 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible cargo shipping between orthogonal stations of a nanoscaffold upon redox input†
Soumen K.
Samanta
,
Anup
Rana
and
Michael
Schmittel
*
Center of Micro- and Nanochemistry and Engineering, Organische Chemie I, Universität Siegen, Adolf-Reichwein-Str. 2, D-57068 Siegen, Germany. E-mail: schmittel@chemie.uni-siegen.de
First published on 19th May 2014
Abstract
The sterically shielded terpyridine 5 was prepared, both as a new ligand and as part of the four-station nanoscaffold 2. Mixing of terpyridine 5, the parent phenanthroline 4 and the shielded phenanthroline 3 in the presence of Zn2+ (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1) furnished quantitatively the inverse HETTAP complex [Zn(4)(5)]2+ by self-sorting, while in the presence of Cu+ the HETPHEN complex [Cu(3)(4)]+ was preferred (89%). Due to the akin coordination preferences of Cu2+ and Zn2+, the above self-sorting was implemented for Cu+/Cu2+ on nanoscaffold 2, the latter equipped with the binding motifs of 3 (PhenAr2) and 5 (TerpyAr2). When 2 was reacted with Cu+ and phenanthroline (4) in a 1:2:2 ratio, only the PhenAr2 stations became involved in complex formation (= Cu1phen). In contrast, upon oxidative formation of Cu2+, ligand 4 was exclusively moved to the TerpyAr2 stations (Cu1terpy). Electrochemical oxidation–reduction prompted the cargo to be shipped reversibly on a subsecond time scale between the two different stations of 2.
:1:1:1) furnished quantitatively the inverse HETTAP complex [Zn(4)(5)]2+ by self-sorting, while in the presence of Cu+ the HETPHEN complex [Cu(3)(4)]+ was preferred (89%). Due to the akin coordination preferences of Cu2+ and Zn2+, the above self-sorting was implemented for Cu+/Cu2+ on nanoscaffold 2, the latter equipped with the binding motifs of 3 (PhenAr2) and 5 (TerpyAr2). When 2 was reacted with Cu+ and phenanthroline (4) in a 1:2:2 ratio, only the PhenAr2 stations became involved in complex formation (= Cu1phen). In contrast, upon oxidative formation of Cu2+, ligand 4 was exclusively moved to the TerpyAr2 stations (Cu1terpy). Electrochemical oxidation–reduction prompted the cargo to be shipped reversibly on a subsecond time scale between the two different stations of 2.
Introduction
Many enzymes use translocation processes1 (ligand and metal) to initiate fascinating biological protocols. Likewise, shifting subcomponents in either supramolecular2 or molecular3 structures has been ingeniously utilised in fascinating abiological devices.4,5 To control reversible motion in multistable devices by switching, the binding of the movable part (Kassoc) has to favour a particular site over the other depending on the given input, such as chemicals,6 redox equivalents7 or light.8As reported by Sauvage and coworkers,9 changing the oxidation state of copper from +I to +II leads to an altered coordination preference: the copper(I) ion prefers tetracoordination, while the copper(II) ion is best accommodated in a penta- or hexacoordination. Remodeling this principle, we present here the reversible shipping of cargo between two different sites on a scaffold by applying electrochemical stimuli (Fig. 1). Our design of reversible cargo shipping relies on two heteroleptic binding algorithms developed in our group, i.e. the HETPHEN and HETTAP concepts,10 instead of using topological constraints, as is amply done in rotaxanes and catenanes.11 Our concepts are based on the use of a bulky 2,9-diaryl substituted phenanthroline (PhenAr2), e.g., ligand 3 (Chart 1),12 whose front-side shielding prevents the formation of the homoleptic complex [M(3)2]n+ with Mn+ = Cu+, Ag+, Zn2+. In presence of a sterically slim counterpart, like the parent phenanthroline (for HETPHEN)12a,b or terpyridine (for HETTAP),12c,d3 and Mn+ will afford quantitatively the heteroleptic complex.
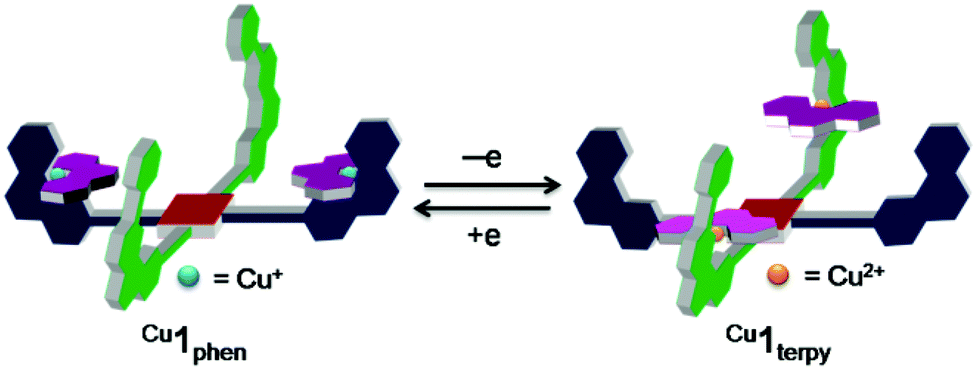 | ||
| Fig. 1 Controlling cargo transport between two stations via self-sorting by oxidation/reduction. Cu1phen = [Cu2(2)(4)2]2+. Cu1terpy = [Cu2(2)(4)2]4+. | ||
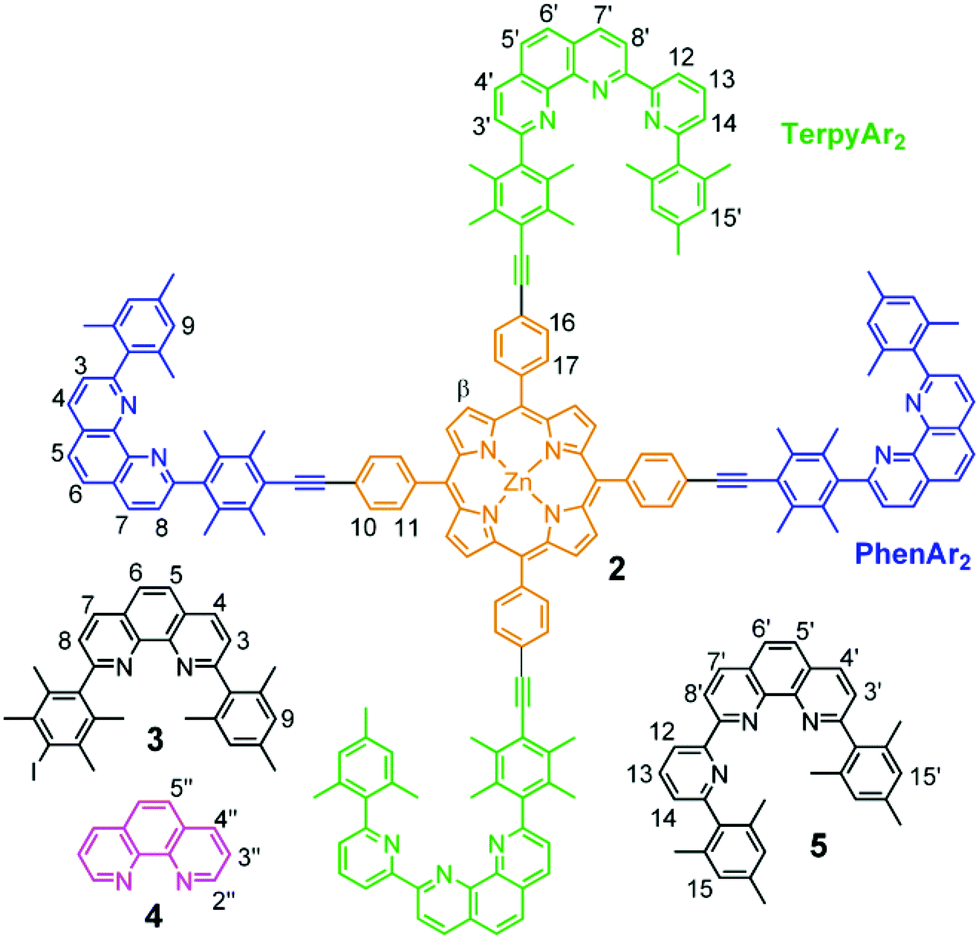 | ||
| Chart 1 Scaffold 2 and ligands 3–5 used in the present study. | ||
For the present study, structural aspects known from the HETTAP and HETPHEN protocols guided our design of the new terpyridine 5 that is sterically shielded by aryl groups (TerpyAr2). The shielding does not prevent, but slows down formation of the homoleptic complex [M(TerpyAr2)2]n+ thus favouring heteroleptic complexation. The new scaffold 2 arises by attaching the binding motifs of both PhenAr2 and TerpyAr2 sites to a zinc(II) porphyrin core. Depending on the oxidation state of the copper ions a switch in the self-sorting13 should lead to cargo shuffling (cargo = copper + ligand 4) between the PhenAr2 and TerpyAr2 stations on the scaffold (Fig. 1). As such, our example is a very rare case of redox initiated self-sorting14 in solution.
