 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixing the spacers in azacryptands: effects on halide recognition†
Greta
Bergamaschi
*a,
Massimo
Boiocchi
b,
Maria Lucia
Perrone
a,
Antonio
Poggi
a,
Ivan
Viviani
a and
Valeria
Amendola
*a
aDipartimento di Chimica, Università di Pavia, via Taramelli 12, I-27100, Pavia, Italy. E-mail: greta.bergamaschi@unipv.it; amendola@unipv.it
bCentro Grandi Strumenti, Università di Pavia, via Bassi 21, I-27100, Pavia, Italy
First published on 13th May 2014
Abstract
In this work, we report on two new asymmetric dicopper cryptates, characterized by alternate furanyl and p-xylyl spacers. The results of the potentiometric, UV-vis and X-ray diffraction studies are discussed. In particular, for one of the cages in the octaprotonated form, the crystal structure of the complex with nitrate is described. From the point of view of the anion binding in water, the new dicopper cryptates display stronger similarities to each other than to the symmetric analogues (i.e. p-xylyl and furan cryptates). The substitution of even only one spacer modifies the distance between the metal ions. This prevents the development of the CT band, typically accompanying the binding of halides by the furan cryptate, and favors the formation of complexes of different stoichiometry with the linear azide anion, not observed for the symmetric cryptates. The obtained results may be very helpful in the design of new molecular receptors based on dimetallic cryptates, in particular for the tuning of the sensitivity towards specific anionic substrates.
Introduction
Azacryptands are macrocyclic polyaza receptors, formed by two tren units linked by molecular spacers.1 These molecular systems display six secondary and two tertiary amino groups, arranged around the receptor's cavity. In the field of anion recognition,2 azacryptands have been studied by many authors as selective receptors for anions. In this context, the interaction with the guest may involve either H-bonds with the protonated amino groups of the cage,2,3 or coordinative interactions with transition metal ions within the cavity,4 in the so-called “cascade complexes”.1In the first case, stable complexes are formed by encapsulation of the anionic species by the poly-protonated azacryptand. Anion recognition generally occurs in acidic aqueous solution, and selectivity is based on the geometric complementarity between the positively charged cavity and the included anions.2,3 In most examples, the azacryptand is in the hexaprotonated form; it presents an expanded ovoid cavity, with the secondary ammonium groups pointing inward, and interacting with the included anion. Some examples are also known of inclusion complexes involving the azacryptand in the octaprotonated form.5 In the case of the encapsulated chloride in the octaprotonated thiophene-based cryptand reported by Hossain et al., the spherical anion is bound and bridged by two in–in protons of the tertiary ammonium groups.5d In this situation, the terminal N atoms are pointing towards the centre of the cavity, whereas the secondary ammonium groups point outside the cage. As a consequence, the ovoid shape of the inclusion complex is severely affected.
In the case of cascade complexes, the two bis-tren units of the azacryptand coordinate one transition metal ion each (e.g. Co(II), Ni(II), Cu(II), etc.). In these complexes, the metal ions are coordinatively unsaturated, with one of the axial positions available for the coordination of a further ligand.4c The shape and size of the cavity, as well as the selectivity, can be tuned by changing the nature (length, rigidity) of the spacers. Noticeably, the spacers may also play an active role in anion recognition. In the case of the dicopper(II) complex of the furan-based azacryptand reported by Fabbrizzi et al., the encapsulation of halides is signalled by the development of a rather intense anion-to-metal charge transfer absorption band in the visible region.6 Experimental studies pointed out the correlation of the anion-to-metal CT band to the interaction of the encapsulated halide with the furan rings of the spacers.
We have now synthesised two asymmetric azacryptands, containing spacers of different length and nature (i.e. furanyl and p-xylyl units), arranged in alternate fashion, in order to verify the effect of the asymmetric substitution on anion binding, in particular on halide recognition. It has to be noted that, with respect to furanyl spacers, p-xylyl moieties are not expected to participate in halide binding, either by stabilizing the inclusion complex, or by signaling the recognition.
The new receptors (Fig. 1) have been investigated in aqueous solution by potentiometric and UV-vis titrations. Here, we compare the binding tendencies of the new cages with those of the symmetrically substituted analogues (i.e. furanyl and p-xylyl azacryptands).
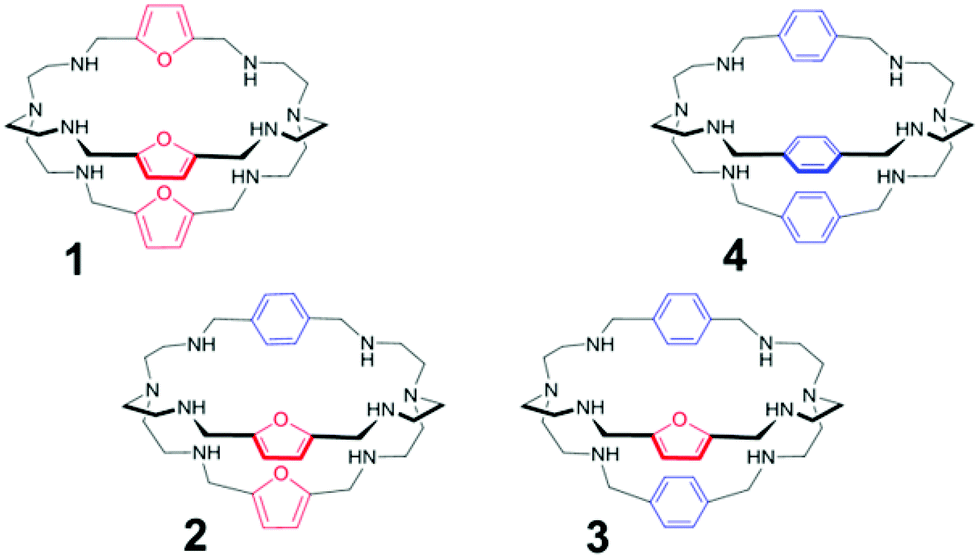 | ||
| Fig. 1 Azacryptands 1–4. | ||
Results and discussion
Synthesis of the cages
The synthesis of symmetric cryptands, e.g.1 and 4, comprises the [2 + 3] Schiff condensation between tren and the dialdehyde of the spacer (i.e. furanyl and p-xylyl, for 1 and 4, respectively), followed by the reduction of the polyimine intermediate with NaBH4.7 On the other hand, the preparation of asymmetric cages requires a multi-step synthesis (see the ESI†), recently described by our group.8 In particular, mono-BOC protected tren9 is first reacted in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with the appropriate dialdehyde (i.e. furanyl for receptor 2 and p-xylyl for receptor 3), yielding the intermediate polyimine macrocycle. After reduction with NaBH4, followed by deprotection, the macrocycle is reacted in a 1:1 ratio with the second dialdehyde, leading to the corresponding diimine cryptand. Reduction with NaBH4 finally gives the desired product. The crude azacryptands are then purified by precipitation as the nitrate salts (see ESI† for details).
