 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium(II) complexes containing functionalised β-diketonate ligands: developing a ferrocene mimic for biosensing applications†
Yeng Ying
Lee
,
D. Barney
Walker
,
J. Justin
Gooding
* and
Barbara A.
Messerle
*
School of Chemistry, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: b.messerle@unsw.edu.au; justin.gooding@unsw.edu.au; Fax: +61 2 93854161; Tel: +61 2 93854653
First published on 26th June 2014
Abstract
Three series of ruthenium complexes with the general formula Ru(bpy)n(β-diketonato)3−n (bpy = 2,2′-bipyridine, n = 0, 1, 2) were prepared and investigated using cyclic voltammetry and UV-vis spectroscopy. Variation of both the number and electronic demand of the β-diketonato ligands resulted in well-defined modulation of the potential at which oxidation of the metal centre occurred. The observed potentials were shown to be in good agreement with calculated ligand electrochemical parameters. A novel ruthenium(II) complex with electrochemical behaviour similar to that of ferrocene was identified.
Introduction
Ferrocene is well established as an electrochemically active tag for investigating and monitoring biological activity in situ.1–9 This is largely due to the fact that, in addition to its favourable electrochemical properties, the ferrocenyl group can be readily functionalised and is considered to be relatively stable in aqueous, aerobic media. However, the ferricenium ion formed following oxidation has been shown to decompose when exposed to chloride ions,10–13 potentially limiting the application of ferrocene as a redox label in sensors designed for long-term analyte monitoring (e.g. implantable devices).The stable and reversible nature of the Ru2+/3+ redox couple suggests that ruthenium complexes could be an attractive alternative to ferrocene for biological sensors and several groups have explored this possibility.14–22 An advantage of utilising ruthenium species is that the potential at which oxidation of the metal centre occurs can be influenced by the electronic demand of the ligands occupying the primary coordination sphere. Large changes in the E1/2 of a Ru2+ centre can be induced either by introducing anionic ligands23,24 and/or by changing the number of π-acceptor coordinating species around the metal.25–30 More subtle ‘tuning’ of the redox potential has previously been demonstrated by attaching either electron-withdrawing or electron-donating groups (EWG or EDG) to the peripheral sites around the coordinating ligands.28,31,32
Here we present the synthesis and electrochemical characterisation of three series of ruthenium complexes (of the general formulae [Ru(bpy)2(β-diketonato)](PF6), [Ru(β-diketonato)3] and [Ru(bpy)(β-diketonato)2], bpy = 2,2′-bipyridine) wherein the number and type of ligand is varied in order to tune the redox potential of the metal centre towards values suitable for biosensing applications. We take advantage of the fact that the potential at which oxidation of these ruthenium complexes occurs can be attenuated by varying the substituents on the β-diketonato ligands and identify several candidates for biosensor integration that have a very similar electrochemical profile to that of ferrocene.
Results and discussion
Synthesis
Three series of ruthenium complexes with β-diketonato ligands were prepared (Scheme 1) where the complexes of Series I each contains a single β-diketonato ligand, Series II, three β-diketonato ligands and Series III, two β-diketonato ligands.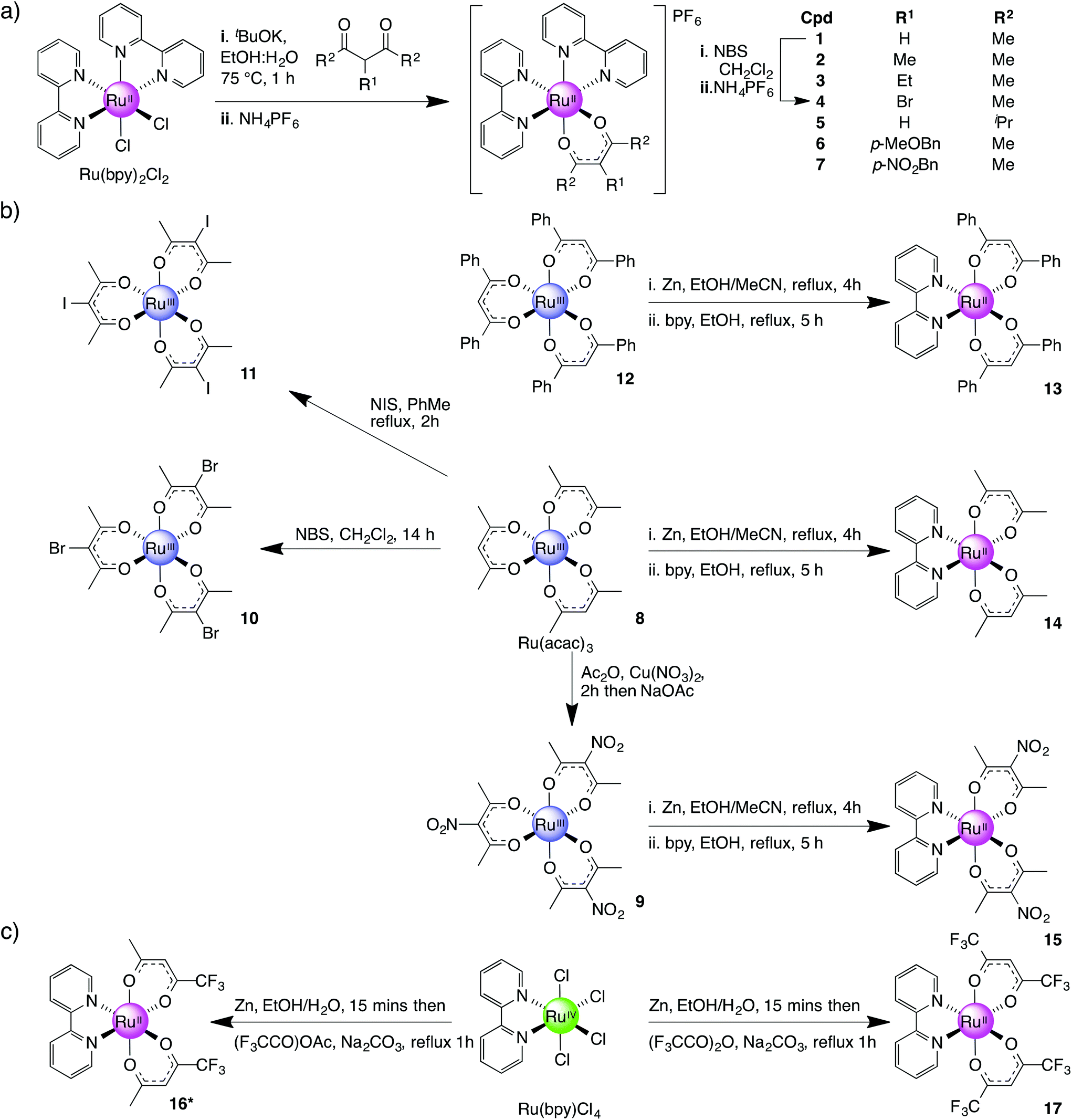 | ||
| Scheme 1 Synthesis of complexes 1–17. | ||
Complex 13 was synthesised in two steps via a bis-acetonitrile (Ru(dbm)2(MeCN)2) (dbm = dibenzoylmethane) intermediate. This intermediate was accessed by the reduction of complex 12 with activated zinc dust in refluxing EtOH followed by cooling and the addition of excess MeCN. The mixture was then refluxed for a further 2 h resulting in a colour change from bright red to orange.45 Treatment of the intermediate (Ru(dbm)2(MeCN)2) with an equimolar amount of bpy in refluxing EtOH gave Ru(dbm)2(bpy) (13) as a dark green solid in 60% yield. The methyl analogue (R2 = Me) was prepared using the same procedure (8 was readily converted to 14 in a 60% overall yield). This method also proved successful for the preparation of nitro-analogue 15 but treatment of the tri-bromo/iodo ruthenium(III) complexes (10, 11) with elemental zinc resulted in dehalogenation of the ruthenium species and instead formed 14 along with other decomposition products.
