 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterocyclic substituted methanides as promising alternatives to the ubiquitous nacnac ligand†‡
David-R.
Dauer
and
Dietmar
Stalke
*
Institut für Anorganische Chemie der Georg-August-Universität Göttingen, Tammannstr. 4, 37077 Göttingen, Germany. E-mail: dstalke@chemie.uni-goettingen.de; Fax: +49 551 39 33373; Tel: +49 551 39 33000
First published on 26th May 2014
Abstract
A series of group 13 complexes containing deprotonated bisheterocyclomethanes have been prepared and structurally as well as spectroscopically characterised. In the case of the parent neutral homo-disubstituted bisheterocyclomethanes bis-(benzoxazol-2-yl)-methane (abbreviated as (NCOC6H4)2CH2) (1) and bis-(benzothiazol-2-yl)-methane (abbreviated as (NCSC6H4)2CH2) (2), two interesting ligand systems were investigated with respect to their coordination abilities toward group 13 metals. Upon deprotonation of the acidic methylene bridge in each case a β-diketiminate-like structure is formed, where the negative charge becomes delocalised about the whole ligand framework. There is a mesomeric stabilisation between the carbanionic and the amidic resonance structure. The ligands mentioned above were treated with AlMe3, AlMe2Cl and GaMe3 to achieve deprotonation of the backbone and coincidental metal complexation. The resulting complexes [Me2Al{(NCOC6H4)2CH}] (3), [Me2Al{(NCSC6H4)2CH}] (4), [ClMeAl{(NCOC6H4)2CH}] (5), [ClMeAl{(NCSC6H4)2CH}] (6), [Me2Ga{(NCOC6H4)2CH}] (7), and [Me2Ga{(NCSC6H4)2CH}] (8) were structurally characterized in the solid state by X-ray single crystal diffraction and in solution by different (heteronuclear) 1D/2D-NMR spectroscopic techniques. Each of those molecules shows a nearly planar arrangement with the group 13 metal cation coordinated by the two ring nitrogen atoms of the conjugated heterocycles. Furthermore, in this work (NCOC6H4)3C(C3H7) (9) could be isolated, which might turn out to be a promising analogue of the omnipresent trisoxazoline ligands in asymmetric catalysis.
Introduction
Referring to the monoanionic β-diketiminate ligand, the so-called nacnac ligand, enabling stabilisation of the main group elements in the low oxidation state, new ligand systems should be exploited mimicking those electronic and steric properties. In the case of nacnac substituted systems it is known that the scaffold tends to deviate from a planar arrangement within metal coordination by means of twisting or bending, and merely the backbone remains in plane. This deviation is caused by the flexible but bulky organic substituents at the imine moieties. Although this feature of the twisting seems to be problematic, it also offers an efficient shielding of the coordinated (low-valent) metal cations, which are sensitive towards reactions with electrophiles or dimerization.1Therefore, the new designed ligand systems shown in Scheme 1 should maintain more rigidity and lead to a more planar alignment in the corresponding metal complexes. Due to this planarity within the six-membered metalla heterocycle C3N2M the orbital overlap between the donor atoms and the chelated metal atom should be increased, so that a more efficient conjugation and hence delocalisation can be obtained.
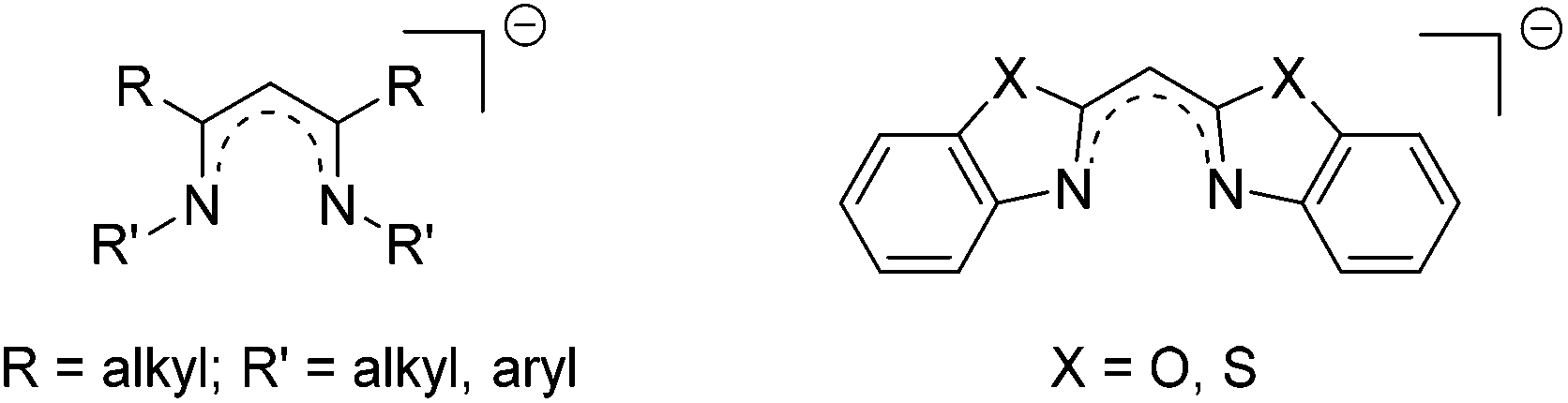 | ||
| Scheme 1 Similarities between the nacnac ligand (left) and the bisheterocyclomethanides (right). | ||
In former publications, we also investigated related compounds containing pyridyl substituents instead of benzoxazole and benzothiazole.2 Several complexes of this bis-(pyrid-2-yl)-methanide with group 1 and 13 metals could be synthesised and structurally characterized by X-ray diffraction: representing the group 1 complexes [([12]crown-4)2Li][Li{(2-NC5H4)2CH}] can be highlighted as a solvent separated ion pair or [(thf)2Li{(2-NC5H4)2CH}] as a monomeric lithiated compound both solely yielding Li–N contacts.3,4 Additionally, some group 13 complexes like [Me2Al{(2-NC5H4)2CH}] and [Me2Ga{(2-NC5H4)2CH}], where formally the (thf)2Li unit is replaced by a Me2Al or Me2Ga moiety, were prepared.5,6 Moreover, this kind of ligand system was varied by replacing the bridging CH unit isoelectronically by N,7,8 P9,10 or As.11,12 The corresponding main group metal complexes were investigated.
