chem., 2001, 40, 472–477 CrossRef CAS.
- F. A. Cotton, C. Lin and C. A. Murillo, Inorg. Chem., 2001, 40, 575–577 CrossRef CAS.
- M. H. Chisholm, N. J. Patmore, C. R. Reed and N. Singh, Inorg. Chem., 2010, 49, 7116–7122 CrossRef CAS PubMed.
- F. A. Cotton, L. M. Daniels, C. Lin and C. A. Murillo, J. Am. Chem. Soc., 1999, 121, 4538–4539 CrossRef CAS.
- F. A. Cotton, C. Lin and C. A. Murillo, Inorg. Chem., 2001, 40, 478–484 CrossRef CAS.
- F. A. Cotton, L. M. Daniels, S. C. Haefner and F. E. Kühn, Inorg. Chim. Acta, 1999, 287, 159–166 CrossRef CAS.
- F. A. Cotton and F. E. Kühn, J. Am. Chem. Soc., 1996, 118, 5826–5827 CrossRef CAS.
- APEX suite of crystallographic software, APEX 2, version 2008.4, Bruker AXS Inc., Madison, WI, 2008 Search PubMed.
- SAINT, version 7.56a, SADABS, version 2008.1, Bruker AXS Inc., Madison, WI, 2008 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435–436 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, M. C. Burla, G. Polidori, M. Camalli and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, Göttingen, Germany, 1998 Search PubMed.
- A. J. C. Wilson, in International Tables for Crystallography, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1992 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and the assignment of different proton species of compounds 2–4, TG-MS spectra of compounds 2 and 3, single-crystal XRD details of compounds 2 and 3 and parameters of CV measurements of compounds 2–4. CCDC 989256 and 989257. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt01560f |
| This journal is © The Royal Society of Chemistry 2014 |