
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Excited state evolution towards ligand loss and ligand chelation at group 6 metal carbonyl centres†
Jennifer C.
Manton
a,
Saeed
Amirjalayer
b,
Anthony C.
Coleman
a,
Suzanne
McMahon
a,
Emma C.
Harvey
a,
Gregory M.
Greetham
c,
Ian P.
Clark
c,
Wybren Jan
Buma
b,
Sander
Woutersen
b,
Mary T.
Pryce
a and
Conor
Long
*a
aSchool of Chemical Sciences, Dublin City University, Dublin 9, Ireland. E-mail: Conor.long@dcu.ie
bUniversity of Amsterdam, Van't Hoff Institute for Molecular Sciences, Science Park 904, 1098 XH Amsterdam, P.O. Box 94157, 1090 GD Amsterdam, The Netherlands
cCentral Laser Facility, Science & Technology Facilities Council, Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, OX11 0QX, UK
First published on 28th July 2014
Abstract
The photochemistry and photophysics of three model “half-sandwich” complexes (η6-benzophenone)Cr(CO)3, (η6-styrene)Cr(CO)3, and (η6-allylbenzene)Cr(CO)3 were investigated using pico-second time-resolved infrared spectroscopy and time-dependent density functional theory methods. The (η6-benzophenone)Cr(CO)3 complex was studied using two excitation wavelengths (470 and 320 nm) while the remaining complexes were irradiated using 400 nm light. Two independent excited states were detected spectroscopically for each complex, one an unreactive excited state of metal-to-arene charge-transfer character and the other with metal-to-carbonyl charge transfer character. This second excited state leads to an arrested release of CO on the pico-second time-scale. Low-energy excitation (470 nm) of (η6-benzophenone)Cr(CO)3 populated only the unreactive excited state which simply relaxes to the parent complex. Higher energy irradiation (320 nm) induced CO-loss. Irradiation of (η6-styrene)Cr(CO)3, or (η6-allylbenzene)Cr(CO)3 at 400 nm provided evidence for the simultaneous population of both the reactive and unreactive excited states. The efficiency at which the unreactive excited state is populated depends on the degree of conjugation of the substituent with the arene π-system and this affects the efficiency of the CO-loss process. The quantum yield of CO-loss is 0.50 for (η6-allylbenzene)Cr(CO)3 and 0.43 for (η6-styrene)Cr(CO)3. These studies provide evidence for the existence of two photophysical routes to CO loss, a minor ultrafast route and an arrested mechanism involving the intermediate population of a reactive excited state. This reactive excited state either relaxes to reform the parent species or eject CO. Thus the quantum yield of the CO-loss is strongly dependent on the excitation wavelength. Time-dependent density functional theory calculations confirm that the state responsible for ultrafast CO-loss has significant metal-centred character while the reactive state responsible for the arrested CO-loss has significant metal-to-carbonyl charge-transfer character. The CO-loss product (η6-allylbenzene)Cr(CO)2 formed following irradiation of (η6-allylbenzene)Cr(CO)3 reacts further with the pendent alkenyl group to form the chelate product (η6,η2-allylbenzene)Cr(CO)2.
Introduction
Our understanding of the photophysics of CO-loss from metal carbonyl complexes has traditionally been informed by the behaviour of homoleptic systems such as Cr(CO)6.1–6 The general description of this process involves the population of excited states which, in a static description would not react to expel CO. The efficient expulsion of CO is explained by considering coupling with non-totally symmetric vibrational modes which reduce the symmetry of the excited state. This results in the excited state becoming repulsive to one CO ligand. As a consequence the resulting energy profile is accelerating with respect to CO loss and in the case of homoleptic metal carbonyls does not exhibit an energy minimum along the CO-loss reaction coordinate. Thus for Cr(CO)6, CO loss occurs on the ultrafast timescale (<100 fs) from an excited state or states which cannot be probed by spectroscopic methods.7,8 This has led to a general, albeit erroneous conclusion that photoinduced CO loss from all metal carbonyl complexes is ultrafast.Recently the development of fast time-resolved spectroscopic methods has provided greater insights into the photophysical processes underpinning CO-loss from metal carbonyl systems.9 It is clear from these studies that not all photoinduced CO-losses from metal carbonyl complexes are ultrafast.10–15 In particular half sandwich complexes of the type (η6-arene)M(CO)3 (M = Cr, Mo, or W) and some Fischer carbene complexes of Cr(CO)5, have been shown to undergo so-called “arrested CO-loss” where CO is ejected on the ps rather than the fs timescale. The excited state or states responsible for CO-loss can be detected by spectroscopic methods.
Fast (ps) time-resolved infrared spectroscopy (ps-TRIR) techniques are now available which can probe early-stage processes in the photoinduced CO-loss reaction by monitoring changes in the mid-infrared region of the spectrum where metal carbonyl stretching modes are observed.9 The high extinctions of the metal carbonyl absorptions make this class of complex particularly suited to such studies. In addition the availability of time-dependent density functional theory (TDDFT) provides a method of describing excited state dynamics at a reasonable computational cost which can assist in explaining the results obtained in ps-TRIR experiments.16–18 For the systems in this investigation these calculations provide a plausible explanation for the arrested CO-loss observed in the ps-TRIR experiments and can also help in the design of systems with specific photophysical and photochemical properties.
The use of derivatives of (η6-arene)Cr(CO)3 complexes where the arene ligand contains pendent coordination sites such as pyridyl, alkenyl or sulphides have been proposed as ultra-fast photoswitches.19–23 For instance when the arene ligand contains a substituent with both pendent alkenyl and pyridyl substituents the photoejection of a CO-ligand presents the possibility of two chelate products, one involving coordination of the pyridyl nitrogen to the metal dicarbonyl centre and the other coordination of the alkenyl substituent. The ability to discriminate between these two chelate products is fundamental to their use as switches.19 However it is essential to firstly investigate the effects that substituents on the arene ligand will have on the fundamental photophysics of these complexes and this will rely on a better understanding of the excited states involved and the excited state dynamics leading to photochemical change.
In this paper we describe the photophysical and photochemical behaviour of three model “half-sandwich” complexes, namely (η6-benzophenone)Cr(CO)3, (η6-styrene)Cr(CO)3, and (η6-allylbenzene)Cr(CO)3. These systems were chosen because they allow us to probe the effect on the photophysics and photochemistry of substituents on the arene ligand. The presence of a substituent on the arene ligand has two main consequences. Firstly the symmetry of the complex is reduced compared to (η6-benzene)Cr(CO)3, and secondly they present additional intramolecular reaction opportunities for trapping processes such as ring-slip or chelate formation. Processes such as these could have important applications in optically driven memory devices where potentially the coordination mode of the arene ligand could be monitored spectroscopically to “read” the state of the memory store while the coordination mode could be altered by UV/Vis. excitation.