In the past decade many main group metal containing β-diketiminate structures were synthesised and fully characterized.13–25 The presented aluminium(III) and gallium(III) complexes 3–8, which are described in the following, can be compared with many analogue compounds of the Dipp-substituted nacnac ligand. These complexes embrace aluminium(III), gallium(III) and indium(III) compounds and, in the case of Al and Ga, also the reduced species in the oxidation state +I.26–28 The former range from the alkyl over the hydride to halide substituted metal centres. The latter, the so-called metallylenes, are rarely known and offer interesting prospects as ligands in catalysis, where they might replace N-heterocyclic carbenes or phosphanes.29,30 Furthermore the dialkylaluminium β-diketiminates show catalytic activities towards ring-opening polymerisation.31
The bisheterocyclomethanides show three optional donor sites: the deprotonated bridging carbanionic center, the ring-chalcogene (oxygen or sulfur) and -nitrogen atoms. Because O and S do not share the same good Lewis-donor abilities of the ring-nitrogen atoms, each of the metallated species shows a nacnac-like coordination, wherein the metal gets chelated by the two heteroaromatic nitrogen atom donors. It should be noted that the presented ligand systems show also analogy to the popular bis-(oxazolin-2-yl)-methanes, which are used in asymmetric catalysis. In contrast to 1 and 2 those ligands consist of saturated not benzannulated heterocycles with chiral centres, which can transfer chiral information in catalysis.32
Results and discussion
Syntheses
The herein discussed bisheterocyclomethanes 1 and 2 mimic the related β-diketiminate ligand. Therefore, formally the imine substituent in the case of nacnac is fixed to the backbone of the ligand by building up an additional five-membered heterocycle containing either oxygen or sulfur atoms as additional donor sites.As depicted in Scheme 2 both ligand systems were synthesised in a cyclocondensation reaction of a suitable linker derived from malonic acid and two equivalents of the corresponding ortho-substituted anilines.
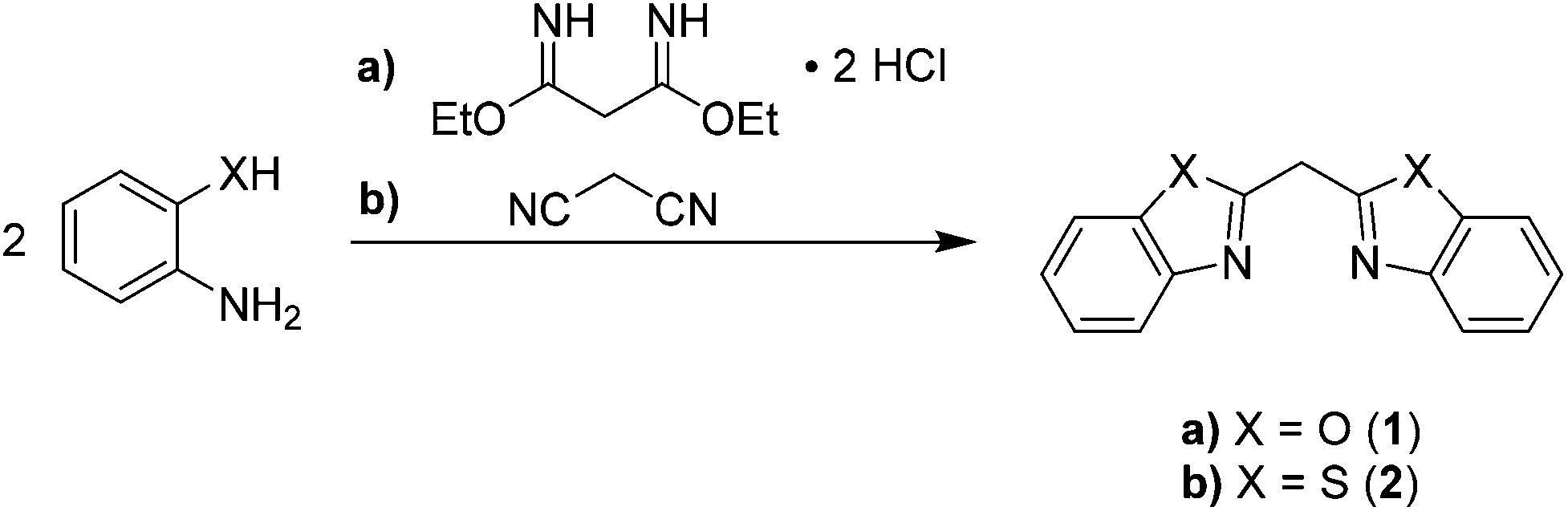 | ||
| Scheme 2 Cyclocondensation reactions for synthesis of the parent ligand systems 1 and 2. | ||
The reaction pathway (a) in Scheme 2 shows that the cyclocondensation reaction of the phenol derivative is more complicated as in the case of the thiophenol (b). Therefore, the former malononitrile has to be further activated before it undergoes a reaction with the phenolic nucleophile. This can be achieved by generating the derived ethylbisimidate dihydrochloride, wherein ammonia as well as ethanol act as leaving groups.33
The syntheses of the closely related [Me2Al{Dipp2nacnac}] and [Me2Ga{Dipp2nacnac}] can be achieved by a facile reaction of nacnacH with AlMe3 and GaMe3, respectively.13,14,31 Referring to this also LH 1 and 2 can be deprotonated at the acidic methylene bridge by release of methane and coordinated by the group 13 metal in a concerted way (Scheme 3). In this reaction protocol many different metallated compounds are accessible and can be compared structurally.