A second approach to preparing Series III complexes via Ru(bpy)(Cl)4 was investigated concurrently. The starting material was obtained in 85% yield by dissolving RuCl3·3H2O in dilute HCl, adding 1.2 equiv. of bpy and then leaving the solution for 21 days at room temperature.46 Subsequent treatment of Ru(bpy)(Cl)4 with activated zinc dust in EtOH–H2O for 15 min followed by the addition of Na2CO3 and either 1,1,1-trifluoropentane-2,4-dione (tfac) or 1,1,1,5,5,5-hexafluoropentane-2,4-dione (hfac) followed by refluxing for 1 h resulted in the formation of complexes 16 and 17 in 25% and 33% yields respectively (Scheme 1c).47 Complex 16 exists as a mixture of three geometrical isomers in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 ratio of cis- and trans-isomers due to its asymmetric β-diketonato ligand. This was confirmed by 1H NMR where the following configurations were observed: trans-CF3-cis-H-[Ru(tfac)2(bpy)], cis-CF3-cis-H-[Ru(tfac)2(bpy)] and cis-CF3-trans-H-[Ru(tfac)2(bpy)]. This is further supported by the presence of four signals in 19F NMR (see Experimental section).
:1:2 ratio of cis- and trans-isomers due to its asymmetric β-diketonato ligand. This was confirmed by 1H NMR where the following configurations were observed: trans-CF3-cis-H-[Ru(tfac)2(bpy)], cis-CF3-cis-H-[Ru(tfac)2(bpy)] and cis-CF3-trans-H-[Ru(tfac)2(bpy)]. This is further supported by the presence of four signals in 19F NMR (see Experimental section).
Whilst this route initially appeared to be a more direct way of preparing Ru(bpy)(β-diketonato)2 complexes compared with preforming the Ru(β-diketonato)3 species first (Scheme 1b) our observation was that the overall yields tended to be significantly lower and in some cases the final product did not form at all. However this method did allow us to furnish sufficient amounts of complexes 16 and 17 for subsequent electrochemical analysis.
Cyclic voltammetry
All complexes in Series I–III were shown to undergo a redox cycle (assumed to be Ru2+/3+) with a peak separation (ΔE) between 59 to 95 mV, and ipa/ipc at near unity indicating that the complexes were both electrochemically and chemically reversible. The E1/2 of the complexes was independent of the scan rate (ν) and the redox processes were diffusion-controlled as ipvs. ν1/2 was found to be linear. Waves were assigned by comparison to those of analogous metal complexes.48| [Ru(bpy)2((R2C(O))2CR1))]PF6 |
E
1/2a/V |
|---|---|
| a E 1/2 vs. FcH+/0 in 0.01 M AgNO3 in MeCN with 0.1 M NBu4PF6, ν = 0.1 V s−1. | |
| 1 (R1 = H, R2 = Me) | 0.228 |
| 2 (R1 = Me, R2 = Me) | 0.128 |
| 3 (R1 = Et, R2 = Me) | 0.181 |
| 4 (R1 = Br, R2 = Me) | 0.292 |
| 5 (R1 = H, R2 = iPr) | 0.201 |
| 6 (R1 = p-MeOBn, R2 = H) | 0.167 |
| 7 (R1 = p-NO2Bn, R2 = H) | 0.194 |
By the same argument, introducing an electron-withdrawing group (EWG) should result in an anodic shift in the E1/2 and this effect was observed for complex 4 (R1 = Br) when compared with 1 (ΔE1/2 = 64 mV).
As expected, complex 5 (R2 = iPr) and complex 1 (R2 = Me) had almost identical electrochemical profiles (ΔE1/2 = 27 mV). The potentials at which complexes 6 and 7 were oxidised are quite similar (167 mV and 194 mV respectively) despite having functional groups with quite different electronic demands in the para position of the benzyl group (OMe vs. NO2). This is not surprising considering that the aromatic ring is not in direct conjugation with the diketonate portion of the ligand bound to the ruthenium(II) ion. Consequently a modest cathodic shift (ΔE1/2 = 61 mV for 6, ΔE1/2 = 34 mV for 7) is observed in both cases due to the slight electron donating effect of the benzylic CH2 group.
| Series II |
E
1/2a/V |
|---|---|
| 8 (acac) | −1.06 |
| 9 (NO2-acac) | −0.389 |
| 10 (Br-acac) | −0.830 |
| 11 (I-acac) | −0.829 |
| 12 (dbm) | −0.906 |
It might be expected that moving from Series II to Series III compounds, wherein one β-diketonato ligand is substituted for a bpy ligand, would result in a set of complexes with an E1/2 closer to the desired redox window (−0.743 V to +0.067 V vs. FcH+/0, −0.343 V to +0.467 V vs. Ag/AgCl).
The replacement of a β-diketone in 12 and 8 by bpy to form 13 and 14 raised the E1/2 by 490 mV and 672 mV respectively. This was attributed to the fact that bpy is a π-acceptor – and the HOMO–LUMO gap of the complex could be increased and may result in a more stabilised Ru2+ complex. The stabilising effect of bpy works in concert with the NO2-acac ligands used to form 15 resulting in an observed E1/2 of −54 mV (vs. FcH+/0). In the same way it was possible to anodically shift the potential at which the ruthenium metal centre is oxidised by replacing the acac ligands with tri- and hexafluorinated 2,4-diones. A comparison of the E1/2s of 16 with 14 demonstrates this effect (ΔE1/2 = 391 mV). The potential at which 17 oxidised was anodically shifted by 482 mV compared to that of 16. This difference corresponds to the number of trifluoromethyl substituents on the ligands: there are four in 17 compared to two in 16. Of the Series III complexes, 15 and 16 were each shown to have an E1/2 very close to that of ferrocene (E1/2 = −54 mV and −87 mV respectively).