:1 ratio with the appropriate dialdehyde (i.e. furanyl for receptor 2 and p-xylyl for receptor 3), yielding the intermediate polyimine macrocycle. After reduction with NaBH4, followed by deprotection, the macrocycle is reacted in a 1:1 ratio with the second dialdehyde, leading to the corresponding diimine cryptand. Reduction with NaBH4 finally gives the desired product. The crude azacryptands are then purified by precipitation as the nitrate salts (see ESI† for details).
Crystal structure of [3H8(NO3)](NO3)7·3H2O
From the slow evaporation of an aqueous solution of 3 with excess nitric acid, single crystals of formula [3H8(NO3)](NO3)7·3H2O, suitable of for X-ray diffraction analysis, were obtained. In Fig. 2, the plot showing the thermal ellipsoids of the [3H8(NO3)]7+ molecular cation, occurring in the crystal of [3H8(NO3)](NO3)7·3H2O, is reported. A simplified view of the [3H8(NO3)]7+ molecular cation, projected down the line passing through the terminal amine groups, is also reported, in order to emphasize that all protonated secondary amines point their proton outside the cage.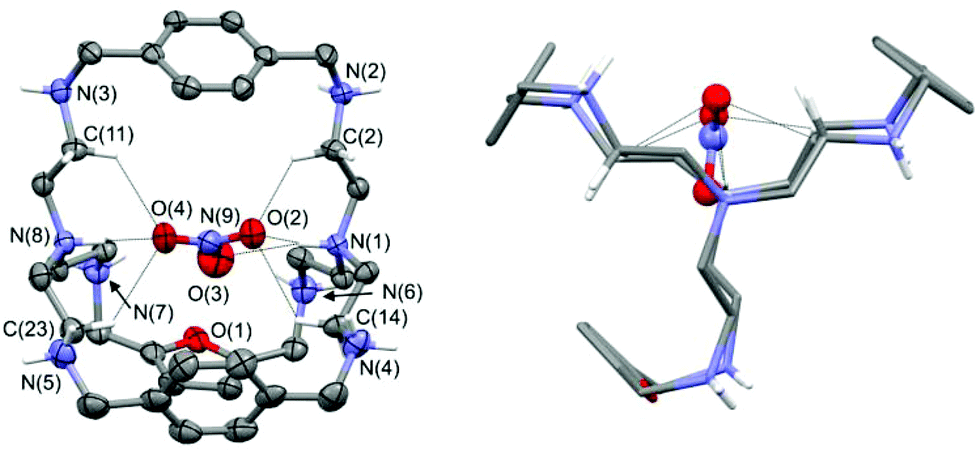 | ||
| Fig. 2 Left: plot showing thermal ellipsoids (20% probability level) of the [3H8(NO3)]7+ molecular cation, occurring in the crystal of [3H8(NO3)](NO3)7·3H2O. Atom names are shown only for N and O atom sites, and for C atoms involved in H-bonds. Some H atoms are also drawn and dashed lines indicate N–H⋯O and C–H⋯O interactions. Right: the simplified view of the [3H8(NO3)]7+ molecular cation projected down the line passing through the terminal amine (tren) groups. | ||
The azacryptand 3, containing two p-xylyl and one furanyl spacers, is in the octaprotonated form and the molecular structure displays an encapsulated nitrate anion, which is H-bonded to the two protonated tertiary amines. Interestingly, all oxygen atoms of the included anion participate in the N–H⋯O interactions. As shown in Fig. 2 (left), oxygen O4 of the included nitrate anion is H-bonded to the protonated bridgehead atom N(8), which acts as a H-donor of a single H-bond. On the other hand, the two remaining oxygens are involved in two H-bonds originating from N(1), which acts as a H-donor of a bifurcated H-bond. Additional C–H⋯O interactions involve some sp3 carbon atoms and the nitrate ion; these interactions favor the trapping of the nitrate ion inside the cage. In particular, the C–H⋯O interactions, having both a C⋯O separation ≤3.2 Å and a H⋯O distance ≤2.7 Å, are shown in Fig. 2. Geometrical features of H-bonds involving the encapsulated nitrate ions are reported in Table 1 (more details are available in the ESI†). The remaining seven nitrate counterions are outside the cage, and they interact with the secondary ammonium groups and additional water molecules (see Fig. S1 and S2†). Because all secondary ammonium groups point their protons outside the cage (see Fig. 2, right), the cavity tends to assume a compressed ovoid shape, with the protonated terminal N tren atoms being drawn towards the centre of the cavity, and the secondary ammonium groups turned towards the outside. The distance between the bridgehead N atoms N(1)–N(8) is 6.67(1) Å, a value shorter than that observed by Ghosh et al. in the crystal structure of the iodide complex of the octaprotonated p-xylyl azacryptand (6.93 Å; CSD refcode JOJKID).5e
| Donor group D | D⋯A (Å) | H⋯A (Å) | D–H⋯A (°) | Acceptor atom A |
|---|---|---|---|---|
| N(1)–H(1N) | 3.16(1) | 2.48(1) | 132.1(4) | O(2) |
| N(1)–H(1N) | 3.02(1) | 2.13(1) | 166.9(5) | O(3) |
| N(8)–H(8N) | 2.82(1) | 2.00(1) | 150.0(4) | O(4) |
| C(2)–H(2A) | 3.20(1) | 2.54(1) | 125.8(5) | O(2) |
| C(11)–H(11B) | 3.11(1) | 2.45(1) | 124.9(5) | O(4) |
| C(14)–H(14A) | 3.18(1) | 2.54(1) | 123.9(5) | O(2) |
| C(23)–H(23B) | 3.19(1) | 2.48(1) | 130.0(6) | O(4) |
Similarly to our molecular complex, the p-xylyl cage has all the protonated secondary amines pointing their protons outside the cage. However, the resulting iodide complex is more symmetric because the cage contains only p-xylyl spacers. The p-xylyl cage exhibits a pseudo three-fold axis passing through the terminal tren groups, and the distances between the centroid of the three aromatic rings define the edges of an almost equilateral triangle (centroid–centroid separations are in the range 8.29–8.58 Å). The center of the triangle is on the middle of the line connecting the two tren groups, and the iodide ion is located at this point. Our octaprotonated cage, having an asymmetric arrangement of the spacers, loses the pseudo three-fold axis. The centroid–centroid separations between the three aromatic rings define an almost isosceles triangle (with two long edges of 8.23 and 8.48 Å, and a short edge of 5.90 Å).
The center of the geometric figure is no longer along the line connecting the two tertiary amines. Nevertheless, the included anion is still near the center of the triangle, with the N(9) nitrate atom placed 0.77 Å from it. Noticeably, the O(3) nitrate oxygen almost lies on the middle of the line connecting the bridgehead N(1) and N(8) atoms: the O(3)–N(1) and O(3)–N(8) separations are 3.02(1) and 3.69(1) Å, respectively, whereas the N(1)–O(3)–N(8) angle is 166.4(4)°. With respect to the symmetric p-xylyl cage, the presence of a furanyl spacer seems to increase the flexibility of the cavity, thus allowing the protonated bridgehead N atoms to come closer.