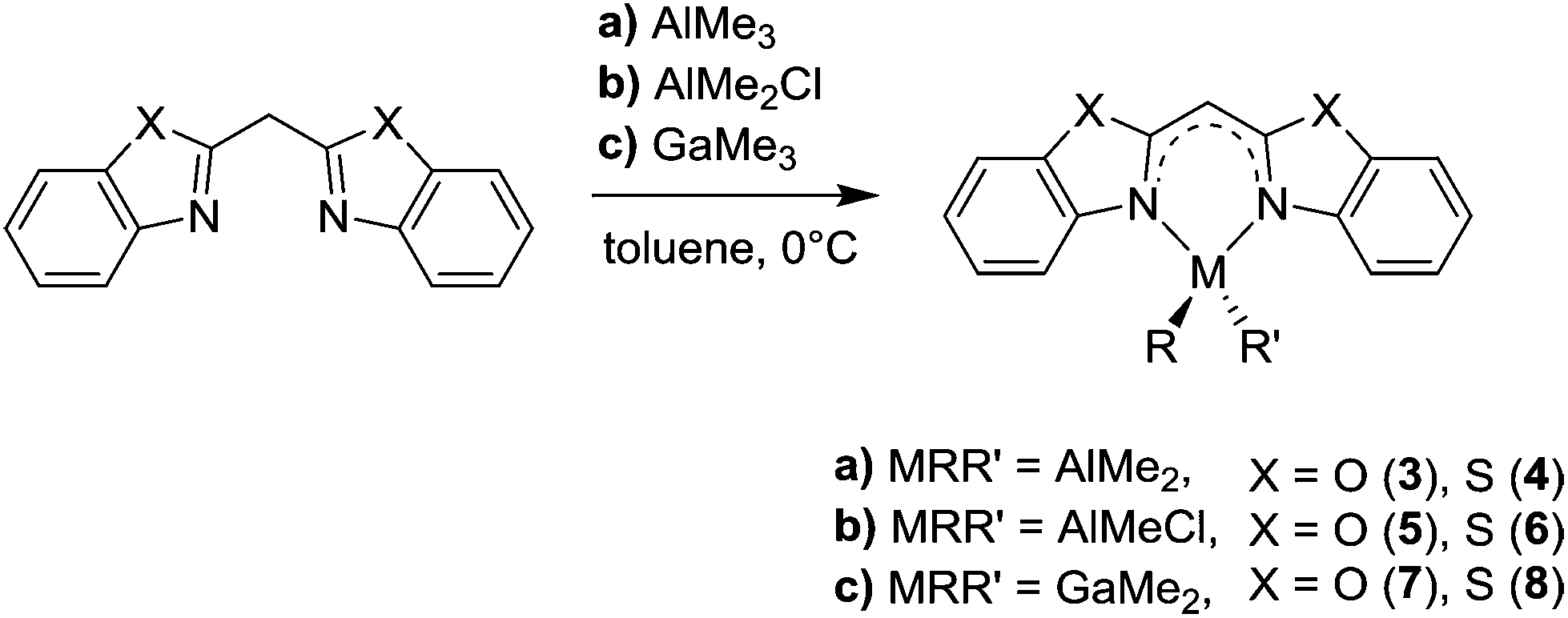 | ||
| Scheme 3 Syntheses of the metallated compounds 3–8. | ||
In this context the effect of the diverging metal coordination (M = Al, Ga) and moreover the related substituents at these metal cations (R = R′ = Me; R = Me, R′ = Cl) on the bonding situation within the different ligand systems should be investigated.
Structural data
In the following part a structural comparison of compounds 1–8, which could be obtained by the earlier mentioned reaction protocols, will be discussed. For a better commensurability also the molecular solid state structures of the parent ligand systems 1 and 2 have been determined by X-ray single crystal diffraction and are shown in Fig. 1 and 2.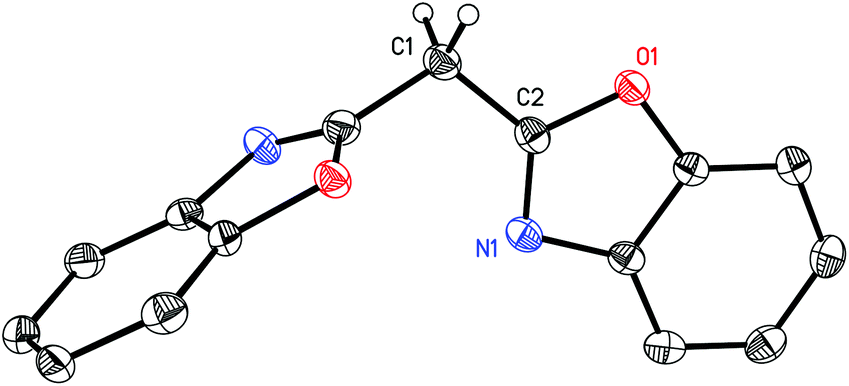 | ||
| Fig. 1 Molecular structure of (NCOC6H4)2CH2 (1). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity except the methylene bridge. Structural data are given in Tables 1 and 2. | ||
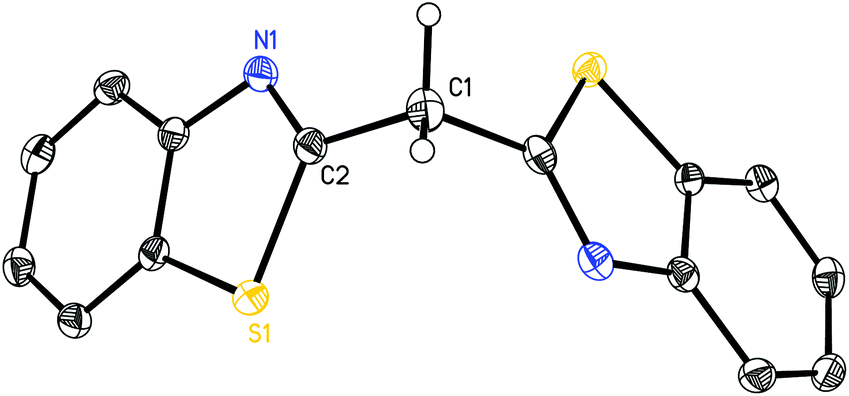 | ||
| Fig. 2 Molecular structure of (NCSC6H4)2CH2 (2). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity except the methylene bridge. Structural data are given in Tables 1 and 2. | ||
Exemplarily for the metallated species the crystal structures of 3, 6 and 8 are depicted in the anisotropic displacement parameter plots in Fig. 3–5. Selected (averaged) bond lengths and angles are listed in Table 1. The metallated species 3–8 have several intrinsic features in common, which have to be highlighted:
(a) In comparison with the deprotonated conjugated ligand system it is obvious that the free ligand LH shows in both cases a considerably larger torsion angle concerning the C–N bonds within the heterocycles.
(b) Upon deprotonation the whole ligand system becomes nearly planar in each molecule and the metal cations are coordinated exclusively by the two ring-nitrogen donor atoms.
(c) Furthermore, the metals are significantly dislocated from the ideal C3N2 planes, ranging from 10.93(27) to 29.57(26) pm, depending on the heteroatom in the backbone of the ligand.
(d) The experimentally determined C–C and C–N distances for 3–8 span the narrow range of 1.380(3) to 1.394(9) Å and 1.323(9) to 1.365(3) Å, respectively. These facts are an indicator of an apparent fully conjugated and delocalised ligand character, as far as the abovementioned bonds are concerned.
(e) The observed N–M–N bite angle inside the formed C3N2M metalla heterocycle is the narrowest one (89.2(2)–96.84(9)°) whereas all the other angles are wider than 120°.