Experimental
The ULTRA laser system used in ps-TRIR studies at the Rutherford Appleton Laboratory is a cryogenically cooled Ti:Sapphire laser amplifier providing 50 fs duration pulses at a 10 kHz repetition rate.9 The 0.8 mJ, 800 nm output is divided to generate 400 nm pump and mid-IR probe beams through second harmonic generation and difference frequency generation (DFG) of signal and idler generated in an optical parametric amplifier, respectively. The pump beam was attenuated to 0.5 μJ at the sample and focused to overlap with the probe beam in the sample. The pump and probe spot sizes in the sample were 100 and 50 μm, respectively. After the sample, the IR probe beam was dispersed onto two linear 128 element MCT detector arrays acquiring spectra at 10 kHz. The signals from these two detector arrays were then averaged. By modulating the pump laser output at 5 kHz, difference spectra could be generated.24 A portion of the IR probe taken before the sample was dispersed on to a second 64 element MCT detector array, which with interpolation was used to generate reference spectra used to remove energy and spectral instabilities of the laser. Samples were contained in flow-through cells with CaF2 windows with a 500 μm Teflon spacer and rastered to avoid photodegradation of the sample solution.The transient IR apparatus in Amsterdam, used UV pump and mid-IR probe pulses generated by a Ti:sapphire laser with a repetition rate of 1 kHz. The UV pump pulse (470, 400 or 320 nm) was generated by SHG. IR probe pulses were generated by DFG of the signal and idler from a BBO-based OPA in AgGaS2. The delay positions were scanned by mechanically adjusting the beam-path of the UV pump using a translation stage. The temporal resolution of 200 fs has been obtained from the FWHM of the pump-probe cross-correlate function. Measurements of the (η6-styrene)Cr(CO)3 and (η6-allylbenzene)Cr(CO)3 systems were taken at the Rutherford Appleton Laboratory while those for the (η6-benzophenone)Cr(CO)3 complex were obtained at the University of Amsterdam. No duplicate measurements were made. The (η6-styrene)Cr(CO)3 and (η6-benzophenone)Cr(CO)3 complexes used in this study were obtained from Ellanova Laboratories and were used as supplied, while (η6-allylbenzene)Cr(CO)3 was prepared using a reported method (mass spectral and 13C NMR data are presented in the ESI†).25,26
UV/Vis. spectra were measured using an Agilent 8453 spectrophotometer, sample solutions were contained in 1 cm quartz cuvettes in spectroscopic grade solvents, CH3CN or n-heptane. All ps-TRIR studies used IR cells fitted with CaF2 windows, in the case of (η6-styrene)Cr(CO)3 or (η6-allylbenzene)Cr(CO)3 studies a flow-through cell was used and the cell position was rastered in two dimensions to avoid photodecomposition of the solution. The ps-TRIR studies on (η6-benzophenone)Cr(CO)3 used a static cell and solution.
All quantum chemical calculations were performed using the Gaussian 09 programme suite (Revision D.01)27 on either the Stokes or Fionn computers at the Irish Centre for High-End Computing. Fragment contributions to valence orbitals were calculated using the AOMix software.28
Results
The UV/Vis. absorption spectra of the complexes used in this study are presented in Fig. 1. IR absorption data in the νCO region are presented in Table 1. Kinetic analysis of the time-dependent behaviour of absorbance features were obtained by exponential curve fitting techniques and the quoted errors correspond to the standard error obtained from these calculations. All ps-TRIR experiments were conducted at 293 K.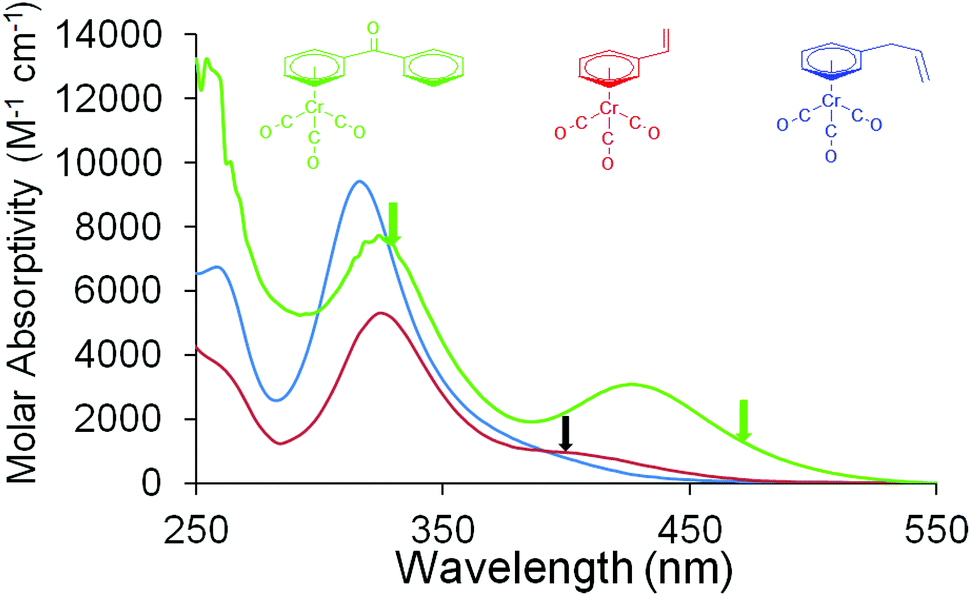 | ||
| Fig. 1 The UV/Vis. absorption spectra of (η6-styrene)Cr(CO)3 (red; 1.90 × 10−4 M in n-heptane), (η6-allylbenzene)Cr(CO)3 (blue, 9.65 × 10−5 M in n-heptane) and (η6-benzophenone)Cr(CO)3 (green, 3 × 10−3 M in acetonitrile) at room temperature, the black arrow indicates the excitation wavelength used in the ps-TRIR experiments on (η6-styrene)Cr(CO)3, and (η6-allylbenzene)Cr(CO)3 while the green arrows indicate the excitation wavelengths used in experiments on (η6-benzophenone)Cr(CO)3; the sample pathlength was 1 cm. | ||
| Complex |
ν
CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (cm−1) a (cm−1) |
ν
COb (cm−1) |
Solvent | ||
|---|---|---|---|---|---|
| a Obtained by FTIR spectroscopy. b Obtained from the initial bleached signal in ps-TRIR experiments. | |||||
| (η6-Benzophenone)Cr(CO)3 | 1977 | 1904 | 1965 | 1897 | Acetonitrile |
| (η6-Styrene)Cr(CO)3 | 1981 | 1917 | 1983 | 1919 | n-Heptane |
| (η6-Allylbenzene)Cr(CO)3 | 1979 | 1911 | 1977 | 1908 | n-Heptane |
Time resolved spectroscopic investigation on (η6-benzophenone)Cr(CO)3