Diffusion coefficient, Do
The diffusion coefficients, Do, of complexes 1–17 in acetonitrile were obtained using the Randles–Sevcik equation:
| i p = 2.69 × 105n3/2ADo1/2Cν1/2 | (1) |
| Series I | D o/10−9 m2 s−1 |
|---|---|
| 1 (R1 = H, R2 = Me) | 1.759 |
| 2 (R1 = Me, R2 = Me) | 1.489 |
| 3 (R1 = Et, R2 = Me) | 1.182 |
| 4 (R1 = Br, R2 = Me) | 1.330 |
| 5 (R1 = H, R2 = iPr) | 1.561 |
| 6 (R1 = p-MeOBn, R2 = H) | 1.495 |
| 7 (R1 = p-NO2Bn, R2 = H) | 1.346 |
| Series III | |
|---|---|
| 13 (dbm) | 0.100 |
| 14 (acac) | 1.354 |
| 15 (NO2-acac) | 0.645 |
| 16 (tfac) | 1.745 |
| 17 (hfac) | 1.369 |
The electrode area on the glassy carbon electrode, A, was determined using the known Do of ferrocene in acetonitrile with 0.1 M NBu4PF6 as a supporting electrolyte (2.24 × 10−5 cm2 s−1).50
Complexes with bulky ligands, and those of higher charge, would be expected to have smaller Do values as they would be expected to migrate more slowly in solution.15,51,52 Additionally, changes in electron density around ruthenium brought on by donor/acceptor groups on β-diketonates will affect the degree to which counterions will attach to the reduced complexes.53 The structurally simplest complex in Series I, 1, has the largest Do while the rest of the complexes are between 1.182 to 1.561 × 10−9 m2 s−1. However, the bulkiest complex of the series did not have the smallest Do. This could be attributed to the interaction between these charged complexes of Series I with acetonitrile and the supporting electrolytes.
In Series III, the Do for 13, which contains the bulkiest ligand, dbm, is about one order of magnitude smaller than that of 8. In the same way complex 17, with two trifluoromethyl groups on each β-diketonato ligand, has a smaller Do than 16 which only has one trifluoromethyl group on each ligand. Complex 15 has a lower Do when compared to 16 and 17, which can possibly be attributed to its interaction with the supporting electrolyte as the nitro group, although formally neutral, has considerable polarisation between its nitrogen and oxygen atoms.
Relationships between the nature of the ligands and E1/2
The ruthenocycle formed by the coordination of acac-type ligands to Ru cations can be considered pseudo-aromatic. As such, the Hammett constant can be used to correlate the combined electronic effects of ‘para’ and ‘meta’ substituents on the β-diketonates with the potential at which the metal centres oxidised on the basis that such complexes can be thought of as having similar geometries and a common redox centre, for which linear free energy relationships can be established.54–56 In this study correlation of the calculated Hammett constants with the recorded E1/2s is limited by the fact that the contribution of the bpy ligands is not included. Consequently, a reasonably linear correlation was observed for Series II and III but not for Series I (see ESI†).Instead a more comprehensive method for probing the relationship between the structural and electronic features of a complex and its redox potential was applied.57 The most commonly utilised additive model was originally developed by Lever58 and has proved to be a useful tool for investigating metal-centred redox processes. The ligand electrochemical parameter, EL(L), used in this model and based on the Ru2+/3+ redox couple, has been widely applied to examine the correlation between ligands and E1/2 of metal complexes. For a metal (M) centre bound to multiple varying ligands (Xx, Yy, Zz), EL(L) is defined as:
| ∑E(L) = xEL(X) + yEL(Y) + zEL(Z). | (2) |
This formula can be used to predict the E1/2 of complexes with an octahedral geometry.58
The ∑E(L) was calculated for all complexes in Series I–III where values for E(L)(L) were available (Table S4). A strongly linear correlation was established between E(L)(L) and E1/2 of the complexes of Series I–III suggesting that the ligand contributions are additive (Fig. 1).
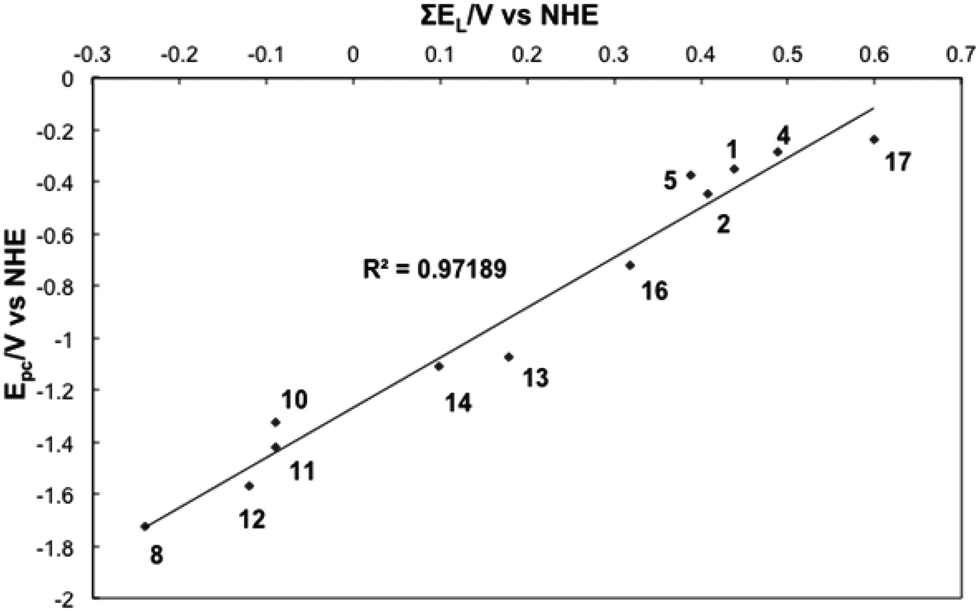 | ||
| Fig. 1 ∑E(L)vs. E1/2 for selected complexes. | ||
Correlation between UV-Vis absorbance and E1/2
UV-vis spectra of complexes 1–7 (Series I) were recorded in MeCN at a concentration of 0.05 mM and the data is summarised in Table 4. In total, there are two strong absorption bands assigned to π→π* intraligand transitions in the UV region and three MLCT bands dπ (Ru2+)→π* (L) (L = bpy, β-diketonate) in the visible region. For complexes 1–3 the data reported here is in agreement with literature values.48,59,60 The UV-vis spectrum for complex 4 (R1 = Br, R2 = Me) shows a slight hypsochromic shift in the MLCT bands (504 vs. 515 nm, 558 vs. 568 nm) when compared with those of 1 that could be indicative of an increased HOMO–LUMO gap. This is in agreement with the electrochemical data as a notably higher potential is required to oxidise the Ru(II) metal centre of 4 (E1/2 = 292 mV) when compared with 1 (E1/2 = 228 mV). Overall, the absorbance spectra of all Series I complexes are dominated by the influence of the anionic β-diketonato ligand and are consequently very similar to each other (see Fig. S4† for spectra).61| Complex | λ/nm (ε/103 M−1 cm−1) |
|---|---|
| 1 (R1 = H, R2 = Me) | 247 (28.4), 295 (53.8), 369 (11.3), 515 (9.3), 568sh (6.1) |
| 2 (R1 = Me, R2 = Me) | 247 (22.9), 296 (46.5), 374 (10), 521 (7.5), 586sh (5.1) |
| 3 (R1 = Et, R2 = Me) | 247 (26.0), 295 (5.9), 373 (11.2), 519 (10.4), 580sh (5.6) |
| 4 (R1 = Br, R2 = Me) | 246 (23.8), 294 (53.6), 370 (10.7), 504 (8.3), 558sh (5.5) |
| 5 (R1 = H, R2 = iPr) | 247 (25.2), 295 (47.8), 370 (10.2), 516 (7.7), 574sh (5.5) |
| 6 (R1 = p-MeOBn, R2 = H) | 246 (25.5), 295 (51.1), 376 (11.2), 518 (8.0), 577sh (5.7) |
| 7 (R1 = p-NO2Bn, R2 = H) | 247 (20.7), 295 (42.1), 373 (8.5), 512 (5.9), 575sh (4.1) |
In contrast to Series I, the UV-vis absorption spectra of complexes in series III are markedly different from each other (Table 5). The absorptions due to the four phenyl rings attached to complex 13 are clearly visible in the UV-vis spectrum at 246 nm. These additional aromatic groups may also account for the broad band observed around 300 nm which could result from a degree of overlapping with the π→π* intraligand transitions of bpy.