Results and discussion
Synthesis
The new compounds 2 and 5 (Scheme 1) were prepared via established protocols. First, 2,4,6-trimethylphenylboronic acid was treated with 2,6-dibromopyridine via Suzuki coupling to furnish compound 6, which was reacted further on with 2-(2,4,6-trimethylphenyl)-[1,10]-phenanthroline to afford ligand 5.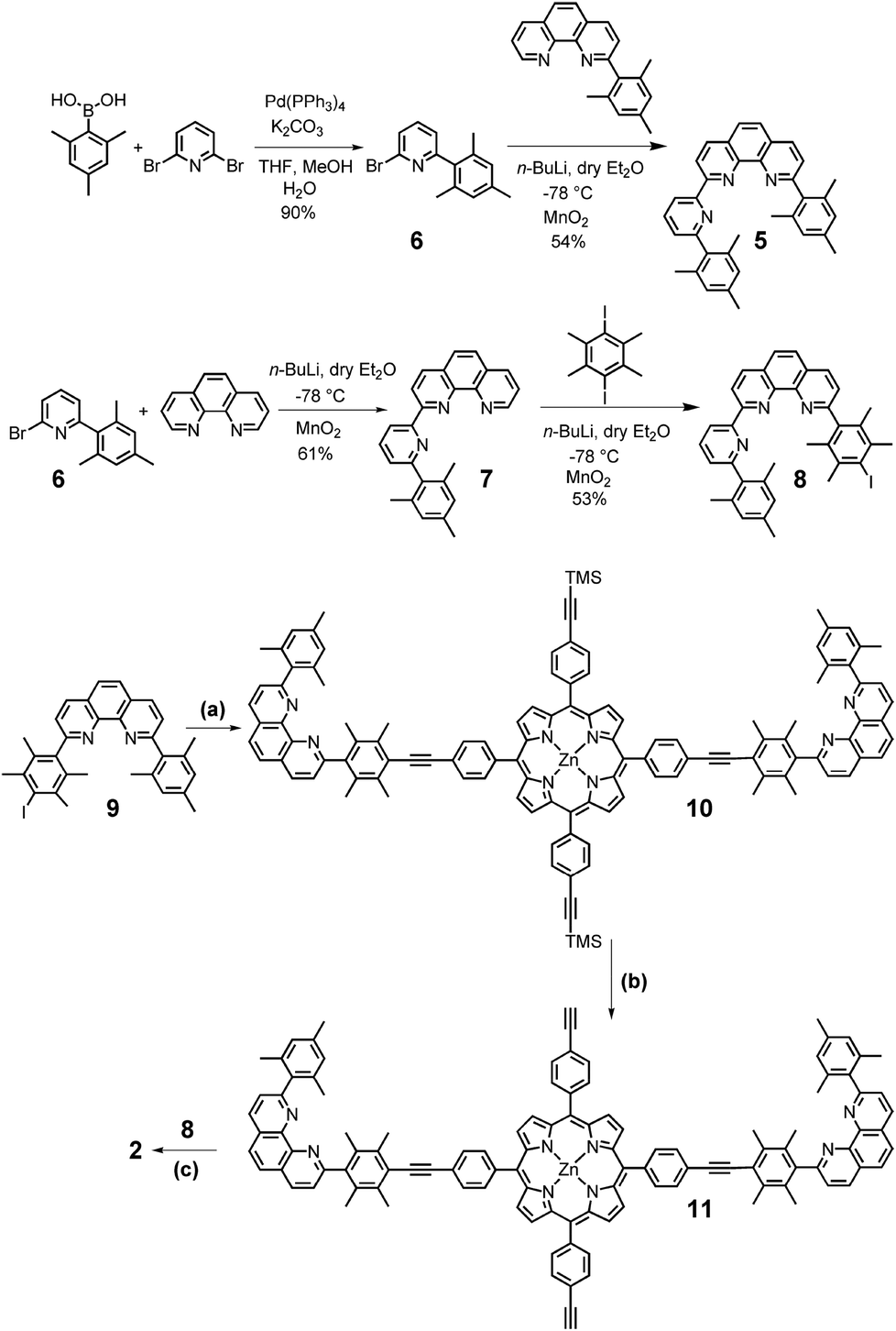 | ||
| Scheme 1 Synthesis of ligands 2, 5 and 8. (a) Zinc(II)-5,15-bis(4-iodophenyl)-10,20-bis(4-trimethylsilylethynylphenyl)porphyrin, Pd(PPh3)4, NEt3, DMF, 90 °C, 12 h, 58%. (b) KOH, THF, MeOH, room temperature, 90%. (c) Pd(PPh3)4, NEt3, DMF, 90 °C, 12 h, 35%. | ||
For the preparation of scaffold 2 with two different kinds of stations, pyridine 6 was subjected to a bromo/lithium exchange and then reacted with [1,10]-phenanthroline to afford the terpyridine-analog 7. A follow-up reaction with 1,4-diiododurene/n-BuLi furnished 8 serving as the direct precursor for the preparation of scaffold 2. To finalise the synthesis, zinc(II)-5,15-bis(4-iodophenyl)-10,20-bis(4-trimethylsilylethynylphenyl)porphyrin was first treated with the known shielded phenanthroline 912d in presence of Pd(PPh3)4 to afford 10, which after deprotection of the trimethylsilyl groups furnished 11. In the final step, 11 was reacted with ligand 8 in a second Sonogashira coupling to provide the desired target 2 (Scheme 1). Scaffold 2 was easily and fully characterised by spectroscopic means.
Complexation properties of 5
Insight into the binding properties of 5 was received by reacting 0.5 equiv. of [Cu(CH3CN)4]PF6 with 5 in CD2Cl2. 1H NMR and ESI-MS of the resulting yellow solution revealed formation of the homoleptic complex [Cu(5)2]+ by showing two different sets of upfield shifted mesityl protons at 5.62 and 6.19 ppm (Fig. 2) and a mass peak at 1051.0 Da. Assuming a coordination number 4 for Cu+, [Cu(5)2]+ may actually form in three isomeric structures, i.e. iso-I, iso-II & iso-III (Charts 2 and 3) due to six nitrogen atoms being available from two ligands 5. In iso-I, Cu+ engages exclusively with the phenanthroline nitrogen atoms (N2, N3, N2′ and N3′), while the pyridyl nitrogen atoms (N1 and N1′) remain unbound. In iso-II, Cu+ is bound to N2, N3, N2′, N1′ whereas N1, N3′ are left uncoordinated. In contrast, iso-III deals with coordination at N2, N1, N2′, N1′ with nitrogens N3, N3′ remaining nonbonded. The unbound pyridyl unit (N1 or N1′) of 5 may undergo rotation about the single C–C bond thereby generating various conformations in iso-I and iso-II of [Cu(5)2]+. For instance, iso-I may have three major conformers – iso-IA (out–out = N1 and N1′ directed away from Cu+ center), iso-IB (out–in), and iso-IC (in–in). Similarly iso-II may have two conformers – iso-IIA (in) and iso-IIB (out). Obviously, in iso-III both pyridyl units are conformationally fixed as they are involved in bonding with Cu+. DFT15 computations suggest that iso-IB has lowest energy. The relative energy of the isomers follows the order: iso-IB (0) < iso-IA (3.94) < iso-IIB (4.04) < iso-IIA (6.62) kcal mol−1.16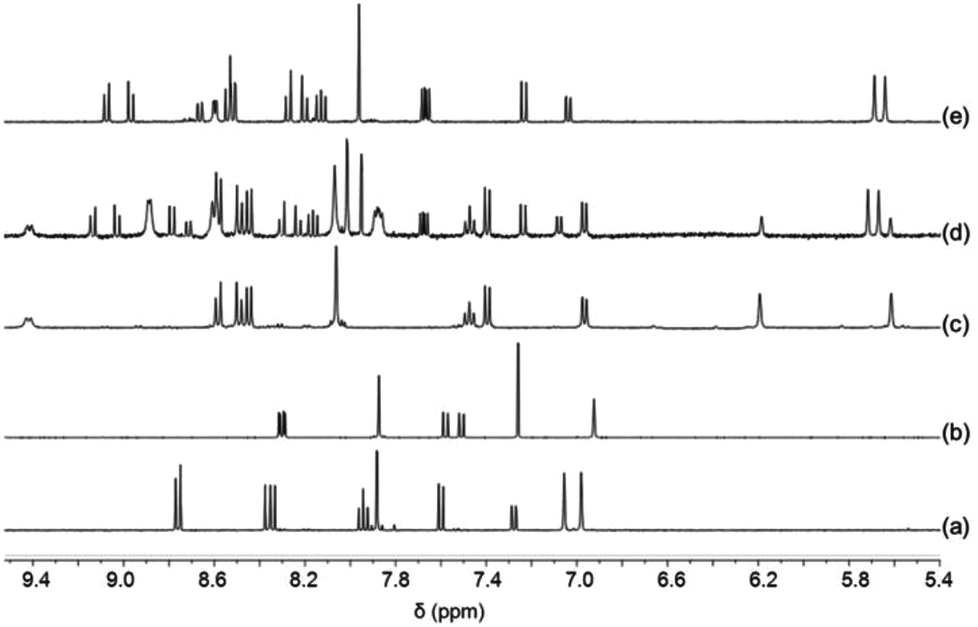 | ||
| Fig. 2 Partial 1H NMR spectra of (a) 5 in CD2Cl2, (b) 3 in CDCl3, (c) [Cu(5)2]PF6 in CD2Cl2, (d) equimolar mixture of 4, 5 and Cu+ in CD2Cl2 and (e) equimolar mixture of 4, 5 and Zn2+ (in CD2Cl2–CD3CN = 3:1). | ||
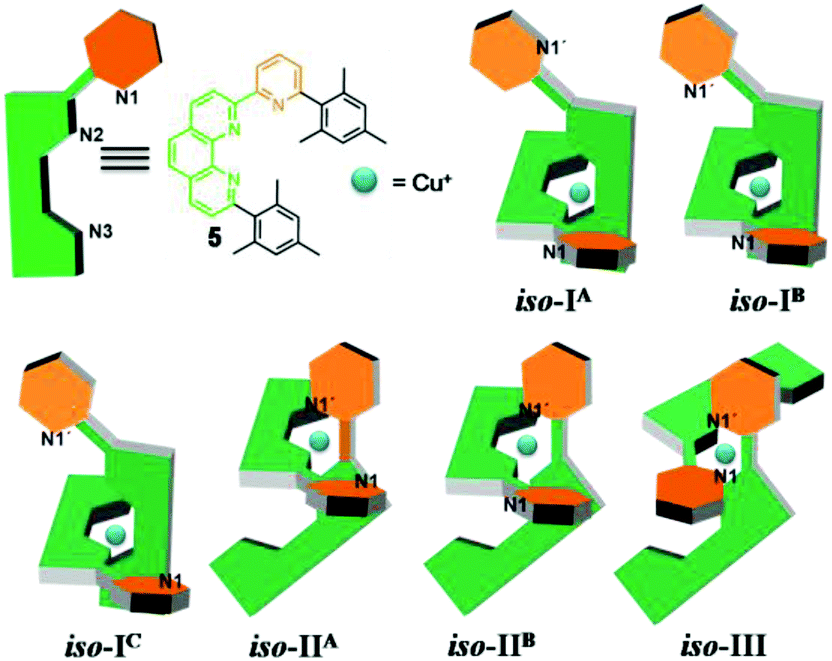 | ||
| Chart 2 Cartoon representations of compound 5 and the three isomers (iso-I, iso-II & iso-III) of the homoleptic complex [Cu(5)2]+. Superscript letters A, B and C denote conformations. | ||
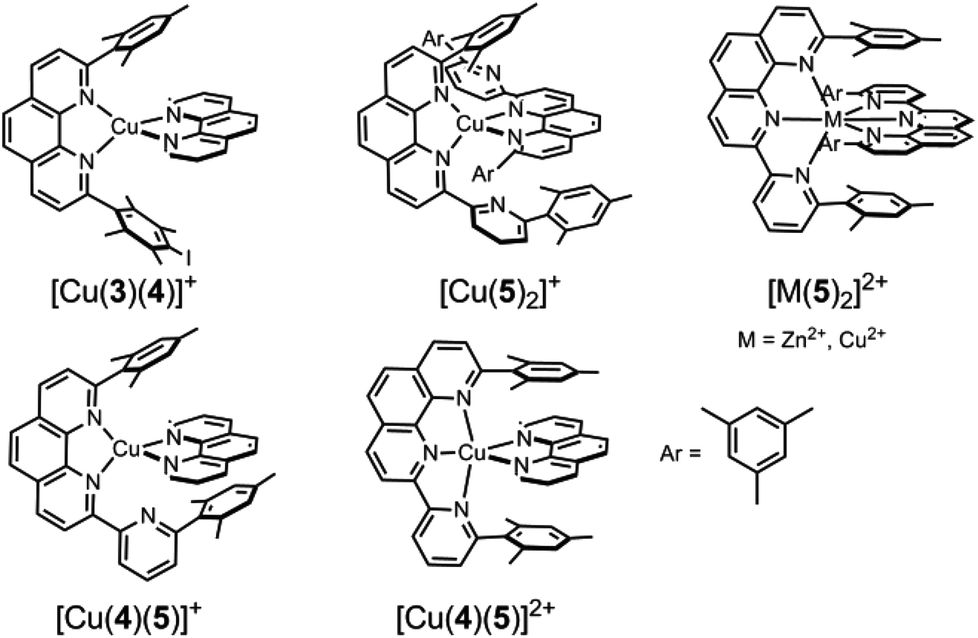 | ||
| Chart 3 Possible complexes resulting from ligands 3, 4, 5 and the metal ions Cu+, Cu2+ and Zn2+. | ||
Experimental information about the coordination mode in [Cu(5)2]+ was obtained from cyclic voltammetry (CV). Complex [Cu(5)2]+ exhibits a high oxidation potential at E1/2 = 634 mVSCE (quasireversible: ΔEp = 164 mV, see ESI†) clearly excluding the possibility of penta- or hexa-coordination. Most likely, steric crowding by the mesityl groups prevents Cu2+ to extend its coordination number from 4 to 5/6.