The pulsed photolysis of (η6-benzophenone)Cr(CO)3 was investigated using ps-TRIR in acetonitrile solution using two excitation wavelengths 470 and 320 nm at the University of Amsterdam. These wavelengths were selected because they span the lowest-energy absorption band of this complex (Fig. 1). Acetonitrile was chosen because this complex is not sufficiently soluble in n-heptane which is the solvent of choice for our ps-TRIR studies. Excitation of (η6-benzophenone)Cr(CO)3 at 470 nm resulted in the depletion of the two parent bands (1965 and 1897 cm−1) within the excitation pulse (200 fs). Two new features were produced at 2008 and 1934 cm−1, again within the excitation pulse (Fig. 2). Over the subsequent 110 ps, the parent absorptions recover to their pre-irradiated state with concomitant decay of the features at 2008 and 1934 cm−1 which correspond to an excited state species. The bands of the excited state occur to the high energy side of each of the parent bands suggesting that the metal atom is less electron rich in the excited state compared to the ground state. This is consistent with a metal-to-ligand charge-transfer excited state (MLCT) in particular a metal-to-arene charge-transfer state (MACT) where the electron density on the arene ligand is greater in the excited state compared to the ground-state. Because the decay of the excited state (k1 = 4.0 (±0.5) × 1011 s−1 measured at 1934 cm−1) (k1 is the observed first order rate constant obtained from single exponential curve fitting to the time dependent behaviour of the IR absorption band) results in the complete recovery of the parent features (k1 = 6.2 (±0.5) × 1011 s−1 measured at 1897 cm−1), this excited state is unreactive i.e. it does not lead to photochemical change. It is clear that the observed rate constant for the decay of the excited state and the recovery of the parent are different. This could be the effect of vibrational relaxation in both the excited state and the recovering parent bands which would occur with different rates. This will result in different effective rate constants. In particular if the relaxation to the ground-state produces a vibrationally “hot” species (i.e. a vibrational quantum level v > 0) then vibrational relaxation to v = 0 must occur before the fundamental absorption is observed in the infrared spectrum.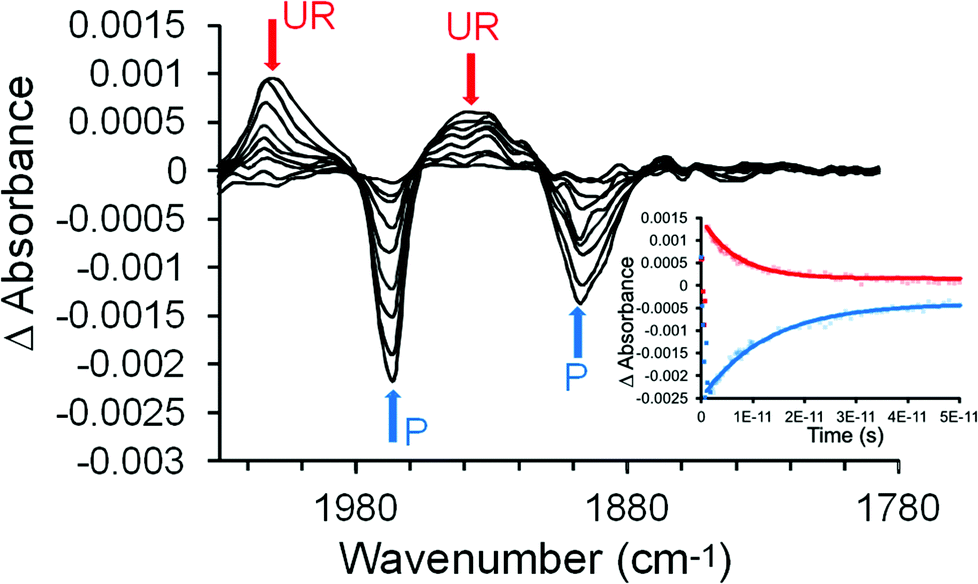 | ||
| Fig. 2 The time-resolved IR spectra obtained at 1.2, 3, 5, 8, 12, 20, 30, 60, 110 ps following 470 nm excitation of (η6-benzophenone)Cr(CO)3 in acetonitrile solution, showing depletion and recovery of the bands of (η6-benzophenone)Cr(CO)3 (P blue arrows) and the decay (red arrows) of the unreactive (UR) MACT excited state species. The inset shows the time-dependent behaviour of the 1965 cm−1 and 2008 cm−1 bands. The single exponential curve fits are superimposed upon the experimental data points. | ||
The transient spectra obtained following irradiation at 320 nm are presented in Fig. 3. These spectra confirm the formation of two discrete excited states, one unreactive (2008 and 1934 cm−1) which was also observed following photolysis at 470 nm, and the other (1948 and 1866 cm−1) which reacts to produce the CO-loss species (1909 and 1843 cm−1). The first order rate constant for this process obtained by monitoring at 1843 cm−1 band is k1 = 1.9 (±0.1) × 1010 s−1. The (η6-benzophenone)Cr(CO)3 parent also partially recovers on this timescale and the residual depletion (at 1965 cm−1) can be used to estimate the quantum yield for the CO-loss process which is approximately 0.43. It should be noted that the residual depletion at 1897 cm−1 is smaller than that at 1965 cm−1 because it is partly cancelled by the CO-loss peak at 1909 cm−1.
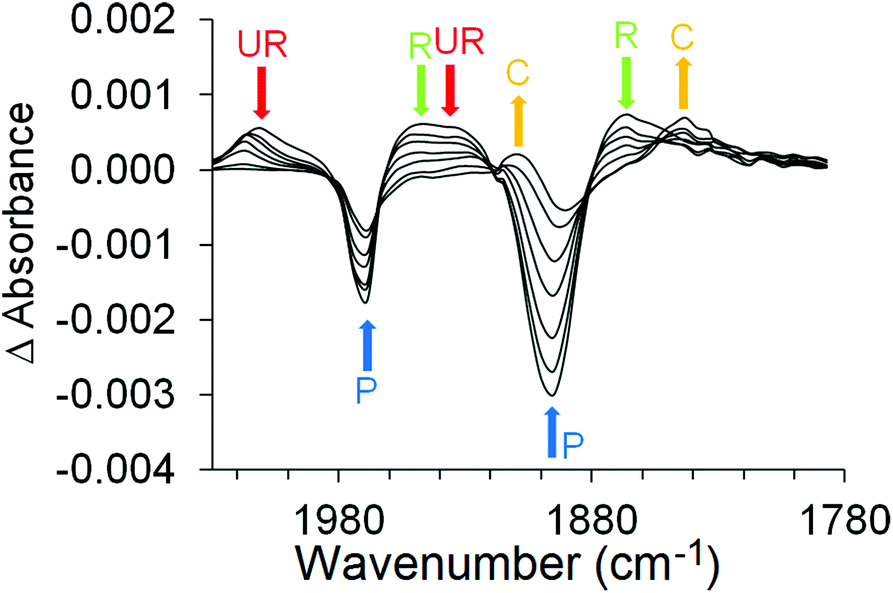 | ||
| Fig. 3 The ps-TRIR spectra obtained at 3, 6, 10, 20, 35, 80, and 300 ps after excitation at 320 nm of (η6-benzophenone)Cr(CO)3 in acetonitrile solution, the arrows indicate the time-dependent behaviour of each band and confirms the population of two discrete excited state, one absorbing at 2008 and 1934 cm−1 (red arrows, UR) and the other absorbing at 1948 and 1866 cm−1 (green arrows, R) the latter one decays to produce the CO-loss species (orange arrows, C) absorbing at 1909 and 1843 cm−1. | ||
ps-TRIR investigation of (η6-styrene)Cr(CO)3
The photochemical behaviour of (η6-styrene)Cr(CO)3 was investigated by ps-TRIR at the Rutherford Appleton Laboratory. The excitation wavelength used was 400 nm which is on the low energy side of the lowest energy absorption maximum at 324 nm in n-heptane solution (Fig. 1). Within the excitation pulse (50 fs) the two bands of the parent absorptions at 1983 and 1919 cm−1 are depleted. Fig. 4 depicts the early spectral changes observed up to 6.7 ps after excitation, while Fig. 5 shows the changes between 9.26 and 160 ps after excitation. Broad and weak absorptions were observed at 2003 and 1894 cm−1. The 2003 cm−1 band can be assigned to an unreactive (UR) excited state but the corresponding low energy absorption of this species is difficult to identify because of the weakness of these absorption features. It is likely that the second band of this species is located around 1950 cm−1. During the first 7 ps after excitation, features develop at 1966 and 1894 cm−1 as indicated by asterisks in Fig. 4 (k1 = 5.0 (±0.8) × 1011 s−1). These new features are produced without significant change to the absorbances of either the parent (P) or the unreactive excited state, labelled UR in Fig. 4. These features correspond to a reactive excited state (R). This state is