| Complex | λ/nm (ε/103 M−1 cm−1) |
|---|---|
| 13 (dbm) | 246 (28.8), 300 (34.8), 323 (31.2), 485 (9.0), 608 (7.5) |
| 14 (acac) | 277sh (24.5), 297 (17.5), 411 (8.8), 617 (4.9) |
| 15 (NO2-acac) | 249 (14.8), 287 (19.7), 297 (21.0), 408 (8.5), 546 (5.14) |
| 16 (tfac) | 248 (15.0), 287sh (27.5), 294 (25.4), 425 (7.7), 559 (6.7) |
| 17 (hfac) | 243 (6.5), 289 (16.2), 337 (1.6), 509 (5.6) |
The spectrum for complex 15 contains significant distortion of the band at around 300 nm where no clear maximum is visible. This could be attributed to an overlap of the nitro group absorption with the t2g→π* (MLCT) band.62 The strongly electron-withdrawing nitro group may also account for the significantly blue-shifted MLCT maxima observed at 546 nm.
Both 16 and 17 contain strongly electron-withdrawing trifluoromethyl groups on the β-diketonato ligands. In line with this, the spectrum of complex 16 contains a MLCT band at 559 nm whereas the corresponding peak for 17 appears at 509 nm (Fig. 2 and 3).
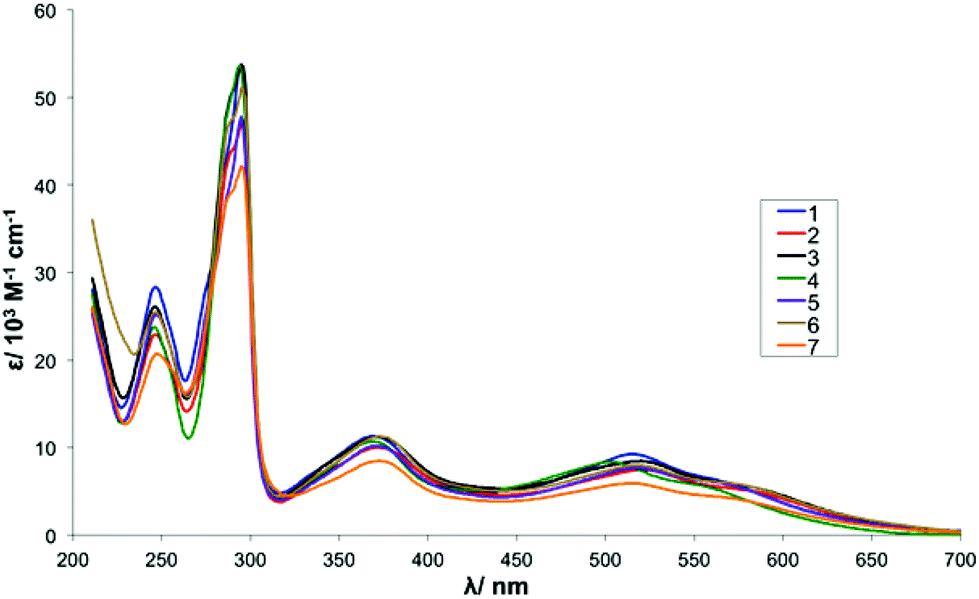 | ||
| Fig. 2 UV-Vis absorbance spectra for complexes 1–7. | ||
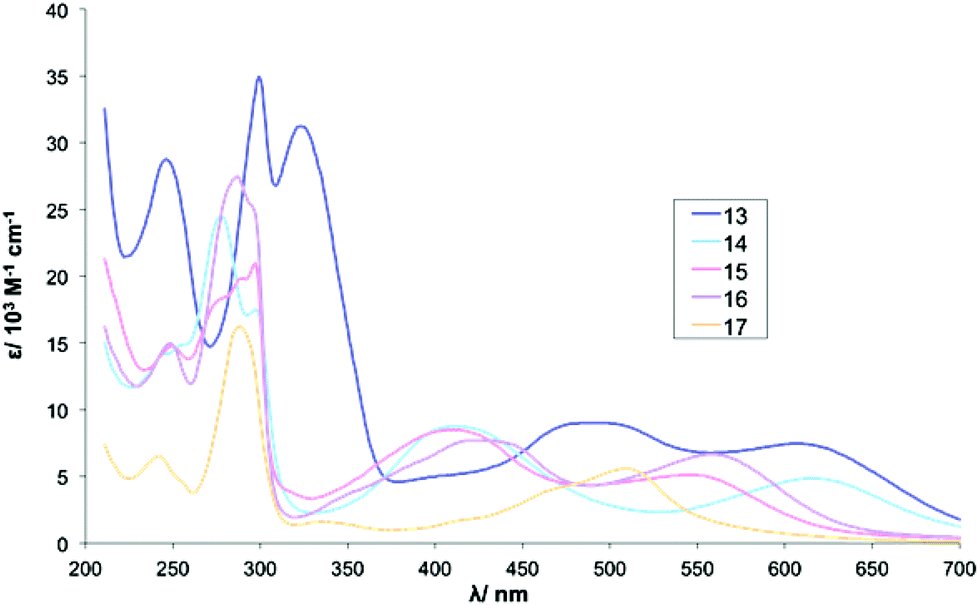 | ||
| Fig. 3 UV-Vis absorbance spectra for complexes 13–17. | ||
Conclusions
In this paper, ruthenium complexes bearing bpy and β-diketonato ligands were prepared, fully characterised and investigated using cyclic voltammetry and UV-vis spectroscopy. Three series of ruthenium complexes were investigated: Series I (1–7) of the general formula [Ru(bpy)2(β-diketonato)](PF6); Series II (8–12) of the general formula Ru(β-diketonato)3 and Series III (13–17) of the general formula Ru(bpy)(β-diketonato)2. In line with previous studies, varying the substituents tethered to the β-diketonato ligand attenuated the E1/2 of the complexes. It was observed that EDG shifted the E1/2 cathodically whereas EWG shifted the E1/2 anodically. Furthermore the effects were shown to be cumulative based on the number of β-diketonato ligands bound to the metal centre. Further correlations were made between E1/2 of complexes and the ligand electrochemical parameter (∑E(L)). The ligand electrochemical parameter was shown to have a linear relationship with E1/2 for all three series.Of the complexes prepared, the E1/2 of complexes 7, 13, 14, 15 and 16 were demonstrated to have E1/2s within the range considered suitable for biological sensors (−0.743 V to +0.067 V vs. FcH+/0). Furthermore, the novel complex 15 and the previously reported complex 16 were demonstrated each to have an E1/2 very close to that of ferrocene (E1/2 = −54 mV and −87 mV vs. FcH+/0, respectively) suggesting that they would be also good candidates for redox labels in electroactive biosensors.