Heteroleptic complexes with ligand 5
When a 1:1:1 mixture of 4, 5 and copper(I) ions was reacted in CD2Cl2, the homoleptic complexes [Cu(4)2]+ and [Cu(5)2]+ as well as the heteroleptic complex [Cu(4)(5)]+ were observed in the ESI-MS, with the latter generating the major peak (at 745.5 Da). The 1H NMR data corroborate the formation of both complexes [Cu(4)(5)]+ and [Cu(5)2]+ (Fig. 2), with peaks at 5.63 & 5.69 ppm being diagnostic for [Cu(4)(5)]+ and those at 5.62 and 6.19 ppm (Fig. 2) for [Cu(5)2]+. Unfortunately, characteristic signals for [Cu(4)2]+ were obscured in the aromatic region.
The DFT optimised structure of [Cu(4)(5)]+ predicts that the metal ion is engaged in coordination with both phenanthroline nitrogens (N2 and N3) of 5, while the pyridyl nitrogen atom (N1) remains nonbonded. Strong π⋯π interactions (C⋯C = 4.033 Å and 4.043 Å) between the two mesityl units of 5 and 4 are observed. Other isomers of [Cu(4)(5)]+ involving N1(pyridyl)→Cu interactions were not located computationally.
When an equimolar mixture of 4, 5 and Cu(I) was oxidised in CV, a broad wave at 540–713 mV was observed corresponding to oxidation of all three copper complexes, [Cu(4)2]+, [Cu(5)2]+ and [Cu(4)(5)]+. In contrast, the cathodic scan generated a single, irreversible peak at Epc = 251 mV assigned to the reduction of [Cu(4)(5)]2+ exclusively. Indeed, the independently prepared complex [Cu(4)(5)]2+ (vide infra) showed a reduction wave at Epc = 245 mV. Apparently, upon oxidation, both homoleptic complexes [Cu(5)2]2+ and [Cu(4)2]2+ reorganised to [Cu(4)(5)]2+ so that each copper(II) ion would realise pentacoordination in the heteroleptic complex.
When a 1:1 mixture of 4 and 5 was treated with 1 equiv. of Zn(OTf)2 in CD2Cl2–CD3CN = 3:1, the heteroleptic complex [Zn(4)(5)]2+ formed quantitatively as documented by spectroscopic data. The ESI mass spectrum shows peaks at 370.0 Da and 888.3 Da diagnostic for the doubly charged [Zn(4)(5)]2+ and singly charged [Zn(4)(5)](OTf)+ after loss of two and one counteranion(s) (OTf−), respectively (Fig. S37, ESI†). No mass peaks of homoleptic complexes are visible. The 1H NMR spectrum displays two diagnostic signals at 5.64 & 5.69 ppm for the heteroleptic species (Fig. 2).
Due to the similarity in its coordination behaviour to that of Zn2+, Cu2+ also afforded cleanly the heteroleptic complex [Cu(4)(5)]2+, as evidenced by ESI-MS and CV. The DFT optimised structure of [Cu(4)(5)]2+ suggests that ligand 5 is tilted toward the phenanthroline unit in a square pyramidal rather than trigonal bipyramidal geometry presumably due to the strong pyridyl→Cu binding (Cu⋯N1 = 2.27 Å). The latter interaction draws the two mesityl groups of 5 toward each other and leaves little space for ligand 4. Upon irreversible reduction of the freshly prepared [Cu(4)(5)]2+ at Epc = 245 mV a follow-up reaction occurs that is visible in the reverse anodic scan: the broad waves at Epa = 538 & 720 mV may be assigned to the oxidation of a mixture of homo- and heteroleptic complexes (alike the complex mixture from 4, 5 and Cu+, vide supra).
Self-sorting
To study self-sorting,17 a mixture of 3, 4, 5 and Cu+ (1:1:1:1) was sonicated in CD2Cl2. Although six copper(I) complexes may be expected, the ESI mass spectrum shows only two peaks at 800.3 Da and 1051.0 Da suggesting the exclusive formation of the complexes [Cu(3)(4)]+ and [Cu(5)2]+, respectively (Fig. S36, ESI†). In full agreement, the 1H NMR spectrum displays diagnostic peaks at 6.00 ppm for the heteroleptic complex [Cu(3)(4)]+ and at 5.62 & 6.19 ppm for the homoleptic complex [Cu(5)2]+ (Fig. 3). Two sharp singlets at 5.64 and 5.69 ppm would have been diagnostic for [Cu(4)(5)]+, but those signals are absent. NMR integration suggests that complexes [Cu(3)(4)]+ and [Cu(5)2]+ are present in 89%:11% along with unused ligands 3, 4 and 5.
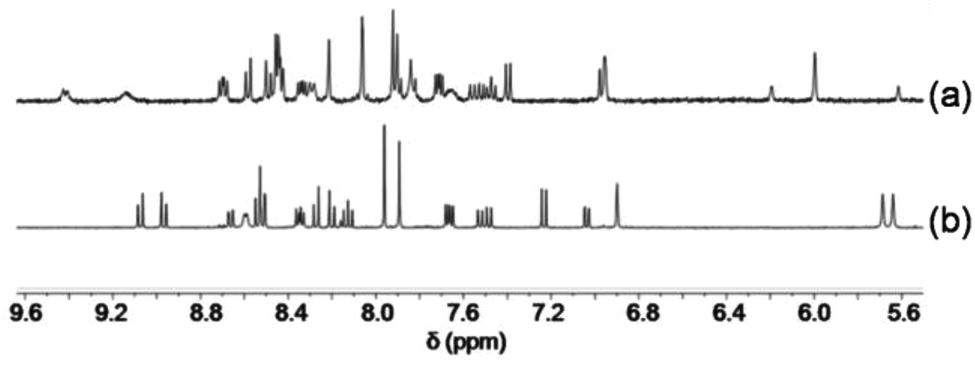 | ||
| Fig. 3 Partial 1H NMR spectra of equimolar mixtures (a) of 3, 4, 5 and Cu+ (CD2Cl2) and (b) of 3, 4, 5 and Zn2+ (CD2Cl2–CD3CN = 3:1). | ||
When 3, 4, 5 and Zn(OTf)2 (1:1:1:1) were sonicated for 30 min in CD2Cl2–CD3CN (3:1), quantitative formation of the heteroleptic complex [Zn(4)(5)]2+ was observed. The 1H NMR spectrum shows singlets at 5.64 & 5.69 ppm that are diagnostic for the heteroleptic complex along with a CHMes signal at 6.94 ppm indicative for the ligand 3 (Fig. 3). The two peaks at 370.0 and 888.3 Da in the ESI mass spectrum correspond to the doubly charged [Zn(4)(5)]2+ and singly charged [Zn(4)(5)](OTf)+, respectively (Fig. S38, ESI†). Due to the analogous d10 configuration of Zn2+ and Cu2+, similar self-sorting phenomena are expected for both metal ions suggesting quantitative complex formation of [Cu(4)(5)]2+ with 3 remaining untouched in solution.
The effect of changing the metal's oxidation state on self-sorting was studied by CV. In the anodic scan of an equimolar mixture of 3, 4, 5 and Cu+, the irreversible peak at Epa = 763 mV is assigned to the oxidation of [Cu(3)(4)]+. The oxidation wave of the homoleptic complex [Cu(5)2]+ (Epa = 720 mV, vide supra) is hardly detectable due to its small amount (11%, see NMR). In the reverse scan, the irreversible reduction wave at Epc = 242 mV indicates that the complex must have undergone a major reorganisation. Apparently, both [Cu(3)(4)]2+ and [Cu(5)2]2+ undergo fast ligand shuffling in solution to afford exclusively the heteroleptic complex [Cu(4)(5)]2+. After reduction of [Cu(4)(5)]2+, the anodic back scan again shows the oxidation waves corresponding to complexes [Cu(3)(4)]+ and [Cu(5)2]+ at Epa = 763 mV. No oxidation wave was observed corresponding to [Cu(4)(5)]+. In fact, both peaks did not show reversibility at any of chosen scan rates (from 50 to 200 mV s−1) at this concentration (c = 0.65 mM). Randles–Sevcik plots yield a linear dependence of the anodic peak intensity ipa with the square root of the scan rate (ν1/2), thus attesting a diffusion-limited electron-transfer process. This study clearly indicates fast metal ligand reorganisation upon changing the oxidation state of copper (Cu+ → Cu2+ → Cu+) or changing from copper(I) to zinc(II) ions both resulting in a self-sorting between heteroleptic complexes.
Self-sorting on scaffold 2
To use the redox-triggered switching of the self-sorting process for cargo shipping between two different stations, the new ligand 2 is used as a nanoscaffold. At the outset, 2, 4 and Cu+ (1:2:2) were reacted in CD2Cl2 to afford the complex Cu1phen = [Cu2(2)(4)2](PF6)2, in which both PhenAr2 stations of 2 are quantitatively occupied by [Cu(4)]+, while the TerpyAr2 stations remain unloaded (Fig. 4). The complex was characterised by 1H NMR, 1H–1H COSY and ESI-MS. 1H NMR (Fig. 5) reveals that protons 9-H are upfield shifted from 6.94 to 6.00 ppm while protons 15′-H remain unchanged at 6.97 ppm (as in compound 2). Other protons of the PhenAr2 stations also undergo major shifts due to formation of the heteroleptic complex while protons of the TerpyAr2 units remain unchanged (see Table S3†). Formation of the complex is ascertained by a peak at 1565.1 Da in the ESI mass spectrum (Fig. S40, ESI†) corresponding to the doubly charged species [Cu2(2)(4)2]2+ after loss of two counteranions (PF6−). Finally, the 1H DOSY trace proves that assembly Cu1phen exists in solution as a single species (Fig. S50, ESI†).