Potentiometric/pH-spectrophotometric studies in water
The asymmetric azacryptands, as the free bases 2 and 3, were first investigated by potentiometric titrations in aqueous solution (0.1 M CF3SO3Na, at 25 °C). The protonation constants, reported in Table 2, were compared with those obtained for the symmetric cages, 1 and 4, under the same experimental conditions. All potentiometric titrations were performed in the pH range of 2.4 to 11.4. Under these conditions, only six protonation constants could be determined despite the eight potential protonation sites. The experimental data were fitted with a non-linear least-squares procedure using the HyperQuad program.10| 1 6b | 2 | 3 | 4 3b | |
|---|---|---|---|---|
| K LH1 | 9.51(1) | 10.09(2) | 10.04(5) | 9.89(4) |
| β LH2 | 18.28(1) | 18.76(2) | 18.94(7) | 18.96(4) |
| β LH3 | 26.43(1) | 26.47(4) | 26.95(8) | 27.13(5) |
| β LH4 | 33.19(1) | 32.98(8) | 33.74(9) | 34.51(5) |
| β LH5 | 38.52(1) | 38.95(7) | 39.78(9) | 39.88(4) |
| β LH6 | 43.23(1) | 43.37(8) | 44.40(9) | 45.03(6) |
| β LH3Cu | 34.82(2) | 34.39(6) | 34.87(4) | n.a. |
| β LCu2 | 25.21(1) | 22.90(5) | 22.15(5) | n.a. |
| β LCuH-1 | 19.44(9) | 14.75(8) | 14.34(7) | n.a. |
| β LCuH-2 | 9.2(1) | 4.24(8) | 3.57(7) | n.a. |
| Log KW | −13.7(1) |
The constants reported in Table 2 correspond to the protonation equilibria of the secondary amines. Noticeably, azacryptand 4 is slightly more basic than the other cages, as shown by the comparison of log βLH6 values. This trend is congruent with the acid–base properties of octaaminocryptands, reported by other authors.2b Analogous measurements were performed in the presence of 2 equiv. of Cu(II) ions (as the triflate salt). For all investigated cryptands, best fitting was obtained by assuming the formation of the following Cu(II) complexes: [CuLH3]5+, [Cu2L]4+, [Cu2L(OH)]3+ and [Cu2L(OH)2]2+ (L = 1–3). The distribution diagrams are reported in Fig. 3 and S3.†
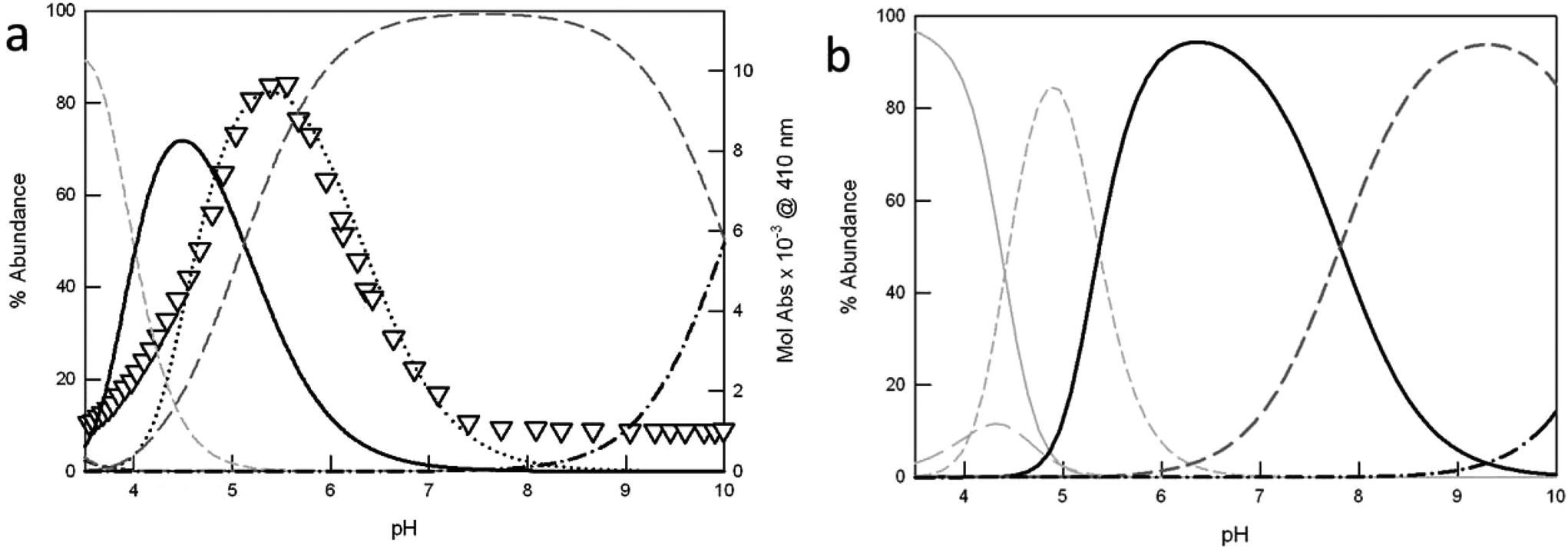 | ||
| Fig. 3 Distribution diagrams (% abundance vs. pH) of the species obtained upon potentiometric titration of azacryptands (a) 1 and (b) 3 (0.2 mM), in the presence of 2 equiv. Cu(CF3SO3)2 (0.1 M CF3SO3Na, at 25 °C). The corresponding protonation and complexation constants are shown in Table 2 (solid light grey = LH66+; long dashed light grey = LH55+; short dashed light grey = [CuLH3]5+; solid black = [Cu2L]4+; long dashed dark grey = [Cu2L(OH)]3+ and dot-dashed black line = [Cu2L(OH)2]2+). In (a), the triangles correspond to the profile of Mol Abs (×10−3, M−1 cm−1) at 410 nm vs. pH, obtained upon pH-spectrophotometric titration of 1 (0.25 mM), in the presence of 2 equiv. Cu(CF3SO3)2 and excess NaCl (5 mM). The plot is superimposed to the distribution curve of the [Cu21Cl]3+ species (black dots), obtained as already reported.6b | ||
In the case of cryptand 1, at pH 2.4 the cage is present in the hexaprotonated form, 1H66+ (>90%).
Upon titration with NaOH, the monometallic complex [Cu1H3]5+ forms, with one of the two tren compartments triply protonated, reaching its maximum concentration (80%) at pH 3.5 (see Fig. 3a). The dicopper complex, [Cu21]4+, is the major species in solution at pH 4.5 (71%). In [Cu21]4+, each Cu(II) ion occupies one of the tren units, adopting the typical trigonal-bipyramidal geometry imparted by tripodal ligands. The apical positions on the metal ions, left free by the receptor, are occupied by water molecules.
