Induced circular dichroism of polyoxometalates via electrostatic encapsulation with chiral organic cations†
Yizhan
Wang
,
Lei
Shi
,
Yang
Yang
,
Bao
Li
* and
Lixin
Wu
*
State Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, P.R. China. E-mail: wulx@jlu.edu.cn; Fax: (+86) 431-8519-3421
First published on 17th April 2014
Abstract
To explore the principle of chiral induction in inorganic clusters, chiral organic cations with two stereocenters, R- and S-BPEA, are used to encapsulate a series of polyoxometalates (POMs) bearing different structures and transition absorption bands in aqueous solution, constructing a series of chiral supramolecular complexes. Due to the induction of chiral organic cations, POMs possessing both chiral and achiral structures show an induced circular dichroism (ICD) effect. ICD signals in the absorption bands corresponding to ligand to metal charge transfer (LMCT) transitions, d–d transitions and intervalence charge transfer (IVCT) transitions are observed for different complexes. Moreover, the ICD of the POMs exhibits a direct correlation with the degree of POM distortion and the distance between the chiral center and the POM surface. The encapsulation of POMs with chiral organic cations via electrostatic interactions provides a facile and effective method for constructing optically pure POM-based materials.
Introduction
Polyoxometalates (POMs), as a kind of nano-sized metal–oxygen cluster with a number of different compositions and a variety of frame structures, have potential in the fields of catalysis, optics, and nanomaterials.1–4 Chirality, as a general natural phenomenon, always attracts great attention due to its mysterious source and wide applications.5,6 In light of the advantages of POMs and the importance of chirality, substantial efforts have been devoted to POM-based chiral structures because of interest in their structural chemistry and in functional materials.7,8 In this context, chiral POMs are expected to show asymmetric catalysis, chiral recognition, chiroptical switching, and nonlinear optical properties.8 However, the preparation of POM-based chiral structures, especially optically pure materials, is still a considerable challenge because most POM clusters possess high symmetry, which leads to difficulty in constructing chiral structures.8 The known POMs that bear chiral structures are usually unstable either in solution or in the solid state, and their enantiomers are easily interconverted and/or difficult to isolate from each other. For example, chiral POMs can be prepared from structural vacancies and alternating bond length distortions,7,9 but such enantiomers often racemize due to water molecule exchange, partial hydrolysis, or fluxional behavior, so that few enantiopure conglomerates have been successfully obtained.To obtain chiral POM architectures, some important synthetic strategies have been developed,7,8 and the strategy used most often is chiral resolution of enantiomeric structures by the addition of chiral supporting agents or crystallization.10–13 But, the key limitation is the requirement of intrinsic chirality of the POMs, and this method cannot be extended to achiral POM systems, of which a large majority have diverse functional properties. An improved route is the direct attachment of a chiral unit (organic moiety or metal complex), as a chirality transfer agent, onto the POMs through covalent bonding, obtaining stable chiral POM structures.14–16 The chirality transfer from the attached chiral ligands induces optical activity in the POM frame-structures, and the resulting chiral POM structures demonstrate interesting self-assembly and enantioselective catalysis.17,18 However, the chiral POM structures prepared by covalent synthesis are only a very small part of the POM family because of the limitation of detailed chemical synthesis. Therefore, it is significant to explore a general strategy for the introduction of chirality into POMs through a simple approach, while retaining the intrinsic properties.
It is known that intermolecular interactions can give rise to an induced circular dichroism (ICD) in the achiral counterpart.19 Induced chirality has proved to be a convenient approach for creating chiral materials from achiral molecules or components.20,21 The replacement of POM counterions by organic cations through electrostatic interactions is a simple and flexible method for POM modification.22 Versatile properties different to those of POMs alone are well developed in the resulting POM complexes.23–25 The combination of electrostatic modification and chiral induction should shed light on the simple preparation of stable, well-defined chiral POM structures.
Although ICD effects in POMs in the presence of chiral cations has long been known, the generality of ICD in POMs with different transition bands is not well developed. Some enantiopure POMs based on the electrostatic coupling of chiral cations have been described.26–29 Pope and co-workers26 used L-brucine to combine chiral P2Mo18O626−, and proved the chiral optical activity of the resulting complexes within the oxygen to molybdenum charge transfer transitions. Nlate and co-workers29 reported chiral dendron-like POM complexes constructed by electrostatic interactions between enantiopure branched ammonium ions and an achiral polyanion, which show optical activity at approximately 280 nm, indicating the transfer of chirality from the organic units to the inorganic cluster. For such systems, studies mainly focus on the LMCT transitions, while the principle of chiral induction with different chiral cations in POM systems is not well understood in detail.
We have explored chiral POM-based materials with asymmetric catalysis30 and chiroptical switching31 properties through electrostatic encapsulation of specific POMs with chiral organic cations. To further extend this synthetic strategy for constructing optically pure POM-based materials, herein, we use the same chiral organic cations to encapsulate POMs with different structures and transition absorption bands, obtaining a series of chiral supramolecular complexes (Scheme 1). The induction of chiral organic cations triggers the ICD effect in achiral and chiral POMs. ICD signals corresponding to ligand to metal charge transfer (LMCT) transitions, d–d transitions and intervalence charge transfer (IVCT) transitions are observed for different complexes. To study the influence of chiral organic cations on the chiroptical activity of the POMs, we synthesize chiral organic cations with different spacer lengths. It is found that the ICD of the POMs has a direct correlation with the distance between the chiral center and the POM surface. These results provide us with a simple and general method for the construction of optically pure POM-based materials.
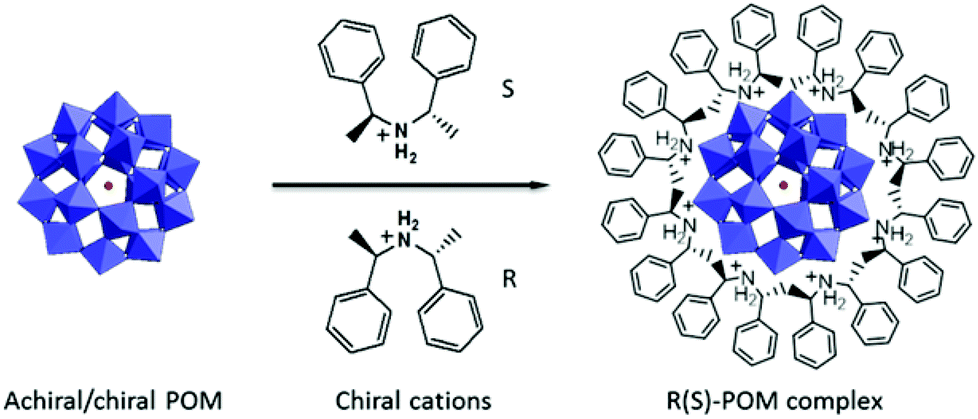 | ||
| Scheme 1 Schematic illustration of the preparation of chiral POM complexes via electrostatic interactions. | ||
Experimental section
Materials and reagents
H3PMo12O40 (PMo12) and H4SiW12O40 (SiW12) were analytical grade and were purchased from Beijing Chemical Reagent Company. K12.5Na1.5[NaP5W30O110]·nH2O (P5W30), K12[EuP5W30O110]·nH2O (EuP5W30), K6P2W18O62 (P2W18), K28Li5H7[P8W48O184]·92H2O (P8W48), Na12K8H4[K8P8W48O184(V4V2O12(H2O)2)2]·∼80H2O (P8W48V12), (NH4)6[P2Mo18O62]·12H2O (P2Mo18), K5[PW11VIVO40]·8H2O (PW11VIV), K6[SiW11VIVO40]·2H2O (SiW11VIV), K7[PW11O39]·12H2O (PW11), K6SiW11O39Co (SiW11Co) and K6CoW12O40 (CoW12) were synthesized according to the published procedures.32–39 (+)-Bis[(R)-1-phenylethyl]amine hydrochloride (R-BPEA) and (−)-bis[(S)-1-phenylethyl]amine hydrochloride (S-BPEA) were purchased from Sigma-Aldrich. (R)- and (S)-N,N,N-trimethyl-1-phenylethanaminium iodide (R-, S-C0) were synthesized according to the published procedures.40 (R)-N,N,N-trimethyl-5-oxo-5-(1-phenylethoxy)pentan-1-aminium bromide (R-C5) and (R)-N,N,N-trimethyl-11-oxo-11-(1-phenylethoxy)undecan-1-aminium bromide (R-C11) were synthesized according to the procedures presented in the ESI.† The ethanol and acetonitrile used in the experiments were chromatographic grade.Measurements
FT-IR spectra were collected on a Bruker Vertex 80v spectrometer equipped with a deuterated triglycine sulfate detector (32 scans) at a resolution of 4 cm−1. Organic elemental analysis was carried out on an Elementar vario MICRO cube. 1H NMR spectra were recorded on a Bruker Ultra-Shield TM 500 MHz spectrometer using tetramethylsilane (TMS) as an internal standard (δ = 0 ppm). The UV-vis spectra were recorded on a Shimadzu 3100 PC spectrometer, and the slit width was set at 2 nm. Circular dichroism (CD) spectra were obtained on a Bio-Logic MOS-450 spectropolarimeter with a step size of 1 nm at a speed of 2 s nm−1 at room temperature.Typical preparation procedure for chiral complexes
POMn− and 0.1 g R- or S-BPEA or Cm (m = 0, 5, 11) were both dissolved in 20 mL deionized water (for P8W48, 0.1 M aqueous LiCl solution was used; for P2Mo18, ethanol was used) at room temperature to form two separate solutions. Then, the aqueous solution of the organic cation was added to the POMn− solution with stirring. The initial molar ratio of R- or S-BPEA or Cm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :POMn− was controlled at about n:1. The mixture was stirred at room temperature for 2 h. The resulting precipitate was filtered and washed several times with deionized water until no precipitate was formed on treating the filtrate with AgNO3 solution. The precipitate was dried under vacuum, giving the complex R-POM or S-POM.
