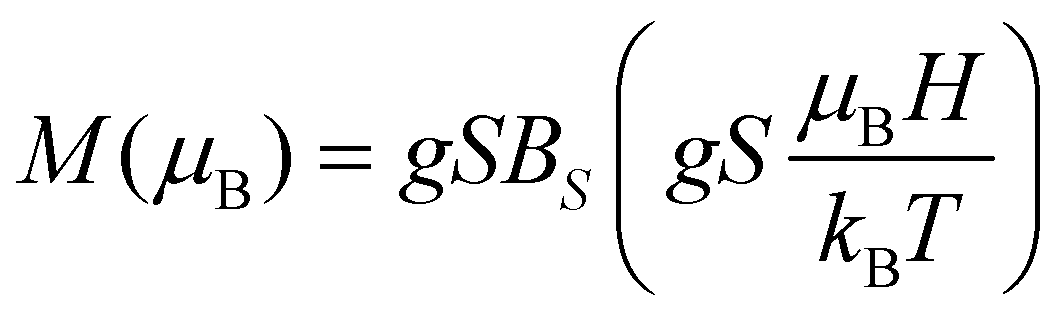DOI:
10.1039/C4DT01518E
(Paper)
Dalton Trans., 2014,
43, 13349-13357
Preparation and properties of a calcium(II)-based molecular chain decorated with manganese(II) butterfly-like complexes†
Received
23rd May 2014
, Accepted 23rd July 2014
First published on 23rd July 2014
Abstract
The room temperature reaction of [Mn2O2(bipy)4](ClO4)3 (bipy = 2,2′-bipyridine) with Ca(CHCl2COO)2 in methanol produced a yellow crystalline material. The X-ray determined structure comprises of a multiple calcium(II) carboxylate bridged chain-like structure which is decorated with [Mn(bipy)2(OH2)]2+ subunits. The redox behaviour for the complex in H2O and MeCN is reported. In the latter solvent the oxidation of the manganese ions appears to be facilitated by the presence of the calcium ions. Magnetic susceptibility and low temperature magnetization measurements show that the Mn moment is isotropic, with g = 1.99(1) and S = 5/2, confirming it is in the 2+ oxidation state. A very weak antiferromagnetic interaction is also detected. Frequency-dependent ac measurements evidence slow magnetic relaxation of the Mn(bipy)2 units. Two relaxation mechanisms are identified: a very slow direct process and a faster one caused by the Resonant Phonon Trapping mechanism. This is the first example of field-induced single ion magnet (SIM) behavior in a mononuclear Mn(II) complex.
Introduction
Nature over billions of years of evolution has harnessed the properties of readily abundant metals for performing indispensable biological functions.1 Intricate molecular machinery was developed to undertake reactions that under normal conditions were either extremely slow or required molecules to be activated in some manner.2 For example, carboxy peptidase is representative of the zinc(II)-based metalloenzyme which facilitates the hydrolysis of C-terminal amino acids from peptide substrates.3 Under normal physiological conditions the rate of the reaction is extremely slow because of the robust nature of the amide bond. The activation of carbon dioxide, again by zinc(II), in human carbonic anhydrase II is very much representative of how a relatively inactive molecule can be converted extremely quickly (106 s−1, pH 9, 25 °C) into the bicarbonate anion.4 In these two cases, evidently the redox chemistry of the metal ion is unimportant, and it is the zinc(II) ion interaction with the substrate that is partly responsible for driving the reaction. The converse is redox-based reactions and the need for the metal ion to alter oxidation state, and the surrounding amino acid ligands in the peptide to facilitate any structural alterations (e.g., bond length changes).5 Some relevant enzyme examples include superoxide dismutase,6 peroxidases,7 catalases,8 oxidases9 and oxygenases.10 Knowledge of the structure of the active site and the periphery amino acid residues for many of the outlined peptide-based structures has helped in understanding the workings of the enzymes.11 There are obviously examples of enzymes for which the structure of the active site is unknown, or clouded by uncertainty because of the low structural resolution from the X-ray data. The water oxidation catalyst (WOC) found in photosystem II is certainly a structure which has been surrounded by controversy.12 The structure determined by Barber and co-workers13 has recently been supplanted by a more detailed analysis to 1.9 Å resolution by Umena and co-workers.14 All the metal ions and ligands are uniquely located in the renowned manganese cluster comprising Mn4CaO5. The cube-like structure contains three manganese ions with the final site taken up by a calcium ion. The additional manganese ion is located outside the cubane. The linking units are oxo bridges. The cluster performs the essential sacrificial oxidation of water to generate dioxygen and protons.15 Because it is not possible to isolate the cluster outside its protective amino acid rich enzyme shell, attempts have been made to produce heterometallic Mn–Ca clusters to mimic the WOC center.16 Some pertinent examples of WOC mimics were produced by the groups of Christou,17 Milios18 and Agapie.19 In efforts to construct heterometallic Mn–Ca clusters our attention focussed on using preorganised [Mn2O2(bipy)4](ClO4)3 (bipy = 2,2′-bipyridine) with the intention of forming an accretion with Ca(CHCl2COO)2. It transpired that a calcium carboxylate-based chain was formed and dotted along its framework are [Mn(II)(bipy)2OH2]2+ subunits. The redox behaviour for the assembly appears to be affected by the presence of the calcium(II) ions, but not because of their Lewis acid property. As well as being able to focus on the electrochemical properties of the assembly, the incorporation of paramagnetic Mn(II) ions into the molecular framework also opened up the opportunity to study its magnetic properties. Magnetic dynamic behaviour of the Mn(II) units is of interest in the context of new mononuclear transition metal compounds with single-ion magnet (SIM) behaviour. Although mixed-valence manganese clusters have been reported to exhibit single-molecule magnet (SMM)20,21 properties, there are only a few recent examples of Mn-based SIMs.22–24 It turned out that the new assembly displayed SIM behavior despite the expected low anisotropy (S = 5/2, L = 0).