Upon further addition of NaOH to the complex solution, the deprotonation of the coordinated water occurs, leading to the stable hydroxide complex, [Cu21(OH)]3+. This species already starts to form at pH 4.1. In the crystal structure reported by Nelson et al. for [Cu21(OH)]3+,11 the hydroxide anion bridges the two metal centers (dCu–Cu = 3.900(1) Å). Due to its high stability, this species predominates in solution (>90%) over the wide pH range of 6.1 to 9.1.
Moving towards basic pH values, the deprotonation of a second water molecule takes place, leading to [Cu21(OH)2]2+.
For receptor 1, potentiometric and pH-spectrophotometric titrations, in the presence of Cu(II) and excess halide anions (X−), demonstrate the formation of stable inclusion complexes, [Cu21(X)]3+, characterized by typical anion-to-metal CT bands in the visible region.6 In the case of chloride, the [Cu21(Cl)]3+ cryptate reaches the maximum abundance (80%) at pH 5.4, as shown by the profile of Mol Abs at 410 nm vs. pH in Fig. 3a (see the black triangles). The band at 410 nm corresponds to the chloride-to-copper(II) CT transition, peculiar of [Cu21(Cl)]3+. In Fig. 3a, the absorbance profile is superimposed to the distribution curve of [Cu21(Cl)]3+, calculated from the constants previously reported by our group.6b Of course, the plot cannot fit the distribution diagram obtained in the absence of chloride, for which the major species in solution at pH 5.4 (i.e. in correspondence of the maximum development of the CT band) is the hydroxyl-bridged complex [Cu21(OH)]3+ (65%). Interestingly, all copper-containing species of the asymmetric ligands 2 and 3 are characterized by lower association constants than the analogous complexes of 1. As a consequence, they start to form at higher pH values (see Fig. 3b and S3† for 3 and 2, respectively). For example, the monometallic [CuLH3]5+ species of 2 and 3 reach their maximum abundance (>80%) at pH ≥ 4.5 (i.e. 4.5 and 4.9, for 2 and 3 respectively), instead of pH 3.5 as for 1. Also the dicopper complexes, [Cu2L]4+, start to form at pH > 4, and represent the principal species (>70%) over the pH interval of 5.3 to 7.2. Very interestingly, the formation of the hydroxide complexes, [Cu2L(OH)]3+ (L = 2–3), occurs only at pH > 6, i.e. two log units higher than for the furanyl cage 1. The substitution of one or more furanyl spacers with p-xylyl units is actually expected to increase the distance between the two included metal ions. This may prevent the formation of a [Cu2L(OH)]3+ complex, with the hydroxide anion bridging the Cu(II) centers. For the [Cu2L(OH)]3+ complex (L = 2, 3), we can hypothesize a structure similar to that reported by Nelson et al. for the p-xylyl-spaced dicopper(II) cryptate, [Cu24(H2O)(OH)](ClO4)3.12 Noticeably, in [Cu24(H2O)(OH)]3+, the distance between the copper ions is 6.1213(12) Å, i.e. about 2.22 Å longer than that measured by the same authors for [Cu21(OH)]3+.11 Under these conditions, OH− cannot bridge the copper ions. Thus, the anion is bound to only one metal center, and a water molecule occupies the apical position of the second metal ion. For the hydroxide complexes of the asymmetric cages, 2 and 3, we can expect a similar situation. However, crystals suitable for X-ray diffraction studies could not be obtained; thus the hypothesis could not be verified. Further addition of NaOH to the solution of [Cu2L(OH)]3+ promotes the formation of the [Cu2L(OH)2]2+ species, with two hydroxide anions bound to the metal centers.
Noticeably, upon pH-spectrophotometric titration of 2 and 3 in the presence of 2 equiv. of Cu(II) and excess halide anions, no new bands attributable to anion-to-metal CT developed in the UV-vis spectrum. This result confirms that the replacement of furanyl with p-xylyl spacers not only disrupts the symmetry of the cage, but also increases the distance between the metal centers (as already pointed out for the hydroxide complex). The longer distance prevents the interactions between the included halide and the oxygen atom of the furan units. The last feature, in particular, is responsible for the lack of the CT band in the absorption spectrum. For both 2 and 3, copper binding is accompanied by the development of a broad band around 270 nm, attributable to the N(amine) → Cu(II) charge transfer. Moreover, two d–d bands develop in the visible region, from 650 to 850 nm, typical of bipyramidal complexes containing the CuN5 chromophore (see Fig. S4† for ligand 2).13
Unfortunately, in the case of 4, potentiometric titrations in the presence of copper ions could not be performed, due to the precipitation of the complex species in basic solution. Thus, we could not determine the association constants for a comparison with those reported in Table 2 for the other ligands.
UV-vis studies with halides in aqueous solution
To assess the binding tendencies of the new dicopper cryptates towards halides, we performed UV-vis titrations in buffered aqueous solution, following the changes in the absorption bands of the complexes upon halide binding. Fig. 4 shows the UV-vis spectra taken upon titration of [Cu23]4+ with NaF (more details are reported in the ESI†).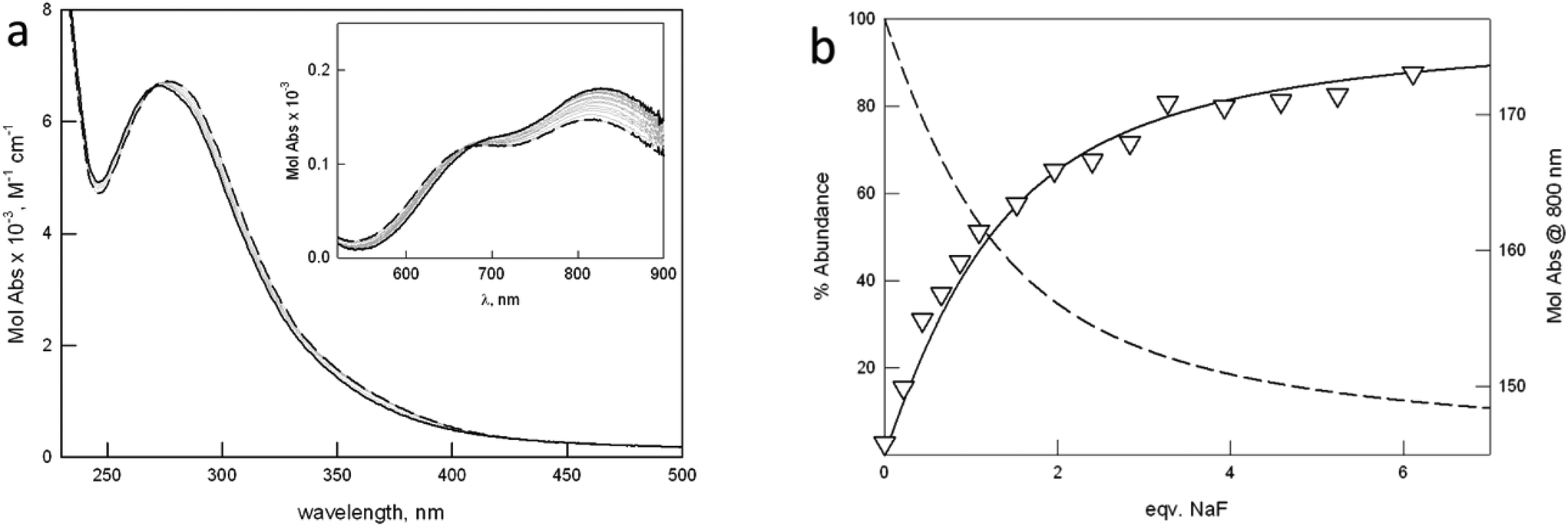 | ||
| Fig. 4 (a) Family of UV-vis spectra taken upon addition of a solution of NaF to [Cu23]4+ (1.1 mM) in 0.1 M MES at pH 5.8. (b) Distribution diagrams (% abundance vs. equiv. of added NaF) of the species calculated for log K11 = 3.17(1) (T = 25 °C). Triangles correspond to the experimental profile of Mol Abs at 800 nm vs. equiv. NaF. | ||
The working pH of 5.8 (0.1 M MES, 25 °C) was chosen in agreement with the following requisites: (i) the initial solution must contain a single major species, i.e. the dicopper cryptate in one of its forms, either [Cu2L]4+ or [Cu2L(OH)]3+, for all investigated receptors; (ii) the CT band typical of the halide-bridged cryptate must be close to maximum intensity. The latter requisite is based on the pH-spectrophotometric titration on the system, [Cu21]4+, in the presence of halide anions.