:POMn− was controlled at about n:1. The mixture was stirred at room temperature for 2 h. The resulting precipitate was filtered and washed several times with deionized water until no precipitate was formed on treating the filtrate with AgNO3 solution. The precipitate was dried under vacuum, giving the complex R-POM or S-POM.
For R-P5W30, yield: 86%. IR (KBr pellet): v = 3600–3300, 3100–2400, 1593, 1499, 1456, 1383, 1207, 1165, 1078, 1018, 982, 933, 912, 789, 762, 739, and 698 cm−1. Elemental analysis (%) calcd for R-P5W30 (C208H260N13KNaP5W30O110, 10434 or C208H260N13NaNaP5W30O110, 10418): C 23.94 or 23.99, H 2.51 or 2.52, N 1.75 or 1.75; found: C 23.53, H 2.51, N 1.71, suggesting the chemical formula: (R-BPEA)13M(K or Na)NaP5W30O110. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.63 (6H, broad), 3.97 (2H, broad), 7.31–7.44 (10H, broad), 9.26 (2H, broad). (For R-BPEA, 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.58 (6H, d, J = 6.8 Hz), 3.90 (2H, m), 7.37–7.42 (10H, m), 10.07 (2H, broad).)
For R-EuP5W30, yield: 91%. IR (KBr pellet): ν = 3600–3300, 3100–2400, 1587, 1498, 1456, 1383, 1159, 1144, 1067, 1022, 979, 941, 914, 792, 764, and 700 cm−1. Elemental analysis (%) calcd for R-EuP5W30 (C192H240N12EuP5W30O110, 10298): C 22.39, H 2.35, N 1.63; found: C 22.06, H 2.19, N 1.76, suggesting the chemical formula: (R-BPEA)12EuP5W30O110. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.64 (6H, d, J = 6.5 Hz), 4.11 (2H, m), 7.40–7.48 (10H, m), 9.32 (2H, broad).
For R-SiW12, yield: 69%. IR (KBr pellet): ν = 3600–3300, 3200–2400, 1604, 1580, 1497, 1457, 1384, 1204, 1063, 1013, 965, 922, 883, 798, 766, and 700 cm−1. Elemental analysis (%) calcd for R-SiW12 (C64H80N4SiW12O40, 3779): C 20.34, H 2.13, N 1.48; found: C 20.24, H 2.18, N 1.46, suggesting the chemical formula: (R-BPEA)4SiW12O40. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.52 (6H, d, J = 6.5 Hz), 4.12 (2H, m), 7.37–7.49 (10H, m), 9.24 (2H, broad).
For R-P2W18, yield: 85%. IR (KBr pellet): ν = 3600–3300, 3100–2400, 1610, 1495, 1456, 1385, 1092, 1022, 957, 910, 793, and 700 cm−1. Elemental analysis (%) calcd for R-P2W18 (C96H120N6P2W18O62, 5721): C 20.15, H 2.11, N 1.47; found: C 19.84, H 2.11, N 1.47, suggesting the chemical formula: (R-BPEA)6P2W18O62. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.53 (6H, d, J = 6.7 Hz), 4.12 (2H, m), 7.38–7.49 (10H, m), 9.25 (2H, broad).
For R-P8W48, yield: 62%. IR (KBr pellet): ν = 3600–3300, 3100–2400, 1593, 1499, 1454, 1383, 1205, 1134, 1078, 1018, 922, 798, 764, 717, and 700 cm−1. Elemental analysis (%) calcd for R-P8W48 ((C16H20N)27Li13P8W48O184, 18217 or (C16H20N)27K13P8W48O184, 18635): C 28.48 or 27.84, H 2.99 or 2.92, N 2.07 or 2.03; found: C 28.22, H 3.14, N 2.06, suggesting the chemical formula: (R-BPEA)27M13P8W48O184. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.43 (6H, broad), 3.70 (2H, broad), 7.27–7.37 (10H, m), 9.32 (2H, broad).
For R-P2Mo18, yield: 76%. IR (KBr pellet): ν = 3600–3300, 3100–2400, 1578, 1497, 1456, 1385, 1078, 1003, 937, 904, 874, 833, 783, and 700 cm−1. Elemental analysis (%) calcd for R-P2Mo18 (C80H104N6P2Mo18O62, 3930.55): C 24.44, H 2.67, N 2.14; found: C 24.58, H 2.61, N 2.24, suggesting the chemical formula: R-BPEA5(NH4)P2Mo18O62. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.53 (6H, d, J = 6.7 Hz), 4.12 (2H, m), 7.09 (0.8H, t) 7.38–7.49 (10H, m), and 9.27 (2H, broad).
For R-CoW12, yield: 70%. IR (KBr pellet): ν = 3600–3300, 3100–2400, 1580, 1499, 1456, 1385, 1067, 1030, 928, 874, 764, and 702 cm−1. Elemental analysis (%) calcd for R-CoW12 (C96H120N6CoW12O40, 4263): C 27.04, H 2.84, N 1.97; found: C 26.73, H 2.82, N 1.94, suggesting the chemical formula: (R-BPEA)6CoW12O40. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.51 (6H, s), 4.06 (2H, broad), 7.36–7.45 (10H, m), and 9.19 (2H, broad).
For R-SiW11Co, yield: 46%. IR (KBr pellet): ν = 3600–3300, 3100–2400, 1593, 1499, 1456, 1414, 1383, 1207, 1078, 995, 947, 901, 802, 771, 762, and 700 cm−1. Elemental analysis (%) calcd for R-SiW11Co (C96H120N6SiW11CoO39, 4091): C 28.18, H 2.96, N 2.05; found: C 27.79, H 2.97, N 2.00, suggesting the chemical formula: (R-BPEA)6SiW11CoO39. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.41 (6H, s), 3.75 (2H, broad), 7.27–7.45 (10H, m).
For R-P8W48V12, yield: 90%. IR (KBr pellet): ν = 3600–3300, 3100–2400, 1589, 1499, 1454, 1383, 1205, 1136, 1088, 1022, 914, 823, 783, and 700 cm−1. Elemental analysis (%) calcd for R-P8W48V12 ((C16H20N)21Na3[K8P8W48O184(V4V2O12(H2O)2)2], 18218 or (C16H20N)21K3[K8P8W48O184(V4V2O12(H2O)2)2], 18266): C 22.15 or 22.09, H 2.37 or 2.36, N 1.61 or 1.61; found: C 22.20, H 2.41, N 1.61. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.55 (6H, broad), 3.97 (2H, broad), 7.37 (10H, broad), 9.29 (2H, broad).