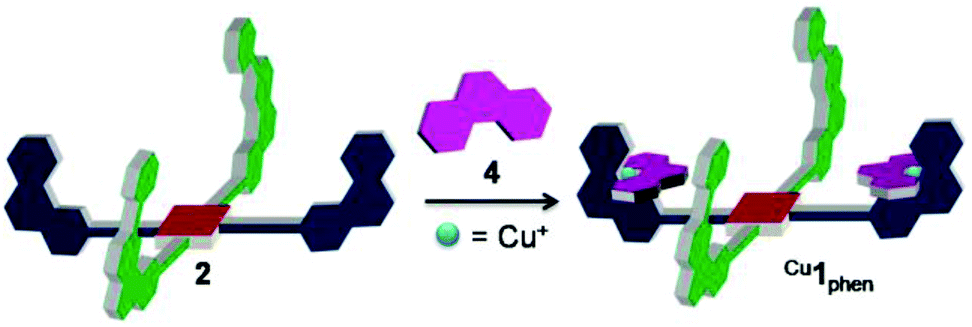 | ||
| Fig. 4 Preparation of complex Cu1phen. | ||
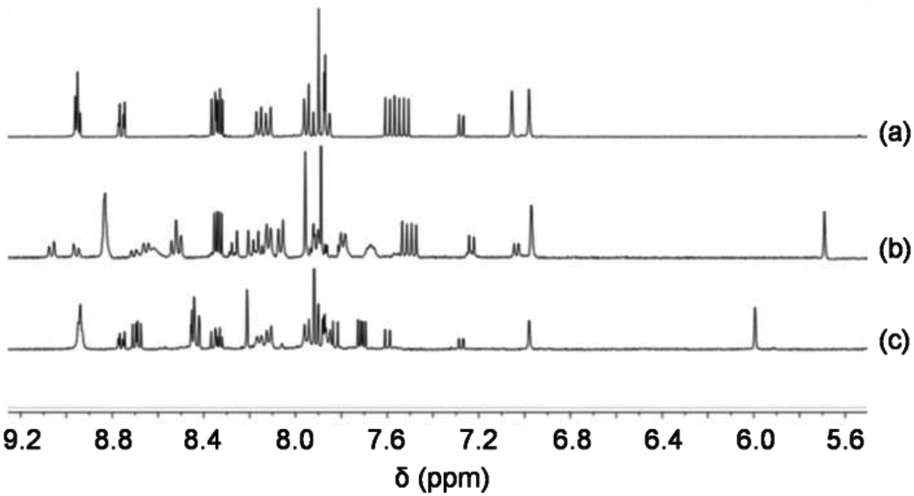 | ||
| Fig. 5 Partial 1H NMR spectra of (a) 2 (CD2Cl2), (b) Zn1terpy (CD2Cl2–CD3CN = 3:1) and (c) Cu1phen (CD2Cl2). | ||
When a 1:2 mixture of 2 and 4 was treated with 2 equiv. of Cu(ClO4)2·6H2O, complex Cu1terpy = [Cu2(2)(4)2](ClO4)4 formed quantitatively as derived from the ESI-MS data. They (Fig. S42, ESI†) show two peaks at 782.7 Da and 1076.8 Da corresponding to the quadruply-charged ([Cu2(2)(4)2]4+) and triply-charged species ([Cu2(2)(4)2](ClO4)3+) after loss of four and three-counter anions (ClO4−), respectively. Strong paramagnetic broadening by Cu2+ prevented us from characterising the complex by 1H NMR. However, expecting a similar coordination behaviour of zinc(II) and copper(II) ions (vide supra for the model study), we reacted Zn2+ with 1 and 4 (2:1:2). As a result, complex Zn1terpy was afforded, in which the [Zn(4)]2+ units quantitatively occupy the TerpyAr2 stations with the PhenAr2 stations remaining unloaded. Diagnostically, the 1H NMR (see Fig. 5, Table S4†) showed protons 15′-H (TerpyAr2) to be shifted upfield to 5.69 ppm while protons 9-H of both PhenAr2 units remain unaltered at 6.91 ppm. A similar behaviour was observed for other protons of the PhenAr2 and TerpyAr2 units. In the ESI mass spectrum (Fig. S41, ESI†), Zn1terpy exhibited three peaks at 783.5 Da, 1094.4 Da and 1716.1 Da that are assigned to quadruply-charged ([Zn2(2)(4)2]4+), triply-charged ([Zn2(2)(4)2](OTf)3+) and doubly-charged ([Zn2(2)(4)2](OTf)22+) species after loss of four, three and two counter anions (OTf−), respectively. Finally, 1H DOSY suggests that Zn1terpy exists in solution as a single species (Fig. S51, ESI†).
Electrochemical cargo shipping
Inspired by the reversible interconversion of two heteroleptic complexes via electrochemical means, both complexes Cu1phen and Cu1terpy were tested for cargo shipping between the two different stations (PhenAr2 and TerpyAr2) on scaffold 2. In the CV, complex Cu1terpy = [Cu2(2)(4)2](ClO4)4 (scan rate = 100 mV s−1) exhibits an irreversible reduction wave at Epc = 246 mV (Fig. 6, right) that is characteristic for a pentacoordinated copper(II) complex (inverse HETTAP complex). On the reverse scan, the oxidation of the reduced species displays an irreversible peak at Epa = 786 mV that is characteristic for oxidation of a copper(I/II) HETPHEN complex (unfortunately, the peak is merged with the first reversible oxidation peak of the zinc(II) porphyrin unit with Epa = 830 mV and E1/2 = 790 mV). Shifting the cathodic switching potential towards negative potential (by ΔE1 = −100 mV) and thus increasing the time for rearrangement increases the anodic current (ipa) at Epa = 786 mV (see ESI†) clearly arguing for ligand translocation from the TerpyAr2 to PhenAr2 stations upon reduction of Cu2+ to Cu+. Anodic peak current (ipa) enhancement at Epa = 786 mV is also observed when using a 5 s delay at the first switching potential (E1 = −100 mV). Moreover, both peaks show hardly any reversible behaviour in the scan rate range (50 to 1000 mV s−1).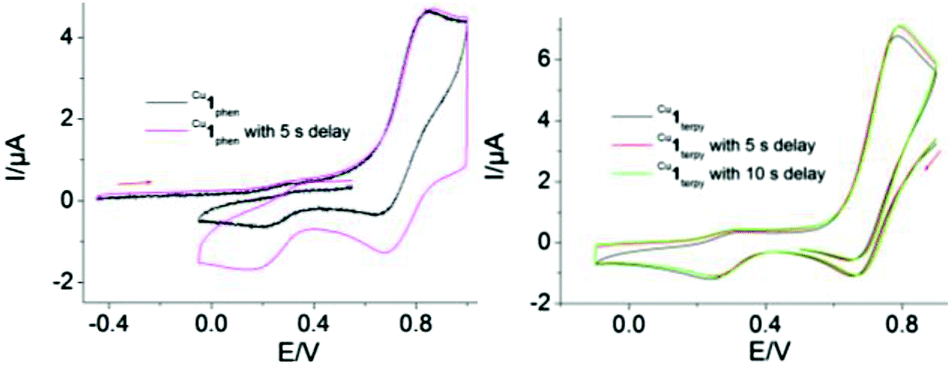 | ||
| Fig. 6 Cyclic voltammogram of (left) Cu1phen (scan rate = 500 mV s−1) and (right) Cu1terpy (scan rate = 100 mV s−1) in dry CH2Cl2. | ||
Similarly, when HETPHEN complex Cu1phen = [Cu2(2)(4)2](PF6)2 is electrochemically oxidised at v = 500 mV s−1, the oxidation wave is irreversible (Epa = 844 mV). The wave overlaps with the first oxidation potential of the zinc(II) porphyrin (por/por˙+; Fig. 6, left). In the reverse reductive scan, an irreversible peak is observed at Epc = 174 mV that is assigned to the reduction of electrochemically produced Cu1terpy indicating metal and ligand translocation from PhenAr2 to TerpyAr2 stations. A time delay of 5 s at the first switching potential (E1 = 900 mV) increases the current of the Cu2+/Cu+ redox couple (E1/2 = 269 mV) at the TerpyAr2 station. Hence, CV studies clearly establish the reversible interconversion between the two stable species Cu1phen and Cu1terpy through cargo transport (Fig. 1).
The CV behaviour can be understood as an EC process. Electron transfer is followed by fast translocation of both metal ion and ligand between the two different stations. Simulation of the mechanism by DigiSim® was possible using a simplified scheme. Due to the fact that the rate constants for both cargo transport processes are hidden in each individual CV trace, one is able to even cross-check the data. Accordingly, the best agreement for Cu2+PhenAr2 → Cu2+TerpyAr2 was K = 10 and k = 10 s−1 and for Cu+TerpyAr2 → Cu+PhenAr2 was K = 20 and k = 5 s−1 (Fig. S49, ESI†). Using the same rate constants, also the redox-mediated shuttling between [Cu(3)(4)]+ + 5 and [Cu(4)(5)]2+ + 3 (Fig. S44†) was successfully simulated, lending further credibility to the above values. Moreover, a concentration-dependent CV study of Cu1phen indicated negligible effects on the reversibility at E1/2 = 263 mV (Fig. S48, ESI†), suggesting that dissociation is rate-limiting. A plausible mechanism for shipping is thus the rate-determining release of the labilised metal ions (after oxidation/reduction) on the subsecond time scale along with their phenanthroline cargo from a particular station followed by fast reassociation at the other station (a bimolecular process), which can be either intra- or intersupramolecular.
Conclusion
In conclusion, we have shown that two heteroleptic complexes can be interconverted by changing the oxidation state of the copper ions. Using this redox-triggered self-sorting, cargo (ligand 4 and copper) may be shipped reversibly between two different stations (PhenAr2 and TerpyAr2) on platform 2. If the redox state at copper is +I, ligand 4 occupies quantitatively the PhenAr2 stations, whereas for copper(II), the TerpyAr2 stations are engaged with the cargo molecules. Electrochemical oxidation and reduction leads to reversible shipping of the cargo between the two different stations. Concentration-dependent CV studies advocate that dissociation of the cargo from the scaffold is the rate determining step.Experimental procedure
Commercial reagents were used without further purification. Solvents were dried with appropriate desiccants and distilled prior to use. 1H and 13C NMR spectra were recorded at 400 MHz or 600 MHz using a deuterated solvent as the lock and residual protiated solvent as internal reference. The following abbreviations are utilised to describe NMR peak patterns: s = singlet, d = doublet, t = triplet, dd = doublet of doublets. The following abbreviations are used to describe peak patterns of IR spectra: s = sharp, m = medium, w = weak. The numbering of the carbon skeleton in molecular formulae as shown in the manuscript does not comply with the IUPAC nomenclature rules; it is only used for assignments of NMR signals. Electrospray ionisation (ESI) mass spectra were recorded on a Thermo-Quest LCQ deca. Melting points were measured on a Büchi SMP-20 and are uncorrected. Infrared spectra were recorded on a Varian 1000 FT-IR instrument. Elemental analysis measurements were made using the EA 3000 CHNS. Cyclic voltammetry (CV) was measured on a Parstat 2273. CV of millimolar solutions was carried out in dry CH2Cl2 (2.0 mL) with 0.1 M n-Bu4NPF6 as electrolyte against a Ag wire as a quasi-reference electrode and triphenylpyrylium tetrafluoroborate (TPP) as internal standard. All CV spectra are calibrated against the standard calomel electrode (SCE).2-(2,4,6-Trimethylphenyl)-6-bromopyridine (6)
2,6-Dibromopyridine (2.16 g, 9.12 mmol), 2,4,6-trimethylphenylboronic acid (1.60 g, 6.10 mmol) and Pd(PPh3)4 (70.0 mg, 60.6 μmol) were dissolved in a degassed solution of MeOH (40 mL), THF (100 mL) and aqueous K2CO3 (2 M, 30 mL), then the solution was refluxed for 12 h at 90 °C. The solvent was evaporated and the residue redissolved in DCM. The organic phase was washed with water thrice and then dried over anhydrous Na2SO4. The product was further purified by column chromatography (SiO2, hexane–EtOAc = 9:1, Rf = 0.3) to afford a liquid. Yield: 90%, 1H NMR (CDCl3, 400 MHz): δ = 2.04 (s, 6 H, CH3), 2.31 (s, 3 H, CH3), 6.92 (s, 2 H, 15′-H), 7.18 (dd, 3J = 7.8 Hz, 4J = 0.8 Hz, 1 H, 14-H), 7.45 (dd, 3J = 7.8 Hz, 4J = 0.8 Hz, 1 H, 12-H), 7.61 (t, 3J = 7.8 Hz, 1 H, 13-H) ppm. 13C NMR (CDCl3, 100 MHz): δ = 20.1, 21.0, 123.7, 125.9, 128.3, 135.6, 136.2, 137.9, 138.5, 141.7, 161.1 ppm. IR (KBr): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3944, 3690, 3055, 2986, 2306, 1571, 1425, 1265, 741 cm−1. ESI-Ms: m/z (%) 277.1 (100) [M + H]+. Anal Calcd for C14H14BrN·0.2H2O: C, 60.10; H, 5.19; N, 5.01. Found: C, 60.06; H, 5.17; N, 5.20.