For all the investigated anions, a single equilibrium in solution was observed, leading to the formation of a 1:1 cryptate:anion complex (1). The corresponding constants are reported in Table 3, as log units.
| [Cu2L]4+ + X− ⇄ [Cu2L(X)]3+ | (1) |
| [Cu21]4+ | [Cu22]4+ | [Cu23]4+ | [Cu24]4+ | |
|---|---|---|---|---|
| a Two constants are determined, a and b, corresponding to eqn (2) (log K21) and (3) (log K11), respectively (see the text). | ||||
| NaF | 2.53(1) | 3.10(1) | 3.17(1) | 3.85(2) |
| NaCl | 3.70(1) | 2.35(3) | 2.23(1) | <2 |
| NaBr | 2.85(1) | <2 | <2 | <2 |
| NaI | <2 | <2 | <2 | <2 |
| NaN3 | 4.26(1) | 3.96(2)a | 3.75(2)a | 5.75(6) |
| 3.05(3)b | 3.98(4)b | |||
| CH3COONa | 2.32(1) | <2 | 2.00(1) | <2 |
The affinity of [Cu21]4+ for halides had already been investigated by our group, by following the formation of the anion-to-metal CT band, at pH 5.2 in 0.05 M MES.6a The values for the binding constants at pH 5.2 were higher than those reported in Table 3, determined at pH 5.8 (0.1 M MES). This can be attributed to the difference in the experimental conditions (i.e. ionic strength, and pH).
The initial spectrum of [Cu21]4+ displays a band at 360 nm (3500 M−1 cm−1), not present in the spectra of the other cages. The band can be assigned to a charge transfer involving the water oxygen atom bridging the metal centers. In the case of the other cryptates [Cu2L]4+ (L = 2–4), the distance between the Cu(II) ions is too long for the formation of a bridge. Halide binding promotes a shift of the N(amine) → Cu(II) CT band and a change in the intensity ratio between the two d–d bands in the visible region, the band at lower energy (around 850 nm) generally increasing in intensity. This last feature depends on the variation of both the symmetry around the metal centers, and the energy of d orbitals, upon anion binding.
As shown in Table 3, the substitution of one or two furanyl spacers with p-xylyl units has a similar effect on the anion binding constants. In particular, for both [Cu22]4+ and [Cu23]4+, the affinity towards halides decreases with the density charge of the anion following the spectrochemical series, i.e. F− > Cl− > Br−, I−. Fig. 5 demonstrates that, in the case of F−, moving from the p-xylyl cryptate to the furan cage, the affinity constants decrease by more than one log unit, with the asymmetric cryptates showing an intermediate value.
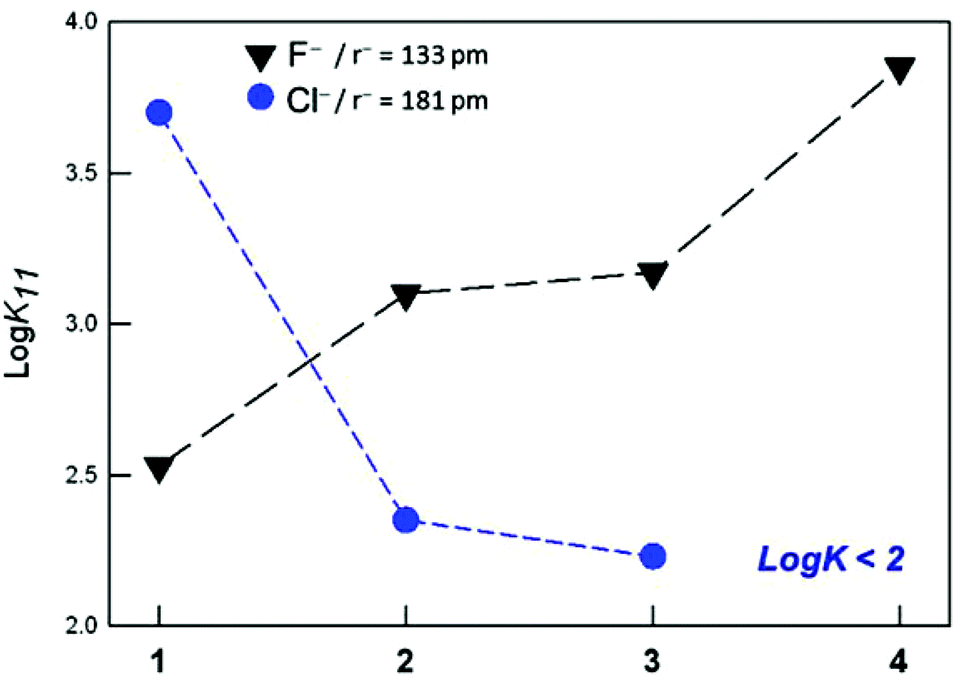 | ||
| Fig. 5 Affinity trend towards F− and Cl− anions (black and blue symbols, respectively), displayed by the investigated dicopper cryptates of 1–4. | ||
For the chloride anion, instead, the affinity trend is reversed, i.e. furan cage > asymmetric cryptates > p-xylyl cage (see Fig. 5). As already mentioned, the high affinity of receptor [Cu21]4+ for chloride depends on both the complementarity of the anion with the receptor's cavity, and the favorable interaction of the included anion with the furanyl spacers.