Results and discussion
Synthesis
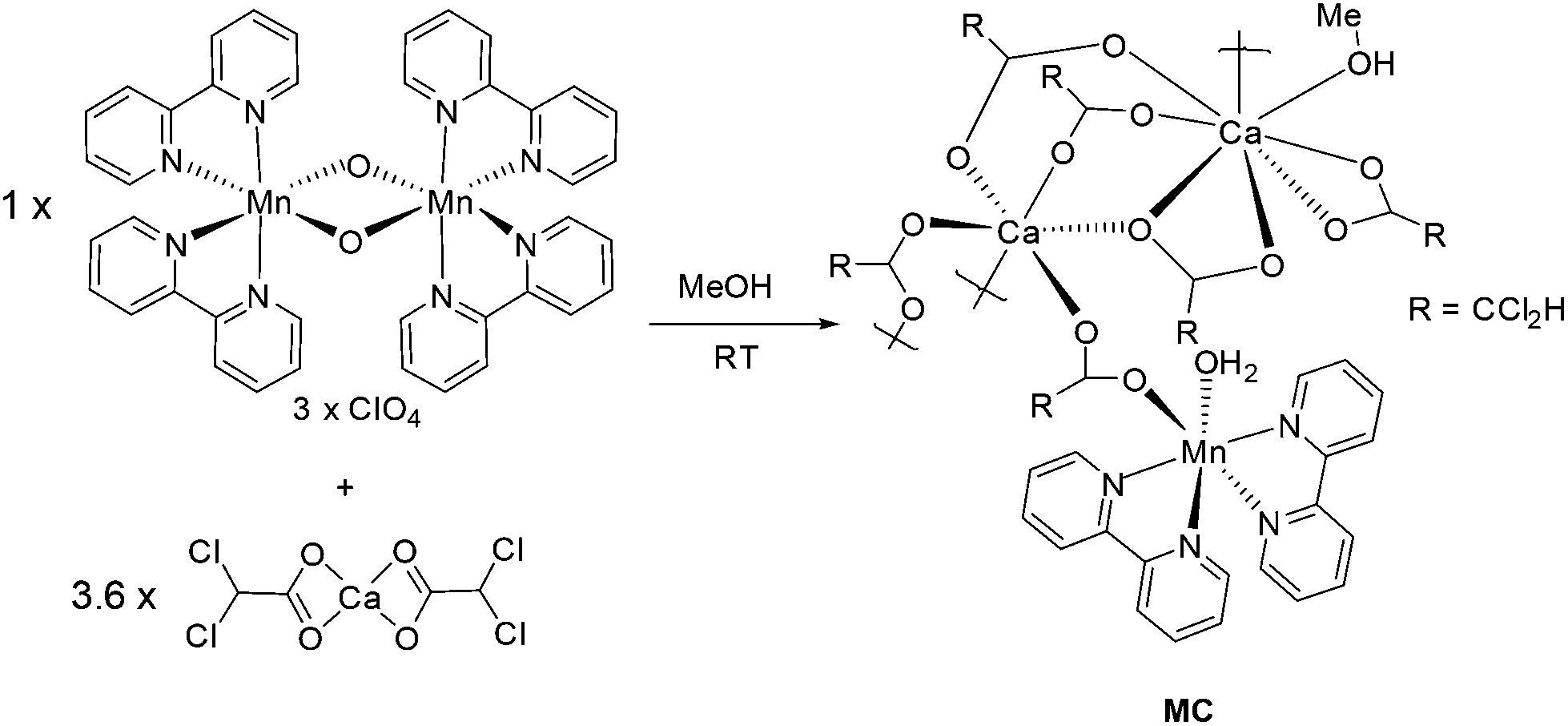
There is no steadfast method for constructing extensive mixed-metal ion structures, especially in which active metal ion(s) should be in close proximity to a potential reactive site. Rather than applying a hit-and-miss approach one of our main molecular design criteria was to utilise the diverse binding motif of the carboxylate group. The functional group has served many other groups in their quest to fabricate supramolecular structures.25 Metal organic frameworks (MOFs) are prime examples for which carboxylates play a key role in their construction.26 It is well recognised that the carboxyl group can bind through several disparate modes when interacting with either a single metal ion or multiple metal sites.27 Especially pertinent is the bridging mode for which the two oxygen atoms link together two ions, and help propagate the formation of clusters and higher-order assemblies. Another bridging motif involves bidentate chelation to a single metal ion and a single oxygen interaction with a proximal metal ion. There is no clear-cut method of forcing a specific binding mode to dominate when a superstructure is engineered from all its individual molecular components. Such explicit crystal engineering is still at the infancy stage, but it is clear that the R group of the carboxyl group must have some effect on controlling packing forces. The chosen R group for this study was the bulky dichloromethane subunit, the intention being to manipulate intermolecular interactions.
In an attempt to have partial control of how the manganese centre assembled, the preorganised mixed valence complex, [Mn(III)(bipy)2O2Mn(IV)(bipy)2](ClO4)3,28 was targeted as a starting material. To introduce simultaneously the calcium(II) and carboxylate group the complex Ca(CHCl2COO)2 was used. The full synthetic procedure is illustrated in Scheme 1 and represents the optimised conditions for preparation of the supramolecular structure, MC. Thus, mixing of [Mn(III)(bipy)2O2Mn(IV)(bipy)2](ClO4)3 with Ca(CHCl2COO)2 in MeOH at room temperature, followed by leaving the solution overnight, resulted in a drastic colour change from dark black to yellow. Upon standing for 24 hours a yellow crystalline material precipitated from the solution. A basic FT-IR of the crystals confirmed the presence of the carboxylate groups (CO stretch 1590 cm−1 and 1390 cm−1), and that the [Mn(bipy)2]2+ was still intact and probably part of the overall complex (C–H stretch 3384 cm−1 and 3009 cm−1). It is apparent from the colour change that one or two of the manganese centres had undergone reduction. The MeOH was presumably the reductant for the reaction.
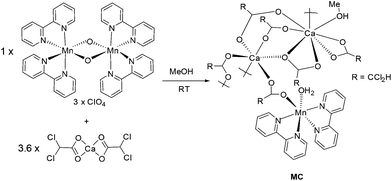 |
| | Scheme 1 The room temperature procedure for preparation of the mixed-metal calcium manganese cluster. | |
X-ray crystal structure determination
Structure of MC.
Crystals of the isolated material MC were of suitable quality to collect X-ray diffraction data and to solve the structure. The crystal system is monoclinic and the space group is P21/n. The basic asymmetric unit structure is shown in Fig. 1, and collected in Table 1 are a selection of pertinent bond lengths. The butterfly-like Mn(bipy)2 group bridges via a carboxyl group to Ca1 and the vacant coordination site is taken up by a water molecule. The bridging carboxylate between the Mn and Ca centres is reminiscent to that observed in the natural WOC cubane cluster. Presumably the water molecule was scavenged from residual water in the MeOH or is from the original bridging oxide. No isotopic labelling experiments were performed to try and answer the conundrum. From a formal charge count just at the manganese centre, the oxidation state appears to be only +1/2. In fact, the Mn–N bond lengths (Table 1) are typical for a bipy-based complex with a formal oxidation state of manganese(II). Magnetic measurements (see later) were more insightful on this matter. The Ca1 centre is six coordinate comprising of five μ-carboxylate units, with the final oxygen being part of a η2-carboxylate coordinated to Ca2. The Ca2 centre is 8-coordinate, but as well as containing bridging carboxylate units the ion also binds two η2-carboxylate subunits. The coordination sphere is completed by a MeOH molecule. The Ca1–Ca2 distance of ∼4.08 A implies that there is no significant interaction between the two ions. It is noted that the water molecule for Mn–OH2 is not in close contact with any of the calcium ions, but is instead hydrogen bonded to a η2-carboxylate group of Ca2. Recalling that the activation of water is often enhanced by secondary bonding interactions such an H-bond, may suggest it has a role in the chemistry of MC. The first coordination sphere of the manganese ion consists of four nitrogen atoms from two bipyridyl ligands, one oxygen atom from a carboxyl group of dichloroacetic acid that bridges Mn to Ca and one more vacant coordination site which is taken up by a water molecule (Fig. 1).