produced from a higher energy non-quantised excited state, i.e. an excited state whose energy at excitation lies above the maximum vibrational quantum state. This excited state could be described as a predissociative state. Over the subsequent 150 ps these features decay (k1 = 2.17 (±0.05) × 1010 s−1) with concomitant partial recovery of the parent and formation of the CO-loss species ((η6-styrene)Cr(CO)2S, S = n-heptane) (k1 = 2.0 (±0.1) × 1010 s−1) labelled C in Fig. 5. The residual bleach of the parent signal allows the quantum yield for CO loss process to be determined at approximately 0.43. This is lower than the quantum yield for CO-loss from (η6-benzene)Cr(CO)3 of 0.7129 but similar to that obtained for the (η6-benzophenone)Cr(CO)3 complex following 320 nm excitation. The smaller quantum yield for the styrene or the benzophenone complex is the result of the partial population of the unreactive excited state at the chosen excitation wavelengths. | ||
| Fig. 4 Stepped offset spectra obtained following 400 nm excitation of (η6-styrene)Cr(CO)3 in n-heptane presenting the IR spectra obtained up to 6.7 ps after excitation showing the formation of two features at 1966 and 1894 cm−1 (indicated by asterisks) of an excited state precursor to CO-loss and also traces of the CO-loss species on this timescale (CO-loss). | ||
 | ||
| Fig. 5 The ps-TRIR spectra obtained at 9.26, 11.96, 14.84, 17.96, 21.34, 25.02, 29.08, 33.6, 38.7, 44.54, 51.4, 59.66, 70.1, 84.26, 106.38, and 160 ps after a 400 nm pulsed excitation of (η6-styrene)Cr(CO)3 in n-heptane solution, showing the decay of the unreactive excited state (UR), the two features of the reactive excited state (R), the two CO-loss product bands (C) and the recovery of the parent bands (P); the inset shows the time-dependent behaviour of the bands indicated by the coloured arrows showing that the parent recovery, the growth of the CO-loss product and the decay of the reactive excited state all occur on the same time-scale. | ||
ps-TRIR investigation of (η6-allylbenzene)Cr(CO)3
The photophysical and photochemical processes induced by 400 nm excitation of (η6-allylbenzene)Cr(CO)3 were also investigated by ps-TRIR spectroscopy at the Rutherford Appleton Laboratory. Within the excitation pulse, the two bands of the parent (η6-allylbenzene)Cr(CO)3 complex at 1977 and 1908 cm−1 were depleted (Fig. 6). Broad and weak features were evident 1 ps after the excitation pulse at 1983 and 1958 cm−1 corresponding to two excited states similar to those observed following irradiation of (η6-styrene)Cr(CO)3 described above. Because of the weakness of the feature at 1983 cm−1, it is difficult to unambiguously locate the low energy band of this species. The 1983 cm−1 feature decays rapidly ((k1 = 2.9 (±0.1) × 1011 s−1) but again because of the weakness of these absorptions it is not possible to confirm if the parent is regenerated by this process. This species is an unreactive excited state similar to that observed following irradiation of (η6-styrene)Cr(CO)3 (see above). A weak depletion was also evident at 1933 cm−1 but this is the result of photolysis of a stable product identified below. Over the initial 6.7 ps two features develop (k1 = 1.0 (±0.1) × 1012 s−1) at 1958 and 1878 cm−1 corresponding to the formation of a reactive excited state. This reactive excited state in turn decays to reform the parent complex and also the CO-loss species (1918 and 1863 cm−1) (see Fig. 6). The quantum yield of this process can be estimated by measuring the residual depletion of the parent absorptions. A quantum yield of 0.50 for irradiation at 400 nm for this system is slightly larger than that obtained for either the styrene or the benzophenone system (0.43). This indicates that a non-conjugating substituent on the arene ligand is less effective at facilitating the population of the unreactive excited state. After approximately 50 ps, additional features develop at 1933 and 1876 cm−1 in a zeroth order process (k0 = 93 M s−1 at 293 K). This corresponds to the formation of the chelate product (η6,η2-allylbenzene)Cr(CO)2 (κ). The time-dependent behaviour of selected bands is presented in Fig. 7. It is clear that the chelate formation is slow, presumably because of the presence of a molecule of solvent in the vacant coordination site on the metal atom. Alkane solvents are known to act as “token” ligands where formally coordinatively unsaturated intermediates are generated photochemically.30–33 These “token” ligands affect the kinetics of the reactions of such photoproducts. By assuming a quantum yield of 0.5 for the CO-loss process the extinction coefficients of the CO-loss absorption band at 1918 cm−1 can be estimated at 5207 M−1 cm−1. This allows an estimation of the zeroth order chelate-formation process presented above. | ||
| Fig. 6 The ps-TRIR spectra obtained 6.74, 9.26, 11.96, 14.84, 17.96, 21.4, 25.02, 29.08, 33.6, 38.7, 44.54, 51.4, 59.66, 70.1, 84.26, 106.38, 160, 250, 350, 500, 750, 1000 ps after 400 nm excitation of (η6-allylbenzene)Cr(CO)3 in n-heptane showing the decay of the reactive excited state (R), reformation of the parent (P), formation of CO-loss species (C) and finally formation of the chelate species (κ); weak features of an unreactive excited state was also observed at 1983 cm−1 but this had decayed before the initial spectrum presented here. | ||