Binding of the azide anion: UV-vis studies in water
In order to better understand the anion binding properties of the new receptors, we performed UV-vis titrations with the linear azide anion. We chose N3− because it forms a well-known stable 1:1 inclusion complex with the [Cu24]4+ cryptate, as demonstrated by the crystal structure reported by Nelson et al.7a The azide complex is also highly stable in aqueous solution, as demonstrated by the affinity constant value calculated at pH 5.8, i.e. log K11 = 5.75(6) (see Table 3). The formation of the [Cu24(N3)]3+ complex is accompanied by the development of a new band in the UV-vis spectrum at about 430 nm (see Fig. S9†), attributable to the charge transfer transition from azide to copper, typical of the μ-1,3 azide linear bridge, as confirmed by the crystal structure.7a The azide anion also forms a 1:1 complex with the other cages. In particular, azide binding to [Cu21]4+ promotes the development of a CT band at higher energy with respect to [Cu24]4+ (i.e. 386 nm),6a probably due to the different binding geometry of the bidentate anion to the copper centers (see Fig. S10,† where the 1:1 stoichiometry is confirmed by the presence of isosbestic points). This spectroscopic feature, as well as the lower value of the binding constant (log K11 = 4.26(1)), demonstrates the worse fitting of the bidentate anion within the furan cage's cavity. In the case of the asymmetric cryptates, a decrease in the log K11 values is observed, with respect to both symmetric receptors: the decrease is especially remarkable (2 log units) on going from 4 to 3. Thus, replacement of just one p-xylyl spacer with a furanyl group seems to drastically reduce the hosting capability of the receptor towards azide, because of the alterations to the receptor's cavity.
Furthermore, for both asymmetric cryptates, the formation of the 1:1 complex with azide is anticipated by another equilibrium, leading to the dimeric species [(Cu2L)⋯(N3)⋯(Cu2L)]7+ (L = 2, 3).
As a consequence, over the course of the titration, the following equilibria are observed (the corresponding constants, log K21 and log K11, are reported in Table 3):
| 2[Cu2L]4+ + N3− ⇄ [(Cu2L)⋯(N3)⋯(Cu2L)]7+ | (2) |
| [(Cu2L)⋯(N3)⋯(Cu2L)]7+ + N3− ⇄ 2[Cu2L(N3)]3+ | (3) |
Fig. 6 shows the family of spectra taken upon titration of [Cu23]4+ with azide (see Fig. S8† for the analogous titration on [Cu22]4+). The formation of [(Cu23)⋯(N3)⋯(Cu23)]7+ is accompanied by the development of a band around 420 nm, attributable to the azide-to-copper transition, involving the bidentate anion and the metal centers of two different [Cu23]4+ units. Interestingly, the maximum of this band is close to that observed for the monomeric [Cu24(N3)]3+ complex, containing a linearly bridging azide anion. Further azide addition promotes the formation of the 1:1 complex (i.e. [Cu23(N3)]3+), according to equilibrium (3), and the shift of the CT band towards higher energies. In the final spectrum, corresponding to the [Cu23(N3)]3+ species, the CT band is centered around 390 nm, as for the monomeric complex of the furan cage.
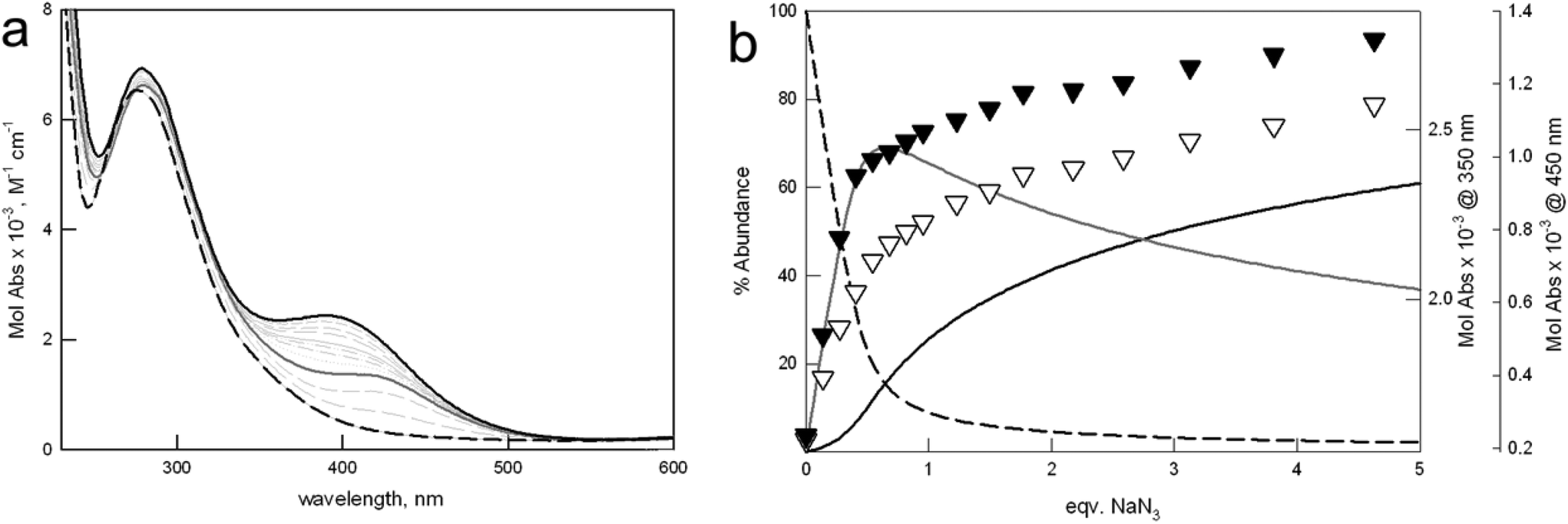 | ||
| Fig. 6 (a) Family of UV-vis spectra taken upon addition of a solution of NaN3 to [Cu23]4+ (1.1 mM) in 0.1 M MES, at pH 5.8. Solid and dashed black lines: initial and final spectrum, corresponding to [Cu23]4+ and [Cu23(N3)]3+ species, respectively. Solid gray line: spectrum taken at 0.5 equiv. NaN3, relative to the [(Cu23)⋯(N3)⋯(Cu23)]7+ complex. (b) Distribution diagram (% abundance vs. equiv. of added NaN3) of the species, calculated for log K11 = 3.75(2), and log K21 = 3.98(4), according to eqn (2) and (3), respectively (T = 25 °C). Solid and dashed black lines: [Cu23]4+ and [Cu23(N3)]3+ species, respectively; solid gray line: [(Cu23)⋯(N3)⋯(Cu23)]7+. Black and white triangles show the profiles of Mol Abs vs. equiv. NaN3 at 350 and 450 nm, respectively. | ||
Noticeably, the formation of dimeric species also occurs in organic solvents upon halide binding to [Cu21]4+,6c and in aqueous mixtures in the recognition of nucleoside monophosphates by dicopper cryptates.6d In the latter case, the formation of the dimer may induce a beneficial relief of steric constraints. This seems to be confirmed also for the asymmetric cryptates, in particular for receptor 3, by the large value of log K21.