 |
| | Fig. 1 X-ray determined molecular structure for MC showing the basic asymmetric unit and the Mn(Ca)2 motif. Grey = carbon, red = oxygen, lime green = chlorine, aqua green = calcium, blue = nitrogen, purple = manganese. | |
Table 1 Selected bond lengths [Å] for MC
| Mn–O(6) |
2.147(2) |
Mn–O(1) |
2.119(2) |
| Mn–N(4) |
2.274(2) |
Mn–N(2) |
2.271(2) |
| Mn–N(1) |
2.260(2) |
Mn–N(3) |
2.260(2) |
| Ca(1)–Ca(2) |
4.0877(8) |
Ca(1)–O(5) |
2.292(2) |
| Ca(1)–O(7) |
2.367(2) |
Ca(1)–O(8) |
2.299(2) |
| Ca(1)–O(9A) |
2.311(2) |
Ca(1)–O(10) |
2.299(2) |
| Ca(1)–O(13) |
2.365(2) |
Ca(2)–Ca(2B) |
3.9621(11) |
| Ca(2)–O(16) |
2.395(2) |
Ca(2)–O(4) |
2.487(2) |
| Ca(2)–O(5) |
2.580(2) |
Ca(2)–O(11) |
2.335(2) |
| Ca(2)–O(12) |
2.333(2) |
Ca(2)–O(14) |
2.537(2) |
| Ca(2)–O(15) |
2.505(2) |
Ca(2)–O(15B) |
2.396(2) |
The crystal packing diagram is revealing in that the fully-bridged calcium(II)-based motif is more clearly seen (Fig. 2A). A chain-like structure with a Ca core is apparent, with Mn(bipy)2 moieties adorning the outer casing. This Ca core is more clearly evident when only the binding groups at the calcium(II) ions are shown (Fig. 2B). The structure comprises a tetramer of calcium ions running in a coordination number algorithm of 6-8-8-6, with each tetramer linked together by two carboxylate groups. A search of the Cambridge Structural Database found only three similar structures based on a polymer-like calcium(II) ion chain.29–31 The apparent charge count around Ca2 is −3 (two η2 carboxylates and two η1 carboxylates), whereas at Ca1 the charge count is −2½ (five η1 carboxylates). The excess charge at the bis-calcium(II) segment is −1½, and is clearly the counter charge required for the manganese(II) ion. At a simplistic level the calcium(II) chain-like segment is an extended anion for the periphery manganese complexes. The Mn(bipy)2 subunits are well separated (intra-chain Mn–Mn = 15.17 Å, inter-chain Mn–Mn = 12.31 Å) in the structure and any interactions between them would expected to be weak. Further crystallographic details are found in the ESI.†
 |
| | Fig. 2 Partial crystal packing diagram for MC displaying the manganese complexes decorating the calcium(II)-based chain (A), and the basic structure demonstrating the 6-8-8-6 coordination mode of the calcium(II) ions (B). The insert picture highlights the bridging motif of the carboxyl group. Colours are the same as in Fig. 1. | |
Magnetic measurements
Static magnetic measurements.
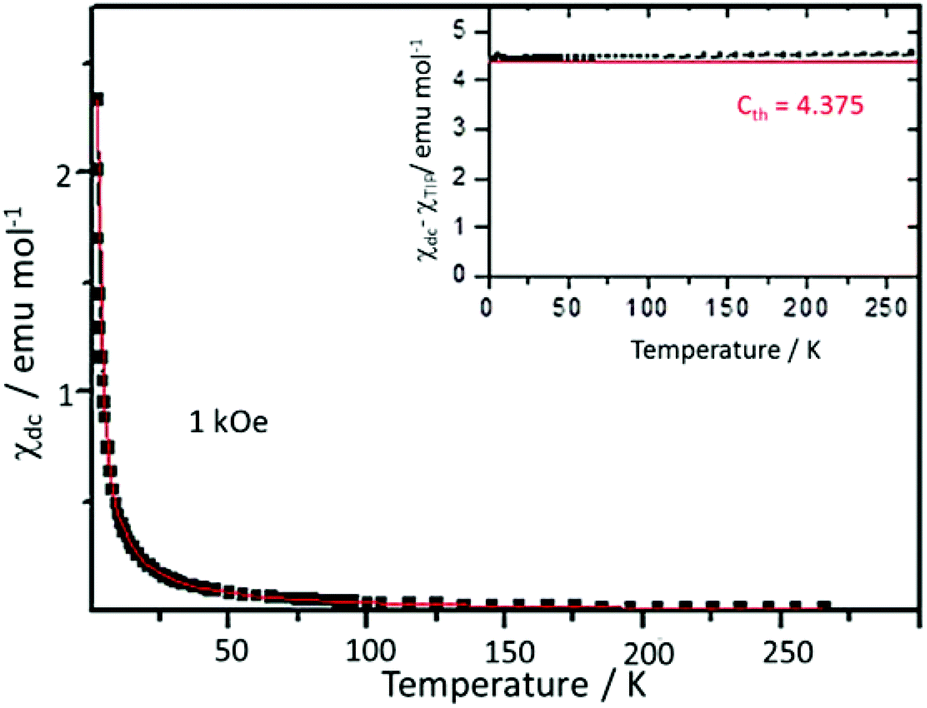
The equilibrium dc magnetic susceptibility of MC was measured from 1.8 K to 265 K with a dc field of H = 1 kOe. The thermal dependence can be described with a Curie–Weiss law, χdc = χTIP + C/(T − θ), where χTIP is a temperature independent term, C is the Curie constant, and θ is the Curie temperature which accounts for magnetic interactions (Fig. 3). The obtained values from fit, χTIP = −0.0018 ± 0.0003 emu mol−1, C = 4.463 ± 0.007 emu K mol−1 and θ = −0.033 ± 0.003 K agree with expected values for a Mn2+ d5 ion.32 Indeed, the expected Curie constant (Cth) for a Mn2+ ion, with L = 0, S = 5/2 isotropic spin and a gyromagnetic factor g = 2.0, is = 4.375 emu K mol−1 and is in very good agreement with experimental results. No deviations from Curie law behaviour are observed down to 1.8 K (inset Fig. 3). The obtained value of the Curie temperature would suggest that possible anti-ferromagnetic interactions between separate Mn2+ ions are almost negligible.