Experimental methods
All manipulations of metal complexes and air sensitive reagents were performed using either standard Schlenk techniques or in a nitrogen/argon filled Braun glove-box. Reagents were purchased from Aldrich Chemical Company Inc. or Alfa Aesar Inc. and were used without further purification unless otherwise stated. For the purposes of air sensitive manipulations and in the preparation of air sensitive complexes, pentane, hexane, dichloromethane and tetrahydrofuran were dispensed from a PuraSolv solvent purification system. Solvents were dried and distilled under an atmosphere of nitrogen using standard procedures and stored under nitrogen in glass ampoules, each fitted with a Youngs© Teflon valve prior to use. Ethanol (EtOH) was distilled from diethoxymagnesium and dimethylformamide (DMF) was first dried over molecular sieves (4 Å) and distilled. Argon (>99.999%) was obtained from Air Liquide and used as received. Nitrogen gas for Schlenk line operation comes from in-house liquid nitrogen boil-off. 1H and 13C NMR spectra were recorded on Bruker DPX300, DMX400, DMX500 and DMX600 spectrometers operating at 300, 400, 500 and 600 MHz (1H) respectively and 75, 100, 125 and 150 MHz (13C) respectively. Unless otherwise stated, spectra were recorded at 25 °C and chemical shifts (δ) are quoted in ppm. Coupling constants (J) are quoted in Hz and have uncertainties of ±0.05 Hz for 1H and ±0.5 Hz for 13C. 1H and 13C NMR chemical shifts were referenced internally to residual solvent resonances. Deuterated solvents were purchased from Cambridge Stable Isotopes and used as received. Microanalyses were carried out at the Campbell Micro-analytical Laboratory, University of Otago, New Zealand or at the Research School of Chemistry, the Australian National University, Canberra, Australia. Mass spectra were acquired using a Thermo LTQ Orbitrap XL located in the Bio-analytical Mass Spectrometry Facility (BMSF) of the Mark Wainwright Analytical Centre, UNSW or on a Micromass ZQ (ESI-MS) mass spectrometer located in the School of Chemistry, UNSW. M is defined as the molecular weight of the compound of interest or cationic fragment for cationic metal complexes.Cyclic voltammetry was performed using an Autolab PGSTAT 12 potentiostat (Eco Chemie, Netherlands). The CV data was processed using Nova Windows software. A conventional three-electrode electrochemical cell comprised of a working electrode (glassy carbon), a counter electrode (platinum wire) and a reference electrode (aqueous or non-aqueous depending on the solvent system of the experiment) was used for all analysis. The aqueous reference electrode was a Ag/AgCl (3 M KCl) electrode (CH Instruments, Inc., TX, USA) while the non-aqueous reference electrode was a Ag/Ag+ electrode (CH Instruments, Inc., TX, USA) which was referenced against ferrocene. The working electrodes used were glassy carbon electrodes (CH Instruments, Inc., TX, USA). The solutions for electrochemical measurements were deoxygenated with nitrogen gas for 10 min prior to measurements and kept under a blanket of nitrogen during the course of measurements.
UV-Vis spectra were recorded on Shimadzu UV-2401PC in dry MeCN (5 × 10−4 M) and reported as λmax/nm (ε/M−1 cm−1).
For characterisation of previously reported compounds see the ESI.† All complexes were isolated as a racemic mixture of Δ and Λ enantiomers.
[Ru(bpy)2(eacac)][PF6], 3
Ru(bpy)2Cl2·2H2O (0.406 g, 0.780 mmol, 1 equiv.) was dissolved in degassed H2O–EtOH (1
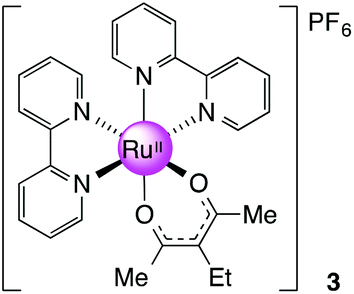 :1) and heated to 75 °C for 30 minutes. 3-ethyl-2,4-pentanedione (eacac, 0.100 g, 0.780 mmol, 1.5 equiv.) was added to the solution followed by t-BuOK (0.131 g, 1.17 mmol, 1.5 equiv.). The mixture was then stirred at 75 °C for 1 h and cooled to room temperature before NH4PF6 (0.699 g, 4.29 mmol, 5.5 equiv.) was added to precipitate the product. The solid was collected and recrystallised from CH2Cl2–hexane to give the crude products. The dark solid was then purified using column chromatography (silica gel, CH2Cl2–MeCN 4:1). Yield: 0.05 g, 15%. 1H NMR (400 MHz, DMSO-d6): δ 0.92 (t, J = 6.8 Hz, 3H), 1.90 (s, 6H), 2.21 (q, J = 6.8 Hz, 2H), 7.21 (m, 2H), 7.66 (m, 2H), 7.74 (m, 2H), 7.83 (m, 2H), 8.16 (m, 2H), 8.62 (m, 4H), 8.75 (m, 2H) ppm. 13C NMR (150.90 MHz, DMSO-d6): δ 15.32, 23.51, 27.02, 109.61, 123.34, 123.45, 125.61, 126.37, 134.60, 136.44, 149.77, 152.73, 157.34, 158.76, 184.99 ppm. MS (ESI): m/z 541.1166 ([M]+ required 541.1172). Anal. Calcd for C27H27F6N4O2PRu: C, 47.30; H, 3.97; N, 8.17; found: C, 46.93; H, 3.94; N, 7.91.