For R-C0-P5W30, yield: 82%. IR (KBr pellet): v = 3038, 2968, 2905, 2400, 1639, 1487, 1458, 1163, 1074, 1013, 980, 930, 910, 789, 742, and 715 cm−1. Elemental analysis (%) calcd for R-C0-P5W30 ((C11H18N)11K3NaP5W30O110, 9377): C 15.50, H 2.13, N 1.64; found: C 15.65, H 2.06, N 1.63, suggesting the chemical formula: (C11H18N)11K3NaP5W30O110. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.74 (3H, broad), 3.21 (9H, broad), 4.87 (1H, broad), 7.44–7.63 (5H, broad).
For R-C5-P5W30, yield: 43%. IR (KBr pellet): v = 3100–2800, 1728, 1487, 1454, 1265, 1165, 1072, 1013, 980, 930, 910, 795, 744, and 702 cm−1. Elemental analysis (%) calcd for R-C5-P5W30 ((C16H26NO2)14NaP5W30O110, 11154): C 24.12, H 3.29, N 1.76; found: C 24.43, H 3.20, N 1.71, suggesting the chemical formula: (R-C5)14NaP5W30O110. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.48 (3H, d, J = 6.6 Hz), 1.55 (2H, m), 1.71 (2H, m), 2.41 (2H, t), 3.22 (9H, s), 3.29 (2H, m), 5.81 (1H, m), 7.29 (1H, m), 7.37 (4H, m).
For R-C11-P5W30, yield: 87%. IR (KBr pellet): v = 2926, 2852, 1732, 1628, 1578, 1475, 1373, 1246, 1165, 1069, 1012, 980, 931, 912, 791, 741, and 700 cm−1. Elemental analysis (%) calcd for R-C11-P5W30 ((C22H38NO2)14NaP5W30O110, 12332): C 29.99, H 4.35, N 1.59; found: C 29.84, H 4.24, N 1.79, suggesting the chemical formula: (C22H38NO2)14NaP5W30O110. 1H NMR (500 MHz, DMSO-d6, TMS, ppm): δ = 1.23 (12H, m), 1.45 (3H, d, J = 6.6 Hz), 1.51 (2H, m), 1.68 (2H, m), 2.31 (2H, t), 3.23 (9H, s), 3.28 (2H, m), 5.80 (1H, m), 7.29 (1H, m), 7.35 (4H, m).
The preparation and characterization of R-, S-PW11, R-, S-PMo12, R-, S-PW11VIV, and R-, S-SiW11VIV can be found in our previous work.30,31
Results and discussion
Preparation and characterization of chiral POM complexes
We select commercially available chiral cationic ammoniums with two stereocenters, (+)-bis[(R)-1-phenylethyl]amine hydrochloride (R-BPEA) and (−)-bis[(S)-1-phenylethyl]amine hydrochloride (S-BPEA), to replace the counterions in the POMs through ion substitution reactions. To confirm the generality of the present method for the construction of chiral POM complexes, we select a series of anionic POMs with LMCT, d–d, and IVCT transitions (Fig. 1). Because the chiral cationic ammoniums show good solubility in water, we perform all the ion substitution reactions in aqueous solution, except for the ion substitution reactions of the P2Mo18 cluster, which are performed in ethanol, due to the possible decomposition of this POM in aqueous solution. After mixing the two aqueous solutions at room temperature, the cationic ammonium and the anionic POM bind together through electrostatic interactions according to the charge ratio, and the resulting R- and S-enantiomers of the chiral complexes (abbreviated as R-POM and S-POM, respectively) precipitate directly in water. Compared with covalent modification and chiral resolution, the present experimental conditions are mild, and the chiral POM complexes can be easily obtained without complicated reaction procedures or long term crystallization.10–16 In contrast to POMs that are soluble in water, all the complexes are no longer miscible with water, but readily dissolve in organic media such as acetonitrile, DMF and DMSO, suggesting that the POMs have been successfully enwrapped by the hydrophobic chiral cations due to the charge neutralization.30 It is known that directly adding small amounts of chiral cations to POM solutions can result in an obvious ICD effect via ion-pairing.26 However, electrostatic encapsulation of POMs by chiral organic cations can improve the solubility of POMs in organic solvents, and possible chiral functional applications of POMs can be realized in these organic solvents.30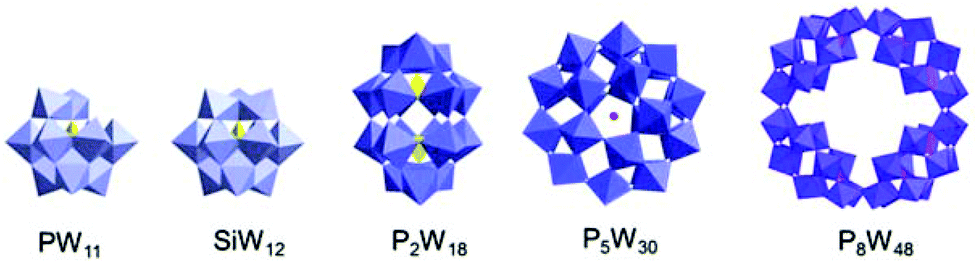 | ||
| Fig. 1 The POM structures of [PW11O39]7−, [SiW12O40]4−, [P2W18O62]6−, [NaP5W30O110]14− and [P8W48O184]40−. | ||
The chiral complexes were characterized using IR, NMR, UV-vis spectroscopy and elemental analysis. Taking the chiral complex composed of the [NaP5W30O110]14− cluster (P5W30) as an example, the characteristic W–O vibrational bands of P5W30 at 933 (νas W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Od), 912 (νas W–Ob–W), and 789 (νas W–Oc–W) cm−1 are observed in the IR spectrum of the complex, confirming that the cluster framework structure has been retained after encapsulation (Fig. S1†). The C–H out-of-plane bending vibration of the mono-substituted benzene group in the chiral organic cation is also observed for the complex, at approximately 762 and 698 cm−1, which implies that the counterions of P5W30 have been replaced by chiral organic cations. There are no obvious differences in the preparation processes or in the IR spectra of R-P5W30 and S-P5W30. The 1H NMR spectra of the two complexes also prove the presence of chiral organic cations (Fig. S2–S3†). Comparing the 1H NMR spectra of R-P5W30 and the corresponding organic cation, we find that the 1H signal of the N–H group of R-P5W30 shifts from 10.07 to 9.26 ppm, while the proton signal of the chiral carbon atom moves from 3.90 to 3.97 ppm, confirming the tight electrostatic interaction between the ammonium group and P5W30. All the other complexes were characterized similarly, and the results are consistent with the expected chemical compositions.
Od), 912 (νas W–Ob–W), and 789 (νas W–Oc–W) cm−1 are observed in the IR spectrum of the complex, confirming that the cluster framework structure has been retained after encapsulation (Fig. S1†). The C–H out-of-plane bending vibration of the mono-substituted benzene group in the chiral organic cation is also observed for the complex, at approximately 762 and 698 cm−1, which implies that the counterions of P5W30 have been replaced by chiral organic cations. There are no obvious differences in the preparation processes or in the IR spectra of R-P5W30 and S-P5W30. The 1H NMR spectra of the two complexes also prove the presence of chiral organic cations (Fig. S2–S3†). Comparing the 1H NMR spectra of R-P5W30 and the corresponding organic cation, we find that the 1H signal of the N–H group of R-P5W30 shifts from 10.07 to 9.26 ppm, while the proton signal of the chiral carbon atom moves from 3.90 to 3.97 ppm, confirming the tight electrostatic interaction between the ammonium group and P5W30. All the other complexes were characterized similarly, and the results are consistent with the expected chemical compositions.