 |
| | Fig. 3 Temperature dependence of the dc magnetic susceptibility of MC (black squares). Experimental data measured in an applied field H = 1 kOe. Fit to a Curie–Weiss law is shown in red. Inset: temperature dependence of the product (χdc − χTIP)T; in red, Cth, expected C value for Mn(II). | |
The isothermal M(H) curves, measured at T = 1.8 K, 2.5 K, 3.5 K and 5.0 K show a saturation trend as a function of magnetic field, H. The magnetisation per formula unit, in Bohr magnetons (μB), as a function of the reduced magnetic field, μBH/kBT, where kB is the Boltzmann constant, is given in Fig. 4. It is observed that all the curves collapse to a universal curve. The saturation magnetization amounts to 5μB f.u.−1 for one Mn2+ ion per formula unit. In the case of non-interacting paramagnetic entities and assuming an isotropic g factor, such a dependency is described by eqn (1):
| |  | (1) |
where
BS is the Brillouin function. Isothermal curves can be fitted to above function for
S = 5/2 and isotropic
gJ = 1.991 ± 0.001.
 |
| | Fig. 4 The magnetisation per formula unit, in Bohr magnetons, as a function of the reduced magnetic field, μBH/kBT. | |
Therefore, down to 1.8 K, an oxidation state of Mn2+ is observed. For this ion, zero field splitting are usually small, of the order of 10−2 cm−1, and so a S = 5/2 g = 2 isotropic state is normally observed. Moreover, no contribution from the Mn(II) magnetic interaction down to 1.8 K can be distinguished.
Dynamic magnetic measurements.
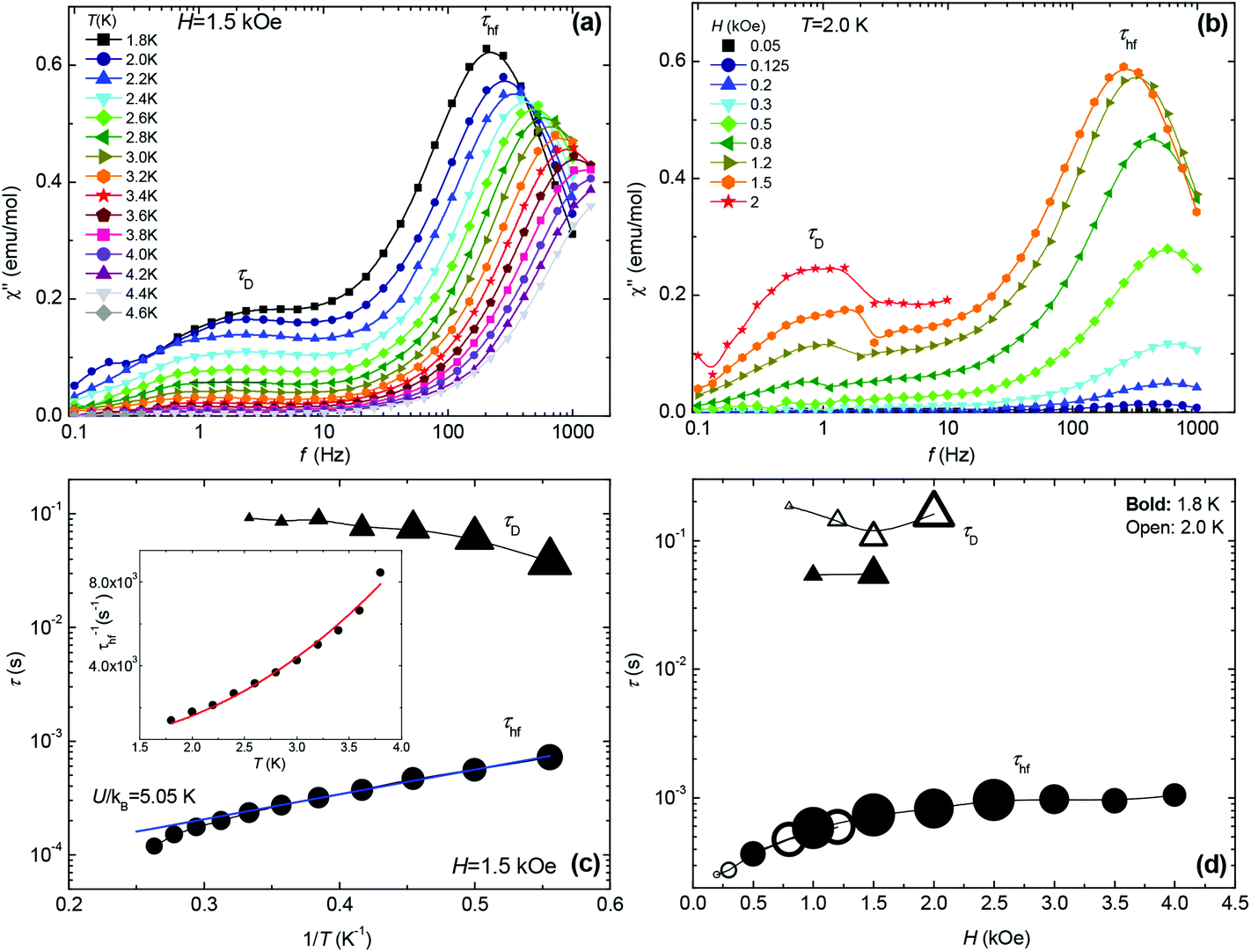
In order to perform a more complete picture of the magnetic behaviour for MC, ac susceptibility measurements in the frequency range 0.1 < f < 2000 Hz were performed as a function of field strength (0–4 kOe) and temperature (1.8–4.6 K). At H = 0 no slow relaxation behaviour is observed. However, under the application of field two different relaxational pathways become accessible, as apparent from the double peaked χ′′(f) data shown in Fig. 5a and b. From these data the relaxation time, τ, at different temperatures and fields is obtained since χ′′(f) has a maximum when the condition 2πfτ = 1 is fulfilled (Fig. 5c and d). The intensity of the χ′′ peak corresponding to the high frequency relaxation process, with relaxation time τhf, is much higher than that of the slow process, τD, which gains intensity with the field. Fig. 5c shows the dependence of the two relaxation times with 1/T, determined from the χ′′(f,T) data at the field H = 1.5 kOe and optimising the τhf peak. The first relaxational process τD ∼ 0.1 s, is almost temperature independent, whereas the second one τhf ∼ 1–8 × 10−4 s, exhibits a ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) τ(1/T) dependence with a slope of ∼5.05 K at low T. The order of magnitude of τD and its field and temperature behaviour suggests this relaxation process may correspond to the ubiquitous direct process reported in many SIMs.33 The other τhf process cannot be ascribed to an Orbach process, since the energy calculated from the τhf(1/T) plot of 5.05 K, is larger than the Zeeman splitting between levels at 1.5 kOe; the ΔZee = gμBHS = 1.02 K and the anisotropy energy for Mn is expected to be very small. Alternatively, the process can be explained in terms of the resonant phonon trapping (RPT) mechanism,34,35 taking place when the phonon-bottleneck (PB) effect sets in. This is not unreasonable since the observed τhf−1(T) = KT2.4 temperature dependence (inset Fig. 5c) is close to the τhf−1 ∝ T2 law theoretically expected for this type of process.36 The slight deviation in the exponent value indicates that in the studied range of temperatures and fields, the MC does not represent a perfect two-level system.37 The slow relaxation through RPT has been previously reported in isotropic (L = 0) Gd(III)37,38 complexes but, to our knowledge, never in Mn examples.