:1) and heated to 75 °C for 30 minutes. 3-ethyl-2,4-pentanedione (eacac, 0.100 g, 0.780 mmol, 1.5 equiv.) was added to the solution followed by t-BuOK (0.131 g, 1.17 mmol, 1.5 equiv.). The mixture was then stirred at 75 °C for 1 h and cooled to room temperature before NH4PF6 (0.699 g, 4.29 mmol, 5.5 equiv.) was added to precipitate the product. The solid was collected and recrystallised from CH2Cl2–hexane to give the crude products. The dark solid was then purified using column chromatography (silica gel, CH2Cl2–MeCN 4:1). Yield: 0.05 g, 15%. 1H NMR (400 MHz, DMSO-d6): δ 0.92 (t, J = 6.8 Hz, 3H), 1.90 (s, 6H), 2.21 (q, J = 6.8 Hz, 2H), 7.21 (m, 2H), 7.66 (m, 2H), 7.74 (m, 2H), 7.83 (m, 2H), 8.16 (m, 2H), 8.62 (m, 4H), 8.75 (m, 2H) ppm. 13C NMR (150.90 MHz, DMSO-d6): δ 15.32, 23.51, 27.02, 109.61, 123.34, 123.45, 125.61, 126.37, 134.60, 136.44, 149.77, 152.73, 157.34, 158.76, 184.99 ppm. MS (ESI): m/z 541.1166 ([M]+ required 541.1172). Anal. Calcd for C27H27F6N4O2PRu: C, 47.30; H, 3.97; N, 8.17; found: C, 46.93; H, 3.94; N, 7.91.
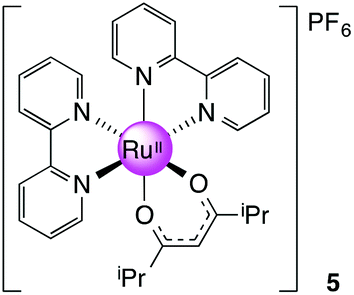
[Ru(bpy)2(dmhd)][PF6], 5
Prepared as for 3 with 0.100 g (0.640 mmol, 1.2 equiv.) of 2,6-dimethyl-3,5-heptanedione (dmhd). Yield: 0.37 g, 60%. 1H NMR (300 MHz, DMSO-d6): δ 0.58 (d, J = 6.8 Hz, 6H), 0.79 (d, J = 6.8 Hz, 6H), 2.25 (qq, J = 6.8, 6.8 Hz, 2H), 5.34 (s, 1H), 7.23 (ddd, J = 7.3, 7.3, 1.3 Hz, 2H), 7.73 (ddd, J = 7.3, 7.3, 1.3 Hz, 2H), 7.83–7.90 (m, 4H), 8.16 (ddd, J = 7.3, 7.3, 1.5 Hz, 2H), 8.50 (dd, J = 7.3, 1.3 Hz, 2H), 8.64 (dd, J = 7.3, 1.3 Hz, 2H), 8.75 (dd, J = 7.3, 1.5 Hz, 2H) ppm. 13C NMR (150.90 MHz, DMSO-d6): δ 19.98, 38.18, 94.27, 123.01, 123.21, 125.40, 126.03, 134.74, 136.35, 149.63, 153.11, 157.46, 158.75, 192.85 ppm. MS (ESI): m/z 569.1547 ([M]+ required 569.1485). Anal. Calcd for C29H31F6N4O2PRu: C, 48.81; H, 4.38; N, 7.85. Found: C, 48.15; H, 4.52; N, 7.70.
[Ru(bpy)2(mbpd)][PF6], 6
Prepared as for 3 with 0.111 g (0.503 mmol, 1.2 equiv.) of mbpd. Yield: 0.20 g, 53%. 1H NMR (400 MHz, DMSO-d6): δ 1.79 (s, 6H), 3.57 (s, 2H), 3.73 (s, 3H), 6.86 (m, 4H), 7.22 (m, 2H), 7.71 (m, 2H), 7.80–7.88 (m, 4H), 8.22 (m, 2H), 8.67 (m, 4H), 8.78 (m, 2H) ppm. 13C NMR (100.64 MHz, DMSO-d6): δ 27.56, 34.70, 54.99, 106.26, 113.76, 123.38, 123.44, 125.57, 126.37, 128.03, 133.26, 134.64, 136.51, 149.74, 152.79, 157.28, 157.36, 158.68, 185.82 ppm. MS (ESI): m/z 633.1420 ([M]+ required 633.1434). Anal. Calcd for C33H31F6N4O3PRu: C, 50.97; H, 4.02, N, 7.20. Found: C, 50.34; H, 4.04; N, 7.24.

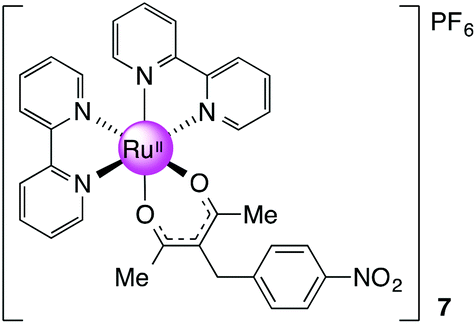
[Ru(bpy)2(nbpd)][PF6], 7
Prepared as for 3 with 0.113 g (0.480 mmol, 1 equiv.) of nbpd. Yield: 0.20 g, 27%. 1H NMR (300 MHz, DMSO-d6): δ 1.79 (s, 6H), 3.81 (s, 2H), 7.23 (m, 2H), 7.28 (m, 2H), 7.72 (m, 2H), 7.84–7.88 (m, 4H), 8.16 (m, 2H), 8.23 (m, 2H), 8.65 (m, 2H), 8.71 (m, 2H), 8.78 (m, 2H) ppm. 13C NMR (100.64 MHz, DMSO-d6): δ 27.74, 35.82, 105.25, 123.42, 123.46, 123.52, 125.61, 126.49, 128.39, 134.73, 136.65, 145.84, 149.77, 150.49, 152.85, 157.29, 158.68, 185.96 ppm. MS (ESI): m/z 648.1188 ([M]+ required 648.1179). Anal. Calcd for C32H28F6N5O4PRu: C, 48.49; H, 3.56, N, 8.84. Found: C, 47.85; H, 3.53; N, 8.80.