Unfortunately, for other pseudo-halides, the affinity constants with the asymmetric cages could not be safely determined due to precipitation. Binding tendencies towards oxo-anions (e.g. nitrate, sulfate and acetate) were also studied, and were found to be very poor: in fact, only acetate is bound by cryptates of 1 and 2, with constants higher than or equal to 2.0 log units.
Conclusions
In this work, we reported both the syntheses and the anion binding properties of two new asymmetric dicopper cryptates, characterized by alternate furanyl and p-xylyl spacers. Our studies pointed out some interesting results. (i) The anion binding tendencies of the two asymmetric cages are very close, even if the number of the furanyl spacers replaced by p-xylyl units is different. (ii) In the case of halides, the new receptors display intermediate binding properties between the symmetric analogues (i.e. p-xylyl and furan cryptates). (iii) The substitution of even only one furanyl unit with a p-xylyl spacer significantly modifies the distance between the metal ions, thus preventing the development of the CT band, typically accompanying the binding of halides by the furan cryptate, [Cu21]4+. As a consequence, the variation in the UV-vis spectrum, occurring upon titration with halides, is similar to that observed for the p-xylyl cage, [Cu24]4+. (iv) In the case of azide, the asymmetric receptors tend to form stable complexes of different stoichiometry, not observed for the symmetric cryptates.This work also confirms some peculiarities of receptor [Cu21]4+. In particular, except for fluoride, all the halides display higher affinity for cryptate [Cu21]4+ than for the other cages. This is not only a matter of geometric complementarity between the cavity of [Cu21]4+ and the included anion. The interaction of the included halide with furanyl spacers plays in fact an active role in the stabilization of the inclusion complex. Noticeably, this stabilizing effect drops when furan rings are replaced by p-xylyl fragments.
In conclusion, with all the investigated anions, the asymmetric receptors display stronger similarities to each other than to the symmetric analogues. This unexpected result may be very helpful in the design of new molecular receptors based on dimetallic cryptates, in particular for the tuning of the sensitivity towards specific anionic substrates.
Experimental section
All reagents were purchased form Aldrich/Fluka and used without further purification. All reactions were performed under dinitrogen. Azacryptands 1 and 4 were prepared following already described procedures.7 The synthesis of the new azacryptands, 2 and 3, is reported in the ESI.† The structures of 2 and 3 were verified by 1H NMR, 13 C NMR, ESI-MS and elemental analysis.Experimental general procedures and details are also included in the ESI.† Mass spectra were acquired on a Thermo-Finnigan ion-trap LCQ Advantage Max instrument equipped with an ESI source. 1H and 13C NMR spectra were taken on a Bruker AVANCE 400 spectrometer (operating at 9.37 T, 400 MHz). UV-vis spectra were run on a Varian Cary 100 SCAN spectrophotometer with quartz cuvettes of the appropriate path length (0.1–1 cm) at 25.0 ± 0.1 °C under inert conditions. In any case, the concentration of the chromophore and the optical pathway were adjusted to obtain spectra with AU ∼ 1.
Potentiometric titrations
All measurements were performed at 25 °C in aqueous solution (0.1 M CF3SO3Na) with a Radiometer TitraLab 90 titration system. Titrations were performed under a dinitrogen atmosphere, in the presence of a double junction pH reference electrode (SCE) filled with aqueous CF3SO3Na 0.1 M. For all the investigated receptors, protonation constants were determined at constant ionic strength in pure water CF3SO3Na 0.1 M. In a typical experiment, 10 mL of a 5 × 10−4 M solution of the ligand, as the free base, was treated with an excess of CF3SO3H. Titrations were run by addition of 10 μL aliquots of carbonate-free standard 0.1 M NaOH, recording 80–100 points for each titration. Complexation constants were determined by carrying out a similar potentiometric titration experiment, with the additional presence of 2 equiv. CuII(CF3SO3)2. Prior to each potentiometric titration, the standard electrochemical potential (E°) of the glass electrode was determined in CF3SO3Na 0.1 M by a titration experiment according to the Gran method.14 Titration data (emf vs. mL of NaOH) were processed with the Hyperquad® package10 to determine the equilibrium constants (reported in Tables 2 and 3).Spectrophotometric titrations
All titrations were performed in aqueous solution, buffered at pH 5.8 by 0.1 M MES at 25.0 ± 0.1 °C. In a typical experiment, a solution of the ligand copper complex was titrated with a 100-fold more concentrated solution of the sodium salt of the envisaged anion. Titration data were processed with a non-linear least-squares procedure (Hyperquad® package),10 in order to determine the equilibrium constants.Crystal structure analysis
Diffraction data for a pale yellow single crystal were collected at room temperature with MoKα X-ray radiation (λ = 0.71073 Å) on a Bruker-AXS diffractometer equipped with a SMART-APEX CCD detector. Crystal data for [3H8(NO3)](NO3)7·3H2O: C34H66N16O28, Mr = 1146.98, T = 293 K, crystal dimensions 0.30 × 0.12 × 0.08 mm3, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 12.5559(13) Å, b = 12.7718(13) Å, c = 17.8881(18) Å, α = 110.689(2)°, β = 91.837(2)°, γ = 102.914(2)°, V = 2596.2(5) Å3, Z = 2, ρcalcd = 1.467 g cm−3, F(000) = 1202, μ MoKα = 0.128 mm−1, θmax = 23°, 17958 measured reflections, 7234 independent reflections (Rint = 0.037), 3387 strong reflections [Io > 2σ(Io)], 754 refined parameters, R1 = 0.1131 (strong data) and 0.1808 (all data), wR2 = 0.3090 (strong data) and 0.3669 (all data), GOF = 1.125, 0.62 and −0.33 max and min electron density residuals.
(no. 2), a = 12.5559(13) Å, b = 12.7718(13) Å, c = 17.8881(18) Å, α = 110.689(2)°, β = 91.837(2)°, γ = 102.914(2)°, V = 2596.2(5) Å3, Z = 2, ρcalcd = 1.467 g cm−3, F(000) = 1202, μ MoKα = 0.128 mm−1, θmax = 23°, 17958 measured reflections, 7234 independent reflections (Rint = 0.037), 3387 strong reflections [Io > 2σ(Io)], 754 refined parameters, R1 = 0.1131 (strong data) and 0.1808 (all data), wR2 = 0.3090 (strong data) and 0.3669 (all data), GOF = 1.125, 0.62 and −0.33 max and min electron density residuals.
Data reduction was performed using the SAINT software.15 Intensities were corrected for Lorentz and polarization effects; absorption effects were empirically evaluated using the SADABS software16 and absorption correction was applied to the data (0.878 and 0.999 min and max transmission factors). The crystal structure was solved by direct methods (SIR 97)17 and refined by full-matrix least-squares procedures on F2 using all reflections (SHELXL 97).18 Anisotropic displacement parameters were used for all non-hydrogen atoms. Hydrogens have been placed at calculated positions and their positions refined according to a riding model. Positions of hydrogens belonging to water solvent molecules remained undetermined.