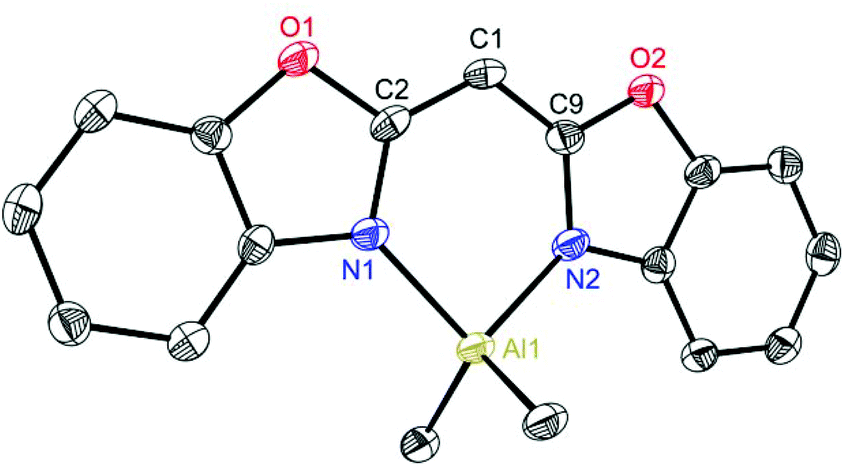 | ||
| Fig. 3 Molecular structure of [Me2Al{(NCOC6H4)2CH}] (3). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Structural data are given in Tables 1 and 2. | ||
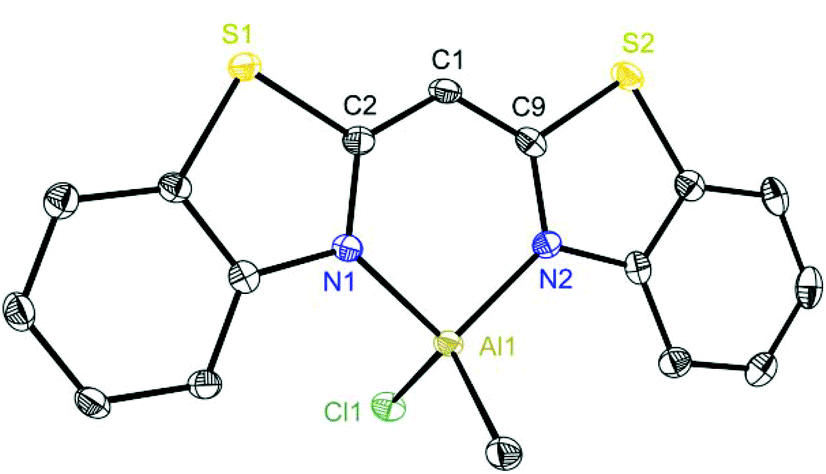 | ||
| Fig. 4 Molecular structure of [ClMeAl{(NCSC6H4)2CH}] (6). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms and the positional disorder are omitted for clarity. Structural data are given in Tables 1 and 2. | ||
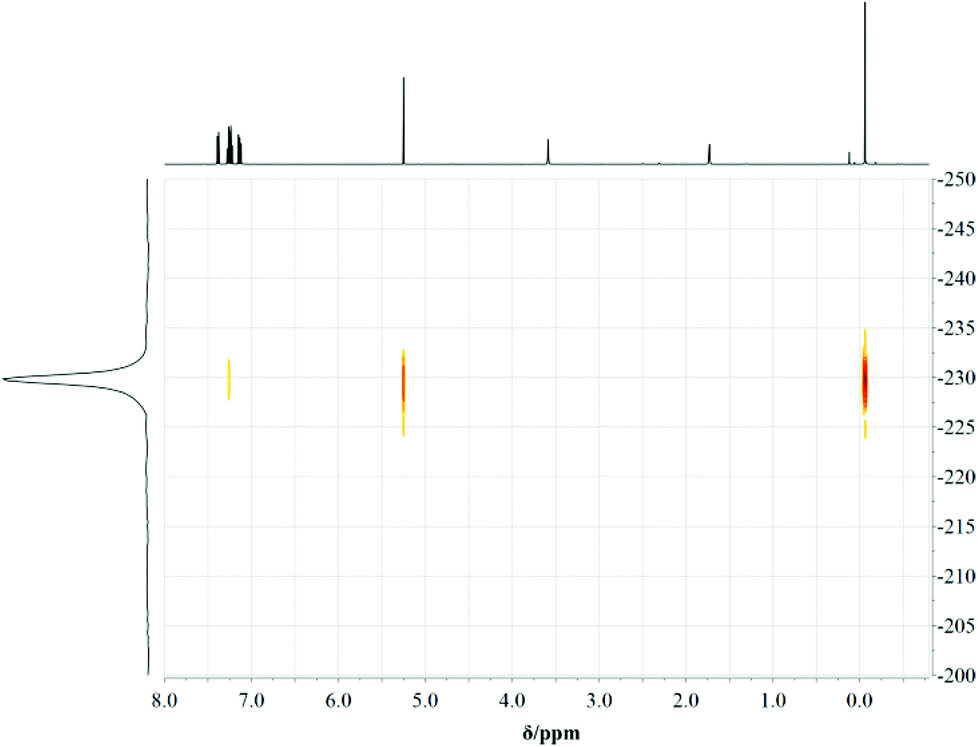 | ||
| Fig. 5 1H, 15N-HMBC-NMR spectrum of 7. | ||
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| M–N(av) | — | — | 1.9176(20) | 1.9233(14) |
| C ip –Cbr(av) | 1.4891(15) | 1.5106(15) | 1.380(3) | 1.390(2) |
| N–Cip(av) | 1.2864(14) | 1.2955(16) | 1.346(3) | 1.3518(19) |
| N–M–N | — | — | 91.67(9) | 94.79(6) |
| C ip –Cbr–Cip | 111.23(9) | 109.59(14) | 119.5(2) | 123.54(14) |
| 5 | 6 | 7 | 8 | |
|---|---|---|---|---|
| br = bridge; ip = ipso. | ||||
| M–N(av) | 1.8930(17) | 1.894(2) | 1.996(6) | 1.9945(13) |
| C ip –Cbr(av) | 1.382(2) | 1.388(3) | 1.394(9) | 1.3909(19) |
| N–Cip(av) | 1.348(3) | 1.365(3) | 1.323(9) | 1.3436(19) |
| N–M–N | 94.33(11) | 96.84(9) | 89.2(2) | 92.99(8) |
| C ip –Cbr–Cip | 120.2(3) | 124.1(2) | 119.7(6) | 124.0(2) |
The most interesting feature within the series of the obtained structures is the deviation of the metal atom from the extended plane, which is made from the N1–C2–C1–C9–N2 array. With this M⋯plane distance the folding angle between both heteroaromatic substituents is associated, which can be seen as a reason for the different deviations. Various data concerning the M⋯plane distances, the torsion and the folding angles are listed in Table 2.