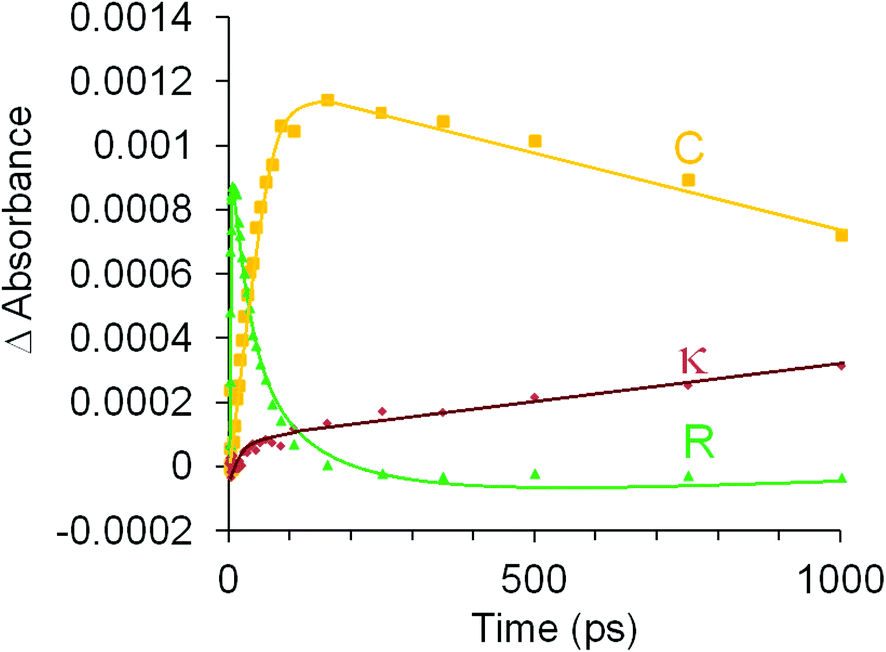 | ||
| Fig. 7 The time-dependence of selected features, following excitation of (η6-allylbenzene)Cr(CO)3. Green triangles indicate the formation and decay of the reactive excited state (R), the orange squares describe the formation and decay of the CO-loss species (C) and the mauve diamonds show the formation of the chelate species (κ). | ||
Attempted isolation of (η6,η2-allylbenzene)Cr(CO)2
A sample of (η6-allylbenzene)Cr(CO)3 was dissolved in cyclohexane (10 mL) and the solution was purged with argon. The solution was then irradiated using λ > 350 nm. During the photolysis aliquots of the solution were extracted for analysis by IR spectroscopy. Initially IR stretching vibrations at 1978 and 1910 cm−1 were observed corresponding to the parent (η6-allylbenzene)Cr(CO)3 complex. During the photolysis these bands decreased in intensity and new bands were formed at 1934 and 1881 cm−1 (see ESI†). These IR bands compare closely to those observed in the ps-TRIR experiments (1933 and 1876 cm−1). During the photolysis the chelate product (η6,η2-allylbenzene)Cr(CO)2 (κ) formed as an air sensitive orange solid. Attempts to spectroscopically characterise this complex failed because of the extreme sensitivity of this material.34Computational results
A similar approach was adopted for the characterisation of each complex in this study. An initial molecular structure was obtained from a simple molecular mechanics calculation. The final optimisation used Becke's three parameter hybrid density functional using the LYP correlation functional (B3LYP)35–38 with a triple zeta basis set and valence polarisation functions (Tzvp).39 Optimisation used a pruned (99590) integration grid and very tight optimisation criteria (Opt = VeryTight). The stability of the Hartree Fock wavefunction and the location of the stationary point on the hypersurface was verified by the usual methods. A suitable structural parameter was selected to represent a specific reaction coordinate (outlined below) and a relaxed potential energy scan was performed along this reaction coordinate. Each step in this scan provides a set of atomic coordinates in the ground-state which were then subjected to time-dependent density functional theory (TDDFT) analysis. This provides estimates of the vertical excitation energy to the lowest twenty singlet excited states (not all are shown in the figures for clarity). A plot of the energy of selected excited states along the reaction coordinate then provides information on the likely dynamic behaviour of the excited states.
Quantum chemical calculations on (η6-benzophenone)Cr(CO)3
The Cr to C distance of one carbonyl ligand was chosen as a suitable reaction coordinate for this study. The Cr–C distance was increased from the equilibrium distance of 1.864 Å in twenty steps of 0.1 Å.40,41 The energies of the twenty lowest energy singlet excited states using each of these sets of coordinates were estimated using TDDFT methods and the results of these calculations are presented in Fig. 8. These plots indicate the presence of three low lying excited states which can be populated using excitation photons in the 490 to 435 nm region. These excited states are bound states with respect to the CO-loss reaction coordinate. This means that they exhibit a minimum along this reaction coordinate and will not eject CO without surmounting an additional thermal barrier. The third excited state has the greatest oscillator strength of the three and this can be optically accessed using 437 nm photons. The fourth singlet excited state (accessible with photons of wavelength less than 365 nm and coloured blue in Fig. 8) presents an accelerating profile to CO-loss (it does not exhibit a minimum) which, in the non-adiabatic description, will cross lower excited states (dashed curve in Fig. 8). These crossing points, i.e. conical intersections or seams, present branching zones on the hypersurface which control the efficiency, rate, and direction of a photochemical process such as CO-loss.5,6 Population of the third singlet excited state with the highest oscillator strength (0.0666 see ESI†) is the most efficient and will be the dominant excited state for short-wavelength excitation. This excited state is a bound state with respect to the CO-loss reaction coordinate, and the crossing with the 4th excited state generates a thermal barrier to CO-loss of approximately 30 kJ mol−1. These calculations suggest that this complex will exhibit wavelength dependent photochemistry, with CO-loss becoming increasingly efficient for higher energy excitations. Analysis of the composition of the orbitals involved in the first three singlet excited states (see ESI†) indicate that the three lowest energy excited states are metal to arene charge-transfer in character, while the fourth excited state has significant metal-based and metal to carbonyl charge-transfer character.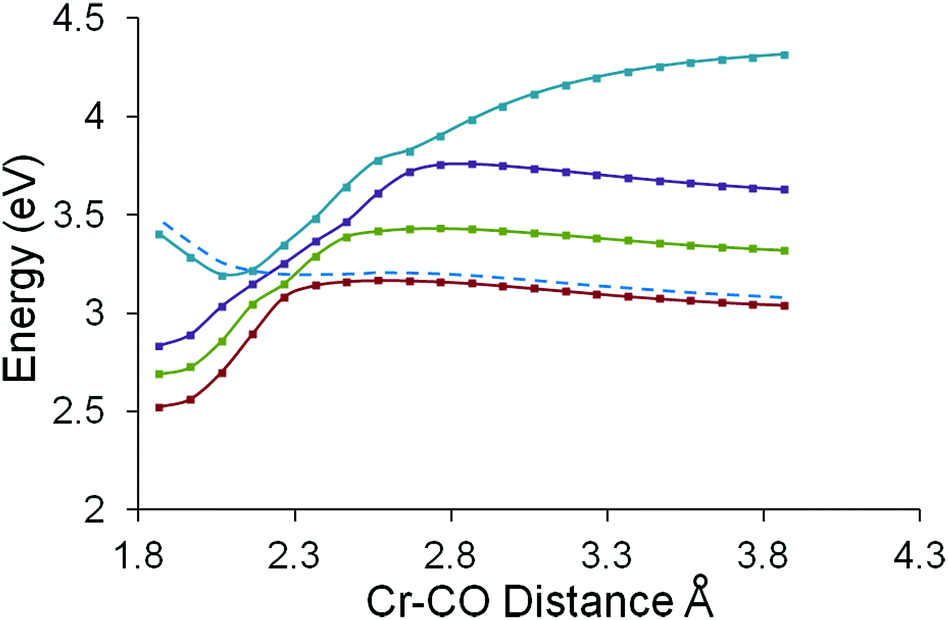 | ||