= 3944, 3690, 3055, 2986, 2306, 1571, 1425, 1265, 741 cm−1. ESI-Ms: m/z (%) 277.1 (100) [M + H]+. Anal Calcd for C14H14BrN·0.2H2O: C, 60.10; H, 5.19; N, 5.01. Found: C, 60.06; H, 5.17; N, 5.20.
2-(6-(2,4,6-Trimethylphenyl)pyrid-2-yl)-9-(2,4,6-trimethylphenyl)-[1,10]-phenanthroline (5)
To a solution of 6 (841 mg, 3.05 mmol) in dry diethyl ether (80 mL), 2.5 M n-BuLi (1.22 mL, 3.05 mmol) in hexane was added dropwise at −78 °C under N2 atmosphere over a period of 15 min. The reaction mixture was stirred at −40 °C for 2 h, then 2-(2,4,6-trimethylphenyl)-[1,10]-phenanthroline (605 mg, 2.03 mmol) was added under N2 atmosphere. The solution turned immediately dark violet. After stirring for another 12 h at room temperature, saturated aqueous NH4Cl (100 mL) was added. The reaction slurry was stirred for 1 h, then the solution was extracted with DCM (3 × 100 mL). After drying over anhydrous Na2SO4, the solvent was removed. The solid orange residue was dissolved in DCM (100 mL) and treated with MnO2 (2.65 g, 30.5 mmol). After stirring for 12 h at room temperature, the solution was filtered through a pad of celite. The solvent was evaporated to dryness. Finally, the compound was purified by column chromatography (SiO2, hexane–ethyl acetate = 9:1, Rf = 0.4). Yield: 54%, mp: 236 °C. 1H NMR (CD2Cl2, 400 MHz): δ = 2.10 (s, 6 H, CH3), 2.26 (s, 6 H, CH3), 2.35 (s, 3 H, CH3), 2.40 (s, 3 H, CH3), 6.98 (s, 2 H, [15 or 15′]-H), 7.04 (s, 2 H, [15′ or 15]-H), 7.25 (dd, 3J = 7.4 Hz, 4J = 1.0 Hz, 1 H, 14-H), 7.62 (d, 3J = 8.4 Hz, 1 H, 4′-H), 7.84 (s, 2 H, 5′-, 6′-H), 7.92 (t, 3J = 7.4 Hz, 1 H, 13-H), 8.29 (d, 3J = 8.4 Hz, 1 H, 3′-H), 8.31 (d, 3J = 8.4 Hz, 1 H, 8′-H), 8.78 (d, 3J = 8.4 Hz, 1 H, 7′-H), 8.81 (dd, 3J = 7.4 Hz, 4J = 1.0 Hz, 1 H, 12-H) ppm. 13C NMR (CD2Cl2, 100 MHz): δ = 19.6, 20.7, 20.9, 21.7, 118.4, 118.8, 118.9, 127.6, 130.5, 130.8, 130.9, 131.1, 131.2, 131.7, 132.4, 132.7, 134.8, 135.6, 136.0, 136.5, 137.4, 138.9, 139.2, 142.5, 149.4, 149.7, 149.9, 160.2, 160.8 ppm. IR (KBr): = 3001, 2943, 2913, 1583, 1541, 1477, 1374, 1355, 1158, 1134, 909, 862, 847, 752, 637, 629 cm−1. ESI-MS: m/z (%) 494.4 (100) [M + H]+. Anal Calcd for C35H31N3·0.2H2O: C, 84.54; H, 6.36; N, 8.45. Found: C, 84.46; H, 6.15; N, 8.82.
2-(6-(2,4,6-Trimethylphenyl)pyrid-2-yl)-[1,10]-phenanthroline (7)
Under N2 atmosphere, 2.5 M n-BuLi (1.45 mL, 3.62 mmol) in hexane was added dropwise to a solution of 6 (1.00 g, 3.62 mmol) in dry diethyl ether (80 mL) at −78 °C over a period of 15 min. The reaction mixture was stirred at −40 °C for 2 h, then [1,10]-phenanthroline (489 mg, 2.72 mmol) was added under N2 atmosphere. The solution turning immediately to a dark violet was stirred for another 12 h at room temperature. Saturated aqueous NH4Cl (100 mL) was then added and the reaction slurry was stirred for 1 h. Thereafter, the solution was extracted with DCM (3 × 100 mL). After drying over anhydrous Na2SO4, the solvent was removed. The solid orange residue was dissolved in DCM (100 mL) and treated with MnO2 (3.15 g, 36.2 mmol). After stirring for 12 h at room temperature, the solution was filtered through a pad of celite. The solvent was evaporated to dryness. Finally, the compound was purified by column chromatography (SiO2, DCM–ethyl acetate = 1:3, Rf = 0.25). Yield: 61%, mp: 165 °C. 1H NMR (CD2Cl2, 400 MHz): δ = 2.01 (s, 6 H, CH3), 2.34 (s, 3 H, CH3), 6.91 (s, 2 H, 15-H), 7.23 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 1 H, 14-H), 7.55 (d, 3J = 8.0 Hz, 1 H, 8′-H), 7.63 (dd, 3J = 8.0 Hz, 3J = 4.4 Hz, 1 H, 3′-H), 7.82 (d, 3J = 8.8 Hz, 1 H, [5′ or 6′]-H), 7.86 (d, 3J = 8.8 Hz, 1 H, [6′ or 5′]-H), 7.94 (t, 3J = 7.6 Hz, 1 H, 13-H), 8.28 (d, 3J = 8.0 Hz, 2 H, 4′-, 7′-H), 8.83 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 1 H, 12-H), 9.21 (dd, 3J = 4.4 Hz, 3J = 1.6 Hz, 1 H, 2′-H) ppm. 13C NMR (CD2Cl2, 100 MHz): δ = 19.6, 20.7, 124.8, 125.0, 127.6, 128.5, 130.5, 130.8, 130.9, 131.7, 132.6, 135.6, 135.9, 136.0, 136.0, 137.4, 137.6, 138.0, 141.4, 146.1, 146.1, 160.3, 160.9 ppm. IR (KBr): = 2967, 2923, 1583, 1521, 1447, 1374, 1365, 1158, 1134, 919, 856 cm−1. ESI-MS: m/z (%) 376.4 (100) [M + H]+. Anal Calcd for C26H21N3·CH2Cl2: C, 70.44; H, 5.04; N, 9.13. Found: C, 70.67; H, 5.15; N, 8.82.
2-(6-(2,4,6-Trimethylphenyl)pyrid-2-yl)-9-(4-iodo-2,3,5,6-tetramethylphenyl)-[1,10]-phenanthroline (8)
Over a period of 15 min, 2.5 M n-BuLi (0.800 mL, 2.00 mmol) in hexane was added dropwise at −78 °C under N2 atmosphere to a solution of 1,4-diiodo-2,3,5,6-tetramethylbenzene (770 mg, 2.00 mmol) in dry diethyl ether (80 mL). The reaction mixture was stirred at −40 °C for 2 h, then 7 (500 mg, 1.33 mmol) was added under N2 atmosphere. The solution turning immediately to dark violet was stirred for another 12 h at room temperature. Saturated aqueous NH4Cl (100 mL) was then added and the reaction slurry was stirred for 1 h. Thereafter, the solution was extracted with DCM (3 × 100 mL). After drying over anhydrous Na2SO4, the solvent was removed. The solid orange residue, redissolved in DCM (100 mL), was treated with MnO2 (1.72 g, 20.0 mmol) for 12 h at room temperature. After passing the solution through a pad of celite, the solvent was removed. Finally, the compound was purified by column chromatography (SiO2, DCM–ethyl acetate = 70:30, Rf = 0.4). Yield: 53%, mp: >213 °C. 1H NMR (CD2Cl2, 400 MHz): δ = 2.09 (s, 6 H, CH3), 2.24 (s, 6 H, CH3), 2.33 (s, 6 H, CH3), 2.40 (s, 3 H, CH3), 6.97 (s, 2 H, 15′-H), 7.28 (dd, 3J = 7.6 Hz, 4J = 0.8 Hz, 1 H, 14-H), 7.60 (d, 3J = 8.4 Hz, 1 H, 4′-H), 7.87 (s, 2 H, 5′-, 6′-H), 7.94 (t, 3J = 7.6 Hz, 1 H, 13-H), 8.34 (d, 3J = 8.4 Hz, 1 H, 3′-H), 8.35 (d, 3J = 8.4 Hz, 1 H, 8′-H), 8.75 (d, 3J = 8.4 Hz, 1 H, 7′-H), 8.77 (dd, 3J = 7.6 Hz, 4J = 0.8 Hz, 1 H, 12-H) ppm. 13C NMR (CD2Cl2, 100 MHz): δ = 19.3, 20.7, 21.6, 21.9, 118.6, 124.6, 124.9, 127.6, 129.7, 130.5, 130.8, 130.9, 132.4, 132.9, 134.5, 135.1, 135.9, 136.2, 136.9, 137.4, 137.6, 138.5, 141.3, 142.3, 143.5, 146.1, 146.1, 160.3, 160.9 ppm. IR (KBr): = 2956, 2916, 1565, 1521, 1454, 1354, 1324, 1158, 1126, 919, 887 cm−1. ESI-MS: m/z (%) 637.4 (100) [M + H]+. Anal Calcd for C36H32IN3·CH3CO2C2H5: C, 62.62; H, 6.76; N, 3.91. Found: C, 62.46; H, 6.45; N, 3.82.