| Compound | Metal dist. from C3N2 plane (pm) | Torsion angle (°) | Folding angle (°) |
|---|---|---|---|
| (NCOC6H4)2CH2 (1) | — | 81.3(9) | — |
| (NCSC6H4)2CH2 (2) | — | 188.98(24) | — |
| [Me2Al{(NCOC6H4)2CH}] (3) | 29.57(26) | 1.0(8) | 9.12(8) |
| [Me2Al{(NCSC6H4)2CH}] (4) | 13.79(29) | 1.2(6) | 1.36(2) |
| [ClMeAl{(NCOC6H4)2CH}] (5) | 18.06(32) | — | 3.69(40) |
| [ClMeAl{(NCSC6H4)2CH}] (6) | 10.93(27) | 0.6(8) | 1.21(2) |
| [Me2Ga{(NCOC6H4)2CH}] (7) | 14.34(3) | — | 3.62(2) |
| [Me2Ga{(NCSC6H4)2CH}] (8) | 20.61(26) | — | 8.90(6) |
To highlight this special aspect the molecular structures of 3 and 8 are shown in Fig. 3 and 6. Both species exhibit the greatest metal distance from the C3N2 plane (29.57(26) and 20.61(26)°, respectively) and also the greatest folding angle of the heteroaromates (9.12(8) and 8.90(6)°, respectively). As a representative of the AlMeCl containing species the molecular structure of 6 is shown in Fig. 4.
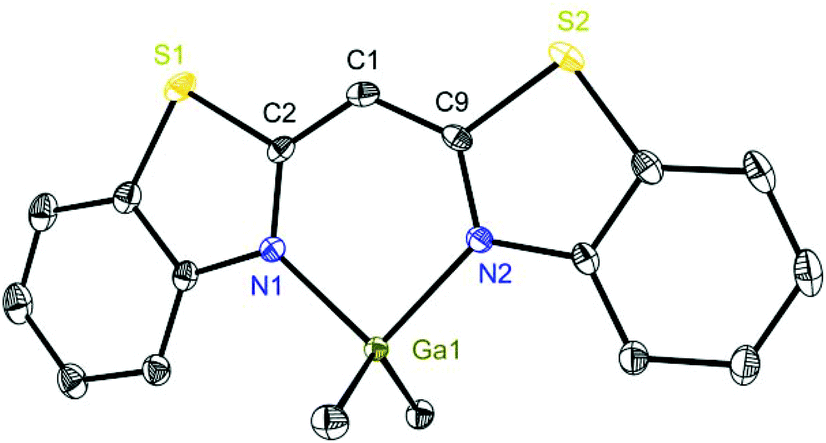 | ||
| Fig. 6 Molecular structure of [Me2Ga{(NCSC6H4)2CH}] (8). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Structural data are given in Tables 1 and 2. | ||
Concerning the series of the synthesised group 13 metal complexes of the bis-(benzothiazol-2-yl)- and bis-(benzoxazol-2-yl)-methanides, one has to emphasize that in the case of the oxygen containing ligand on average the bite angle is significantly smaller than in the sulfur analogues (89.2(2)–94.33(11)° vs. 92.99(8)–96.84(9)°) and the angle around the bridging carbon atom in the backbone is also smaller (119.5(2)–120.2(3)° vs. 123.54(14)–124.1(2)°).
The aluminium species show a correlation between two important structural features: the narrower the N–M–N angle, the greater the metal distance from the C3N2 plane and also the greater the folding angle of the heteroaromatic residues. However, in the case of the gallium complexes this tendency is the other way round. Because of these observations there are reasons to believe that the ligand derived from 1 coordinates more properly gallium than aluminium and the ligand derived from 2vice versa.
By comparing the structural values of the parent ligands with those of the metallated species given in Table 2, it can be considered that there is a change in hybridisation of the central bridging carbon atom upon metallation from sp3 to sp2. As an evidence for this statement the angular sum around the bridging carbon atom can be taken into account ranging from 359.9(2) to 360.1(2)°, which displays a nearly ideal trigonal planar coordination geometry for C1 as expected for a sp2-hybridised carbon atom. In both cases the benzothiazole as well as the benzoxazole containing ligand, a shortening of the Cipso–Cbridge bond (starting from 1.5106(15) and 1.4891(15) Å to approx. 1.39 Å) and a widening of the Cipso–Cbridge–Cipso angle (from 109.59(14) and 111.23(9)° to approx. 120°) were observed. Including the corresponding elongated N–Cipso distances in the deprotonated species 3–8, an efficient delocalisation of the lone pair is anticipated. As shown in Scheme 4 there are different mesomeric resonance structures which can be proposed: (a) a carbanionic, (b) an amidic and (c) a completely delocalised canonical form.