ICD of O→M (W or Mo) LMCT transitions in POM complexes
CD spectra show the differences in the absorptions of left-handed and right-handed circularly polarized light due to chirality.41 The chiroptical activities of the chiral organic cations were investigated by CD spectroscopy at room temperature in solution. As expected, R-BPEA and S-BPEA show positive and negative Cotton effects at 218 nm at low concentration (3 × 10−4 mol L−1) in acetonitrile, and their spectra are distinct mirror images (Fig. S4–S5†), demonstrating the enantiopurity of the two chiral cations. On increasing the concentration, in acetonitrile, the chiral organic cations exhibit a B band centered at 256 nm, corresponding to the π→π* transition, with vibrational fine structure due to the presence of the phenyl group. Meanwhile, CD signals associated with the transition band appear at approximately 240–275 nm.42Molecules display ICD within their absorption bands when in association with chiral inducing components.19 By comparing the absorption bands of the POMs and the organic cations, the chiroptical activities at wavelengths larger than 275 nm can be definitively attributed to the ICD of the POM clusters. For the complexes showing different chiroptical activity at wavelengths less than 275 nm, the observed CD signals may result from two situations: ICD of the POMs, or CD of chiral organic cations located in different environments.43 Normally, it is difficult to distinguish the sources of activity at wavelengths less than 275 nm due to overlap of the transition bands of the organic cations and the POMs in this spectral region. Therefore, in the following studies we mainly focus on the ICD bands in the spectral region outside the absorption bands (>275 nm) of the organic cations.
We firstly investigate the chiroptical activity of R-P5W30 and S-P5W30 in solution. The CD spectra of the complexes in acetonitrile are mirror images of each other, demonstrating the enantiopurity of the two complexes. This is not unusual because R- and S-BPEA only change their counterions during the encapsulation process, and no racemization takes place. In the wavelength range from 190 to 400 nm, the complexes show different Cotton effects to those of the chiral organic cations alone (Fig. 2). By referring to the absorption of this POM at 275–325 nm, the obvious opposite CD signals at 293 nm ([θ] = 7.2 × 105 deg cm2 dmol−1) can be definitively assigned to the ICD of the POMs in the two complex enantiomers. Because the characteristic absorption bands of P5W30 in this region are derived from O→W LMCT transitions,44 the ICD can be ascribed to the transfer of chirality from R- or S-BPEA to the LMCT band. No obvious changes in the CD and UV-vis spectra of the two complexes are observed over several weeks, indicating that they are enantiomerically stable, which is of great importance in the chiral application of POMs in solution. The P5W30 cluster is thought to possess a chiral frame structure. Due to the alternating short and long bond length distortions within the two W10O10 rings in the cluster, the symmetry of P5W30 is reduced from D5h to chiral D5.9 However, this POM usually exists as a racemic mixture in solution, as well as in the solid state. The resolution and chiroptical properties of P5W30 have rarely been reported. The obvious chiroptical activities of complexes based on P5W30 within the LMCT transition bands may result from two main factors: one is that perturbation of the chiral organic cations affects the enantiomer equilibrium of the racemic POMs (intrinsic ICD); and the other is that the chiral counterions cause ICD in a chromophore of the POM due to intermolecular interactions (extrinsic ICD).19 To the best of our knowledge, this is the first time that the chiroptical activity of P5W30 is presented. Moreover, when the sodium ion located on the fivefold axis within P5W30 is replaced by Eu3+, the obtained europium substituted P5W30 also features alternating short and long bond length distortions. Yet, the complexes R-EuP5W30 and S-EuP5W30 also exhibit obvious ICD at the wavelengths corresponding to the LMCT transitions of the POMs, and the CD signals of R-EuP5W30 and S-EuP5W30 are similar to those of R-P5W30 and S-P5W30 (Fig. S6–S7†).
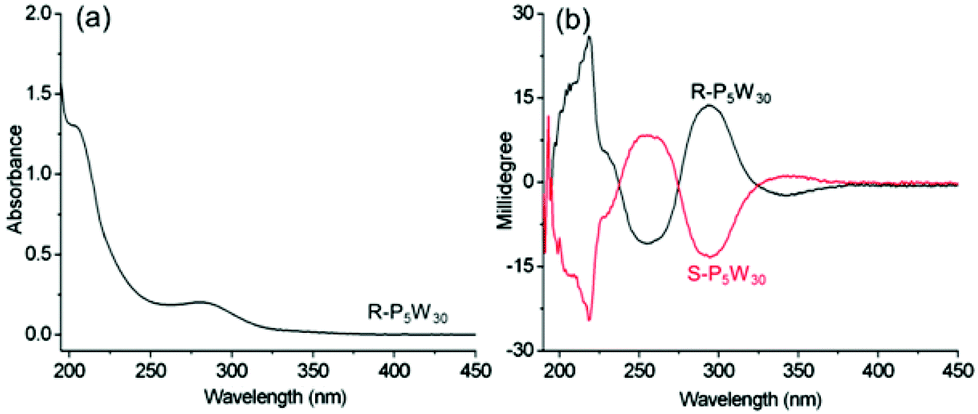 | ||
| Fig. 2 (a) The UV-vis spectrum of R-P5W30 and (b) CD spectra of R-P5W30 and S-P5W30 in CH3CN (c = 0.2 mg mL−1, l = 1 mm). | ||
The present method using ion substitution reactions is a general method for the construction of various chiral POM complexes. To study the ICD properties within the LMCT transition band, we extend the present system to some more tungsten-based POMs, PW11, SiW12, P2W18, and P8W48. After mixing the aqueous solutions of the POMs and the chiral organic cations at room temperature, we can easily obtain the chiral POM complexes R-, S-PW11, R-, S-SiW12, R-, S-P2W18, and R-, S-P8W48 as precipitates (Fig. S8–S11†). Although the cluster structures, morphologies and compositions are different from one another, all the POMs exhibit the O→W LMCT transition band. R-SiW12 and R-PW11, with Keggin-type structures, show absorption bands centered at 262 and 258 nm, respectively, as shown in Fig. 3. R-P2W18 and R-P8W48 also exhibit absorption bands at approximately 250–350 nm, which correspond to the O→W LMCT transition bands.
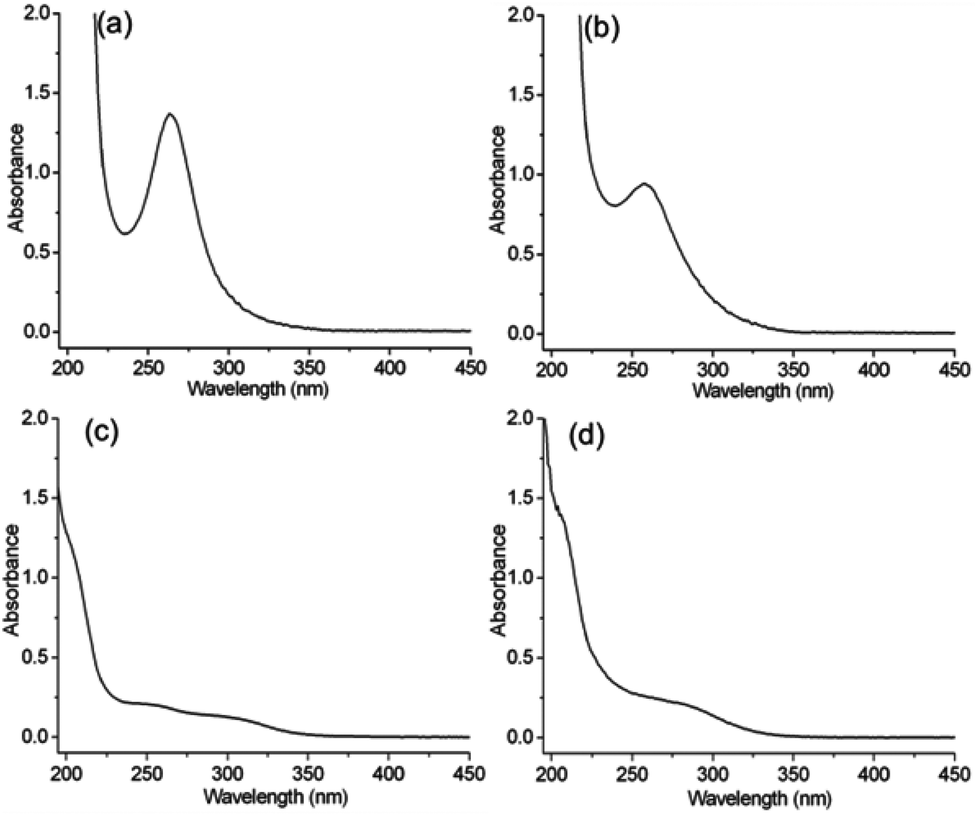 | ||
| Fig. 3 The UV-vis spectra of (a) R-SiW12, (b) R-PW11 (c = 1 mg mL−1, l = 1 mm), (c) R-P2W18 and (d) R-P8W48 (c = 0.2 mg mL−1, l = 1 mm) in CH3CN. | ||
The chiroptical activities within the LMCT transition bands of all four complexes in solution are shown in Fig. 4. The CD spectra of the R-POM and S-POM complexes in acetonitrile show mirror symmetry at approximately 190–400 nm, which clearly indicates that all the complexes are enantiopure. Cotton effects at approximately 218 nm, comparable with the corresponding bands for the organic ammonium cations at almost the same wavelength, are observed in the spectra of all the complexes. The mirror symmetric CD signals at approximately 290, 313, 305, and 298 nm in the spectra of R-, S-SiW12, R-, S-PW11, R-, S-P2W18 and R-, S-P8W48, respectively, appear at the absorption band positions of the O→W LMCT transitions of the tungstate POMs, and can be definitively assigned to the ICD of the POMs in the complexes. Though the SiW12 and P2W18 clusters possess higher symmetry, the electrostatic interactions with the chiral organic cations still trigger ICD of the achiral POMs in the complexes, indicating the strong chiral induction abilities of the chiral organic cations used. Clearly, due to the induction of chiral organic cations, both the chiral and achiral POM clusters in the complexes show chiroptical activities within the LMCT transitions.