Zinc(II) porphyrin 10
Zinc(II)-5,15-bis(4-iodophenyl)-10,20-bis(4-trimethylsilylethynylphenyl)porphyrin (250 mg, 223 μmol) and phenanthroline 9 (203 mg, 446 μmol) were placed in a round-bottomed flask. Then, Pd(PPh3)4 (25.7 mg, 22.3 μmol), dry NEt3 (10 mL) and dry DMF (30 mL) were added under N2 atmosphere. The reaction mixture was allowed to stir at 90 °C for 12 h. Then, it was evaporated to dryness, dissolved in DCM (50 mL), and washed with water (3 × 50 mL). After drying over anhydrous Na2SO4, the solvent was evaporated and the residue was purified by column chromatography (SiO2, hexane–EtOAc = 80:20, Rf = 0.4). The compound was further purified by size exclusion chromatography over biobeads SX-3 isolating the first moving band in toluene. Yield: 58%, mp: >300 °C. 1H NMR (400 MHz, CD2Cl2): δ = 0.38 (s, 18 H, TMS-H), 2.04 (s, 12 H, CH3), 2.12 (s, 12 H, CH3), 2.31 (s, 6 H, CH3), 2.54 (s, 12 H, CH3), 6.92 (s, 4 H, 9-H), 7.52 (d, 3J = 8.4 Hz, 2 H, 7-H), 7.59 (d, 3J = 8.4 Hz, 2 H, 4-H), 7.87 (d, 3J = 8.4 Hz, 4 H, 11-H), 7.88 (s, 4 H, 5-,6-H), 7.93 (d, 3J = 8.4 Hz, 4 H, 17-H), 8.10 (d, 3J = 8.4 Hz, 4 H, 10-H), 8.15 (d, 3J = 8.4 Hz, 4 H, 16-H), 8.36 (d, 3J = 8.4 Hz, 2 H, 8-H), 8.54 (d, 3J = 8.4 Hz, 2 H, 3-H), 8.92 (d, 3J = 4.0 Hz, 4 H, β-H), 8.94 (d, 3J = 4.8 Hz, 4 H, β-H) ppm. 13C NMR (100 MHz, CD2Cl2): δ = 0.1, 19.6, 20.5, 21.0, 27.6, 93.9, 94.0, 95.4, 95.5, 105.0, 111.8, 119.9, 120.6, 122.4, 124.8, 125.0, 126.1, 126.4, 127.2, 127.2, 128.4 (2C), 130.2, 131.9, 132.0, 132.0, 132.6, 134.2, 135.7, 135.8, 136.0, 136.0, 137.5, 137.6, 141.4, 142.1, 142.8, 146.0, 149.8, 149.9, 149.9, 160.2, 160.8 ppm. IR (KBr): = 2947, 2928, 2815, 2320, 2343, 1609, 1556, 1490, 1416, 1380, 1356, 1185, 1099, 1025, 998, 884 cm−1. ESI-MS: m/z (%) 1774.5 (100) [M + H]+. Anal Calcd for C120H100N8Si2Zn·2H2O: C, 79.55; H, 5.79; N, 6.18. Found: C, 79.92; H, 6.19; N, 6.22.
Zinc(II) porphyrin 11
KOH (96.0 mg, 1.71 mmol) was added to a solution of zinc(II) porphyrin 10 (310 mg, 0.175 mmol) in THF (30 mL). Then, MeOH (15 mL) and H2O (5 mL) were added, and the reaction mixture was stirred for 12 h at room temperature. After evaporating the solvent, the residue was redissolved in DCM and washed with water (3 × 100 mL). After drying over anhydrous Na2SO4, DCM was evaporated and the compound was used without further purification. Yield: 90%, mp: >300 °C. 1H NMR (400 MHz, CD2Cl2): δ = 2.04 (s, 12 H, CH3), 2.14 (s, 12 H, CH3), 2.33 (s, 6 H, CH3), 2.53 (s, 12 H, CH3), 3.61 (s, 2 H, ethynyl-H), 6.92 (s, 4 H, 9-H), 7.54 (d, 3J = 8.4 Hz, 2 H, 7-H), 7.58 (d, 3J = 8.4 Hz, 2 H, 4-H), 7.86 (d, 3J = 8.4 Hz, 4 H, 11-H), 7.90 (s, 4 H, 5-, 6-H), 7.98 (d, 3J = 8.4 Hz, 4 H, 17-H), 8.11 (d, 3J = 8.4 Hz, 4 H, 10-H), 8.19 (d, 3J = 8.4 Hz, 4 H, 16-H), 8.37 (d, 3J = 8.4 Hz, 2 H, 8-H), 8.56 (d, 3J = 8.4 Hz, 2 H, 3-H), 8.90 (d, 3J = 4.0 Hz, 4 H, β-H), 8.95 (d, 3J = 4.8 Hz, 4 H, β-H) ppm. 13C NMR (100 MHz, CD2Cl2): δ = 19.4, 20.7, 21.6, 27.4, 93.1, 94.0, 95.1, 95.6, 105.3, 111.1, 119.9, 120.6, 122.4, 124.8, 125.2, 126.7, 126.8, 127.2, 127.3, 128.6, 128.7, 130.5, 131.8, 132.1, 132.3, 132.6, 134.6, 135.8, 135.9, 136.2, 136.3, 137.6, 137.8, 141.4, 142.5, 142.8, 146.1, 149.8, 149.9, 150.1, 160.2, 160.8 ppm. IR (KBr): = 2998, 2935, 2820, 2345, 2323, 1619, 1565, 1490, 1397, 1353, 1189, 1050, 1025, 945, 895 cm−1. ESI-MS: m/z (%) 1630.3 (100) [M + H]+. Anal Calcd for C116H96N8Zn2: C, 80.48; H, 5.05; N, 6.53. Found: C, 80.67; H, 5.29; N, 6.32.
Zinc(II) porphyrin 2
Zinc(II) porphyrin 11 (110 mg, 67.4 μmol) and phenanthroline 8 (85.5 mg, 135 μmol) were put into a round-bottomed flask. Then, Pd(PPh3)4 (15.6 mg, 13.5 μmol), dry NEt3 (10 mL) and dry DMF (30 mL) were added to the reaction mixture under N2 atmosphere. The reaction mixture was allowed to stir at 90 °C for 12 h. The reaction mixture was evaporated to dryness, dissolved in DCM (50 mL), and washed with water (3 × 50 mL). After drying over anhydrous Na2SO4, the solvent was evaporated and the residue was purified by column chromatography (SiO2, DCM–MeOH = 99:1, Rf = 0.1). The compound was further purified by size exclusion chromatography over biobeads SX-3 isolating the first moving band in toluene. Yield: 35%, mp: >300 °C. 1H NMR (400 MHz, CD2Cl2): δ = 2.00 (s, 12 H, CH3), 2.03 (s, 12 H, CH3), 2.09 (s, 12 H, CH3), 2.16 (s, 12 H, CH3), 2.33 (s, 6 H, CH3), 2.35 (s, 6 H, CH3), 2.41 (s, 12 H, CH3), 2.60 (s, 12 H, CH3), 6.95 (s, 4 H, 9-H), 6.98 (s, 4 H, 15′-H), 7.28 (dd, 3J = 7.6 Hz, 4J = 0.8 Hz, 2 H, 14-H), 7.52 (d, 3J = 8.0 Hz, 2 H, [4 or 7]-H), 7.56 (d, 3J = 8.0 Hz, 2 H, [7 or 4]-H), 7.60 (d, 3J = 8.4 Hz, 2 H, 4′-H), 7.86 (d, 3J = 8.4 Hz, 4 H, [17 or 16]-H), 7.87 (s, 4 H, 5′-, 6′-H), 7.90 (s, 4 H, 5-, 6-H), 7.95 (d, 3J = 8.4 Hz, 4 H, [11 or 10]-H), 7.94 (t, 3J = 7.6 Hz, 2 H, 13-H), 8.12 (d, 3J = 8.4 Hz, 4 H, [10 or 11]-H), 8.16 (d, 3J = 8.4 Hz, 4 H, [16 or 17]-H), 8.33 (d, 3J = 8.0 Hz, 2 H, [3 or 8]-H), 8.34 (d, 3J = 8.0 Hz, 2 H, [8 or 3]-H), 8.34 (d, 3J = 8.4 Hz, 2 H, 3′-H), 8.36 (d, 3J = 8.4 Hz, 2 H, 8′-H), 8.76 (d, 3J = 8.4 Hz, 2 H, 7′-H), 8.77 (dd, 3J = 7.6 Hz, 4J = 0.8 Hz, 2 H, 12-H), 8.95 (d, 3J = 4.4 Hz, 4 H, β-H), 8.96 (d, 3J = 4.4 Hz, 4 H, β-H) ppm. 13C NMR (100 MHz, CD2Cl2): δ = 20.0, 20.1, 20.6, 20.8, 21.5, 21.6, 28.0, 28.1, 94.4, 94.6, 97.2, 97.4, 112.5, 112.7, 120.2, 120.3, 120.9, 121.0, 122.7, 123.4, 124.0, 124.4, 124.9, 125.2, 126.3, 126.6, 126.9 (2C), 127.0, 127.5, 127.6, 127.6, 128.8 (2C), 129.1, 129.2, 130.7, 132.4, 133.1 (2C), 134.7, 136.1, 136.2, 136.4, 136.4, 136.5, 136.6, 136.8, 138.0, 138.3, 138.4, 138.6, 138.6, 139.1 (2C), 141.5, 141.9, 141.9, 142.3, 142.3, 143.0, 146.4, 146.4, 146.6, 150.1, 150.1, 150.2, 150.6, 160.3, 160.9, 160.9 ppm. IR (KBr): = 2936, 2916, 2836, 2380, 2343, 1619, 1556, 1490, 1425, 1356, 1326, 1165, 1099 cm−1. ESI-MS: m/z (%) 1322.3 (100) [M + 2H]2+, 881.9 (50) [M + 3H]3+, 661.7 (30) [M + 4H]4+. Anal. Calcd for C186H146N14Zn·1.5CH2Cl2: C, 81.30; H, 5.42; N, 7.08. Found: C, 81.45; H, 5.19; N, 7.22.
Homoleptic complex [Cu(5)2](PF6)
In an NMR tube, ligand 5 (0.373 mg, 0.756 μmol) and [Cu(CH3CN)4]PF6 (0.141 mg, 0.378 μmol) were dissolved in CD2Cl2 to afford the desired complex in quantitative yield. mp: >300 °C. 1H NMR (CD2Cl2, 400 MHz): δ = 0.70 (s, 6 H, CH3), 0.79 (s, 6 H, CH3), 1.97 (s, 12 H, CH3), 2.00 (s, 6 H, CH3), 2.24 (s, 6 H, CH3), 5.61 (s, 4 H, [15 or 15′]-H), 6.19 (s, 4 H, [15′ or 15]-H), 6.97 (dd, 3J = 7.6 Hz, 3J = 1.2 Hz, 2 H, 14-H), 7.40 (d, 3J = 8.0 Hz, 2 H, 4′-H), 7.47 (t, 3J = 7.6 Hz, 2 H, 13-H), 8.06 (s, 4 H, 5′-, 6′-H), 8.44 (d, 3J = 8.0 Hz, 2 H, 3′-H), 8.49 (d, 3J = 8.6 Hz, 2 H, 8′-H), 8.58 (d, 3J = 8.6 Hz, 2 H, 7′-H), 9.41 (dd, 3J = 7.6 Hz, 3J = 1.2 Hz, 2 H, 12-H) ppm. 13C NMR (CD2Cl2, 100 MHz): δ = 19.6, 19.8, 20.3, 21.0, 21.0, 122.5, 124.7, 126.1, 126.6, 126.9, 127.1, 127.2, 128.3, 128.4, 128.6, 130.1, 133.5, 135.6, 135.8, 136.0, 137.2, 137.3, 137.6, 137.6, 137.8, 144.1, 145.1, 153.5, 154.5, 159.7, 160.0 ppm. IR (KBr): = 2965, 2903, 1613, 1585, 1454, 1402, 1339, 1138, 1104, 845, 810, 759 cm−1. ESI-MS: m/z (%) 1051.0 (100) [Cu(5)2]+. Anal Calcd for C70H62CuF6N6P: C, 70.31; H, 5.23; N, 7.03. Found: C, 70.36; H, 5.45; N, 7.15.