| Fig. 8 The adiabatic behaviour of the four lowest energy singlet excited states for (η6-benzophenone)Cr(CO)3, showing that the lowest three excited states are bound with respect to this Cr–CO reaction coordinate, while the fourth excited state (3.4 eV, 365 nm) presents an accelerating profile to CO-loss (dashed curve) in a non-adiabatic description. | ||
Quantum chemical calculations on (η6-styrene)Cr(CO)3
The excited state behaviour of this system along two reaction coordinates was investigated. The two reaction coordinates chosen to represent the ring-slip process or the CO-loss process. The first reaction coordinate was chosen to investigate whether the unreactive excited state identified in the ps-TRIR studies could lead to a change in the hapticity of the styrene ligand. In these calculations the distance between the chromium atom and the terminal vinyl carbon atom was varied between 4.39 Å and 2.79 Å in steps of 0.1 Å. The results of these calculations are presented in Fig. 9. These calculations confirm that none of the excited states accessible following 400 nm excitation present accelerating profiles towards a reduced hapticity or ring-slip intermediate.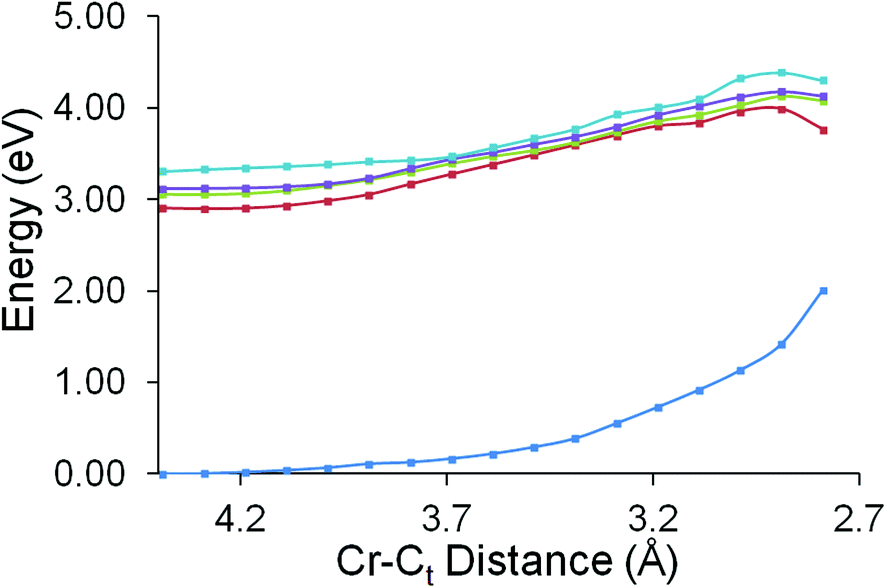 | ||
| Fig. 9 The behaviour of the ground state energy and the four lowest energy singlet excited states in (η6-styrene)Cr(CO)3, as the Cr–C(terminal-vinyl) distance was reduced from 4.39 to 2.79 Å in steps of 0.1 Å, showing that none of the excited states presents an accelerating profile towards a ring-slip or reduced hapticity species. | ||
The CO-loss reaction coordinate was then examined. Here the Cr–CO distance was varied from 1.86 Å to 3.86 Å in 0.1 Å steps (Fig. 10). These calculations show that the in the non-adiabatic description the second singlet excited state crosses the first excited state at a Cr–CO distance greater than the ground-state equilibrium distance and the resulting profile (the red dashed curve in Fig. 10) presents a small barrier to CO-loss. This explains why the CO-loss process is arrested and also why the precursor excited state to CO-loss can be observed in the ps-TRIR experiments.
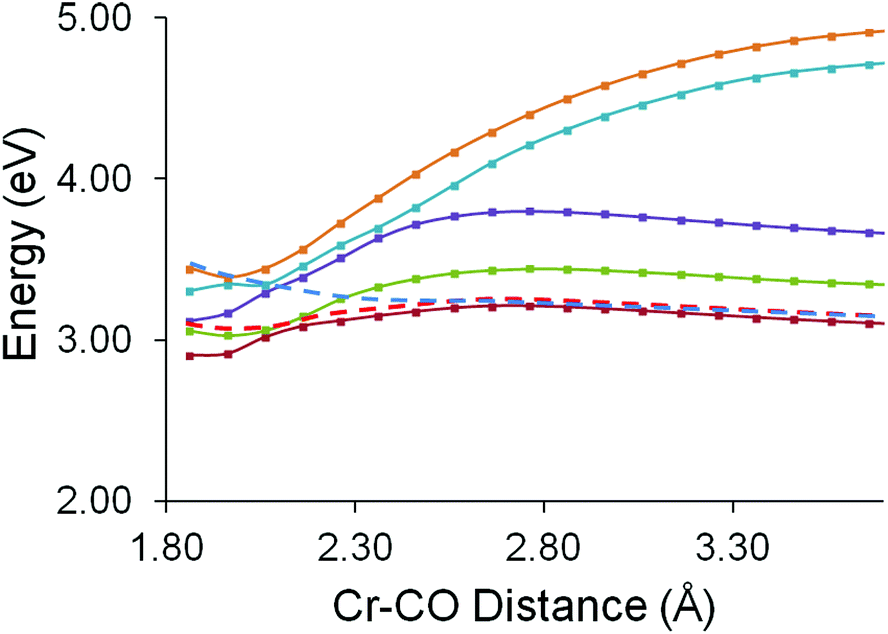 | ||
| Fig. 10 The behaviour of the five lowest energy singlet excited states of (η6-styrene)Cr(CO)3 as one Cr–CO distance is varied from 1.86 to 3.86 Å, the red dashed curve represents the non-adiabatic description of the profile formed when the second excited state crosses the a higher energy excited state which is unbound with respect to CO-loss (blue dashed curve). | ||
Discussion
The absorbance profile of (η6-benzophenone)Cr(CO)3 in the UV/Vis. region presents the opportunity to examine wavelength-dependent features of the photophysics and photochemistry of this complex over a wide span of excitation wavelengths. Two excitation wavelengths were chosen for this investigation, 470 nm which is to the low energy side of the lowest energy absorption maximum and 320 nm which is close to the second λmax. of the complex in acetonitrile. Irradiation at 470 nm resulted in a depletion of the parent ground-state absorptions and the formation of a single excited state characterised by bands at 2008 and 1934 cm−1 (Fig. 2). Over the subsequent 110 ps these bands decay with concomitant and complete recovery of the parent bands. Consequently the excited state formed under these conditions does not react to produce a photochemical change and can be described as an unreactive excited state. It should be noted that for the more symmetrical (η6-benzene)Cr(CO)3 complex, excitation at 400 nm results predominantly in the population of a reactive excited state which subsequently expels CO over the course of 150 ps. The population of the unreactive excited state in the (η6-benzophenone)Cr(CO)3 system is the result of a lowering of the symmetry of the complex making a transition which is formally symmetry forbidden in the case of (η6-benzene)Cr(CO)3, allowed in the case of (η6-benzophenone)Cr(CO)3. Consequently this will have the effect of lowering the quantum yield for the overall CO-loss process for (η6-benzophenone)Cr(CO)3 (0.43 compared to 0.71 for (η6-benzene)Cr(CO)3). TDDFT calculations confirm that the unreactive excited state has substantial metal-to-arene charge-transfer character.Excitation of (η6-benzophenone)Cr(CO)3 using 320 nm photons initially produced two excited state species (Fig. 3), the first has bands identical to the unreactive excited state formed following excitation at 470 nm, and a second set of bands at 1948 and 1866 cm−1. This second excited state decays to both regenerate the parent but also reacts to form the CO-loss species (η6-benzophenone)Cr(CO)2(S) (S = acetonitrile). The nature of this second reactive excited state was investigated by TDDFT methods and these calculations indicate that this excited state has metal-to-carbonyl charge-transfer character with a significant contribution of metal-centred character (see ESI†). No evidence was obtained in this pico-second study to support a hapticity change of the coordinated benzophenone ligand.