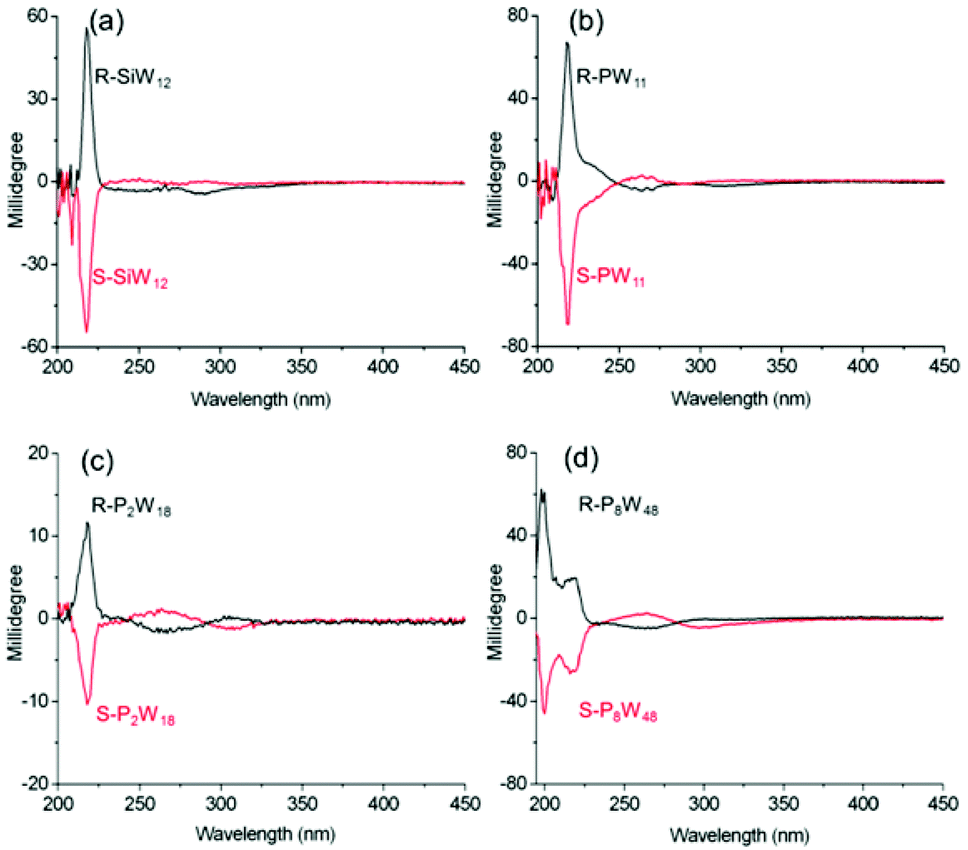 | ||
| Fig. 4 The CD spectra for (a) R-, S-SiW12, (b) R-, S-PW11 (c = 1 mg mL−1, l = 1 mm), (c) R, S-P2W18 and (d) R, S-P8W48 (c = 0.2 mg mL−1, l = 1 mm) in CH3CN. | ||
Meanwhile, for the whole complex, not only the intensity of the electrostatic interaction, but also the degree of POM distortion, has a key effect on the optical activity. The optical activity of chiral systems is often measured through the anisotropic factor (g-factor), which is defined by eqn (1):45
| g = Δε/ε | (1) |
| Complex | [θ]max (deg cm2 dmol−1) | g max = Δε/εa |
|---|---|---|
| a Δε = 3.03 × 10−5θ c−1d−1, where c is the concentration in mol L−1, d is the path length in cm, and θ is the ellipticity in mdeg.43 | ||
| R-P5W30 | 7.2 × 105 (293 nm) | 3.0 × 10−3 (302 nm) |
| R-EuP5W30 | 3.5 × 105 (306 nm) | 2.1 × 10−3 (317 nm) |
| R-SiW12 | −8.8 × 103 (290 nm) | −1.7 × 10−4 (293 nm) |
| R-P2W18 | 1.8 × 104 (305 nm) | 1.4 × 10−4 (309 nm) |
| R-PMo12 | −0.4 × 103 (400 nm) | −0.5 × 10−4 (412 nm) |
| R-P2Mo18 | 1.6 × 105 (336 nm) | 2.2 × 10−3 (343 nm) |
| R-C0-P5W30 | 9.5 × 104 (306 nm) | 5.2 × 10−4 (311 nm) |
To confirm the structural distortion dependence of the induced optical activities, we also characterize molybdate-based POMs, PMo12 and P2Mo18, by preparing the complexes R-, S-PMo12 and R-, S-P2Mo18 (Fig. S12–S14†). Due to the presence of alternating Mo–O bond lengths, both POMs have chiral structures. At the same time, P2Mo18 exhibits a larger distortion than PMo12.9 However, P2Mo18 exists as a racemic mixture in solution and in the solid state because of rapid racemization and partial hydrolysis. For R-PMo12, the absorption band at 300–450 nm can be assigned as the O→Mo LMCT transition (Fig. 5a–b). We also observe ICD signals corresponding to this transition (400 nm: [θ] = 0.4 × 103 deg cm2 dmol−1) in the CD spectra of the R-, S-PMo12 complexes. Similarly, more obvious ICD signals with multiple Cotton effects are observed for the P2Mo18 complexes with chiral structures (336 nm: [θ] = 1.6 × 105 deg cm2 dmol−1) (Fig. 5c–d). The g-factor of R-P2Mo18 is also much larger than that of R-PMo12 (Table 1). This indicates that the intrinsic chirality of P2Mo18 due to the larger degree of alternating short and long bond length distortions can contribute significantly to the net optical activity. The CD and UV-vis spectra of R-P2Mo18 and S-P2Mo18 show no obvious changes over several weeks, indicating that the induced optical activity of P2Mo18 is very stable in solution, which provides a possibility for further functional applications of the chiral structure.30 The ICD of the P2Mo18 complexes can also be observed in methanol and in the solid state (Fig. S15†).