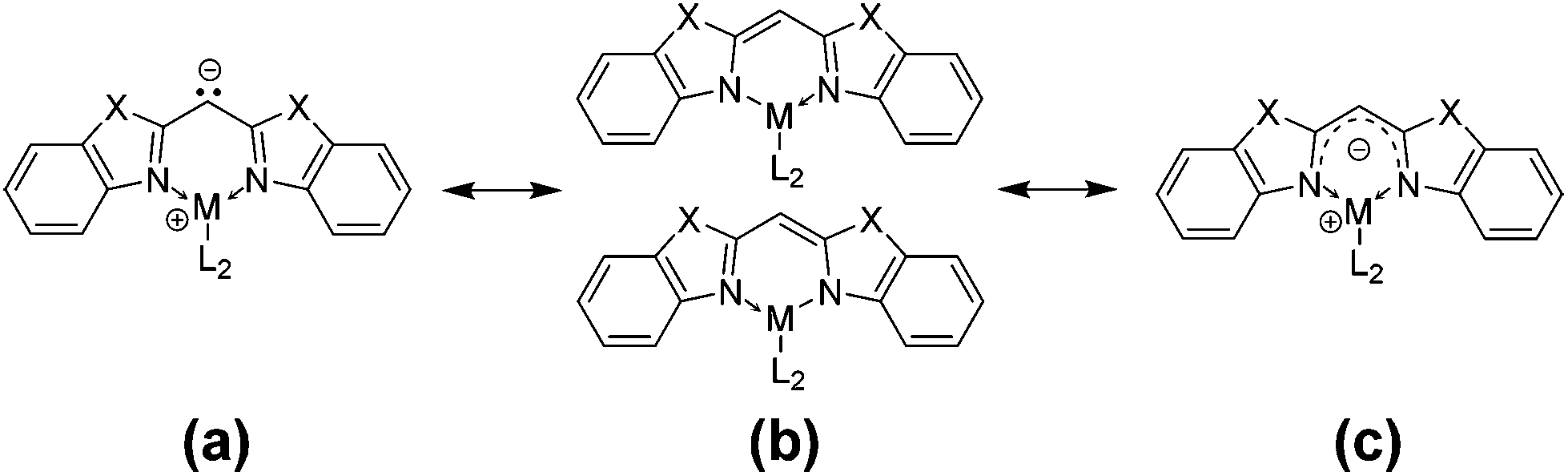 | ||
| Scheme 4 Mesomeric resonance structures of the bisheterocyclomethanides. | ||
Regarding the abovementioned structural changes, species (a) has a certain eligibility as in previous studies within [MeZnCH(2-NC5H4)2]2 the central deprotonated methanide carbon atom is coordinated by the electropositive zinc atom.4 Nevertheless case (c) seems to explain the bonding situation best within the compounds 3–8, because the C–C and C–N bond lengths lie in between a distinct single and double bond character (C(sp2)–C(sp3): 1.510 Å, C(sp2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(sp2): 1.335 Å, C(sp2)–N(sp3): 1.43 Å, C(sp2)N(sp2): 1.29 Å).34 Measurements of 1H, 15N-HMBC-NMR data gave evidence that complexes 3, 4, 7 and 8 show the same structure in solution at room temperature (at the NMR time scale) and in the solid state. The coordination geometry concerning the nacnac-like chelating ability can be proven by the apparent cross peaks, which clearly display 3J-coupling between the protons of the AlMe2 or GaMe2 unit and the nitrogen donor atoms, exemplarily shown in Fig. 5 for compound 7.
C(sp2): 1.335 Å, C(sp2)–N(sp3): 1.43 Å, C(sp2)N(sp2): 1.29 Å).34 Measurements of 1H, 15N-HMBC-NMR data gave evidence that complexes 3, 4, 7 and 8 show the same structure in solution at room temperature (at the NMR time scale) and in the solid state. The coordination geometry concerning the nacnac-like chelating ability can be proven by the apparent cross peaks, which clearly display 3J-coupling between the protons of the AlMe2 or GaMe2 unit and the nitrogen donor atoms, exemplarily shown in Fig. 5 for compound 7.
Conspicuously within the series from the AlMe2 derivatives via AlMeCl towards GaMe2 the N–M distance and the N–M–N angle alter as expected. In comparison with 3 and 4 the N–M distance decreases in 5 and 6 because of the more electronegative, electron withdrawing chloro substituent in the latter cases. Concomitantly the N–M–N angles widen.
Furthermore, the dimethylgallium compounds 7 and 8 feature increasing N–M bond lengths and smaller bite angles due to the bigger covalent radius of the coordinated gallium cation (Al: 1.21 Å, Ga: 1.22 Å).35
In addition to the above discussed structures 1–8 the interesting side product 9 could be isolated from the synthesis of 1 in moderate yields (29%) and structurally and spectroscopically characterised. In a first attempt to synthesise 1 two equivalents of 2-chlorobenzoxazole were attempted to be lithiated in the 2-position and subsequently coupled via addition of one equivalent of diethylcarbonate. Surprisingly the coupling product 9 shown in Fig. 7 was obtained. It represents a side product of the used nBuLi with 2-chlorobenzoxazole, where the butyl carbanion is formally substituted three times by a benzoxazole heterocycle.
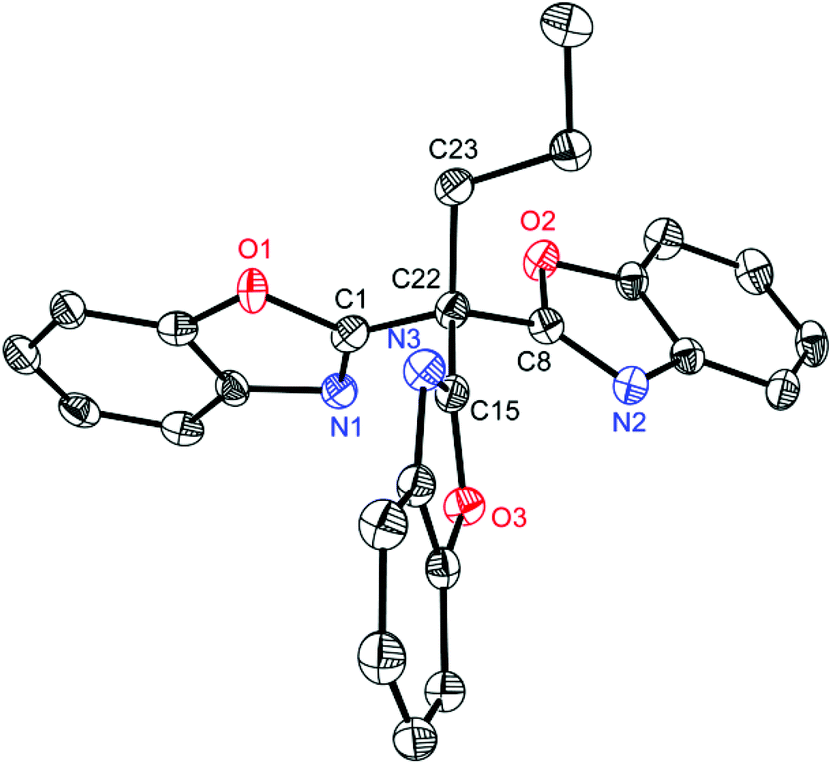 | ||
| Fig. 7 Molecular structure of (NCOC6H4)3C(C3H7) (9). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms and the positional disorder are omitted for clarity. Selected bond lengths and angles C22–C1(av) 151.1(10) pm, C22–C8 150.51(16) pm, C22–C15 151.20(16) pm, C22–C23 155.04(17) pm; C1–C22–C8(av) 110.0(8)°, C1–C22–C15(av) 105.3(14)°, C1–C22–C23(av) 109.9(9)°, C8–C22–C15 113.07(10)°, C8–C22–C23 108.69(10)°, C15–C22–C23 109.91(10)°. | ||
Compound 9 crystallizes in the monoclinic space group C2/c and the central carbon atom C22 shows a nearly ideal tetrahedral coordination environment (105.3(14)–113.07(10)°). The involved C–C bond lengths outgoing from C22 to the benzoxazole moieties lie in a narrow range from 1.5051(16) to 1.5120(16) Å, whereas the C22–C23 distance is significantly longer (1.5504(17) Å). This shortening is expected for an average C(sp3)–C(sp3) (1.544 Å) to an average C(sp3)–C(sp2) (1.510 Å).34
This kind of tripodal, tridentate ligand features interesting coordination abilities because of the presence of soft as well as hard donor atoms within the scaffold and can be regarded as an analogue of trispyrazolylmethanes or even more of trisoxazoline ligands. The latter ones are known to be valuable auxiliary ligands in asymmetric catalysis.