τ(1/T) dependence with a slope of ∼5.05 K at low T. The order of magnitude of τD and its field and temperature behaviour suggests this relaxation process may correspond to the ubiquitous direct process reported in many SIMs.33 The other τhf process cannot be ascribed to an Orbach process, since the energy calculated from the τhf(1/T) plot of 5.05 K, is larger than the Zeeman splitting between levels at 1.5 kOe; the ΔZee = gμBHS = 1.02 K and the anisotropy energy for Mn is expected to be very small. Alternatively, the process can be explained in terms of the resonant phonon trapping (RPT) mechanism,34,35 taking place when the phonon-bottleneck (PB) effect sets in. This is not unreasonable since the observed τhf−1(T) = KT2.4 temperature dependence (inset Fig. 5c) is close to the τhf−1 ∝ T2 law theoretically expected for this type of process.36 The slight deviation in the exponent value indicates that in the studied range of temperatures and fields, the MC does not represent a perfect two-level system.37 The slow relaxation through RPT has been previously reported in isotropic (L = 0) Gd(III)37,38 complexes but, to our knowledge, never in Mn examples.
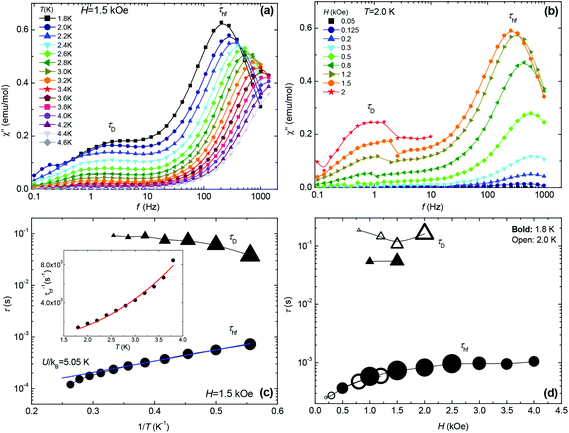 |
| | Fig. 5 Top panels: Imaginary component of the ac susceptibility as a function of frequency, (a) at different temperatures, constant field H = 1.5 kOe; (b) at different fields, constant temperature T = 2.0 K. Bottom panels: (c) relaxation times of the two observed processes as a function of 1/T at 1.5 kOe, and Inset: τhf−1vs. temperature data and fit to the function τhf−1 = KT 2.4 (K = 292 s−1 K−2); (d) field-dependence of the two relaxation times at 1.8 K and 2.0 K. The size of the symbols is proportional to the χ′′ peak intensity. | |
Mass spectrometry analysis.
In solution we might expect the extended structure for MC to disassemble into smaller oligomeric fragments. An electrospray mass spectrum for the complex dissolved in MeOH was collected in attempt to identify possible ions. The low mass region contains molecular ions that appear to incorporate the calcium(II) and manganese(II) ions and the 2,2-dichloroacetate ligand. At higher mass the spectrum is dominated by several clusters of ion peaks (Fig. 6) that appear to contain the basic [Mn(bipy)2]2+ ion. Collected in Table 2 are a series of prospective molecular ions and their masses, noting that water and methanol adducts are feasible. Several ions are identified which support structures where the carboxylate ligand and calcium(II) ion(s) are associated with the manganese centre. The mass spectrometry results would support the hypothesis that in a donor organic solvent calcium-manganese clusters are still intact.
 |
| | Fig. 6 Selected region of the electrospray mass spectrum for MC collected in MeOH. | |
Table 2 Feasible molecular species from dissolution of MC in MeOH as determined by electrospray mass spectrometry
| Molecular ion species |
Mass |
| L = 2,2-dichloroacetate, bipy = 2,2′-bipyridine. Mass of singly positively charged ion by proton loss. Protons removed to obtain singly positively charged ion. |
| {[Mn(bipy)2]2+L}+ + (H2O)n + (MeOH)n |
494
|
| 512 (n = 1), 530 (n = 2) |
| 526 (n = 1), 558 (n = 2) |
| 590 (n = 3) |
| {[Mn(bipy)2]2+LCa2+}3+ + (H2O)n (−2H+)b + (MeOH)n (−2H+)b |
534 (532)a |
| 550 (n = 1), 568 (n = 2) |
| 564 (n = 1), 596 (n = 2) |
| 628 (n = 3) |
| {[Mn(bipy)2]2+L(Ca2+)2}5+ + (H2O)n (−4H+)b + (MeOH)n (−4H+)b |
574 (570)a |
|
588 (n = 1), 606 (n = 2) |
| 600 (n = 1), 632 (n= 2) |
|
664 (n = 3)
|
| {[Mn(bipy)2]2+(L)2Ca2+}2+ + (H2O)n (−H+)b + (MeOH)n (−H+)b |
663 (662)a |
| 680 (n = 1), 698 (n = 2) |
| 694 (n = 1), 726 (n = 2) |
| 758 (n = 3) |
| {[Mn(bipy)2]2+(L)2(Ca2+)2}4+ + (H2O)n (−3H+)b + (MeOH)n (−3H+)b |
703 (700)a |
|
718 (n = 1), 736 (n = 2) |
|
732 (n = 1), 764 (n = 2) |
| 796 (n = 3) |
| {[Mn(bipy)2]2+(L)3(Ca2+)2}3+ + (H2O)n (−2H+)b + (MeOH)n (−2H+)b |
816 (814)a |
| 832 (n = 1), 850 (n = 2), 868(n = 3) |
| 846 (n = 1), 878 (n = 2) |
| 910 (n = 3) |
UV-visible spectroscopy.