Positional disorder affects two of the seven nitrate counterions placed outside the cage. The nitrate ion defined by the N(15), O(20), O(21) and O(22) atom sites is disordered over two alternative positions (A and B), mutually exclusive and half populated. A partly populated O4w atom site is placed near the half populated O22B atom site; O4w hosts the oxygen of an additional water molecule when the O22B site is empty. The nitrate ion defined by the N(16), O(23), O(24) and O(25) atom sites shows two alternative positions for the O(25) site (A and B), mutually exclusive and half populated. Soft restraints on the geometrical features and on the Uij components have been applied in the final least-square cycles of refinements of the disordered atom sites.
The selected single crystal showed poor X-ray diffraction quality; intensities of reflections with θ values greater than 23° were unobservable and the final agreement indexes were high. The poor X-ray diffraction quality was mainly related to the disorder affecting the nitrate counterions. However, the accuracy of the geometrical details of the octaprotonated cage and of the enclosed nitrate anion was considered suitable to the aims of this work.
CCDC 992963 contains the supplementary crystallographic data for this paper.
Acknowledgements
The financial support of the Italian Ministry for Education, Universities and Research is gratefully acknowledged – Project PRIN2010 010CX2TLM_009 Integrated supramolecular technologies for chemical information processing: advanced molecular devices and materials (InfoChem).Notes and references
- (a) J.-M. Lehn, Pure Appl. Chem., 1980, 52, 2441 CAS; (b) J.-M. Lehn, Pure Appl. Chem., 1977, 49, 857 CrossRef CAS; (c) R. J. Motekaitis, A. E. Martell, J.-M. Lehn and E. Watanabe, Inorg. Chem., 1982, 21, 4253 CrossRef CAS.
- (a) K. Bowman-James, A. Bianchi and E. García-España, Anion Coordination Chemistry, Wiley-VCH, Weinheim, 2011 Search PubMed; (b) P. Mateus, N. Bernier and R. Delgado, Coord. Chem. Rev., 2010, 254, 1726 CrossRef CAS PubMed; (c) J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, in Monographs in Supramolecular Chemistry, ed. J. F. Stoddart, Royal Society of Chemistry, Cambridge, 2006 Search PubMed.
- (a) R. Alberto, G. Bergamaschi, H. Braband, T. Fox and V. Amendola, Angew. Chem., Int. Ed., 2012, 51, 9772 CrossRef CAS PubMed; (b) V. Amendola, G. Alberti, G. Bergamaschi, R. Biesuz, M. Boiocchi, S. Ferrito and F.-P. Schmidtchen, Eur. J. Inorg. Chem., 2012, 3410 CrossRef CAS.
- (a) J.-M. Lehn, S. H. Pine, E. Watanabe and A. K. Willard, J. Am. Chem. Soc., 1977, 99, 6766 CrossRef CAS; (b) V. Amendola, D. Esteban-Gomez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343 CrossRef CAS PubMed; (c) V. Amendola, M. Bonizzoni, D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli, F. Sancenón and A. Taglietti, Coord. Chem. Rev., 2006, 250, 1451 CrossRef CAS PubMed.
- (a) I. Ravikumar, P. S. Lakshminarayanan, E. Suresh and P. Ghosh, Beilstein J. Org. Chem., 2009, 5(1), 41 CAS; (b) P. S. Lakshminarayanan, D. K. Kumar and P. Ghosh, Inorg. Chem., 2005, 44(21), 7540 CrossRef CAS PubMed; (c) Md. A. Hossain, M. A. Saeed, G. Gryn'ova, D. R. Powell and J. Leszczynski, CrystEngComm, 2010, 12, 4042 RSC; (d) M. A. Saeed, F. R. Fronczek and Md. A. Hossain, Chem. Commun., 2009, 6409 RSC; (e) P. S. Lakshminarayanan, I. Ravikumar, E. Suresh and P. Ghosh, Cryst. Growth Des., 2008, 8(8), 2842–2852 CrossRef CAS.
- (a) V. Amendola, E. Bastianello, L. Fabbrizzi, C. Mangano, P. Pallavicini, A. Perotti, A. Manotti Lanfredi and F. Ugozzoli, Angew. Chem., 2000, 112, 3039–3042 ( Angew. Chem., Int. Ed. , 2000 , 39 , 2917–2920 ) CrossRef; (b) G. Alibrandi, V. Amendola, G. Bergamaschi, R. Dollenz, L. Fabbrizzi, M. Licchelli and C. Lo Vecchio, Chem. – Eur. J., 2013, 19, 3729–3734 CrossRef CAS PubMed; (c) V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi, A. Poggi and M. Zema, Inorg. Chim. Acta, 2008, 361, 4038–4046 CrossRef CAS PubMed; (d) V. Amendola, G. Bergamaschi, A. Buttafava, L. Fabbrizzi and E. Monzani, J. Am. Chem. Soc., 2010, 132, 147–156 CrossRef CAS PubMed.
- (a) M. G. B. Drew, J. Hunter, D. J. Marrs, J. Nelson and C. Harding, J. Chem. Soc., Dalton Trans., 1992, 3235–3242 RSC; (b) Q. Lu, J.-M. Latour, C. J. Harding, N. Martin, D. J. Marrs, V. McKee and J. Nelson, J. Chem. Soc., Dalton Trans., 1994, 1471 RSC; (c) F. Arnaud-Neu, S. Fuangswasdi, B. Maubert, J. Nelson and V. McKee, Inorg. Chem., 2000, 39, 573 CrossRef CAS.
- V. Amendola, G. Bergamaschi, M. Boiocchi, R. Alberto and H. Braband, Chem. Sci., 2014, 5, 1820–1826 RSC.
- (a) P. Tecilla, U. Tonellato and A. Veronese, J. Org. Chem., 1997, 62, 7621–7628 CrossRef CAS; (b) J. Middleton Boon, T. N. Lambert, B. D. Smith, A. D. Beatty, V. Ugrinova and S. N. Brown, J. Org. Chem., 2002, 67, 2168–2174 CrossRef PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS ; http://www.hyperquad.co.uk/index.htm.
- C. J. Harding, V. Mc Kee, J. Nelson and Q. Lu, J. Chem. Soc., Chem. Commun., 1993, 1768 RSC.
- A. D. Bond, S. Derossi, F. Jensen, F. B. Larsen, C. J. McKenzie and J. Nelson, Inorg. Chem., 2005, 44, 5987–5989 CrossRef CAS PubMed.
- K. G. Ragunathan and P. K. Bharadwaj, J. Chem. Soc., Dalton Trans., 1992, 1653–1656 RSC.
- F. J. C. Rossotti and H. Rossotti, J. Chem. Educ., 1965, 42, 375–378 CrossRef CAS.
- Bruker, SAINT Software Reference Manual. Version 6, Bruker AXS Inc., Madison, Wisconsin, USA, 2003 Search PubMed.
- G. M. Sheldrick, SADABS Siemens Area Detector Absorption Correction Program, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. Moliterni, G. Polidori and R. J. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 992963. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C4DT00886C |
| This journal is © The Royal Society of Chemistry 2014 |