Furthermore, 9 shows structural similarities to the class of scorpionates, which are also tridentate ligands and form facial coordination geometries upon building metal complexes.36 As common popular examples the monoanionic trispyrazolylborates and the neutral isoelectronic trispyrazolylmethanes37 have to be mentioned, which are used in bioinorganic chemistry to mimic the active sites in metallo proteins in model complexes.38
Conclusion
In summary, a variety of group 13 metal complexes containing bisheterocyclomethanides as ligand systems could successfully be synthesised and structurally evaluated. In each case the metal cation is coordinated in a distorted tetrahedral fashion by the ring-nitrogen atoms acting as Lewis-bases and the ligand became almost perfectly planar. Depending on the considered combination of the heterocycle (benzoxazole or benzothiazole) and the chelated metal (Al or Ga) some significant correlations between the folding angle of the scaffold and the metal distance from the C3N2 plane were observed. The metal coordination in all cases takes place exclusively by donation of nitrogen atoms within the heterocyclic moieties. The chalcogenes did not participate in coordination neither in the solid state nor in solution as proven in the appropriate crystal structures and the 1H, 15N-HMBC-NMR experiments.These empirical results gave rise to the occurrence of preferred ligand–metal pairs in presumably the most stable complexes: whereas 1 seems to suit gallium more properly, 2 fits aluminium the most.
Moreover, the negative charge arising upon deprotonation seems to be delocalised about the whole ligand backbone as is indicated by the observed N–Cipso and Cipso–Cbridge bond lengths. In this context the carbanionic canonical formula can be neglected, because the linking carbon atoms show clear sp2-hybridisation and no involvement in metal coordination.
Referring to the title of this work one can conclude that bisheterocyclomethanides as new ligand systems can definitely compete with the popular β-diketiminate ligand and offer promising opportunities for investigating and exploring a new interesting research area.
Experimental section
General procedures
All manipulations were carried out under an atmosphere of N2 or Ar by Schlenk techniques. All solvents used within metallation reactions were distilled from Na or K before use. The starting materials were purchased commercially and used as received. Ethylbisimidate dihydrochloride was prepared according to a literature method.331H, 13C, 15N and 27Al NMR spectroscopic data were recorded on a Bruker Avance 500 MHz, a Bruker Avance 400 MHz and a Bruker Avance 300 MHz spectrometer and referenced to the deuterated solvent (thf-d8). Elemental analyses (C, H, N and S) were carried out on a Vario EL3 at the Mikroanalytisches Labor, Institut für Anorganische Chemie, University of Göttingen. All EI-MS spectra (70 eV) were recorded on a Finnigan MAT 95.Ligand syntheses
Both ligand systems were synthesised by cyclocondensation reaction of a suitable linker derived from malonic acid and the corresponding ortho-substituted anilines.Metallation reactions
To a solution of the corresponding ligand 1 or 2 (1.0 eq.) in toluene a slight excess of the pure organometallic reactant AlMe3, AlMe2Cl or GaMe3 (1.1 eq.) was slowly added at 0 °C. The reaction mixture was stirred overnight and allowed to warm to rt. In the case of 5 and 6 the resulting precipitate was filtered off, and the reaction mixture of 3 and 4 afforded a clear solution. Afterwards the volume of the solution was reduced to a few milliliters and the resulting concentrated solution was stored at −32 °C in a fridge. Overnight crystals suitable for X-ray diffraction experiments could be obtained. The crystals thus formed were filtered, washed twice with pre-cooled toluene or hexane (0 °C) and finally dried in a vacuum. The given yields below are just based on the received crystals unless stated otherwise. No further improvement of the yields was applied to the remaining solutions because of impurities upon repeated precipitation.X-ray crystallographic studies
Single crystals were selected from a Schlenk flask under an argon or nitrogen atmosphere and covered with perfluorated polyether oil on a microscope slide, which was cooled with a nitrogen gas flow using the X-TEMP2 device.39 An appropriate crystal was selected using a polarized microscope, mounted on the tip of a MiTeGen©MicroMount or glass fiber, fixed to a goniometer head and shock cooled by the crystal cooling device.The data for 1–9 were collected from shock-cooled crystals at 100(2) K. The data of 1, 2, 5, 6, 8 and 9 were collected on an Incoatec Mo Microsource40 and compound 7 was collected on an Incoatec Ag Microsource each equipped with mirror optics and an APEX II detector with a D8 goniometer. The data of 3 and 4 were measured on a Bruker TXS-Mo rotating anode with mirror optics and an APEX II detector with a D8 goniometer. All diffractometers were equipped with a low-temperature device and used either MoKα radiation of λ = 71.073 pm or AgKα radiation of λ = 56.086 pm. The data were integrated with Saint41 and an empirical absorption correction (Sadabs)42 was applied. The structures were solved by direct methods (Shelxs-97) and refined by full-matrix least-squares methods against F2 (Shelxl-97).34,43 All non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were refined isotropically on calculated positions using a riding model with their Uiso values constrained to equal 1.5 times the Ueq of their pivot atoms for terminal sp3 carbon atoms and 1.2 times for all other carbon atoms. Disordered moieties were refined using bond length restraints and isotropic displacement parameter restraints.
Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre. The CCDC numbers, crystal data and experimental details for the X-ray measurements are listed in the ESI.‡
Acknowledgements
Thanks to the Danish National Research Foundation DNRF funded Center for Materials Crystallography (CMC) for partial support and the Land Niedersachsen for providing a fellowship in the GAUSS PhD program.The authors thank Prof. Dr. Dr. h.c. mult. H. W. Roesky for the numerous subject-specific discussions and Dr. Regine Herbst-Irmer for the support in crystallographic issues.
Notes and references
- W. W. Schoeller, Inorg. Chem., 2011, 50, 2629–2633 CrossRef CAS PubMed.
- For review: F. Baier, Z. Fei, H. Gornitzka, A. Murso, S. Neufeld, M. Pfeiffer, I. Rüdenauer, A. Steiner, T. Stey and D. Stalke, J. Organomet. Chem., 2002, 661, 111–127 CrossRef CAS.