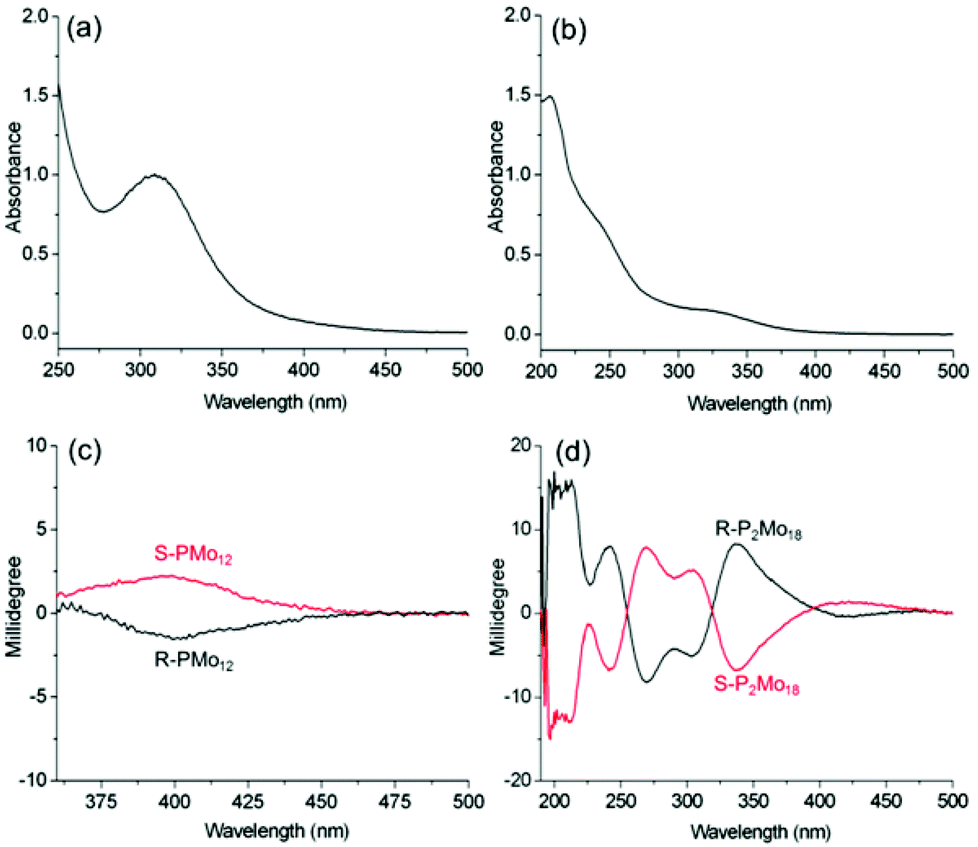 | ||
| Fig. 5 UV-vis spectra of (a) R-PMo12 (c = 1 mg mL−1, l = 1 mm) and (b) R-P2Mo18 (c = 0.2 mg mL−1, l = 1 mm); CD spectra of (c) R-, S-PMo12 (c = 1 mg mL−1, l = 10 mm) and (d) R-, S-P2Mo18 (c = 0.2 mg mL−1, l = 1 mm) in CH3CN. | ||
ICD of d–d transitions in POM complexes
In addition to the LMCT transition bands, some POMs, such as the cobalt substituted polyoxotungstates CoW12 and SiW11Co, also possess d–d transition bands (Fig. S16–S18†). Besides the absorption band at 257 nm attributed to the O→W LMCT transition, R-CoW12 and R-SiW11Co show Co(II) d–d transitions at 482–550 and 626 nm, as shown in Fig. 6. While the CoW12 and SiW11Co complexes show obvious ICD signals in the LMCT region (Fig. S19†), at high concentration, R-, S-CoW12 and R-, S-SiW11Co also display obvious ICD signals corresponding to the d–d transitions of Co(II). The molar ellipticities of R-CoW12 and R-SiW11Co reach 214 deg cm2 dmol−1 (626 nm) and 180 deg cm2 dmol−1 (550 nm), respectively. The strong chiral induction effect of the organic cations in the complexes causes the POMs to have obvious induced optical activities in the LMCT and d–d transition bands. No matter whether the cobalt atom is located at the center or the surface of the cluster, the ICD signals always appear. Apparently, the electrostatic interactions between the organic cations and the POMs can trigger structural perturbation of the cluster framework, leading to the generation of optical activity in the cobalt ions in the distorted POM structures.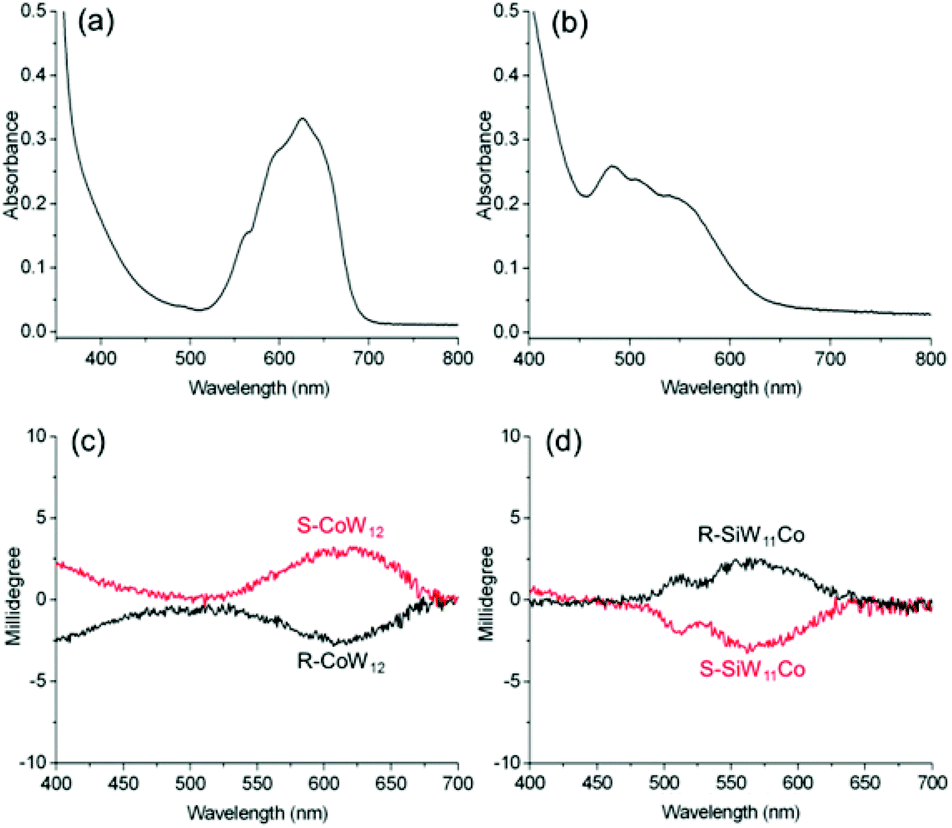 | ||
| Fig. 6 UV-vis spectra of (a) R-CoW12 and (b) R-SiW11Co; and CD spectra of (c) R-, S-CoW12 and (d) R-, S-SiW11Co in CH3CN, where c = 5 mg mL−1 and l = 10 mm. | ||
ICD of IVCT transitions in POM complexes
In addition, to confirm the generality of induced optical activity in POM complexes, we also study the ICD effect on the IVCT transition band, a characteristic absorption band of POMs in their reduced state.47 Many POMs have the ability to undergo stepwise, multielectron redox reactions, while their cluster structures remain intact. Such a reduction is accompanied by the generation of IVCT transitions in the visible region, through photochemical and electrochemical approaches or through the addition of reductants. The chiroptical activities resulting from the IVCT transitions of POMs could be significant for realizing chiroptical switches through reversible redox changes of POMs.31We choose the reduced POMs PW11VIV and SiW11VIV, which bear one-electron reduced vanadium ions, because the obvious IVCT transitions are known to appear in the visible region. For chiral R-, S-PW11VIV and R-, S-SiW11VIV, the presence of V4+ ions causes both complexes to show V4+→W6+ IVCT transitions and V4+ d–d transitions. For R-SiW11VIV in acetonitrile, the strong absorption band at approximately 500 nm can be definitively attributed to the IVCT transition, while the V4+ d–d transition is observed at approximately 680 nm as a shoulder band (Fig. 7). Chiroptical activities are also observed in the CD spectra of the POMs. Both R-SiW11VIV and S-SiW11VIV exhibit mirror symmetric Cotton effects; the induced optical activity associated with the IVCT transition appears at ca. 560 nm, and the d–d transition appears at ca. 680 nm. For R-PW11VIV and S-PW11VIV, similar induced optical activity of the POM is observed. Therefore, it is evident that encapsulating POMs with chiral organic cations through electrostatic interactions is a generally applicable method for constructing chiroptically active POM-based structures.