Excitation of (η6-styrene)Cr(CO)3 with 400 nm photons results in the bleach of the parent absorptions and the formation of two bands of the unreactive excited state within the excitation pulse. Over the first 6.7 ps the two bands of the reactive excited state grow-in. It is clear from these time-resolved spectra that the reactive excited state is not formed from the unreactive excited state, and consequently must be formed from a predissociative excited state which is not observable using spectroscopic techniques. Examination of the spectrum obtained at 6.7 ps after excitation indicates the presence of a small yield of the CO-loss species as indicated Fig. 4. This suggests that the CO-loss species is formed both from the predissociative state and also from the reactive excited state albeit more slowly. Thus the crossing of the predissociative state with the lower energy reactive excited state represents a branching space on the hypersurface leading either to ultrafast CO-loss (blue dashed curve in Fig. 10) or formation of the reactive excited state which acts as an intermediate in the slower CO-loss process over a small thermal barrier (red dashed curve in Fig. 10). TDDFT calculations along a ring-slip reaction coordinate failed to locate any minima on any excited state profile other than the η6-complex. This suggests that ring-slip or hapticity change in the (η6-styrene)Cr(CO)3 complex cannot be induced by population of an optically accessible excited state. Furthermore the experimental results as previously discussed for the benzophenone analogue do not support a photoinduced hapticity change in these systems.
Time-resolved studies on (η6-allylbenzene)Cr(CO)3 again provided evidence for the population of two excited states following pulsed irradiation at 400 nm. One excited state is unreactive, it is produced in low yield and reforms the parent very rapidly. TDDFT calculations indicate that this excited state has substantial metal-to-arene charge-transfer character. The apparent low yield of this excited state contrasts with the greater yields of the lowest energy metal-to-arene charge-transfer excited states in either the (η6-benzophenone)Cr(CO)3 or (η6-styrene)Cr(CO)3 systems. This reflects the lack of conjugation to the arene π-system from the allyl substituent in (η6-allylbenzene)Cr(CO)3. As in the case of the (η6-styrene)Cr(CO)3 system the reactive excited state is not formed within the excitation pulse but forms over the initial 6.7 ps following photon absorption from a non quantised higher energy excited state. This state then reacts to either regenerate the parent or expel CO. Consequently the majority of CO is lost in an arrested process. The subsequent chelate formation occurs via a slow zeroth order process notwithstanding the proximity of the pendant olefin functional group (see Scheme 1). The chelate product was produced in low-temperature steady state photolysis at −20 °C as an air sensitive solid.
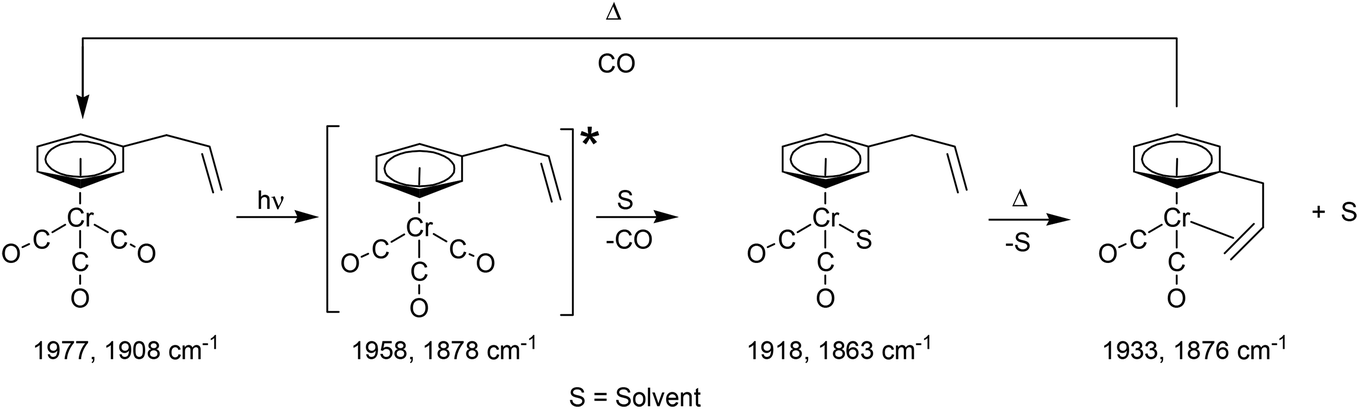 | ||
| Scheme 1 | ||
Concluding comments
These results provide a fuller picture of the photophysical processes proceeding the ejection of CO from (η6-arene)Cr(CO)3 complexes. Substituents on the arene ring reduce the overall symmetry from C3v for (η6-benzene)Cr(CO)3 to C1 for the substituted arene complexes. This means that electronic transitions which are formally forbidden in (η6-benzene)Cr(CO)3 become allowed in the substituted arene complexes. The most important consequence of this is an increase in the importance of MACT excited states in the photophysics of these complexes. These MACT states are unreactive and their presence reduces the efficiency at which reactive excited states can be populated thus lowering the quantum yield for the predominant photoreaction, i.e. CO-loss. Early ps-TRIR spectra of both (η6-styrene)Cr(CO)3 or (η6-allylbenzene)Cr(CO)3 reveal the rapid formation of a MCCT excited state over the first 7 ps following excitation. This MCCT state can be observed spectroscopically and must therefore be a bound state on the excited state hypersurface. TDDFT calculations confirm this and while the state is bound with respect to the CO-loss reaction coordinate, the thermal barrier to CO-loss is sufficiently small to allow CO-loss to occur on the ps timescale. This leads to a general picture for the excited state behaviour along the CO-loss reaction coordinate as outlined in Fig. 11. Thus if half-sandwich complexes of this type are to be used as molecular optical switches it is clear that to maintain the high quantum yields of CO loss required, substituents on the arene ligand must be carefully chosen to minimise the contribution of unreactive excited states. Furthermore because of the presence of MACT excited states careful consideration of the appropriate excitation wavelengths must be made to maximise the quantum yield of the desired photoproduct. | ||
| Fig. 11 A schematic representation of the low-lying excited states of (η6-arene)Cr(CO)3 complexes showing the lowest energy MACT excited state which is not formally optically accessible for (η6-benzene)Cr(CO)3 but becomes accessible for arenes containing substituents which conjugate with the arene π-system, and the predissociative excited state (MC) which relaxes to the spectroscopically active MCCT state. | ||
Acknowledgements
The authors thank the Central Laser Facility for granting access to the ULTRA system under EU Access-no. 92004 and to the Amsterdam Laboratory under EU access-LLAMS-1961. We would also like to thank the Irish Research Council RS/2012/341 (SM) and the Environmental Protection Agency (EPA grant 2008-ET-MS-3-S2) (MTP and JCM) for financial support.Notes and references
- C. Long, in Photophysics of Organometallics, ed. A. J. Lees, Springer, Berlin/Heidelberg, 2010, vol. 29, pp. 37–71 Search PubMed.