- H. Gornitzka and D. Stalke, Angew. Chem., Int. Ed. Engl., 1994, 33, 693–695 CrossRef.
- H. Gornitzka, C. Hemmert, G. Bertrand, M. Pfeiffer and D. Stalke, Organometallics, 2000, 19, 112–114 CrossRef CAS.
- For review: L. Mahalakshmi and D. Stalke, Struct. Bonding, 2002, 103, 85–113 CrossRef CAS.
- H. Gornitzka and D. Stalke, Organometallics, 1994, 13, 4398–4405 CrossRef CAS.
- M. Pfeiffer, F. Baier, T. Stey, D. Leusser, D. Stalke, B. Engels, D. Moigno and W. Kiefer, J. Mol. Model., 2000, 6, 299–311 CrossRef CAS.
- M. Pfeiffer, A. Murso, L. Mahalakshmi, D. Moigno, W. Kiefer and D. Stalke, Eur. J. Inorg. Chem., 2002, 3222–3234 CrossRef CAS.
- A. Steiner and D. Stalke, J. Chem. Soc., Chem. Commun., 1993, 444–446 RSC.
- A. Steiner and D. Stalke, Angew. Chem., 1995, 107, 1908–1910 CrossRef.
- H. Gornitzka and D. Stalke, Eur. J. Inorg. Chem., 1998, 311–317 CrossRef CAS.
- A. Steiner and D. Stalke, Organometallics, 1995, 14, 2422–2429 CrossRef CAS.
- M. Stender, B. E. Eichler, N. J. Hardman, P. P. Power, J. Prust, M. Noltemeyer and H. W. Roesky, Inorg. Chem., 2001, 40, 2794–2799 CrossRef CAS PubMed.
- B. Qian, D. L. Ward and M. R. Smith III, Organometallics, 1998, 17, 3070–3076 CrossRef CAS.
- S. Singh, H.-J. Ahn, A. Stasch, V. Jancik, H. W. Roesky, A. Pal, M. Biadene, R. Herbst-Irmer, M. Noltemeyer and H.-G. Schmidt, Inorg. Chem., 2006, 45, 1853–1860 CrossRef CAS PubMed.
- Z. Yang, H. Zhu, X. Ma, J. Chai, H. W. Roesky, C. He, J. Magull, H.-G. Schmidt and M. Noltemeyer, Inorg. Chem., 2006, 45, 1823–1827 CrossRef CAS PubMed.
- S. Nagendran and H. W. Roesky, Organometallics, 2008, 27, 457–492 CrossRef CAS.
- Y. Ding, H. W. Roesky, M. Noltemeyer, H.-G. Schmidt and P. P. Power, Organometallics, 2001, 20, 1190–1194 CrossRef CAS.
- A. Jana, H. W. Roesky, C. Schulzke, A. Döring, T. Beck, A. Pal and R. Herbst-Irmer, Inorg. Chem., 2009, 48, 193–197 CrossRef CAS PubMed.
- Z. Yang, H. Zhu, X. Ma, J. Chai, H. W. Roesky, C. He, J. Magull, H.-G. Schmidt and M. Noltemeyer, Inorg. Chem., 2006, 45, 1823–1827 CrossRef CAS PubMed.
- Y. Cheng, D. J. Doyle, P. B. Hitchcock and M. F. Lappert, Dalton Trans., 2006, 4449–4460 RSC.
- N. Kuhn, J. Fahl, S. Fuchs, M. Steimann, G. Henkel and A. H. Maulitz, Z. Anorg. Allg. Chem., 1999, 625, 2108–2114 CrossRef CAS.
- N. Kuhn, S. Fuchs and M. Steimann, Eur. J. Inorg. Chem., 2001, 359–361 CrossRef CAS.
- N. Kuhn, S. Fuchs and M. Steimann, Z. Anorg. Allg. Chem., 2002, 628, 458–462 CrossRef CAS.
- C. E. Radzewich, I. A. Guzei and R. F. Jordan, J. Am. Chem. Soc., 1999, 121, 8673–8674 CrossRef CAS.
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 112, 4444–4446 CrossRef.
- X. Li, X. Cheng, H. Song and C. Cui, Organometallics, 2007, 26, 1039–1043 CrossRef CAS.
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991–1992 RSC.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed.
- D. Li, Y. Peng, C. Geng, K. Liu and D. Kong, Dalton Trans., 2013, 42, 11295–11303 RSC.
- B. D. Ward and L. H. Gade, Chem. Commun., 2012, 48, 10587–10599 RSC.
- H. B. Ammar, J. Le Nôtre, M. Salem, M. T. Kaddachi and P. H. Dixneuf, J. Organomet. Chem., 2002, 662, 63–69 CrossRef.
- P. Müller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, in Crystal Structure Refinement – A Crystallographer's Guide to SHELXL, ed. P. Müller, IUCr Texts on Crystallography, Oxford University Press, Oxford, UK, 2006, vol. 8 Search PubMed.
- Cambridge Structural Database, taken from “CSD Elemental Radii”, http://www.ccdc.cam.ac.uk/Lists/ResourceFileList/Elemental_Radii.xlsx, last modified: 21st June 2012.
- S. Trofimenko, Chem. Rev., 1993, 93, 943–980 CrossRef CAS.
- D. L. Reger, Comments Inorg. Chem., 1997, 21, 1–28 CrossRef.
- W. Kaim and B. Schwederski, Bioanorganische Chemie – Zur Funktion chemischer Elemente in Lebensprozessen, B. G. Teubner Verlag, Wiesbaden, 2005 Search PubMed.
- (a) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef; (b) T. Kottke, R. J. Lagow and D. Stalke, J. Appl. Crystallogr., 1996, 29, 465–468 CrossRef CAS; (c) D. Stalke, Chem. Soc. Rev., 1998, 27, 171–178 RSC.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef CAS.
- SAINT v7.68A in Bruker APEX v2011.9, Bruker AXS Inst. Inc., Madison, USA, 2008 Search PubMed.
- G. M. Sheldrick, SADABS 2008/2, Universität Göttingen, Germany, 2008 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467–473 CrossRef.
Footnotes |
| † Dedicated to Professor Frank T. Edelmann on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Tables of data collection parameters, bond lengths and angles for compounds 1–9, 2D-NMR spectra of 3, 4 and 8, additional crystal structures of 4, 5 and 7, nine X-ray files in CIF format. CCDC 995110–995118. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt01008f |
| This journal is © The Royal Society of Chemistry 2014 |