The yellow crystals for MC were readily soluble in deionised water (pH ∼ 6), and the electronic spectrum is shown in Fig. 7. It is likely, as evidenced by ESI mass spectrometry, that the crystal superstructure is disassembled when dissolved in water to produce the basic dimers. Two main electronic absorption bands are observed at 280 nm and 233 nm with the latter being slightly less intense. The molar absorption coefficients (εmax) for these two bands are appreciably large and so are assigned primarily to π–π* transitions associated with the 2,2′-bipyridine ligand. There is an evident tail in the absorption profile from around 300 nm which can be more readily seen in the insert of Fig. 7. We discount this tail is because of Rayleigh scattering from nano-particulates in solution owing to a poor fit to a 1/λ4 dependency. Considering that Laporte forbidden d–d electronic transitions for the d5 Mn2+ ion are likely too weak to be observed, the low-energy profile is assigned to the tail of a metal-to-ligand charge transfer (MLCT) transition.
 |
| | Fig. 7 Room temperature UV-visible spectrum for MC in deionised water (pH ∼ 6). Insert shows an expansion of the region shown by the arrow. | |
Electrochemistry.
There are several reports, particularly by Sawyer et al.,39 describing the electrochemical behaviour of manganese complexes of the type [Mn(bipy)3]2+, but less literature material covers the redox response for [Mn(bipy)2X2]n+ where X represents a spectator or solvent ligand.40 As a starting point for comparison purposes we investigated the cyclic voltammetry for [Mn(bipy)2Cl2] and MC in buffered H2O containing 0.2 M KCl (pH 4) at a glassy carbon working electrode (Fig. 8). The positive potential window was relatively uneventful with no real sign of oxidation of the manganese centre in the two complexes. The only point to note is the small reduction wave observed at around +0.4 V vs. Ag/AgCl which is absent if the potential is not swept above 1 V. This wave is presumably associated with the di-μ-oxo-dimanganese(III,IV or IV,IV) species which is known to be produced following oxidation of manganese(II) polyimine complexes. Holding the potential at +1.3 V for several minutes resulted in an increase in size of the wave as more dimer was produced. The scanning to highly reducing potentials revealed for both complexes two closely-spaced peaks at −1.47 V and −1.55 V vs. Ag/AgCl. Two clear reverse oxidation waves are seen at −1.18 V and −0.97 V for [Mn(bipy)2Cl2] and −1.16 V and −0.99 for MCvs. Ag/AgCl. The reverse parts of the waves are clearly located at less negative potentials than expected if the redox process was fully reversible. Full reversibility is not produced at pH 7 or 10, but at the latter pH the peak separation is 290 mV and ipc/ipa = 1.4. Addition of an electron to either complex is ligand-based and it appears that protonation of ligand-based radical anion is partial cause for the electrochemical irreversibility.
 |
| | Fig. 8 Cyclic voltammograms for [Mn(bipy)2Cl2] (red) and MC (black) in distilled H2O containing 0.2 M KCl (pH 4) at a glassy carbon electrode (scan rate = 50 mV s−1, reference = Ag/AgCl). Insert shows an expansion after cycling to 1.3 V. a = background electrolyte scan. | |
The redox chemistry of MC in dry MeCN at positive potentials is more resolved than the aqueous case and contains at least two waves in the forward sweep (Fig. 9). For comparison purposes a cyclic voltammogram for [Mn(II)(bipy)2Cl2] under near identical conditions is also presented. Clearly the onset of oxidation for MC (region a insert) occurs at lower potential than the simple [Mn(II)(bipy)2Cl2] case. The second wave associated with MC (b and c insert) is quasi-reversible (E1/2 = +0.85 V, ΔE = 80 mV vs. Fc+/Fc) and is comparable to that seen for the control complex. It should be noted that E1/2 = +0.90 V vs. Fc+/Fc for [Mn(bipy)3]2+ measured under the same conditions, and is associated with the reversible Mn(II)/Mn(III) couple. There is again indication that the dimer complex is formed (d insert) after cycling to high oxidation potential. In the negative potential scan segment of the cyclic voltammogram an irreversible ligand-based reduction is seen at −1.92 V vs. Fc+/Fc. This wave is shifted to a more negative potential by ca. 150 mV when compared to [Mn(II)(bipy)2Cl2] and [Mn(bipy)3]2+. There is a clear effect on the metal-centred and ligand-based redox chemistry by the presence of the proximal calcium(II) entity in the complex of MC. Reduction of formal charge at the manganese(II) ion coupled with electron density build-up at the bipy sites would account for the observed redox perturbations. There are several reports on the how redox-inactive metal ions modulate the reduction potentials of heterometallic manganese-oxido clusters. Recent work by Agapie et al.41 found that potential shifts were related to the metal ion Lewis acid strength. The Lewis acid was directly associated with the oxido ligand in the manganese cluster, and as electron density was removed from the cluster its reduction became more facile. The converse is the initial oxidation of the lower oxidation state manganese centres must be more difficult. For MC the calcium(II) ion is not directly associated with the manganese(II) centre, and a Lewis acid strength argument does not appear to be valid. From the X-ray structure for MC there was the reasoning that the manganese centre was less positive than the expected formal +2 charge; magnetic measurements do not support such an idea since all behaviour is consistent with a Mn2+ ion. Solution species are by their nature more dynamic and it is feasible that the cyclic voltammogram is representative of a structure that is not typical of the solid state. It is worth recalling that the “charging up” of the Mn4Ca cluster in PSII is photochemically driven (Kok model) starting with three manganese(III) ions and a manganese(IV) ion. The highly oxidising P680+ can readily drive via the redox-active tyrosine the reaction to create the redox equivalent to oxidise water; the thermodynamic ease of charging up the cluster is not that important. Here we seem to be observing a Ca2+ ion effect which is facilitating the oxidation of the manganese ion.
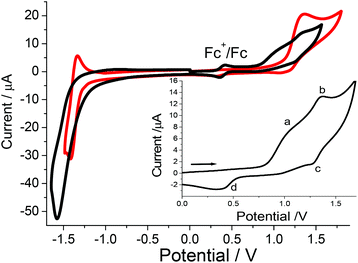 |
| | Fig. 9 Cyclic voltammogram for MC (black) and [Mn(II)(bipy)2Cl2] (red) in dry MeCN containing 0.2 M TBATFB against a silver wire and using ferrocene as the internal standard. Insert shows a positive potential only wave containing no ferrocene for MC. Scan rate = 50 mV s−1, working electrode = glassy carbon. | |
Conclusions
By way of a self-assembly process it is straightforward to prepare a calcium(II) carboxylate chain decorated at precise locations with the butterfly-like Mn(bipy)2 units. It is evident that the molecular system is not structurally related to the Mn4CaO5 WOC catalyst found in Photosystem II, but opens a possible different design route to WOC catalysts not based entirely on the cubane cluster. It is conceivable to imagine decorating around a calcium chain structure, manganese ring-like motifs to form a rod-axle (rotaxane-like) array containing preorganised water channels. We plan to explore this idea by the modification of carboxylate units used in the synthesis and the manganese precursor complexes. In addition, it has been shown that Mn(bipy)2 on the calcium(II) chain behaves as a Single-Ion Magnet, showing field-induced slow magnetic relaxation via two different pathways. This is the first example of a mononuclear manganese(II) complex exhibiting SIM behaviour.