- S. Villaume, A. Strich, C. Daniel, S. A. Perera and R. J. Bartlett, Phys. Chem. Chem. Phys., 2007, 9, 6115–6122 RSC.
- N. Ben Arnor, S. Villaume, D. Maynau and C. Daniel, Chem. Phys. Lett., 2006, 421, 378–382 CrossRef PubMed.
- A. Rosa, E. J. Baerends, S. J. A. van Gisbergen, E. van Lenthe, J. A. Groeneveld and J. G. Snijders, J. Am. Chem. Soc., 1999, 121, 10356–10365 CrossRef CAS.
- E. J. Baerends and A. Rosa, Coord. Chem. Rev., 1998, 177, 97–125 CrossRef CAS.
- C. Pollak, A. Rosa and E. J. Baerends, J. Am. Chem. Soc., 1997, 119, 7324–7329 CrossRef CAS.
- X. L. Xie and J. D. Simon, J. Phys. Chem., 1989, 93, 4401–4404 CrossRef CAS.
- J. D. Simon and X. L. Xie, J. Phys. Chem., 1986, 90, 6751–6753 CrossRef CAS.
- G. M. Greetham, Q. Cao, I. P. Clark, P. S. Codd, M. W. George, R. C. Farrow, T. Matousek, A. W. Parker, M. Pollard, D. A. Robinson, Z. J. Xin and M. Towrie, Appl. Spectrosc., 2010, 64, 1311–1319 CrossRef CAS PubMed.
- C. Long, J. Phys. Chem. A, 2012, 116, 6845–6850 CrossRef CAS PubMed.
- M. W. George, C. Long, M. T. Pryce, X.-Z. Sun and K. Q. Vuong, Organometallics, 2012, 31, 268–272 CrossRef CAS.
- I. P. Clark, M. W. George, G. M. Greetham, E. C. Harvey, C. Long, J. C. Manton, H. McArdle and M. T. Pryce, J. Phys. Chem. A, 2012, 116, 962–969 CrossRef CAS PubMed.
- I. P. Clark, M. W. George, G. M. Greetham, E. C. Harvey, C. Long, J. C. Manton and M. T. Pryce, J. Phys. Chem. A, 2011, 115, 2985–2993 CrossRef CAS PubMed.
- A. C. Coleman, N. M. Boyle, C. Long, R. Augulis, A. Pugzlys, P. H. M. van Loosdrecht, W. R. Browne, B. L. Feringa, K. L. Ronayne and M. T. Pryce, Dalton Trans., 2010, 39, 2201–2203 RSC.
- I. P. Clark, M. W. George, G. M. Greetham, E. C. Harvey, C. Long, J. C. Manton and M. T. Pryce, J. Phys. Chem. A, 2010, 114, 11425–11431 CrossRef CAS PubMed.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS PubMed.
- P. Li, S. Amirjalayer, F. Hartl, M. Lutz, B. De Bruin, R. Becker, S. Woutersen and J. N. H. Reek, Inorg. Chem., 2014, 53, 5373–5383 CrossRef CAS PubMed.
- J. Torres-Alacan, U. Das, A. C. Filippou and P. Vohringer, Angew. Chem., Int. Ed., 2013, 52, 12833–12837 CrossRef CAS PubMed.
- C. B. Duke III, R. G. Letterman, J. O. Johnson, J. W. Barr, S. Hu, C. R. Ross II, C. E. Webster and T. J. Burkey, Organometallics, 2014, 33, 485–497 CrossRef.
- E. J. Heilweil, J. O. Johnson, K. L. Mosley, P. P. Lubet, C. E. Webster and T. J. Burkey, Organometallics, 2011, 30, 5611–5619 CrossRef CAS.
- T. T. To, E. J. Heilweil, C. B. Duke, K. R. Ruddick, C. E. Webster and T. J. Burkey, J. Phys. Chem. A, 2009, 113, 2666–2676 CrossRef CAS PubMed.
- T. T. To, E. J. Heilweil, C. B. Duke and T. J. Burkey, J. Phys. Chem. A, 2007, 111, 6933–6937 CrossRef CAS PubMed.
- T. T. To, C. E. Barnes and T. J. Burkey, Organometallics, 2004, 23, 2708–2714 CrossRef CAS.
- M. Towrie, D. C. Grills, J. Dyer, J. A. Weinstein, P. Matousek, R. Barton, P. D. Bailey, N. Subramaniam, W. M. Kwok, C. S. Ma, D. Phillips, A. W. Parker and M. W. George, Appl. Spectrosc., 2003, 57, 367–380 CrossRef CAS PubMed.
- C. A. L. Mahaffy and P. L. Pauson, Inorg. Synth., 1990, 28, 136–147 CrossRef CAS PubMed.
- I. U. Khand, C. A. L. Mahaffy and P. L. Pauson, J. Chem. Res., Synop., 1978, 352–353 CAS.
- J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. P. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS.
- M. S. Wrighton and J. L. Haverty, Z. Naturforsch., B: Chem. Sci., 1975, 30, 254–258 Search PubMed.
- J. M. Kelly, C. Long and R. Bonneau, J. Phys. Chem., 1983, 87, 3344–3349 CrossRef CAS.
- J. K. Burdett, J. M. Grzybowski, R. N. Perutz, M. Poliakoff, J. J. Turner and R. F. Turner, Inorg. Chem., 1978, 17, 147–154 CrossRef CAS.
- R. N. Perutz and J. J. Turner, J. Am. Chem. Soc., 1975, 97, 4800–4804 CrossRef CAS.
- J. K. Burdett, M. A. Graham, R. N. Perutz, M. Poliakoff, A. J. Rest, J. J. Turner and R. F. Turner, J. Am. Chem. Soc., 1975, 97, 4805–4808 CrossRef CAS.
- M. T. Pryce, Ph.D. Thesis, Dublin City University, 1994.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- W. T. Yang, R. G. Parr and C. T. Lee, Phys. Rev. A, 1986, 34, 4586–4590 CrossRef CAS.
- A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS PubMed.
- B. D. Dunietz, A. Dreuw and M. Head-Gordon, J. Phys. Chem. B, 2003, 107, 5623–5629 CrossRef CAS.
- A. Dreuw, B. D. Dunietz and M. Head-Gordon, J. Am. Chem. Soc., 2002, 124, 12070–12071 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular orbital fragment compositions, excited state energies, kinetic analyses for all complexes in this study, and analytical data for (η6-allylbenzene)Cr(CO)3. See DOI: 10.1039/c4dt01544d |
| This journal is © The Royal Society of Chemistry 2014 |