Heteroleptic complex [Zn(4)(5)](OTf)2
In an NMR tube, compound 5 (0.550 mg, 1.11 μmol), Zn(OTf)2 (0.405 mg, 1.11 μmol) and 4 (0.201 mg, 1.11 μmol) were dissolved in CD2Cl2–CD3CN = 3:1 furnishing the desired complex in quantitative yield. mp: >300 °C. 1H NMR (CD2Cl2–CD3CN = 3:1, 400 MHz): δ = 0.93 (s, 6 H, CH3), 1.00 (s, 6 H, CH3), 1.82 (s, 3 H, CH3), 1.87 (s, 3 H, CH3), 5.64 (s, 2 H, [15 or 15′]-H), 5.69 (s, 2 H, [15′ or 15]-H), 7.04 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 1 H, 14-H), 7.23 (d, 3J = 8.4 Hz, 1 H, 4′-H), 7.67 (dd, 3J = 8.4 Hz, 3J = 8.4 Hz, 2 H, 3′′-H), 7.96 (s, 2 H, 5′′-H), 8.13 (t, 3J = 7.6 Hz, 1 H, 13-H), 8.20 (d, 3J = 8.8 Hz, 1 H, [5′ or 6′]-H), 8.27 (d, 3J = 8.8 Hz, 1 H, [6′ or 5′]-H), 8.52 (dd, 3J = 8.4 Hz, 4J = 1.6 Hz, 2 H, 4′′-H), 8.54 (d, 3J = 8.4 Hz, 1 H, 3′-H), 8.60 (dd, 3J = 8.4 Hz, 4J = 1.6 Hz, 2 H, 2′′-H), 8.66 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 1 H, 12-H), 8.97 (d, 3J = 8.6 Hz, 1 H, 8′-H), 9.08 (d, 3J = 8.6 Hz, 1 H, 7′-H) ppm. 13C NMR (CD2Cl2–CD3CN = 3:1, 100 MHz): δ = 19.2, 19.3, 20.2, 20.3, 121.9, 122.7, 126.5, 128.0 (2C), 128.1 (2C), 128.3, 128.6, 128.9, 129.4, 130.0, 130.0, 134.0, 134.2, 134.8, 135.6, 135.9, 139.1, 139.2, 140.3, 140.3, 141.3, 141.4, 142.5, 149.4, 149.6, 151.1, 151.2, 161.2, 161.6 ppm. IR (KBr): = 3001, 2943, 2913, 1583, 1541, 1477, 1374, 1355, 1158, 1134, 909, 862, 847, 752, 637, 629 cm−1. ESI-MS: m/z (%) 370.0 (100) [Zn(4)(5)]2+, 888.3 (30) [Zn(4)(5)]+. Anal. Calcd for C49H39F6N5O6S2Zn: C, 56.73; 3.79; N, 6.75; S, 6.18. Found: C, 56.47; 3.99; N, 6.58; S, 6.43.
Heteroleptic complex [Cu(4)(5)](ClO4)2
Compound 5 (3.45 mg, 6.99 μmol), Cu(ClO4)2·6H2O (2.59 mg, 6.99 μmol) and 4 (1.26 mg, 6.99 μmol) were dissolved in CD2Cl2–CD3CN = 3:1 furnishing the desired complex as characterised by ESI-MS: m/z (%) 368.7 (100) [Cu(4)(5)]2+, 836.8 (30) [Cu(4)(5)](ClO4)+.
Complex Cu1phen = [Cu2(2)(4)2]2+
In an NMR tube, platform 2 (2.42 mg, 0.916 μmol), [Cu(CH3CN)4]PF6 (0.683 mg, 1.83 μmol) and ligand 4 (0.330 mg, 1.83 μmol) were dissolved in CD2Cl2 affording complex Cu1phen in quantitative yield. Mp: >300 °C. 1H NMR (CD2Cl2, 400 MHz): δ = 1.53 (s, 6 H, CH3), 1.65 (s, 12 H, CH3), 1.66 (s, 12 H, CH3), 1.78 (s, 12 H, CH3), 2.12 (s, 12 H, CH3), 2.28 (s, 12 H, CH3), 2.30 (s, 6 H, CH3), 2.59 (s, 12 H, CH3), 6.00 (s, 4 H, 9-H), 6.97 (s, 4 H, 15′-H), 7.27 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 2 H, 14-H), 7.60 (d, 3J = 8.4 Hz, 2 H, 4′-H), 7.71 (dd, 3J = 8.2 Hz, 3J = 8.2 Hz, 4 H, 3′′-H), 7.83 (d, 3J = 8.4 Hz, 2 H, [4 or 7]-H), 7.86 (d, 3J = 8.0 Hz, 4 H, [10 or 11]-H), 7.89 (d, 3J = 8.4 Hz, 2 H, [7 or 4]-H), 7.91 (s, 4 H, 5′-, 6′-H), 7.92 (s, 4 H, 5′′-H), 7.94 (t, 3J = 7.6 Hz, 2 H, 13-H), 7.95 (d, 3J = 8.0 Hz, 4 H, [16 or 17]-H), 8.12 (d, 3J = 8.0 Hz, 4 H, [17 or 16]-H), 8.16 (d, 3J = 8.0 Hz, 4 H, [11 or 10]-H), 8.21 (s, 4 H, 5-, 6-H), 8.34 (d, 3J = 8.4 Hz, 2 H, 3′-H), 8.36 (d, 3J = 8.0 Hz, 2 H, 8′-H), 8.42 (dd, 3J = 8.2 Hz, 4J = 1.6 Hz, 4 H, 4′′-H), 8.45 (dd, 3J = 8.2 Hz, 4J = 1.6 Hz, 4 H, 2′′-H), 8.69 (d, 3J = 8.4 Hz, 2 H, [3 or 8]-H), 8.70 (d, 3J = 8.4 Hz, 2 H, [8 or 3]-H), 8.76 (d, 3J = 8.0 Hz, 2 H, 7′-H), 8.77 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 2 H, 12-H), 8.94 (d, 3J = 4.0 Hz, 4 H, β-H), 8.95 (d, 3J = 4.0 Hz, 4 H, β-H) ppm. 13C NMR (CD2Cl2, 100 MHz) δ = 19.5, 19.6, 20.2, 20.4, 20.4, 20.6, 21.2, 21.3, 94.1, 94.2, 95.7, 95.9, 105.3, 111.5, 120.3, 120.3, 120.4, 121.0, 122.8, 124.7, 124.8, 124.9, 125.1, 125.4, 126.3, 126.3, 126.4, 126.4, 126.6, 126.7, 126.9, 127.0, 127.1, 127.3, 127.4, 128.2, 128.3, 128.6, 128.6, 128.8, 129.0, 130.4, 131.5, 132.3, 134.8, 134.9, 135.0, 135.2, 135.3, 136.1, 136.2, 136.3, 136.4, 136.5, 136.7, 137.0, 137.3, 137.7, 137.7, 137.8, 138.1, 142.6, 143.2, 143.4, 144.1, 147.8, 149.1, 150.2, 150.3, 150.3, 150.3, 151.1, 151.3, 156.6, 156.6, 159.5, 159.6 ppm. IR (KBr): = 2975, 2847, 2306, 2196, 1604, 1595, 1422, 1376, 1340, 1258, 1224, 1203, 1153, 1062, 1030, 850, 797 cm−1, ESI-MS: m/z (%) 1565.1 (100) [Cu2(2)(4)2]2+. Anal. Calcd for C210H162Cu2F12N18P2Zn: C, 73.75; H, 4.77; N, 7.37. Found: C, 73.49; H, 4.87; N, 7.52.
Complex Zn1terpy = [Zn2(2)(4)2]4+
In an NMR tube, platform 2 (3.65 mg, 1.38 μmol), Zn(OTf)2 (1.00 mg, 2.76 μmol) and ligand 4 (0.498 mg, 2.76 μmol) were dissolved in CD2Cl2–CD3CN = 3:1 furnishing complex Zn1terpy in quantitative yield. Mp: >300 °C. 1H NMR (CD2Cl2–CD3CN = 3:1, 400 MHz): δ = 0.94 (s, 12 H, CH3), 1.02 (s, 12 H, CH3), 1.83 (s, 6 H, CH3), 1.88 (s, 12 H, CH3), 1.94 (s, 12 H, CH3), 1.97 (s, 12 H, CH3), 2.28 (s, 6 H, CH3), 2.49 (s, 12 H, CH3), 5.69 (s, 4 H, 15′-H), 6.91 (s, 4 H, 9-H), 7.05 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 2 H, 14-H), 7.24 (d, 3J = 8.4 Hz, 2 H, 4′-H), 7.48 (d, 3J = 8.0 Hz, 2 H, [4 or 7]-H), 7.53 (d, 3J = 8.0 Hz, 2 H, [7 or 4]-H), 7.68 (dd, 3J = 8.4 Hz, 3J = 8.4 Hz, 4 H, 3′′-H), 7.79 (d, 3J = 8.0 Hz, 4 H, [16 or 17]-H), 7.89 (s, 4 H, 5, 6-H), 7.91 (d, 3J = 8.4 Hz, 4 H, [10 or 11]-H), 7.92 (s, 4 H, 5′′-H), 8.06 (d, 3J = 8.4 Hz, 4 H, [11 or 10]-H), 8.12 (d, 3J = 8.0 Hz, 4 H, [17 or 16]-H), 8.16 (t, 3J = 7.6 Hz, 2 H, 13-H), 8.19 (d, 3J = 8.8 Hz, 2 H, [5′ or 6′]-H), 8.27 (d, 3J = 8.8 Hz, 2 H, [6′ or 5′]-H), 8.33 (d, 3J = 8.0 Hz, 2 H, [3 or 8]-H), 8.35 (d, 3J = 8.0 Hz, 2 H, [8 or 3]-H), 8.51 (d, 3J = 8.4 Hz, 2 H, 3′-H), 8.53 (dd, 3J = 8.4 Hz, 4J = 1.6 Hz, 4 H, 4′′-H), 8.62 (dd, 3J = 8.4 Hz, 4J = 1.6 Hz, 4 H, 2′′-H), 8.69 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 2 H, 12-H), 8.83 (d, 3J = 4.8 Hz, 4 H, β-H), 8.85 (d, 3J = 4.8 Hz, 4 H, β-H), 8.96 (d, 3J = 8.4 Hz, 1 H, 8′-H), 9.06 (d, 3J = 8.4 Hz, 1 H, 7′-H) ppm. 13C NMR (CD2Cl2–CD3CN = 3:1, 100 MHz) δ = 18.5, 18.6, 19.0, 19.8, 20.4, 20.4, 20.6, 27.2, 93.4, 93.6, 96.1, 96.2, 105.0, 111.0, 119.4, 120.0, 122.0, 122.1, 122.5, 124.3, 124.5, 125.9, 126.2, 126.4, 126.5, 127.0, 127.0, 127.1, 127.4, 127.8, 128.0, 128.2 (2C), 128.7, 128.9, 129.2, 129.6, 129.9, 130.2, 131.7, 132.5, 133.6, 133.9, 134.2, 134.5, 135.6 (2C), 136.2 (2C), 136.4, 137.4, 137.5, 137.5, 137.6, 138.2, 139.4, 139.8, 140.0, 140.7, 140.8, 141.1, 141.5, 141.7, 142.3, 142.7, 143.5, 146.1, 148.1, 148.7, 149.5, 149.6, 149.7, 150.5, 160.0, 160.7, 161.3, 161.7 ppm. IR (KBr): = 2921, 2200, 1601, 1478, 1277, 1259, 1030, 996, 797, 638 cm−1. ESI-MS: m/z (%) 783.5 (50) [Zn2(2)(4)2]4+, 1094.4 (100) [Zn2(2)(4)2](OTf)3+, 1716.1 (20) [Zn2(2)(4)2](OTf)22+. Anal Calcd for C214H162F12N18O12S4Zn3·CH2Cl2: C, 67.69; H, 4.33; N, 6.61; S, 3.36. Found: C, 67.56; H, 4.18; N, 6.47; S, 3.56.