Experimental
All chemicals were purchased from commercial sources and used as received unless otherwise stated. The calcium(II) salt of dichloroacetic acid (DCA) was prepared by the reaction between CaCO3 and dichloroacetic acid in a ratio 1:2. The complex [Mn2O2(bipy)4](ClO4)3 was prepared according to the literature.28 IR data (solid sample): 3384, 3009, 1590, 1541, 1472, 1439, 1390, 1221, 1157, 1102, 1016, 960, 826, 761, 700, 646 cm−1.
Cyclic voltammetry experiments were performed using a fully automated HCH Instruments Electrochemical Analyzer and a three-electrode set-up consisting of a glassy carbon working electrode, a platinum wire counter electrode and an Ag/AgCl reference electrode. Cyclic voltammograms were collected in either deoxygenated MeCN containing tetrabutylammonium tetrafluoroborate (0.2 M) as background electrolyte, or in deionised water containing KCl (0.2 M). Oxidation potentials were reproducible to within ±15 mV.
Variable temperature magnetic susceptibility and magnetization measurements were performed using a Quantum Design SQUID-Based Magnetometer MPMS-XL5. Data were collected on polycrystalline samples in the 1.8–260 K range. Isothermal magnetization curves were obtained at 1.8 K, 2.5 K, 3.5 K and 5 K in the range of 0 T ≤ μ0H ≤ 5 T. Measurements on powdered samples were performed with the addition of Daphne oil, introduced to fix the grains at low temperatures.
Preparation of [Mn(bipy)2Ca2(DCA)6(H2O)(CH3OH)] (MC)
[Mn2O2(bipy)4](ClO4)3 (0.3 g, 0.28 mmol) and Ca(CHCl2COO)2 (0.3 g, 1 mmol) were mixed in CH3OH (10 ml). The reaction was stirred overnight to afford a yellow solution. After filtration the solution was allowed to stand undisturbed for 24 h, and the resulting large block yellow crystals were collected by filtration. IR data: 3384, 3009, 1590, 1541, 1472, 1439, 1390, 1221, 1157, 1102, 1016, 960, 826, 761, 700, 646 cm−1. Elemental analysis calc. for C33H28Ca2Cl12MnN4O14: C, 31.33%; H, 2.23%; N, 4.43%. Found: C, 31.12%; H, 2.21%; N, 4.38%.
Acknowledgements
This work was mainly funded by the FP7-PEOPLE-2009-IRSES Nr. 246902 grant, and partially funded by the Spanish MINECO project MAT11/23791 and the DGA project E34 (co-funded by the Fondo Social Europeo) and the European Union FEDER. Authors would like to acknowledge the use of Servicio General de Apoyo a la Investigación-SAI, University of Zaragoza. The EPSRC sponsored Mass Spectrometry Service at Swansea is thanked for collecting mass spectra.
Notes and references
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239 CrossRef CAS; C. Andreini, I. Bertini, G. Cavallaro, G. L. Holliday and J. M. Thornton, J. Biol. Inorg. Chem., 2008, 13, 1205 CrossRef PubMed.
- D. J. Evans and C. J. Pickett, Chem. Soc. Rev., 2003, 32, 268 RSC; P. K. Mascharak, Coord. Chem. Rev., 2002, 225, 201 CrossRef CAS; R. Kramer, Coord. Chem. Rev., 1999, 182, 243 CrossRef.
- B. W. Matthews, Acc. Chem. Res., 1988, 21, 333 CrossRef CAS; S. M. Fabiane, M. K. Sohi, T. Wan, D. J. Payne, J. H. Bateson, T. Mitchell and B. J. Sutton, Biochemistry, 1998, 37, 12404 CrossRef PubMed.
- Md. I. Hassana, B. Shajee, A. Waheed, F. Ahmada and W. S. Sly, Bioorg. Med. Chem., 2013, 21, 1570 CrossRef CAS PubMed; J. P. Colman, J. Biol. Chem., 1967, 242, 5212 Search PubMed; S. Lindskog and J. E. Colman, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 2505 CrossRef.
- J. V. Bannister, W. H. Bannister and G. Rotilio, Crit. Rev. Biochem., 1987, 22, 111 CrossRef CAS.
- D. P. Riley, Chem. Rev., 1999, 99, 2573 CrossRef CAS PubMed.
- J. A. L. da Silva, J. J. F. da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2013, 257, 2388 CrossRef CAS PubMed; T. L. Poulos, Arch. Biochem. Biophys., 2010, 500, 3 CrossRef PubMed; H.-P. Hersleth, U. Ryde, P. Rydberg, C. H. Görbitz and K. K. Andersson, J. Inorg. Biochem., 2006, 100, 460 CrossRef PubMed.
- M. Costas, K. Chen and L. Que Jr., Coord. Chem. Rev., 2000, 200–202, 517 CrossRef CAS; G. Smulevich, C. Jakopitsch, E. Droghetti and C. Obinger, J. Inorg. Biochem., 2006, 100, 568 CrossRef PubMed.
- L. Rulíŝek and U. Ryde, Coord. Chem. Rev., 2013, 257, 445 CrossRef PubMed.
- P. He and G. R. Moran, J. Inorg. Biochem., 2011, 105, 1259 CrossRef CAS PubMed; N. R. Rose, M. A. McDonough, O. N. F. King, A. Kawamura and C. J. Schofield, Chem. Soc. Rev., 2011, 40, 4364 RSC.
-
(a) T. L. Poulos, Chem. Rev., 2014, 114, 3919 CrossRef CAS PubMed;
(b) D. D. Ulmer and B. L. Vallee, Adv. Chem., 1971, 10, 187 Search PubMed.
- A. Guskov, A. Gabdulkhakov, M. Broser, C. Glöckner, J. Hellmich, J. Kern, J. Frank, F. Müh, W. Saenger and A. Zouni, ChemPhysChem, 2010, 11, 1160 CrossRef CAS PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55 CrossRef CAS PubMed.