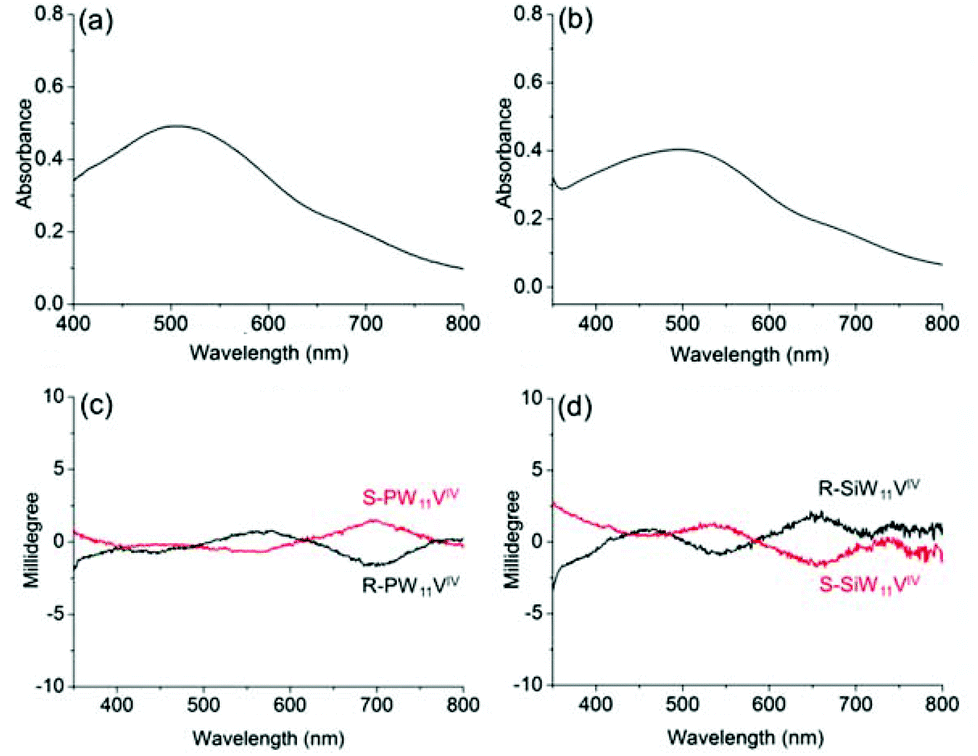 | ||
| Fig. 7 UV-vis spectra of (a) R-PW11VIV and (b) R-SiW11VIV; and the corresponding CD spectra of (c) R-, S-PW11VIV and (d) R-, S-SiW11VIV in CH3CN, where c = 3 mg mL−1 and l = 10 mm. | ||
To further verify the optical activity of the IVCT transition, we also examine the ICD of chiral complexes of P8W48V12 (Fig. S20–S21†). Because of the V4+ ions present in the complexes, broad IVCT transition bands in the visible region are clearly observed (Fig. 8). Correspondingly, the Cotton effect for this transition is clearly observed in the CD spectrum, and the chiral signal extends to a much longer wavelength region. Therefore, the chirality of the organic cations can induce the POMs to show CD signals within the IVCT transition, even up to the near-infrared region, which is meaningful for achieving POM-based chiral photochromic materials.
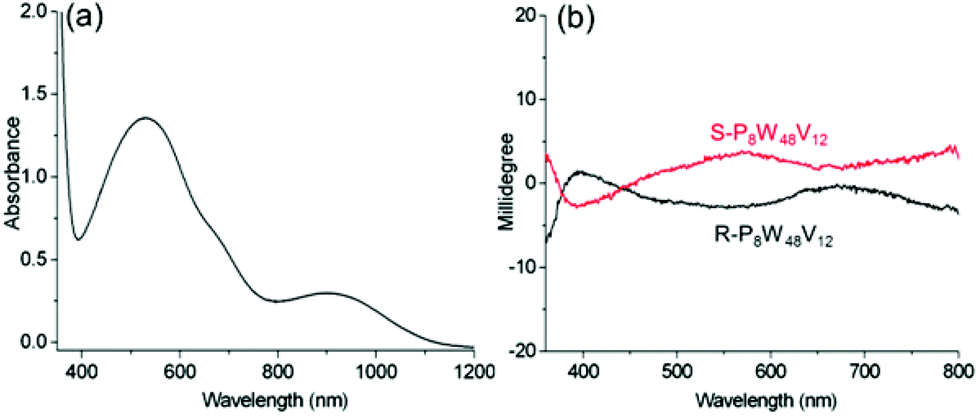 | ||
| Fig. 8 (a) UV-vis spectrum of R-P8W48V12; and (b) CD spectra of R-, S-P8W48V12 in CH3CN, where c = 3 mg mL−1 and l = 10 mm. | ||
Chiral induction ability of organic cations
For chiral POM-based materials, the close relationship between the nature of the organic cations and the POM properties has already been demonstrated.48,49 In order to evaluate the influence of different chiral organic cations on the ICD of the POMs in the complexes, we design and synthesize three kinds of organic cations with single chiral centers. The alkyl chain spacer between the chiral atom and the cationic head of the cations is controlled at zero (C0), five (C5) and eleven (C11) CH2 groups. With these molecules, we prepare chiral P5W30 complexes, R-C0,5,11-P5W30, and S-C0-P5W30 (Fig. S22–S31†). As shown in Fig. 9, R- and S-C0-P5W30 show positive and negative Cotton effects at approximately 308 nm associated with the LMCT transition of P5W30, indicative of the presence of ICD when the single chiral center is very close to the POMs. However, the g-factor of R-C0-P5W30 is nearly an order of magnitude smaller than that of R-P5W30, which implies that organic cations with two chiral centers have stronger induction abilities than those with only one chiral center for the same POMs. In contrast to R-C0-P5W30, in the case of R-C5-P5W30, we only observe weak ICD signals in the LMCT region, and the negative Cotton effect disappears (Fig. 9b). On increasing the spacer length between the chiral center and the ammonium head, the ICD corresponding to the LMCT region eventually disappears in R-C11-P5W30. These results indicate that the distance between the POM surface and the chiral center has a direct correlation with the induced optical activity of the inorganic clusters in the complexes. By adjusting the distance, one can effectively control the chiral induction of POMs.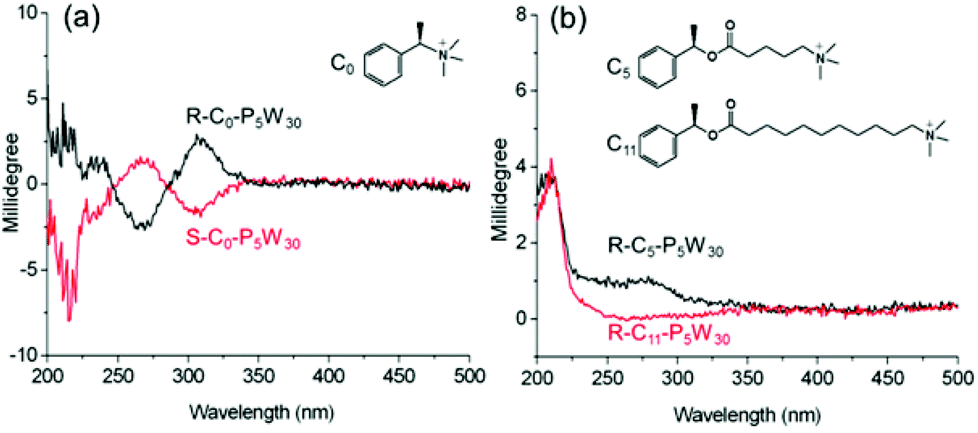 | ||
| Fig. 9 CD spectra of (a) R-, S-C0-P5W30, and (b) R-C5-P5W30 and R-C11-P5W30 in CH3CN, where c = 1 × 10−5 mol L−1 and l = 2 mm. | ||
Conclusions
The replacement of the counterions in POMs with chiral organic cations through electrostatic interactions is an effective and general way for preparing stable chiral structures of POM complexes. The chiral complexes can be easily obtained simply by mixing the organic cations and polyanions together in aqueous solution at room temperature, without strict experimental conditions. The induction of chiral organic cations triggers not only the POMs with chiral structures, but also the achiral POMs in the complexes, to show induced optical activities. At the same time, the chiral induction of the organic cations results in ICD in the absorption spectral regions corresponding to LMCT, d–d, and IVCT transitions of the POMS in different complexes. Moreover, the degree of POM distortion has a key effect on the induced optical activities. Finally, the chiral induction of the POMs can be effectively controlled by adjusting the distance between the chiral center and the POM surface. Following the current understanding, the present study provides a general strategy for the construction of chiral structures of inorganic clusters, which are meaningful for preparing chiral POM materials.Acknowledgements
The authors acknowledge financial support from the National Basic Research Program (2013CB834503), the National Natural Science Foundation of China (91227110 and 21221063), the Ministry of Education of China (20120061110047), and the Open Project of State Key Laboratory of Polymer Physics and Chemistry of Chinese Academy of Science.Notes and references
- D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
- Y. Wang and I. A. Weinstock, Chem. Soc. Rev., 2012, 41, 7479–7496 RSC.