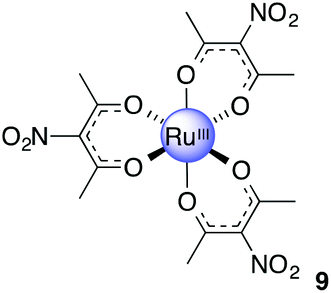
Ru(acac-NO2)3, 9
Acetic anhydride (30 mL) was added to finely ground Cu(NO3)2·3H2O (2.35 g, 9.73 mmol) to give a light blue suspension. The contents were stirred at 0 °C for 15 min after the flask was fitted with a calcium chloride drying tube. 8 (1.20 g, 3.01 mmol) was added to the cold deep blue solution. The mixture was stirred at 0 °C for 2 h and a further 2 h at room temperature. Ice (100 g), deionised water (100 g) and anhydrous sodium acetate (2.14 g, 26.1 mmol) were added to the now reddish brown mixture. The colour immediately turned greenish blue. The solution was left to stir for 18 h. The contents were filtered to give bright red powder. The solid was washed with water to give product as a bright red powder. Yield: 1.12 g, 70%. 1H NMR (400 MHz, CDCl3): δ −3.55 (s, 18H) ppm. MS (ESI): m/z 556.9813 ([M + Na]+ required 556.9826). Anal. Calcd for C15H18N3O12Ru: C, 33.78; H, 3.40; N, 7.88. Found: C, 33.01; H, 3.54; N, 7.65.
Ru(NO2-acac)2(bpy), 15
(a) Compound 9 (0.574 g, 1.08 mmol) was stirred in EtOH with activated zinc dust (0.5 g) for 1 h, during which time the colour changed from bright red to brown. MeCN (5 mL) was added to the brown mixture and refluxed for 4 h. The mixture was filtered through a bed of celite on which a brown layer remained. The crude product was subjected to silica gel column chromatography to first elute unreacted 9 with CH2Cl2 followed by EtOAc to flush the product out from the column as an orange fraction. Solvent was removed to give the title product as an orange solid. Yield: 0.46 g, 92%. 1H NMR (400 MHz, DMSO-d6): δ 2.09 (s, 6H), 2.11 (s, 6H), 2.73 (s, 6H) ppm. 13C NMR (100.64 MHz, DMSO-d6): δ 3.83, 26.59, 26.83, 128.46, 139.13, 183.15, 184.52 ppm. MS (ESI): m/z 495.0052 ([M + Na]+ 495.0060).
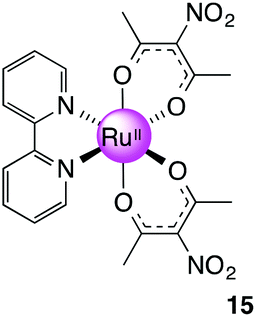
(b) The Ru(acac-NO2)2(MeCN)2 intermediate isolated in (a) (0.200 g, 0.424 mmol) and bpy (0.0660 g, 0.423 mmol) were added to a Schlenk flask followed by EtOH (∼15 mL). The reaction was refluxed for 5 h before solvent was removed under reduced pressure to give a dark brown solid. The solid was purified by column chromatography (silica gel, MeCN–CH2Cl2 1:5) to give a dark brown solid. Yield: 0.030 g, 13%. 1H NMR (500 MHz, DMSO-d6): δ 1.76 (s, 6H), 2.28 (s, 6H), 7.52 (m, 2H), 7.90 (m, 2H), 8.59 (m, 2H), 8.68 (m, 2H) ppm. 13C NMR (150.90 MHz, DMSO-d6): 26.57, 27.11, 122.74, 125.21, 128.44, 134.66, 152.07, 159.82, 182.14, 184.49 ppm. MS (ESI): m/z 569.0209 ([M + Na]+ required 569.0217). Anal. Calcd for C20H20N4O8Ru: C, 44.04; H 3.70; N 10.27. Found: C, 44.17; H, 3.77; N, 10.04.
Acknowledgements
Financial support from the University of New South Wales and the Australian Research Council (LP100200593) is gratefully acknowledged. We also would like to thank staff of the NMR Facility, Mark Wainwright Analytical Centre, UNSW for their assistance.Notes and references
- N. Metzler-Nolte and M. Salmain, in Ferrocenes, John Wiley & Sons, Ltd, 2008, pp. 499–639 DOI:10.1002/9780470985663.ch13.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5986 CrossRef CAS PubMed.
- M. F. R. Fouda, M. M. Abd-Elzaher, R. A. Abdelsamaia and A. A. Labib, Appl. Organomet. Chem., 2007, 21, 613–625 CrossRef CAS.
- M. Kind and C. Woell, Prog. Surf. Sci., 2009, 84, 230–278 CrossRef CAS PubMed.
- I. Willner and E. Katz, Angew. Chem., Int. Ed., 2000, 39, 1181–1218 CAS.
- Y. Wan, Y. Su, X. Zhu, G. Liu and C. Fan, Biosens. Bioelectron., 2013, 47, 1–11 CrossRef CAS PubMed.
- S. Martic, M. Labib, P. O. Shipman and H.-B. Kraatz, Dalton Trans., 2011, 40, 7264–7290 RSC.
- T. Joshi, G. J. Barbante, P. S. Francis, C. F. Hogan, A. M. Bond, G. Gasser and L. Spiccia, Inorg. Chem., 2012, 51, 3302–3315 CrossRef CAS PubMed.
- S. M. Khor, G. Liu, J. R. Peterson, S. G. Iyengar and J. J. Gooding, Electroanalysis, 2011, 23, 1797–1804 CrossRef CAS.
- R. Prins, A. R. Korwagen and A. G. T. G. Kortbeek, J. Organomet. Chem., 1972, 39, 335–344 CrossRef CAS.
- D. Ge and R. Levicky, Chem. Commun., 2010, 46, 7190–7192 RSC.
- R. Prins, A. R. Korswagen and A. G. T. G. Kortbeek, J. Organomet. Chem., 1972, 39, 335–344 CrossRef CAS.
- H. Ju and D. Leech, Phys. Chem. Chem. Phys., 1999, 1, 1549–1554 RSC.
- S. M. Zakeeruddin, D. M. Fraser, M. K. Nazeeruddin and M. Grätzel, J. Electroanal. Chem., 1992, 337, 253–283 CrossRef CAS.
- D. M. Fraser, S. M. Zakeeruddin and M. Grätzel, J. Electroanal. Chem., 1993, 359, 125–139 CrossRef CAS.
- C. Zhang, T. Haruyama, E. Kobatake and M. Aizawa, Anal. Chim. Acta, 2000, 408, 225–232 CrossRef CAS.
- S. Warren, T. McCormac and E. Dempsey, Bioelectrochemistry, 2005, 67, 23–35 CrossRef CAS PubMed.
- T. V. Federova, A. S. Vilesov, S. A. Kurzeev, E. V. Stepanova, E. O. Landesman and O. V. Koroleva, Appl. Biochem. Microbiol., 2006, 42, 550–557 CrossRef.
- E. V. Ivanova, V. S. Sergeeva, J. Oni, C. Kurzawa, A. D. Ryabov and W. Schuhmann, Bioelectrochemistry, 2003, 60, 65–71 CrossRef CAS.