- J. P. McEvoy, J. A. Gascon, V. S. Batista and G. W. Brudvig, Photochem. Photobiol. Sci., 2005, 4, 940 Search PubMed; J. Dasgupta, G. M. Ananyev and G. C. Dismukes, Coord. Chem. Rev., 2008, 252, 347 CrossRef CAS PubMed; G. M. Ananyev, L. Zaltsman, C. Vasko and G. C. Dismukes, Biochim. Biophys. Acta, 2001, 1503, 52 CrossRef.
- Y. J. Park, J. W. Ziller and A. S. Borovik, J. Am. Chem. Soc., 2011, 133, 9258 CrossRef CAS PubMed; D. Shevela, S. Koroidov, M. M. Najafpour, J. Messinger and P. Kurz, Chem. – Eur. J., 2011, 17, 5415 CrossRef PubMed; M. M. Najafpour, T. Ehrenberg, M. Wiechen and P. Kurz, Angew. Chem., Int. Ed., 2010, 49, 2233 CrossRef PubMed; I. J. Hewitt, J.-K. Tang, N. T. Madhu, R. Clérac, G. Buth, C. E. Anson and A. K. Powell, Chem. Commun., 2006, 2650 RSC; C. P. Horwitz, J. T. Warden and S. T. Weintraub, Inorg. Chim. Acta, 1996, 246, 311 CrossRef; S. M. Gorun, R. T. Stibrany and A. Lill, Inorg. Chem., 1998, 37, 836 CrossRef; I. Gil de Muro, M. Insausti, L. Lezama, M. K. Urtiaga, M. I. Arriortua and T. Rojo, Dalton Trans., 2000, 3360 RSC.
- A. Mishra, W. Wernsdorfer, K. A. Abboud and G. Christou, Chem. Commun., 2005, 54 RSC; A. Mishra, J. Yano, Y. Pushkar, V. K. Yachandra, K. A. Abboud and G. Christou, Chem. Commun., 2007, 1538 RSC.
- V. Kotzabasaki, R. Inglis, M. Siczek, T. Lis, E. K. Brechin and C. J. Milios, Dalton Trans., 2011, 40, 1693 RSC.
- E. Y. Tsui, J. S. Kanady and T. Agapie, Inorg. Chem., 2013, 52, 13833 CrossRef CAS PubMed.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS; J. R. Friedman, M. P. Sarachik, J. Tejada and R. Ziolo, Phys. Rev. Lett., 1996, 76, 3830 CrossRef; G. E. Kostakis, A. M. Ako and A. K. Powell, Chem. Soc. Rev., 2010, 39, 2238 RSC.
- A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117 CrossRef CAS PubMed; Q. Wu, Y.-G. Li, Y. H. Wang, R. Clérac, Y. Lu and E.-B. Wang, Chem. Commun., 2009, 5743 RSC; O. Roubeau and R. Clérac, Eur. J. Inorg. Chem., 2008, 4325 CrossRef.
- R. Ishikawa, R. Miyamoto, H. Nojiri, B. K. Breedlove and M. Yamashita, Inorg. Chem., 2013, 52, 8300 CrossRef CAS PubMed.
- J. Vallejo, A. Pascual-lvarez, J. Cano, I. Castro, M. Julve, F. Lloret, J. Krzystek, G. De Munno, D. Armentano, W. Wernsdorfer, R. Ruiz-García and E. Pardo, Angew. Chem., Int. Ed., 2013, 125, 14325 CrossRef.
- A. Grigoropoulos, M. Pissas, P. Papatolis, V. Psycharis, P. Kyritsis and Y. Sanakis, Inorg. Chem., 2013, 52, 12869 CrossRef CAS PubMed.
- B. H. He, M. L. Tong and X. M. Chen, Coord. Chem. Rev., 2005, 249, 545 CrossRef PubMed.
- A. J. Fletcher, K. M. Thomas and M. J. Rosseinsky, J. Solid State Chem., 2005, 178, 2491 CrossRef CAS PubMed; S. Qui and G. Zhu, Coord. Chem. Rev., 2009, 253, 2891 CrossRef PubMed; S. T. Meek, J. A. Greathouse and M. D. Allendorf, Adv.
Mater., 2011, 23, 249 CrossRef PubMed.
-
F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry: A Comprehensive Text, John Wiley & Sons, 4th edn, 1980 Search PubMed.
- A. Prescimone, J. Sanchez-Bentez, K. V. Kamenev, J. E. Warren, A. R. Lennie, M. Murrie, S. Parsons and E. K. Brechin, Z. Naturforsch., B: Chem. Sci., 2010, 65, 221 Search PubMed; S. R. Cooper and M. J. Calvin, J. Am. Chem. Soc., 1977, 99, 6623 CrossRef CAS.
- S. Natarajan, B. R. Srinivasan, J. K. Sundar, K. Ravikumar, R. V. Krishnakumar and J. Suresh, J. Chem. Sci., 2012, 124, 781 CrossRef CAS.
- L.-C. Yu, Z.-F. Chen, H. Liang, C.-S. Zhou and Y. Li, J. Mol. Struct., 2005, 750, 35 CrossRef CAS PubMed.
- S. H. Dale and M. R. J. Elsegood, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, m540 Search PubMed.
- M.-X. Li, G.-Y. Xie, Y.-D. Gu, J. Chen and P.-J. Zheng, Polyhedron, 1995, 14, 1235 CrossRef CAS.
- E. Bartolomé, J. Bartolomé, S. Melnic, D. Prodius, S. Shova, A. Arauzo, J. Luzón, F. Luis and C. Turta, Dalton Trans., 2013, 42, 10153 RSC.
- R. Schenker, M. N. Leuenberger, G. Chaboussant, D. Loss and H. U. Güdel, Phys. Rev. B: Condens. Matter, 2005, 72, 184403 CrossRef.
- D. L. Huber, Phys. Rev. B, 1965, 139, A1684.15.08.2013 Search PubMed.
- A. C. Anderson and J. E. Robichaux, Phys. Rev. B: Solid State, 1971, 3, 1410 CrossRef.
- M. Orendáč, L. Sedláková, E. Čižmár, A. Orendáčová, A. Feher, S. A. Zvyagin, J. Wosnitza, W. H. Zhu, Z. M. Wang and S. Gao, Phys. Rev. B: Condens. Matter, 2010, 81, 214410 CrossRef.
- A. Arauzo, A. Lazarescu, S. Shova, E. Bartolomé, R. Cases, J. Bartolomé and C. Turta, Dalton Trans., 2014, 43, 12342 RSC.
- K. D. Magers, C. G. Smith and D. T. Sawyer, Inorg. Chem., 1978, 17, 515 CrossRef CAS.
- Y. Sato and N. Tanaka, Bull. Chem. Soc. Jpn., 1968, 41, 2064 CrossRef CAS.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray data for MC. CCDC 1004697. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt01518e |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a