- F. Li and L. Xu, Dalton Trans., 2011, 40, 4024–4034 RSC.
- J. Lu, J. Lin, X. Zhao and R. Cao, Chem. Commun., 2012, 48, 669–671 RSC.
- Y. Li, T. Wang and M. Liu, Soft Matter, 2007, 3, 1312–1317 RSC.
- H. Cao, X. Zhu and M. Liu, Angew. Chem., Int. Ed., 2013, 52, 4122 CrossRef CAS PubMed.
- B. Hasenknopf, K. Micoine, E. Lacôte, S. Thorimbert, M. Malacria and R. Thouvenot, Eur. J. Inorg. Chem., 2008, 5001 CrossRef CAS.
- D. Du, L. Yan, Z. Su, S. Li, Y. Lan and E. Wang, Coord. Chem. Rev., 2013, 257, 702 CrossRef CAS PubMed.
- L. Yan, X. López, J. J. Carbó, R. Sniatynsky, D. C. Duncan and J. M. Poblet, J. Am. Chem. Soc., 2008, 130, 8223–8233 CrossRef CAS PubMed.
- H. Naruke, J. Iijima and T. Sanji, Inorg. Chem., 2011, 50, 7535–7539 CrossRef CAS PubMed.
- Y. Hou, X. Fang and C. L. Hill, Chem. – Eur. J., 2007, 13, 9442–9447 CrossRef CAS PubMed.
- J. Zhang, J. Hao, Y. Wei, F. Xiao, P. Yin and L. Wang, J. Am. Chem. Soc., 2010, 132, 14–15 CrossRef CAS PubMed.
- H. Tan, W. Chen, D. Liu, Y. Li and E. Wang, CrystEngComm, 2010, 12, 4017–4019 RSC.
- U. Kortz, M. G. Savelieff, F. Y. A. Ghali, L. M. Khalil, S. A. Maalouf and D. I. Sinno, Angew. Chem., Int. Ed., 2002, 41, 4070–4073 CrossRef CAS.
- X. Fang, T. M. Anderson and C. L. Hill, Angew. Chem., Int. Ed., 2005, 44, 3540–3544 CrossRef CAS PubMed.
- M. Lu, J. H. Kang, D. G. Wang and Z. H. Peng, Inorg. Chem., 2005, 44, 7711–7713 CrossRef CAS PubMed.
- M. Carraro, A. Sartorel, G. Scorrano, C. Maccato, M. H. Dickman, U. Kortz and M. Bonchio, Angew. Chem., Int. Ed., 2008, 47, 7275–7279 CrossRef CAS PubMed.
- K. Micoine, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, Angew. Chem., Int. Ed., 2009, 48, 3466–3468 CrossRef CAS PubMed.
- S. Allenmark, Chirality, 2003, 15, 409–422 CrossRef CAS PubMed.
- J. Kumaki, S. Sakurai and E. Yashima, Chem. Soc. Rev., 2009, 38, 737–746 RSC.
- D. B. Amabilino and J. Veciana, Top. Curr. Chem., 2006, 265, 253–302 CrossRef CAS.
- H. Li, H. Sun, W. Qi, M. Xu and L. Wu, Angew. Chem., Int. Ed., 2007, 46, 1300–1303 CrossRef CAS PubMed.
- Y. Yang, L. Yue, H. Li, E. Maher, Y. Li, Y. Wang, L. Wu and V. W.-W. Yam, Small, 2012, 8, 3105–3110 CrossRef CAS PubMed.
- W. Qi, Y. Wang, W. Li and L. Wu, Chem. – Eur. J., 2010, 16, 1068–1078 CrossRef CAS PubMed.
- T. Noguchi, C. Chikara, K. Kuroiwa, K. Kaneko and N. Kimizuka, Chem. Commun., 2011, 47, 6455–6457 RSC.
- J. Garvey and M. T. Pope, Inorg. Chem., 1978, 17, 1115–1118 CrossRef CAS.
- S. C. Termes and M. T. Pope, Transition Met. Chem., 1978, 3, 103–108 CrossRef CAS.
- K. Nomiya, R. Kobayashi, M. Miwa and T. Hori, Polyhedron, 1984, 3, 1071–1076 CrossRef CAS.
- C. Jahier, M. Cantuel, N. D. McClenaghan, T. Buffeteau, D. Cavagnat, F. Agbossou, M. Carraro, M. Bonchio and S. Nlate, Chem. – Eur. J., 2009, 15, 8703–8708 CrossRef CAS PubMed.
- Y. Wang, H. Li, W. Qi, Y. Yang, Y. Yan, B. Li and L. Wu, J. Mater. Chem., 2012, 22, 9181–9188 RSC.
- Y. Wang, H. Li, C. Wu, Y. Yang, L. Shi and L. Wu, Angew. Chem., Int. Ed., 2013, 52, 4577–4581 CrossRef CAS PubMed.
- I. Creaser, M. C. Heckel, R. J. Neitz and M. T. Pope, Inorg. Chem., 1993, 32, 1573–1578 CrossRef CAS.
- C. Graham and R. G. Finke, Inorg. Chem., 2008, 47, 3679–3686 CrossRef CAS PubMed.
- R. Contant and A. Tézé, Inorg. Chem., 1985, 24, 4610–4614 CrossRef CAS.
- A. Müller, M. T. Pope, A. M. Todea, H. Bögge, J. Slageren, M. Dressel, P. Gouzerh, R. Thouvenot, B. Tsukerblat and A. Bell, Angew. Chem., Int. Ed., 2007, 46, 4477–4480 CrossRef PubMed.
- L. E. Briand, G. M. Valle and H. J. Thomas, J. Mater. Chem., 2002, 12, 299–304 RSC.
- P. J. Domaille, J. Am. Chem. Soc., 1984, 106, 7677–7687 CrossRef CAS.
- N. Haraguchi, Y. Okaue, T. Isobe and Y. Matsuda, Inorg. Chem., 1994, 33, 1015–1020 CrossRef CAS.
- L. C. W. Baker and T. P. McCutcheon, J. Am. Chem. Soc., 1956, 78, 4503–4510 CrossRef CAS.
- Z. Ei-Hachemi, G. Mancini, J. M. Ribó and A. Sorrenti, J. Am. Chem. Soc., 2008, 130, 15176–15184 CrossRef PubMed.
- N. Berova, L. D. Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- H. E. Smith, Chem. Rev., 1998, 98, 1709–1740 CrossRef CAS PubMed.
- J. Gawroński and J. Grajewski, Org. Lett., 2003, 5, 3301–3303 CrossRef PubMed.
- Y. Nagaoka, S. Shiratori and Y. Einaga, Chem. Mater., 2008, 20, 4004–4010 CrossRef CAS.
- A. Guerrero-Martínez, B. Auguié, J. L. Alonso-Gómez, Z. Džolić, S. Gómez-Graña, M. Žinić, M. M. Cid and L. M. Liz-Marzán, Angew. Chem., Int. Ed., 2011, 50, 5499–5503 CrossRef PubMed.
- Q. Tang, S. Liu, Y. Liu, S. Li, F. Ma, J. Li, S. Wang and C. Liu, Dalton Trans., 2013, 42, 8512–8518 RSC.
- T. Yamase, Chem. Rev., 1998, 98, 3 CrossRef PubMed.
- C. Jahier, M.-F. Coustou, M. Cantuel, N. D. McClenaghan, T. Buffeteau, D. Cavagnat, M. Carraro and S. Nlate, Eur. J. Inorg. Chem., 2011, 727–738 CrossRef CAS.
- C. Jahier and S. Nlate, Eur. J. Inorg. Chem., 2012, 833–840 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The synthetic procedures for R- and S-C0, R-C5 and R-C11, IR, NMR, UV-vis and CD spectra of the R- and S-POM complexes, and CD spectra of the chiral organic cations. See DOI: 10.1039/c4dt00866a |
| This journal is © The Royal Society of Chemistry 2014 |