Complex Cu1terpy = [Cu2(2)(4)2](ClO4)4
Compound 2 (3.78 mg, 1.43 μmol), Cu(ClO4)2·6H2O (0.530 mg, 1.43 μmol) and 4 (0.258 mg, 1.43 μmol) were dissolved in CD2Cl2–CD3CN = 3:1 furnishing the desired complex as characterised by ESI-MS: m/z (%) 782.7 (100) [Cu2(2)(4)2]4+, 1076.8 (50) [Cu2(2)(4)2](ClO4)3+.
Acknowledgements
We acknowledge generous funding by the DFG and the Universität Siegen.Notes and references
- (a) L. B. Jilaveanu, C. R. Zito and D. Oliver, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7511 CrossRef CAS PubMed; (b) M. Schliwa and G. Woehlke, Nature, 2003, 422, 759 CrossRef CAS PubMed; (c) M. A. Geeves and K. C. Holmes, Annu. Rev. Biochem., 1999, 68, 687 CrossRef CAS PubMed.
- (a) S. J. Cantrill, A. R. Pease and J. F. Stoddart, J. Chem. Soc., Dalton Trans., 2000, 3715 RSC; (b) T. Murahashi, K. Shirato, A. Fukushima, K. Takase, T. Suenobu, S. Fukuzumi, S. Ogoshi and H. Kurosawa, Nat. Chem., 2012, 4, 52 CrossRef CAS PubMed; (c) C. Wang, Z. Li, D. Cao, Y.-L. Zhao, J. W. Gaines, O. A. Bozdemir, M. W. Ambrogio, M. Frasconi, Y. Y. Botros, J. I. Zink and J. F. Stoddart, Angew. Chem., Int. Ed., 2012, 51, 5460 CrossRef CAS PubMed.
- (a) J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611 CrossRef CAS; (b) A. M. Brouwer, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291, 2124 CrossRef CAS PubMed; (c) V. Amendola, L. Fabbrizzi, C. Mangano and P. Pallavicini, Acc. Chem. Res., 2001, 34, 488 CrossRef CAS PubMed; (d) Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H.-R. Tseng, J. A. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745 CrossRef CAS PubMed; (e) B. Colasson, N. L. Paul, Y. L. Mest and O. Reinaud, J. Am. Chem. Soc., 2010, 132, 4393 CrossRef CAS PubMed.
- V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed; B. L. Feringa and W. R. Browne, Molecular Switches 1+2, Wiley-VCH, Weinheim, 2nd edn, 2011 Search PubMed.
- (a) J.-P. Collin, C. Dietrich-Buchecker, P. Gavina, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477 CrossRef CAS PubMed; (b) J. D. Badjic, C. M. Ronconi, J. F. Stoddart, V. Balzani, S. Silvi and A. Credi, J. Am. Chem. Soc., 2006, 128, 1489 CrossRef CAS PubMed; (c) T. Song and H. Liang, J. Am. Chem. Soc., 2012, 134, 10803 CrossRef CAS PubMed; (d) B. Lewandowski, G. D. Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189 CrossRef CAS PubMed.
- (a) R. T. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150 CrossRef PubMed; (b) M. C. Jiménez, C. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284 CrossRef; (c) M. Haga, T. Takasugi, A. Tomie, M. Ishizuya, T. Yamada, M. D. Hossain and M. Inoue, Dalton Trans., 2003, 2069 RSC; (d) M. Alvarez-Pérez, S. M. Goldup, D. A. Leigh and A. M. Z. Slawin, J. Am. Chem. Soc., 2008, 130, 1836 CrossRef PubMed; (e) G. Haberhauer, Angew. Chem., Int. Ed., 2010, 49, 9286 CrossRef CAS PubMed; (f) S. K. Samanta and M. Schmittel, J. Am. Chem. Soc., 2013, 135, 18794 CrossRef CAS PubMed.
- (a) N. Armaroli, V. Balzani, J.-P. Collin, P. Gaviña, J.-P. Sauvage and B. Ventura, J. Am. Chem. Soc., 1999, 121, 4397 CrossRef CAS; (b) A. Carella, C. Coudret, G. Guirado, G. Rapenne, G. Vives and J.-P. Launay, Dalton Trans., 2007, 177 RSC; (c) F. Durola and J.-P. Sauvage, Angew. Chem., Int. Ed., 2007, 46, 3537 CrossRef CAS PubMed; (d) Y.-L. Zhao, W. R. Dichtel, A. Trabolsi, S. Saha, I. Aprahamian and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 11294 CrossRef CAS PubMed; (e) L. Fang, C. Wang, A. C. Fahrenbach, A. Trabolsi, Y. Y. Botros and J. F. Stoddart, Angew. Chem., Int. Ed., 2011, 50, 1805 CrossRef CAS PubMed; (f) T. Avellini, H. Li, A. Coskun, G. Barin, A. Trabolsi, A. N. Basuray, S. K. Dey, A. Credi, S. Silvi, J. F. Stoddart and M. Venturi, Angew. Chem., Int. Ed., 2012, 51, 1611 CrossRef CAS PubMed; (g) M. Nishikawa, S. Kume and H. Nishihara, Phys. Chem. Chem. Phys., 2013, 15, 10549 RSC.
- (a) N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152 CrossRef CAS PubMed; (b) T. Muraoka, K. Kinbara, Y. Kobayashi and T. Aida, J. Am. Chem. Soc., 2003, 125, 5612 CrossRef CAS PubMed; (c) T. Muraoka, K. Kinbara and T. Aida, Nature, 2006, 440, 512 CrossRef CAS PubMed; (d) S. Kume and H. Nishihara, Dalton Trans., 2008, 3260 RSC; (e) A. Coskun, D. C. Friedman, H. Li, K. Patel, H. A. Khatib and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 2493 CrossRef CAS PubMed; (f) M. C. Basheer, Y. Oka, M. Mathews and N. Tamaoki, Chem. – Eur. J., 2010, 16, 3489 CrossRef CAS PubMed; (g) Z. Li, J. Liang, W. Xue, G. Liu, S. H. Liu and J. Yin, Supramol. Chem., 2014, 26, 54 CrossRef CAS.
- (a) A. Livoreil, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 9399 CrossRef CAS; (b) J. P. Collin, F. Durola, J. Lux and J.-P. Sauvage, Angew. Chem., Int. Ed., 2009, 48, 8532 CrossRef CAS PubMed; (c) H. Li, A. C. Fahrenbach, A. Coskun, Z. Zhu, G. Barin, Y.-L. Zhao, Y. Y. Botros, J.-P. Sauvage and J. F. Stoddart, Angew. Chem., Int. Ed., 2011, 50, 6782 CrossRef CAS PubMed.
- M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815 RSC.
- (a) H. Sleiman, P. Baxter, J.-M. Lehn and K. Rissanen, J. Chem. Soc., Chem. Commun., 1995, 715 RSC; (b) J. Frey, C. Tock, J.-P. Collin, V. Heitz, J.-P. Sauvage and K. Rissanen, J. Am. Chem. Soc., 2008, 130, 11013 CrossRef CAS PubMed; (c) S. Durot, F. Reviriego and J.-P. Sauvage, Dalton Trans., 2010, 39, 10557 RSC; (d) R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434 CrossRef CAS PubMed.
- (a) M. Schmittel and A. Ganz, Chem. Commun., 1997, 999 RSC; (b) M. Schmittel, U. Lüning, M. Meder, A. Ganz, C. Michel and M. Herderich, Heterocycl. Commun., 1997, 3, 493 CAS; (c) M. Schmittel, V. Kalsani, R. S. K. Kishore, H. Cölfen and J. W. Bats, J. Am. Chem. Soc., 2005, 127, 11544 CrossRef CAS PubMed; (d) M. Schmittel and S. K. Samanta, J. Org. Chem., 2010, 75, 5911 CrossRef CAS PubMed; (e) S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555 RSC.
- (a) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784 CrossRef CAS PubMed; (b) K. Osowska and O. Š. Miljanić, Synlett, 2011, 1643 CAS; (c) M. L. Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651 RSC; (d) S. Saha and P. Ghosh, J. Chem. Sci., 2012, 124, 1229 CrossRef CAS PubMed.
- (a) K. Parimal, E. H. Witlicki and A. H. Flood, Angew. Chem., Int. Ed., 2010, 49, 4628 CrossRef CAS PubMed; (b) K. Osowska and O. Š. Miljanić, J. Am. Chem. Soc., 2011, 133, 724 CrossRef CAS PubMed; (c) A.-M. Stadler, C. Burg, J. Ramírez and J.-M. Lehn, Chem. Commun., 2013, 49, 5733 RSC.
- (a) DFT computations were performed with the B3LYP functional, 6-31G* basis set for all main group elements and LANL2DZ basis set for copper implemented in Gaussian 09; ; (b) M. J. Frisch, et al., Gaussian 09, Revision B.01 Search PubMed.
- In parenthesis, the relative energies are given with respect to the lowest energy structure. Structures iso-IC and iso-III could not be optimised in DFT computations due to severe steric crowding between the mesityl groups.
- In another experiment when 3 and 5 were mixed with Cu+ or Zn2+ at a 1:1:1 ratio, there was no evidence for the formation of the heteroleptic species neither by 1H NMR nor ESI-MS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 1H–1H COSY, 13C NMR, ESI-MS, cyclic voltammograms, DOSY and DFT optimised structures. See DOI: 10.1039/c4dt00967c |
| This journal is © The Royal Society of Chemistry 2014 |