- I. L. Medintz, D. Farrell, K. Susumu, S. A. Trammell, J. R. Deschamps, F. M. Brunel, P. E. Dawson and H. Mattoussi, Anal. Chem., 2009, 81, 4831–4839 CrossRef CAS PubMed.
- M.-Y. Wei, S.-D. Wen, X.-Q. Yang and L.-H. Guo, Biosens. Bioelectron., 2009, 24, 2909–2914 CrossRef CAS PubMed.
- N. A. Yusof, Asian J. Chem., 2011, 23, 1153–1157 CAS.
- W.-W. Yang, Y.-W. Zhong, S. Yoshikawa, J.-Y. Shao, S. Masaoka, K. Sakai, J. Yao and M. Haga, Inorg. Chem., 2012, 51, 890–899 CrossRef CAS PubMed.
- R. Cerón-Camacho, S. Hernández, R. L. Lagadec and A. D. Ryabov, Chem. Commun., 2011, 47, 2823–2825 RSC.
- H. Weizman and Y. Tor, J. Am. Chem. Soc., 2002, 124, 1568–1569 CrossRef CAS PubMed.
- M. Vrábel, M. Hocek, L. Havran, M. Fojta, I. Votruba, B. Klepetářá, R. Pohl, L. Rulíšek, L. Zendlová, P. Hobza, I. Shih, E. Mabery and R. Mackman, Eur. J. Inorg. Chem., 2007, 1752–1769 CrossRef.
- J. J. Rack, E. S. Krider and T. J. Meade, J. Am. Chem. Soc., 2000, 122, 6287–6288 CrossRef CAS.
- A. Endo, M. Kajitani, M. Mukaida, K. Shimizu and G. P. Sato, Inorg. Chim. Acta, 1988, 150, 25–34 CrossRef CAS.
- N. L. Frank and T. J. Meade, Inorg. Chem., 2003, 42, 1039–1044 CrossRef CAS PubMed.
- M. Haga, T. Matsumura-Inoue, K. Shimizu and G. P. Satô, J. Chem. Soc., Dalton Trans., 1989, 371–373 RSC.
- L. Wolf, E. Butter and H. Weinelt, Z. Anorg. Allg. Chem., 1960, 306, 87–93 CrossRef CAS.
- A. Endo, K. Shimizu and G. P. Satô, Chem. Lett., 1985, 1985, 581–584 CrossRef.
- S. Munery, J. Jaud and J. Bonvoisin, Inorg. Chem. Commun., 2008, 11, 975–977 CrossRef CAS PubMed.
- M. S. Chan and A. C. Wahl, J. Phys. Chem., 1985, 89, 5829–5831 CrossRef CAS.
- S. Munery, J. Jaud and J. Bonvoisin, Inorg. Chem. Commun., 2008, 11, 975–977 CrossRef CAS PubMed.
- J.-H. Olivier, J. Harrowfield and R. Ziessel, Chem. Commun., 2011, 47, 11179–11188 Search PubMed.
- J. P. Collman, Angew. Chem., Int. Ed., 1965, 4, 132–138 CrossRef.
- J. P. Collman, R. A. Moss, H. Maltz and C. C. Heindel, J. Am. Chem. Soc., 1961, 83, 531–534 CrossRef CAS.
- R. W. Kluiber, J. Am. Chem. Soc., 1960, 82, 4839–4842 CrossRef CAS.
- P. R. Singh and R. Sahai, Aust. J. Chem., 1967, 20, 639–648 CrossRef CAS.
- P. R. Singh and R. Sahai, Aust. J. Chem., 1967, 20, 649–655 CrossRef CAS.
- J. P. Collman, R. L. Marshall, W. L. I. Young and S. D. Goldby, Inorg. Chem., 1962, 1, 704–710 CrossRef CAS.
- I. R. Baird, S. J. Rettig, B. R. James and K. A. Skov, Can. J. Chem., 1999, 77, 1821–1833 CrossRef CAS.
- A. K. Gupta and R. K. Poddar, Indian J. Chem. Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2000, 39, 457–460 Search PubMed.
- T. Kobayashi, Y. Nishina, K. Shimizu and G. P. Satô, Chem. Lett., 1988, 1137–1140 CrossRef CAS.
- R. A. Krause, Inorg. Chim. Acta, 1977, 22, 209–213 CrossRef CAS.
- A. M. El-Hendawy, A. H. Al-Kubaisi and H. A. Al-Madfa, Polyhedron, 1997, 16, 3039–3045 CrossRef CAS.
- A. M. El-Hendawy, A. H. Al-Kubaisi and H. A. Al-Madfa, Polyhedron, 1997, 16, 3039–3045 CrossRef CAS.
- E. Eskelinen, M. Haukka, T.-J. J. Kinnunen and T. A. Pakkanen, J. Electroanal. Chem., 2003, 556, 103–108 CrossRef CAS.
- N. G. Tsierkezos, J. Solution Chem., 2007, 36, 289–302 CrossRef CAS.
- A. Endo, Y. Hoshino, K. Hirakata, Y. Takeuchi, K. Shimizu, Y. Furushima, H. Ikeuchi and G. P. Satô, Bull. Chem. Soc. Jpn., 1989, 62, 709–716 CrossRef CAS.
- H. Ikeuchi, K. Naganuma, M. Ichikawa, H. Ozawa, T. Ino, M. Sato, H. Yonezawa, S. Mukaida, A. Yamamoto and T. Hashimoto, J. Solution Chem., 2007, 36, 1243–1259 CrossRef CAS.
- A. Endo, Bull. Chem. Soc. Jpn., 1983, 56, 2733–2738 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- G. S. Patterson and R. H. Holm, Inorg. Chem., 1972, 11, 2285–2288 CrossRef CAS.
- S. J. Slattery, W. D. Bare, D. L. Jameson and K. A. Goldsby, J. Chem. Soc., Dalton Trans., 1999, 1999, 1347–1352 RSC.
- A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2007, 1473–1482 CrossRef CAS.
- A. B. P. Lever, Inorg. Chem., 1990, 29, 1271–1285 CrossRef CAS.
- A. Wu, J. Masland, R. D. Swartz, W. Kaminsky and J. M. Mayer, Inorg. Chem., 2007, 46, 11190–11201 CrossRef CAS PubMed.
- G. M. Bryant, J. E. Fergusson and H. K. J. Powell, Aust. J. Chem., 1971, 24, 257–273 CrossRef CAS.
- Y. Takahashi, H. Arakawa, H. Sugihara, K. Hara, A. Islam, R. Katoh, Y. Tachibana and M. Yanagida, Inorg. Chim. Acta, 2000, 310, 169–174 CrossRef CAS.
- K. Kuroda, K. Yoshitani, K. Kunigita, Y. Kamiiba and K. Watanabe, Bull. Chem. Soc. Jpn., 1976, 49, 2445–2450 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt01459f |
| This journal is © The Royal Society of Chemistry 2014 |